

Abstract
Summary
Clinical sequencing aims to identify somatic mutations in cancer cells for accurate diagnosis and treatment. However, most widely used clinical assays lack patient-matched control DNA and additional analysis is needed to distinguish somatic and unfiltered germline variants. Such computational analyses require accurate assessment of tumor cell content in individual specimens. Histological estimates often do not corroborate with results from computational methods that are primarily designed for normal–tumor matched data and can be confounded by genomic heterogeneity and presence of sub-clonal mutations. Allele-frequency-based imputation of tumor (All-FIT) is an iterative weighted least square method to estimate specimen tumor purity based on the allele frequencies of variants detected in high-depth, targeted, clinical sequencing data. Using simulated and clinical data, we demonstrate All-FIT’s accuracy and improved performance against leading computational approaches, highlighting the importance of interpreting purity estimates based on expected biology of tumors.
Availability and implementation
Freely available at http://software.khiabanian-lab.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Clinical sequencing assays aim to identify somatic mutations in cancer cells for accurate diagnosis and treatment of patients through therapeutic targeting of driver alterations and tumor mutational signatures (Garraway, 2013). Although in most scientific settings, patient-matched tumor and germline DNA samples are sequenced for this purpose, most implementations for clinical sequencing lack control DNA data and often only tumor specimens undergo genomic profiling (Frampton et al., 2013). Moreover, because of recent rulings on qualified coverage for genomic diagnostic assays (United States Food and Drug Administration, 2017), tumor-only sequencing is poised to become one of the most utilized methods for genomic profiling of cancer patients in clinical settings.
Most specimens that are collected in the clinic are formalin-fixed, paraffin-embedded and contain a mixture of tumor cells as well as surrounding non-tumor cells, which include stromal and hematopoietic populations. Hybrid-capture, high-depth sequencing (i.e. >500× depth of coverage) permits identification of genomic alterations with high statistical confidence in measuring variant allele frequencies (VAFs), especially compared with whole-genome and whole-exome sequencing methods (Damodaran et al., 2015; Shaw and Maitra, 2019). Detected variants in tested samples may arise from germline mutations present in all cells, somatic alterations present in all cancer cells and somatic alterations present in a subset of cancer cells or occasionally in a subpopulation of non-tumor cells (Severson et al., 2018). The power to detect corresponding rare somatic clones depends on sequencing depth and relative abundance of each cell population that harbors them. In the absence of patient-matched control DNA, the single nucleotide polymorphism database as well as variants detected in healthy individuals within public or private cohorts are often used to identify and remove germline variants (Hiltemann et al., 2015). However, this approach fails to capture rare germline alterations that are specific to each patient, leading to possible misclassification of germline mutations as somatic. Therefore, determining whether a detected mutation is germline or truly somatic as well as resolving variant clonality and loss of heterozygosity (LOH), require additional analyses based on an accurate estimation of specimen’s tumor content or purity. LOH events are shown to be pertinent for assessing treatment efficacy (den Brok et al., 2017; McGranahan et al., 2017; Pawlyn et al., 2018; Sade-Feldman et al., 2017). Yet, without accurate estimates of purity, even the availability of patient-matched germline data may not help resolve evidence of LOH in the tumor, which can occur by deletion of the wild-type copy or by duplication of the mutant allele with the loss of the wild-type (copy-neutral LOH). Moreover, accurate estimates of specimen purity help resolve genomic diversity in both tumor and non-tumor cell populations, which adds to the complexity of interpreting a tumor’s mutational landscape and confounds distinguishing sub-clonal tumor alterations from those in a subpopulation of non-tumor cells (Ptashkin et al., 2018; Riedlinger et al., 2019; Severson et al., 2018). In particular, characterizing clonal mutations, which are present in all cancer cells and are postulated as the best candidates for targeted treatment versus mutations that are sub-clonal and are present only in a subpopulation of cells (Amirouchene-Angelozzi et al., 2017), is contingent upon correct estimation of the tumor content in sequenced specimen.
