

Priming human BM-MSCs with engineered HGF-MSCs in vivo significantly improves therapeutic potential for myocardial infarction.
Abstract
The clinical use of human bone marrow–derived mesenchymal stem cells (BM-MSCs) has been hampered by their poor performance after transplantation into failing hearts. Here, to improve the therapeutic potential of BM-MSCs, we developed a strategy termed in vivo priming in which BM-MSCs are primed in vivo in myocardial infarction (MI)–induced hearts through genetically engineered hepatocyte growth factor–expressing MSCs (HGF-eMSCs) that are encapsulated within an epicardially implanted 3D cardiac patch. Primed BM-MSCs through HGF-eMSCs exhibited improved vasculogenic potential and cell viability, which ultimately enhanced vascular regeneration and restored cardiac function to the MI hearts. Histological analyses further demonstrated that the primed BM-MSCs survived longer within a cardiac patch and conferred cardioprotection evidenced by substantially higher numbers of viable cardiomyocytes in the MI hearts. These results provide compelling evidence that this in vivo priming strategy can be an effective means to enhance the cardiac repair of MI hearts.
INTRODUCTION
Human mesenchymal stem cells (hMSCs) have been considered as one of the most promising cell sources for cell-based cardiac regeneration therapy because of their proven safety and notable paracrine effects to secrete numerous antiapoptotic and angiogenic growth factors, which enabled them to be a more competitive agent for clinical applications (1–3). However, unlike promising results obtained from preclinical models of myocardial infarction (MI), recent multiple meta-analyses have debated whether the therapeutic potential of hMSC treatment is sufficient (4, 5). While these clinical trials successfully demonstrated the feasibility and safety of hMSC treatment, the researchers were unable to show significant functional benefit. The overall efficacy of hMSC treatments from multiple meta-analyses has been relatively modest, with an improvement in the left ventricular ejection fraction (LVEF) of approximately 3 to 4% (4, 5). These results generated a consensus for the need for innovative ways to improve the therapeutic potential of hMSC treatment in failing hearts (3).
In response, diverse approaches have been attempted to enhance the therapeutic efficacy of hMSCs in treating MI. For instance, genetically engineered hMSCs overexpressing a number of antiapoptotic proteins (6, 7), growth factors, or prosurvival genes—such as vascular endothelial growth factor (VEGF) (8), insulin-like growth factor 1 (IGF-1) (9), and hepatocyte growth factor (HGF) (10)—showed increased survival and retention in vivo resulting in improved cardiac function and myocardial angiogenesis in MI-induced hearts. However, these approaches require genetic modification and, therefore, are incompatible with clinical applications.
Another strategy to bolster the therapeutic potential of hMSCs is priming/preconditioning the hMSCs—which exposes them to physical treatments (e.g., hypoxia and heat shock), pharmacological agents, growth factors, distinct types of biomaterials, modified culture conditions, or other various molecules, including microRNAs—in vitro before transplantation into the hearts (11–15). Previous studies of the underlying mechanisms of the priming effects reported that hMSCs receiving these priming applications had short-term memory and remembered a priming stimulus, even after relocating to new environments, and shifted their phenotype in therapeutically desirable directions (14, 16).
However, it appears that the priming application only provides a short-term benefit. Although there have been no studies directly evaluating how long the priming/preconditioning effect persists in the heart, the results of previous studies on ischemic preconditioning (IPC), which is one of the major intrinsic cardioprotective processes induced by repeated episodes of short periods of ischemia, imply that the effects of priming/preconditioning in the heart is not likely to last long (17–19). Several studies examining the underlying mechanisms of IPC suggested that the protective effects conferred by IPC began within minutes after the IPC stimulus and was maintained for only 4 to 6 hours (17–19). Consequently, for hMSCs to be used more effectively for comprehensive cardiac repair, an innovative method that can maintain the priming effect of hMSCs more consistently and effectively must be developed.
Therefore, in the present study, we sought to develop a strategy, namely, in vivo priming, which could prime hMSCs in intact hearts in vivo. To induce and maintain the beneficial effects of priming persistently in situ, we loaded MSCs isolated from human bone marrow (BM-MSCs) together with genetically engineered HGF-MSCs (HGF-eMSCs) that continuously secrete HGF within a three-dimensional (3D) cardiac patch, which was implanted in the epicardium of MI-induced hearts. Regarding patch generation, to recapitulate the cardiac tissue–specific microenvironment as closely as possible, we used heart-derived extracellular matrix (hdECM) hydrogel. Our hypothesis was that BM-MSCs can be constantly primed by HGF secreted from HGF-eMSCs within the cardiac patches and those empowered BM-MSCs would demonstrate an enhanced therapeutic potential for cardiac repair. Subsequently, we demonstrated that the primed BM-MSCs had a higher survival rate compared with unprimed BM-MSCs in the patches while they were attached to the MI hearts, which led to a significant improvement in cardiac function and an enhancement of vessel formation after MI. The results of this study carry significant implications for future stem cell therapy in cardiac repair.
