

Abstract
Despite the growing availability of sophisticated bioinformatic methods for the analysis of single-cell RNA-seq data, few tools exist that allow biologists without extensive bioinformatic expertise to directly visualize and interact with their own data and results. Here, we present Cerebro (cell report browser), a Shiny- and Electron-based standalone desktop application for macOS and Windows which allows investigation and inspection of pre-processed single-cell transcriptomics data without requiring bioinformatic experience of the user. Through an interactive and intuitive graphical interface, users can (i) explore similarities and heterogeneity between samples and cell clusters in two-dimensional or three-dimensional projections such as t-SNE or UMAP, (ii) display the expression level of single genes or gene sets of interest, (iii) browse tables of most expressed genes and marker genes for each sample and cluster and (iv) display trajectories calculated with Monocle 2. We provide three examples prepared from publicly available datasets to show how Cerebro can be used and which are its capabilities. Through a focus on flexibility and direct access to data and results, we think Cerebro offers a collaborative framework for bioinformaticians and experimental biologists that facilitates effective interaction to shorten the gap between analysis and interpretation of the data.
Availability and implementation
The Cerebro application, additional documentation, and example datasets are available at https://github.com/romanhaa/Cerebro. Similarly, the cerebroApp R package is available at https://github.com/romanhaa/cerebroApp. All components are released under the MIT License.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Transcriptomics data of single cells (scRNA-seq) are generated with unprecedented frequency due to the recent availability of fully commercialized workflows and improvements in throughput and costs (Svensson et al., 2018). Though sophisticated bioinformatic tools are being developed (Deng et al., 2019; Pliner et al., 2019; Zhang et al., 2019), appropriate analysis and interpretation of data from scRNA-seq experiments largely relies on deep understanding of the biological context and experimental conditions behind the preparation of input cells. However, direct interaction with their own datasets is often out of reach for biologists without bioinformatic expertise. Existing visualization software for scRNA-seq data, such as Loupe Cell Browser by 10× Genomics or iSEE (Rue-Albrecht et al., 2018), often either provide a limited amount of results or require the user to be proficient enough to execute (at least a few) commands in the terminal. Cerebro aims to overcome the technical hurdles and allow direct and interactive exploration of pre-processed scRNA-seq results.
2 Materials and methods
The key features of Cerebro include: (i) visualization of two-dimensional (2D) and three-dimensional (3D) projections such as t-SNE or UMAP; (ii) overview panels for samples and clusters; (iii) tables of most expressed genes and marker genes for each sample and cluster; (iv) tables of enriched pathways in marker genes of samples or clusters and (v) visualization of expression of user-specified genes and gene sets from MSigDB (Liberzon et al., 2011; Subramanian et al., 2005). All these elements are designed to be interactive. Plots can be exported to PNG and/or PDF, while tables can be saved to CSV and Excel format. The core of Cerebro is the cerebroApp R package [built with Shiny (https://CRAN.R-project.org/package=shiny)], which can be installed as a standalone application [built with Electron (https://electronjs.org/)]. Alternatively, the Cerebro user interface is available through the cerebroApp R package or as a Docker container. Input data needs to be prepared using the cerebroApp R package. Currently, cerebroApp offers functionality to export a Seurat object (both Seurat v2 and v3 are supported) to the Cerebro format in a single step (Butler et al., 2018). However, through existing conversion functions available in the Seurat framework, results generated with other analysis frameworks, such as scanpy (AnnData format), can be exported for visualization in Cerebro as well. cerebroApp also provides functions to perform a set of (optional) analyses, e.g. pathway enrichment analysis based on marker gene lists of samples or clusters through Enrichr (Chen et al., 2013; Kuleshov et al., 2016), gene set enrichment analysis for samples and clusters using the gene set variation analysis method (Hänzelmann et al., 2013), and extraction of single-cell trajectories calculated with Monocle 2 (Qiu et al., 2017). Parallel processing in these functions ensures time-efficient execution. For human and mouse datasets, marker genes will be intersected with the gene ontology term ‘cell surface’ (GO: 0009986) to highlight potential markers for experimental enrichment of the respective cell community. The exported .crb file is then loaded into Cerebro and shows the information from the Seurat object (Fig. 1). Full-size versions of the examples of the graphical interface of Cerebro shown in Fig. 1 can be found in Supplementary Figs S1–S6 as well as in the Cerebro GitHub repository.
Fig. 1.