Histological approximations of tumor purity are not always available or when they are, they do not provide the required confidence for these analyses. For this reason, various computational algorithms have been developed that utilize patient-matched germline sequencing or process-matched normal control data to infer tumor content of tumor specimens as well as genome-wide ploidy within the tumors (Yadav and De, 2015). A few methods have been developed to simultaneously estimate tumor purity and average ploidy from somatic DNA aberrations; however, the mathematical models used in some of the most utilized methods are optimized for SNP array platforms (Carter et al., 2012; Van Loo et al., 2010). Other approaches such as CNAnorm (Gusnanto et al., 2012) and Control-FREEC (Boeva et al., 2012) assess copy-number and tumor ploidy of a specimen by correcting its contamination with normal cells, normalizing and scaling the data across tumor genomes. Nevertheless, these approaches operate under the assumption that most tumors are composed of a single clone, which causes the estimates to be inaccurate for heterogenous tumors with multiple subpopulations. Other algorithms such as Pyclone (Roth et al., 2014) and EXPANDS (Andor et al., 2014) utilize tumor purity and based on a clustering of somatic mutations with similar cellular prevalence, they predict clonal population size and mutations specific to each subpopulation. Regardless of the strengths or weaknesses of these methods, they are primarily designed for normal–tumor pairs, and when applied to high-depth, tumor-only data from targeted genomic regions, they produce results that are confounded by the limited scope of sequencing as well as genomic heterogeneity, aneuploidy and presence of sub-clonal mutations in cancer cell populations. Therefore, there is a need for strategies that take advantage of the power of tumor-only clinical sequencing assays for high-confidence measurements of VAF and focal copy-number variations (CNVs). To this end, we developed All-FIT (Allele-Frequency-based Imputation of Tumor Purity), a weighted least square method that through iterative steps estimates specimen purity and its associated confidence intervals (CIs) using detected variants’ VAF and CNV. We evaluate All-FIT’s performance using a comprehensive set of simulated datasets and compare its results with those from ABSOLUTE (Carter et al., 2012), the leading algorithm for estimating purity from VAF and CNV measurements in matched normal–tumor data. Finally, we apply All-FIT to high-depth sequencing of 1861 specimens from patients with solid tumors and show that histological estimates of purity often do not correspond to observed VAF of detected variants, especially when their biological nature is considered. Specifically, we demonstrate the concordance of our estimates with expected biology of tumors by focusing on specimens from a wide range of cancer types, highlighting the prevalence of LOH that affect these commonly mutated tumor suppressors.
2 Materials and methods
2.1 All-FIT weighted least square approach of imputing tumor purity
All-FIT considers all detected variants as its input and requires their observed VAF (f), their total sequencing depth (d) and their loci’s chromosomal copy-number or ploidy (Y). We assume the positions of all mutations have chromosomal copy-number of 2 in normal cells. Because the germline-versus-somatic status of the detected variants is yet to be determined, we need to evaluate the likelihood of Y somatic and Y germline mutational models with their corresponding mutated allele’s copy-number cm (1cmY) for each variant. For a given purity (p), we calculate cancer cell fraction (CCF) for somatic mutations as the ratio of observed VAF and the expected VAF. Although CCF for unfiltered germline heterozygous mutations is always equal to 1, CCF for LOH, copy-neutral LOH and amplification events at the loci of germline mutations can be calculated and generalized as a function of p (Fig. 1). Intuitively, at correct estimate of p, CCFs are equal to one for the variants’ likeliest mutational model. Therefore, we postulate that if detected variants are clonal, the parsimonious estimate of tumor content is the value that optimizes
here, i and j count N variants and M mutational models, respectively, and Wij is the Akaike Information Criterion (AIC) weight of mutational model j for variant i, calculated based on binomially distributed variant depths. To calculate Wij, we follow the approach previously implemented in the LOHGIC algorithm (Khiabanian et al., 2018), where AIC weights are based on binomial likelihoods of observing a variant with a VAF of f and focal ploidy of Y at total sequencing depth of d across 2Y possible mutational models.
Fig. 1.
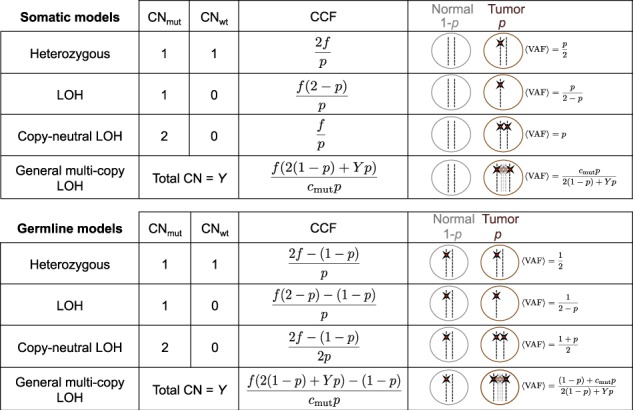
Modeling mutational status and the number of mutated alleles for somatic and germline mutations with their respective expected VAF and CCF. Adapted from Khiabanian et al. (2018)
Since most tumors are heterogeneous in nature and contain both clonal and sub-clonal alterations, we need to evaluate variants’ clonality, which is not possible when p is unknown. Therefore, we propose to first use all detected variants to obtain an estimate for purity that can guide the identification and exclusion of unfiltered germline heterozygous and sub-clonal somatic variants. Next, using the remaining putatively clonal events, we optimize L to estimate p and its CIs (Fig. 2a). All-FIT is implemented in Python 3 and is freely available at http://software.khiabanian-lab.org, along with the scripts to generate simulations.
Fig. 2.

Schematic view of All-FIT’s implementation and the analyzed datasets. (a) All-FIT follows three steps for estimating p, first assuming all variants are clonal, then removing germline heterozygous variants, and finally excluding sub-clonal somatic variants. (b) Three simulated datasets are generated to test the effect of sequencing depth and number of variants, the impacts of sub-clonal somatic variants as well as high-ploidy somatic variants. We also apply All-FIT to 1861 patient specimens, particularly 199 tumor specimens harboring hot-spot TP53 mutations and 73 colon cancer specimens harboring pathogenic APC mutations. (c) If only a group of variants with observed VAFs of 0.35 exists in a specimen, these variants cannot be distinguished between three different somatic mutational models of heterozygous, under LOH, or under copy-neutral LOH, without knowing specimen’s tumor content. If a second group of variants is detected with observed VAFs of 0.70, p = 0.70 can classify variants with observed VAFs of 0.35 and 0.70 as heterozygous and copy-neutral LOH somatic mutations, respectively. Detection of additional variants with VAFs of 0.50 and 0.85 improves confidence in estimating purity
2.2 Simulated data
To evaluate All-FIT’s performance, we generated 10 000 simulated sets of variants for known values of p (range: 0.1–0.9), including a mixture of clonal and sub-clonal mutations with varying ploidy values (Dataset 1). The number of variants for each simulated set ranged from 20 to 100 for which we considered equal probability to be under eight mutational possibilities for somatic and germline variants that follow heterozygous, LOH, copy-neutral LOH and high-ploidy models. These sets included at least one somatic heterozygous mutation; at most 25% of somatic heterozygous mutations were assigned to be sub-clonal. We randomly assigned to each variant a sequencing depth, d (uniformly distributed between 300 and 1000×). Each variant’s VAF was randomly generated from a binomial distribution using d as the number of trials and the expected VAF of assigned mutational model as the success probability.
To assess the broad utility of All-FIT, we also generated two other datasets with similar conditions. Dataset 2 was enriched with sub-clonal mutations assumed to be somatic heterozygous variants with cm = 1. To enrich simulated sets with sub-clonal mutations, we increased the number of somatic heterozygous variants to at least 25% of total number of variants and required at most 67% of them to be sub-clonal. Dataset 3 was enriched with high-ploidy mutations (i.e. variants with 3 ≤ Y ≤ 8 and cm ≥ 1), where we simulated approximately 100 sets for each percentage of high-ploidy mutations, ranging from 0% (absence of high-ploidy variants) to 99% (almost all variants in the sample are high-ploidy changes). Figure 2b provides a brief overview of the three types of simulated datasets used in All-FIT, along with the two study cases from patient data described in Supplementary Methods.
3 Results
Intuitively, All-FIT imputes p by choosing the value that classifies most clonal variants into their respective most likely mutational models. Therefore, it requires variants with various models to break ambiguous inferences to estimate p with high statistical confidence (i.e. small CI). For example, if we assume a ploidy of 2 in both normal and tumor cells for a variant with observed VAF of 0.35, the variant can be considered as a somatic mutation that is heterozygous when p = 0.70, under LOH when p = 0.52 or under copy-neutral LOH when p = 0.35. If all variants in a simulated set have observed allele frequencies of about 0.35, it will be impossible to infer a purity estimate that distinguishes mutational models. If the dataset also includes variants with observed VAF of 0.70, the ambiguity between three possible purity estimates can be broken because at p = 0.70, variants with observed VAF of 0.35 are classified as somatic heterozygous mutations while variants with observed VAF of 0.70 are classified as somatic mutations with copy-neutral LOH (Fig. 2c). Germline heterozygous variants with VAF of ∼0.50 play minimal role in breaking the ambiguity between different purity models, as their CCF is always equal to one, while sub-clonal somatic variants can confound this approach as by definition, their CCFs are never equal to one.
All-FIT provides graphical presentations of for individual variants (1 to N) and presents estimated p and its CI at each step, demonstrating how All-FIT imputes p often with a higher certainty by removing germline heterozygous and sub-clonal somatic variants. Using the estimated purity, All-FIT calculates the expected VAF from the likeliest mutational model to which each variant belongs, and based on an implementation of the LOHGIC algorithm, it presents the concordance between the expected and observed VAFs. It also shows the distribution of CCFs, demonstrating the clonal distribution of variants at the estimated p. Ideally, at correct purity, CCF distribution should peak at one with sub-clonal populations detected at lower fractions. Nevertheless, some variants may exist at fractions that are larger than 1, due to ambiguity in their most likely mutational model (Fig. 3).
Fig. 3.

Representation of All-FIT’s results for detected variants. (a) The contribution of each variant to the sum of likelihoods. (b) The sum of likelihoods across all variants, along with its 2 curve (brown dashed line). The intersections of the 2 curve and the line tangent to the log of the sum of likelihoods at its minimum (grey dotted line) indicate the CI around the estimated p. (c) The distribution of observed allele frequencies. Dashed lines represent the expected allele frequencies of included mutational models for the estimated p. (d) The distribution of CCFs for the estimated p. (e–h) Results after excluding unfiltered germline heterozygous variants. (i-l) Results after excluding sub-clonal somatic variants. The legend box shows the mutated gene, the amino acid change and the VAF of each detected variant. This patient was found to have truncating mutations in a number of tumor suppressors involved in germline tumor syndromes including ATM, BRCA1, DICER1, MSH2, NF1 and PTCH1. All-FIT estimated specimen purity at 0.48 (CI: 0.44–0.50). The application of LOHGIC indicated that these ATM, BRCA1, DICER1 and PTCH1 mutations were somatic without LOH, and therefore these were likely passenger mutations. There were two NF1 truncating mutations that were inferred to be somatic and likely resulted in loss of function of neurofibromin. There were also two MSH2 truncating mutations; one inferred to be germline and the other somatic (Color version of this figure is available at Bioinformatics online.)