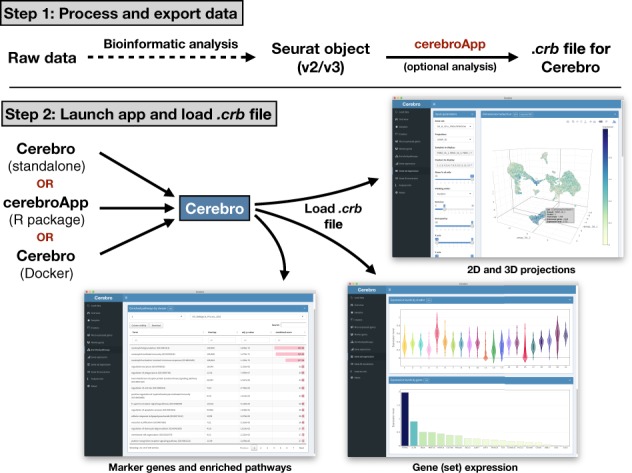
Schematic workflow of Cerebro. In the first step, the raw data are processed and analyzed (barcode extraction, alignment, etc.) using existing tools such as Cell Ranger, stored in a Seurat object, and exported to a .crb file using functions of the cerebroApp package. Subsequently, the .crb file can then be loaded into Cerebro for visualization. Currently, Cerebro can be launched as a standalone application, from the cerebroApp R package, or from the dedicated Docker container
3 Usage scenario
To illustrate the proposed workflow, we analyzed three publicly available scRNA-seq datasets and provide the resulting .crb files in the Cerebro GitHub repository (Russell et al., 2018; Yu et al., 2019). For example, the ‘pbmc_10k_v3’ dataset contains ∼10k human PBMCs from a healthy donor (link to dataset in Supplementary Material) following the basic Seurat (v2 and v3) and basic scanpy (Wolf et al., 2018) workflows. First, we loaded the feature matrix and created a Seurat/AnnData object, filtered cells based on numbers of transcripts and expressed genes, log-transformed transcript counts and normalized each cell to 10 000 transcripts. We then identified variable genes, scaled the expression matrix and regressed out numbers of transcripts, performed cell cycle and principal component analysis, identified clusters and described their relationship in a cluster tree. We also generated 2D and 3D projections using the t-SNE and UMAP algorithms. Then, we used cerebroApp to calculate the percentage of mitochondrial and ribosomal gene expression, obtain the most expressed genes and differentially expressed genes (marker genes) for each sample and cluster, perform pathway enrichment analysis using the identified marker genes, perform gene set enrichment analysis on all 5501 curated C2 genes sets available in the MSigDB, and finally export a .crb file that can be loaded into Cerebro.
Based on the combined information from pathway enrichment (in particular the Enrichr results from the Human Gene Atlas), marker genes and expression of additional genes and gene sets, we were able to retrieve expected cell types commonly found in PBMC samples (dendritic cells, NK cells, B cells, megakaryocytes, monocytes, CD4+ and CD8+ T cells) and assign a cell type to each cluster. If desired, these cell groups could be further discriminated by checking the expression of additional marker genes and gene sets.
4 Conclusion
By providing access to comprehensive information on expression profiles of samples and clusters, we hope that Cerebro will accelerate data interpretation and ultimately knowledge acquisition. Notably, the proposed workflow also provides analytical flexibility by enabling the addition of custom analyses and results to the Seurat object. Since the code is completely open-source, it is possible (and people are encouraged) to modify and adapt Cerebro to display other results and data types. While cerebroApp currently only supports to prepare Seurat objects for visualization in Cerebro, export methods for object types of other popular scRNA-seq analysis frameworks, such as SingleCellExperiment or AnnData [used by scanpy (Wolf et al., 2018)] can be added in the future. Furthermore, Seurat already provides functionality to import data from other frameworks, including the two mentioned above, and therefore serves as a gateway for the majority of datasets. An example of how to export data analyzed in scanpy for visualization in Cerebro is provided in the Cerebro GitHub repository. Due to the nature of Shiny apps, Cerebro can be easily adapted to be hosted on web servers.
5 Software availability
The current standalone version of Cerebro is available for Windows and macOS and can be downloaded from the GitHub repository: https://github.com/romanhaa/Cerebro/releases. Alternatively, users of Windows, macOS and Linux can install (and find the source code of) cerebroApp R package — which provides the same functionality as the standalone version — from: https://github.com/romanhaa/cerebroApp. Analysis of the example datasets was carried out in a Docker container to ensure reproducibility. The container was built using a recipe file stored in the Cerebro GitHub repository and is available through the Docker Hub under the name ‘romanhaa/cerebro’.
Supplementary Material
Acknowledgements
Roman Hillje is a PhD student at the European School of Molecular Medicine (SEMM). We thank our colleagues and early version users for their constructive feedback, and AIRC for supporting this work.
Funding
This work has been funded by the Fondazione AIRC per la Ricerca sul Cancro (grant number IG-2017-20162 to P.G.P.) and the European Research Council (ERC) (grant number 341131 to P.G.P.).
Conflict of Interest: None declared.
References
- Butler A. et al. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol., 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y. et al. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y. et al. (2019) Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat. Methods, 16, 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S. et al. (2013) GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics, 17, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V. et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A. et al. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner H.A. et al. (2019) Supervised classification enables rapid annotation of cell atlases. Nat. Methods, 16, 983–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X. et al. (2017) Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods, 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rue-Albrecht K. et al. (2018) iSEE: interactive SummarizedExperiment Explorer. F1000Research, 7, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A.B. et al. (2018) Extreme heterogeneity of influenza virus infection in single cells. eLife, 7, e32303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V. et al. (2018) Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc., 13, 599–604. [DOI] [PubMed] [Google Scholar]
- Wolf F.A. et al. (2018) SCANPY: large-scale single-cell gene expression data analysis. Genome Biol., 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z. et al. (2019) Single-cell transcriptomic map of the human and mouse bladders. J. Am. Soc. Nephrol., 30, 2159–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.W. et al. (2019) Probabilistic cell type assignment of single-cell transcriptomic data reveals spatiotemporal microenvironment dynamics in human cancers. bioRxiv, 521914. doi: 10.1101/521914.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.