3.1 Purity estimation in simulated data
We used simulated datasets to assess the accuracy of All-FIT. For Dataset 1, comprised of both clonal and sub-clonal mutations with varying ploidy, All-FIT’s purity estimates corroborated with simulated values in 86% of cases with Pearson’s correlation coefficient (r) of 0.99 (Fig. 4a). All-FIT could provide estimates independent of the total number of detected variants. However, permutated subsampling of variants in simulated samples in Dataset 1 indicated that its accuracy improved when more than nine variants were present where the difference between the estimated and simulated values of purity was consistently ≤0.02 (Fig. 5a and Supplementary Methods). On the other hand, All-FIT’s performance was highly influenced by sequencing depth. Specifically, we reduced alternate and total depth for variants in simulated samples in Dataset 1 by factors ranging from 0.1 to 1.0 (Supplementary Methods), which showed that sequencing at 120–400× is required to limit mean estimation difference from simulated value to <0.07 (Fig. 5b). When we removed all germline variants from Dataset 1 to simulate presence of matched-normal DNA, All-FIT’s accuracy improved to 92%, indicating the robustness of the method by only considering somatic alterations. In contrast, ABSOLUTE’s estimated p agreed with simulated p in only 35% of cases with r = 0.69 (Fig. 4b). It should be noted that assessing All-FIT’s estimated values within their 2 CIs provided additional predictive power compared with ABSOLUTE, which produces a single purity estimate without a CI (Supplementary Methods).
Fig. 4.
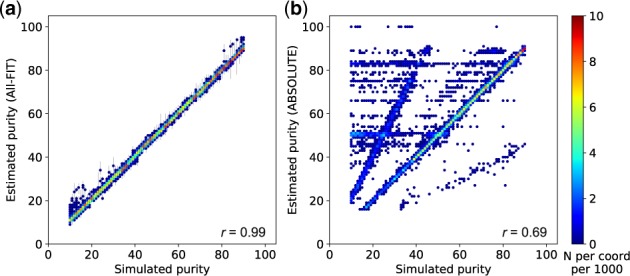
Relationship between simulated purity (ground truth) and estimated purity, respectively from (a) All-FIT and (b) ABSOLUTE using Dataset 1. The scale-bar represents the number of simulated samples overlapping at each coordinate per 1000
Fig. 5.
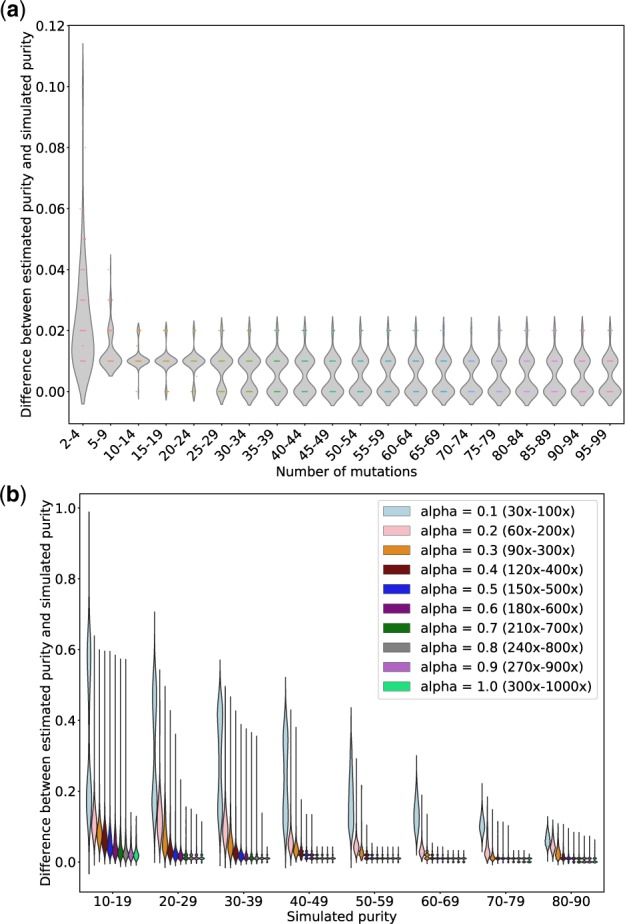
The impact of number of mutations and sequencing depth on All-FIT’s accuracy. (a) Using simulations from Dataset 1, 100 samples are chosen, and each is subsampled repeatedly into 100 permuted sets with varying number of variants ranging from 2 to 99. The x-axis represents the number of variants, and the y-axis represents the median difference between estimated and simulated purities across permutations for each sample (without considering the purity CIs). These results show that the accuracy of All-FIT is independent of the number of variants, when there are at least 10 variants per sample. (b) Using simulations from Dataset 1, 10000 simulated sets are regenerated by changing the variants’ total depth with factors ranging from 0.1 to 1.0; we repeat this permutated subsampling 100 times for each simulated sample at all values of alpha. The x-axis represents the simulated purity (with 1100–1300 samples for each purity categories), and the y-axis represents the median difference between estimated and simulated purities across permutations for each sample. These results show that All-FIT’s accuracy is dependent on the sequencing depth given specimen purity
Next, we increased genomic heterogeneity in simulated data by varying the number of sub-clonal mutations. We classified the simulated sets into four categories based on the percentage of sub-clonal mutations: <25, 25–50, 50–75, and >75%. All-FIT’s accuracy was not affected by the presence of sub-clonal mutation, when the simulated sets were comprised of <25% of sub-clonal mutations. However, when the percentage of sub-clonal mutations varied from 25 to 75%, All-FIT increasingly misestimated p by about one-half, and when >75% of the mutations were designated to be sub-clonal, All-FIT estimated purity as one-half of simulated values for most cases (Fig. 6a). Nonetheless, the correlation coefficient remained >0.76 regardless of the proportion of sub-clonal variants, and it further improved to 0.84 when we removed germline variants, excluding the 38 simulated sets that had >75% of sub-clonal mutations (Supplementary Fig. S1). On the other hand, correlation coefficient for ABSOLUTE’s estimates was lower than those of All-FIT, with its best performance of r = 0.75 when only <25% of variants were sub-clonal. It was also susceptible to misestimating purity at one-half of the simulated p, even with a few sub-clonal variants present (Fig. 6b).
Fig. 6.

Presence of sub-clonal mutations reduces the correlation coefficient between simulated purity and estimated purity using Dataset 2. (a) All-FIT accurately imputes p in simulated sets when percentage of sub-clonal mutations is <50%, beyond which, it increasingly underestimates purity at one-half of the simulated value. (b) ABSOLUTE often fails to predict p correctly even when the percentage of sub-clonal mutations is <25%. The scale-bar represents the number of overlapping samples at each coordinate per 1000 for first three panels from the right, and per 10 for the leftmost panel. All-FIT shows high correlation when <50% of variants are sub-clonal, but overall, its correlation is relatively higher than those of ABOSLUTE, regardless of the percentage of sub-clonal mutation (Supplementary Fig. S1)
We also tested our algorithm with a simulated dataset enriched with variants at high-ploidy, where accuracy of All-FIT decreased to 79% while retaining the Pearson’s r = 0.99 (Supplementary Results and Supplementary Fig. S2).
3.2 Purity estimation in patient data
To test our method with patient data, we applied All-FIT to 1861 solid tumor specimens (Supplementary Methods), which were sequenced using the FoundationOne assay (Foundation Medicine, Inc., Cambridge, MA). Corroborating previous studies, the correlation between our computational estimates and histological values (i.e. pathological purity) was low (Pearson’s r = 0.28; Fig. 7). As others have also noted, this discrepancy is probably due to the limitation in making a histological estimate of tumor content while extracting high-quality DNA from the same specimen. Therefore, histological estimates are often made from the adjacent tumor sections, which may fail to reflect the tumor content of sequenced specimen (Carter et al., 2012; Favero et al., 2015).
Fig. 7.
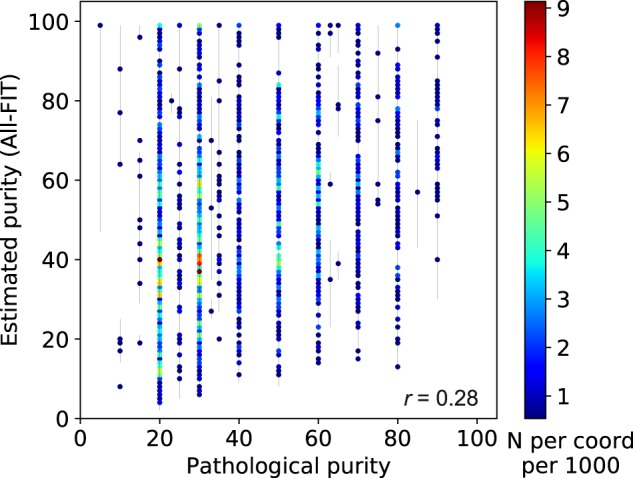
Relationship between pathological purity and estimated purity from 1861 tumor specimens. There is limited corroboration between computational and histological estimates (r = 0.28). The scale-bar represents the number of overlapping specimens at each coordinate per 1000
Therefore, since all purity estimates must be interpreted in the context of tumor biology, we selected 199 tumor specimens that harbored only one hot-spot mutation in the TP53 gene (Supplementary Methods) with detected VAF >0.10. These mutations are commonly seen in all tumor types and are known to be pathogenic. Cells with these mutations are also anticipated to have lost their wild-type copy while duplicating the mutated allele (copy-neutral LOH) (Alexandrova et al., 2017), thus the purity of these specimens will be equal to the observed VAF of these mutations, if they are the drivers of tumor growth (Fig. 1). Therefore, these driver mutations can be used as ‘anchors’ for estimating tumor purity based on their expected biological role in cancer cells. In this analysis, we observed an improved corroboration between the estimated and anchor p values (r = 0.51). However, All-FIT overestimated purity when most variants were detected at VAF of ∼0.40–0.45 (Fig. 8a). This inconsistency could arise from not satisfying All-FIT’s requirement that variants from multiple mutational models should be present. For instance, in one specimen, five variants were detected, three of which had observed VAF of 0.43–0.52 and the other two were at ∼0.70. All-FIT estimated tumor purity to be 0.83 (CI: 0.80–0.85) while the anchor purity from the TP53 mutation was 0.43. If the assumption of copy-neutral LOH for this mutation was correct, this observation implied the absence of detected somatic heterozygous variants in the specimen. As expected, when we manually added a variant with VAF = 0.23, our purity estimate was corrected to 0.48 although with a large CI and an unbroken ambiguity (CI: 0.45–0.66 joint with 0.81–0.83).
Fig. 8.

Relationship between estimated purity and allele frequency of anchor mutations. (a) All-FIT’s purity estimates corroborate with the observed VAFs of hot-spot TP53 mutations detected in 199 solid tumor specimens, which are expected to be under copy-neutral LOH and indicative of the tumor content (r = 0.51). (b) All-FIT’s purity estimates corroborate with VAFs of pathogenic APC mutations detected in 73 colon cancer specimens (r = 0.60). The horizontal lines at each point represent the expected tumor content based on LOH and copy-neutral LOH models for single APC mutation or based on heterozygous model for two APC mutations. The scale-bar represents the number of overlapping specimens at each coordinate per 100
In addition, as was observed in the analysis of simulated datasets, sub-clonal alterations could confound All-FIT’s results. For example, for a specimen in which the TP53 mutation was detected with a VAF of 0.73, All-FIT estimated p to be 0.31 (CI: 0.29–0.32). This discrepancy was possibly due to the detection of sub-clonal variants with VAF <0.30, which All-FIT incorrectly considered as part of the clonal population, resulting in its underestimated tumor purity. Conversely, presence of unfiltered germline variants also affected All-FIT’s estimations. This was particularly seen in a specimen in which one variant was detected at 0.07, two were detected at ∼0.90 and the remaining 13 were detected at ∼0.50. Although the TP53 mutation had a VAF of 0.46, All-FIT estimated p to be 0.98 (CI: 0.93–0.99), as it lacked statistical power to distinguish germline and somatic variants with high confidence.
We also investigated 73 colon cancer specimens harboring pathogenic substitutions or insertions/deletions in the APC gene. These pathogenic mutations could either undergo somatic LOH or somatic copy-neutral LOH if there is one APC mutation, or they could be individually somatic heterozygous if there are two APC mutations. There are two possible purity estimates from both one APC mutation and two APC mutations; purity with somatic LOH or somatic copy-neutral LOH with one APC mutation and purity based on each individual allele frequency with two APC mutations. In both scenarios, we considered the average of these two estimates to assess All-FIT’s results, between which we observed a correlation coefficient of 0.6 (Fig. 8b). Overall, our purity estimates showed improved corroboration with anchor purity compared with those from the histological values of these specimens (Supplementary Fig. S3).
3.3 Clinical application to inform treatment strategy
Clinical hypothesis testing is a routine part of practicing medicine. Diagnosis and prognosis of cancer patients relies on patient clinical history, histology and radiology data, lab results and more recently genomic data. All clinical data put together and none alone, combined with prior information on similar cases and available therapeutics, can inform treatment design. To address key clinical questions and complement decision-support systems, we have implemented All-FIT and LOHGIC to analyze clinical sequencing data for presentation at Rutgers Cancer Institute’s multidisciplinary Molecular Tumor Board (MTB; Hirshfield et al., 2016), especially to guide the evaluation and treatment of patients with pathogenic mutations detected in tumor-suppressor genes or familial cancer genes (Jalloul et al., in preparation). The case presented in Figure 3 from a 38-year-old patient with advanced, mixed histology, ovarian cancer particularly demonstrates the utility of this approach. This patient was found to have truncating mutations in a number of tumor suppressors involved in germline tumor syndromes (Supplementary Fig. S4). Our analysis showed that the majority of detected mutations were somatic without LOH. However, one truncating mutation in MSH2 was inferred to be germline, corroborated with previous reports for its pathogenic role in Lynch syndrome. In addition, we found evidence of microsatellite instability in the mutational signature (Germano et al., 2018). Therefore, the patient was recommended by MTB to have genetic counseling to confirm that the MSH2 mutation was germline. MTB also recommended treatment with immune checkpoint inhibitor therapy, which has resulted in tumor going to remission. Put together, careful interpretation of clinical data has the potential to guide physicians by bringing high-depth DNA sequencing to the forefront of clinical practice.
4 Discussion
The complexity in cellular populations that exists within a tumor specimen is routinely summarized by the single qualitative measure of tumor purity. Since interpreting histologic as well as sequencing results relies on accurate estimates of a specimen’s tumor content, various computational solutions have been implemented to address the inaccuracies in pathological estimates (Yadav and De, 2015). These methods aim to simultaneously infer tumor purity as well as global chromosomal copy-number and often require sequencing data from pairs of tumor and normal samples. However, due to the increase use of tumor-only sequencing in precision oncology, there is a need for computational methods that can infer tumor purity of clinical specimens from detected variants, which may include somatic as well as unfiltered germline alterations. Despite the limited breadth of clinical assays that often interrogate only a few hundred genes, their high-depth of sequencing often provides sufficient power to distinguish mutational models and even to infer LOH events based on VAFs.
Here, we introduced All-FIT, which offers a solution to the problem of inferring variant clonality by imputing tumor purity from deep-sequenced specimens without control DNA. It computes AIC weights and CCFs for a range of somatic and germline mutational models. Through an iterative process, All-FIT estimates purity by minimizing a weighted least squared function with respect to p and provides statistical CIs for its estimates (Fig. 2a). All-FIT reports its results at each step to ensure completeness and to enable visual assessment by the user. Our application of All-FIT to patient data corroborated previously noted discordance between purity estimates and histological observations (Fig. 7), which can be further confirmed by determining whether the mutated allele’s copy-number of each variant exceeds its own chromosomal copy-number (Supplementary Fig. S5).
There are several caveats to our method. All-FIT requires the detection of variants from various mutational models. Our analysis of simulated and clinical data showed that the presence of at least one somatic heterozygous variant is necessary for imputing correct purity. All-FIT’s purity estimates were also confounded when most detected variants were comprised of sub-clonal mutations as they violated All-FIT’s main assumption that variants were clonally present in all cancer cells. Therefore, All-FIT may be more suited for analyzing specimens containing tumor cell populations that have completed clonal expansions and evolutionary sweeps, and are expected to be dominated by clonal populations with clonal alterations (Fittall and Van Loo, 2019). Moreover, because genomic landscape of different tumor types and their evolution under various therapeutic approaches are expected to be distinct (Loh and Khiabanian, 2019; McGranahan and Swanton, 2017), purity estimates should be agnostic to variants’ pathogenicity; therefore, All-FIT does not incorporate their biological role (i.e. driver versus passenger status). All-FIT can be used regardless of the tumor evolutionary models as long as alterations that are present in most cancer cells are detected.
All-FIT reaches optimal performance when detected variants follow different mutational models. As we observed in our analysis of patient data, specimens with limited number of variants were particularly prone to misestimated purity. Yet, our subsampling simulations showed that with at least 10 present variants, the highest difference between simulated and estimated purities was only 0.02, and with 5–9 present variants the difference did not exceed 0.04 (Fig. 5a).
The depth of sequencing can also impact All-FIT’s results. Our subsampling simulations showed that when simulated purity was <0.20, sequencing depth of at least 270–900× was needed for All-FIT to estimate purity with ≤0.13 difference from simulated value. In contrast, when simulated purity was >0.79, the difference between simulated and estimated purities was consistently ≤0.12, even when sequencing depth was 30–100×. Therefore, although All-FIT is designed for deep sequencing assays, it may perform well for whole-exome sequencing data albeit with large CIs for its estimates (Fig. 5b).
In all our analyses, we considered somatic and germline mutational models with equal probability; however, LOH and high-ploidy alterations may not be detected as frequently as heterozygous events. Nonetheless, All-FIT showed high accuracy and correlation with simulated ground truth even when germline variants were removed, resulting in slightly improved accuracy and smaller CIs on estimations. Finally, All-FIT is restricted to sequencing data from hybrid-capture-based assays, since interpreting VAFs from amplicon-based assays could be complicated by PCR efficiency. It also requires specimens to have adequate admixture from surrounding normal tissues, which in simulated data ranged from 10 to 90%. Although All-FIT showed better performance relative to ABSOLUTE, the structure of simulated data may have worked unfavorably toward ABSOLUTE as it is not leveraged to impute tumor purity from single nucleotide variants.
5 Conclusion
In this work, we demonstrated the robustness of a computational method for imputing tumor purity without sequencing matched-normal samples. Our method is mainly applicable to clinical deep sequencing, which is increasingly becoming a standard approach for genomic profiling of patients in clinical settings. All-FIT can also be potentially used to estimate the abundance of tumor DNA in liquid biopsy assays. With knowledge of a specimen’s tumor content, we can infer variant clonality and predict LOH events, leading to more tailored treatment for each patient.
Supplementary Material
Acknowledgements
We would like to thank Nahed Jalloul and other members of the Khiabanian Lab and the staff, physicians, and pathologists of the Division of Precision Medicine Oncology at Rutgers Cancer Institute of New Jersey.
Funding
J.-W.L. is a pre-doctoral fellow of the New Jersey Commission on Cancer Research [DFS18PPC017]. C.G. was a participant in the 2018 Rutgers DIMACS REU program supported by the National Science Foundation [CCF-1559855]. H.K. is supported by a grant from National Cancer Institute [R01CA233662]. G.R., S.G. and H.K. acknowledge support from Rutgers Cancer Institute of New Jersey Bioinformatics Shared Resource [P30CA072720-5917 ]as well as Rutgers Office of Advanced Research Computing [National Institutes of Health 1S10OD012346-01A1].
Conflict of Interest: G.R. reported serving on a scientific advisory board and as consultant to Personal Genome Diagnostics. S.G. reported serving on a scientific advisory board and as consultant for Inspirata Inc, holding patents on digital imaging technology licensed to Inspirata Inc, holding equity in Inspirata Inc, serving on an advisory board for Novartis Pharmaceuticals, and serving as a consultant for Roche. No other disclosures were reported.
References
- Alexandrova E.M. et al. (2017) p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis., 8, e2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amirouchene-Angelozzi N. et al. (2017) Tumor evolution as a therapeutic target. Cancer Discov., 7, 805. [DOI] [PubMed] [Google Scholar]
- Andor N. et al. (2014) EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics, 30, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V. et al. (2012) Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S.L. et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol., 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaran S. et al. (2015) Clinical tumor sequencing: opportunities and challenges for precision cancer medicine. Am. Soc. Clin. Oncol. Educ. Book, 35, e175–e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Brok W. et al. (2017) Homologous recombination deficiency in breast cancer: a clinical review. JCO Precis. Oncol., 1–13. [DOI] [PubMed] [Google Scholar]
- Favero F. et al. (2015) Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol., 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittall M., Van Loo P. (2019) Translating insights into tumor evolution to clinical practice: promises and challenges. Genome Med., 11, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton G.M. et al. (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol., 31, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L.A. (2013) Genomics-driven oncology: framework for an emerging paradigm. J. Clin. Oncol., 31, 1806–1814. [DOI] [PubMed] [Google Scholar]
- Germano G. et al. (2018) The clinical impact of the genomic landscape of mismatch repair-deficient cancers. Cancer Discov., 8, 1518–1528. [DOI] [PubMed] [Google Scholar]
- Gusnanto A. et al. (2012) Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics, 28, 40–47. [DOI] [PubMed] [Google Scholar]
- Hiltemann S. et al. (2015) Discriminating somatic and germline mutations in tumor DNA samples without matching normals. Genome Res., 25, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshfield K.M. et al. (2016) Clinical actionability of comprehensive genomic profiling for management of rare or refractory cancers. Oncologist, 21, 1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiabanian H. et al. (2018) Inference of germline mutational status and evaluation of loss of heterozygosity in high-depth, tumor-only sequencing data. JCO Precis. Oncol., 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh J., Khiabanian H. (2019) Leukemia’s clonal evolution in development, progression, and relapse. Curr. Stem Cell Rep., 5, 73–81. [Google Scholar]
- McGranahan N., Swanton C. (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell, 168, 613–628. [DOI] [PubMed] [Google Scholar]
- McGranahan N. et al. (2017) Allele-specific HLA loss and immune escape in lung cancer evolution. Cell, 171, 1259–1271 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyn C. et al. (2018) Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: a role for PARP inhibition? Leukemia, 32, 1561–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashkin R.N. et al. (2018) Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol., 4, 1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedlinger G. et al. (2019) Association of JAK2-V617F mutations detected by solid tumor sequencing with coexistent myeloproliferative neoplasms. JAMA Oncol., 5, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A. et al. (2014) PyClone: statistical inference of clonal population structure in cancer. Nat. Methods, 11, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M. et al. (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun., 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson E.A. et al. (2018) Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood, 131, 2501–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw K.R.M., Maitra A. (2019) The status and impact of clinical tumor genome sequencing. Annu. Rev. Genomics Hum. Genet. [DOI] [PubMed] [Google Scholar]
- United States Food and Drug Administration. (2017). FDA announces approval, CMS proposes coverage of first breakthrough-designated test to detect extensive number of cancer biomarkers. https://www.fda.gov/news-events/press-announcements/fda-announces-approval-cms-proposes-coverage-first-breakthrough-designated-test-detect-extensive (1 November 2019, date last accessed).
- Van Loo P. et al. (2010) Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA, 107, 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V.K., De S. (2015) An assessment of computational methods for estimating purity and clonality using genomic data derived from heterogeneous tumor tissue samples. Brief. Bioinform., 16, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.