RESULTS
Cellular characteristics of HGF-eMSCs
The engineered HGF-eMSCs were indistinguishable from BM-MSCs. They displayed the homogeneous spindle-like shaped cell morphology representative of hMSCs (fig. S1A) and expressed several markers specific for hMSCs—such as CD44, CD73, CD90, and CD105—without expression of CD34, CD11b, CD19, CD45, and human leukocyte antigen–DR (HLA-DR) (fig. S1B). The engineered HGF-eMSCs demonstrated high proliferative potential exhibited by linear population doubling levels (PDLs), which were observed until 60 days of culture (fig. S1C). HGF-eMSCs stably secreted human HGF protein up to 20 passages, determined by human HGF enzyme-linked immunosorbent assay (ELISA) analysis (fig. S1D). The results from HGF-MSC karyotyping demonstrated the genetic stability of the HGF-eMSCs. It further demonstrated that HGF-MSCs had a normal human karyotype without chromosomal abnormalities (fig. S1E).
In vitro characterization of BM-MSCs primed via HGF-eMSCs
To test our main hypothesis of whether the therapeutic effect of BM-MSCs could be sufficiently enhanced by priming with HGF-eMSCs, we generated an in vitro coculture model with BM-MSCs and HGF-eMSCs (Fig. 1A). BM-MSCs were seeded at the bottom of cell culture dishes, and HGF-eMSCs encapsulated by using 2% hdECM were placed in Transwells for priming the BM-MSCs. Using the in vitro coculture model, we performed several in vitro experiments (Fig. 1, B to F). To test the feasibility of future clinical use of HGF-eMSCs, we irradiated the HGF-eMSCs at 100 grays (Gy) before performing any in vitro experiments (fig. S2). The irradiation of the HGF-eMSCs markedly reduced their proliferation rate both in culture (fig. S2) and within the hdECM patch (fig. S3).
Fig. 1. In vitro characterization of primed BM-MSCs by HGF-eMSC coculture.

(A) Schematic diagram showing the in vitro coculture model of BM-MSCs with HGF-eMSCs. (B) qRT-PCR analysis of relative mRNA expression of multiple angiogenic factors in the unprimed BM-MSCs, primed BM-MSCs with HGF-eMSCs at 3 days after cocultures, or BM-MSCs receiving HGF cytokine (50 ng/ml) for 3 days. The y axis represents relative mRNA expression of target genes to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). †P < 0.05 compared to unprimed BM-MSC group; n = 3 per group. (C) Measurement of secreted VEGF from unprimed or primed BM-MSCs by using ELISA. P < 0.05 compared to unprimed BM-MSC group. (D) Matrigel plug assay. The 10% hMSC-CM was treated to human umbilical cord endothelial cells (HUVECs) on Matrigel to examine the vasculogenic potential. Representative images of tubes formed on Matrigel and quantification summary. n = 3 per group. (E) Endothelial cell (EC) migration assay. The HUVECs were incubated with 10% CM harvested from hMSC cultures (hMSC-CM) or control media to evaluate the migration capability. Representative images under an inverted microscope and quantification summary are shown. *P < 0.05 compared to each group; n = 5 per group. (F) Treatment with CM from primed BM-MSCs increased cell survival after serum starvation for 5 days determined by the annexin V and cholecystokinin-8 (CCK-8) assay. *P < 0.05 compared to unprimed CM treated group; n = 3 per group. PI, propidium iodide; OD, optical density.
Initially, to explore the priming effects of HGF-eMSCs toward BM-MSCs in vitro, we performed quantitative real-time polymerase chain reaction (qRT-PCR) analyses with BM-MSCs cocultured with HGF-eMSCs for 3 days. The qRT-PCR results showed that the BM-MSCs cultured together with HGF-eMSCs significantly up-regulated the expression of several angiogenesis-related genes—such as VEGFa, fibroblast growth factor 2 (FGF-2), and angiopoietin 1 (Ang1)—compared to unprimed BM-MSCs, as well as BM-MSCs receiving HGF cytokine (50 ng/ml) for 3 days (Fig. 1B). Our additional experiments examining the duration of in vitro priming effect in BM-MSC by HGF-eMSC exhibited that elevated expressions of those angiogenic/vasculogenic genes in the primed BM-MSCs were fairly rapidly decreased over time (fig. S4). In addition to the augmented expression of several genes related to angiogenesis, we observed that conditioned media (CM) collected from BM-MSCs primed with HGF-eMSCs contained higher concentrations of VEGF compared to CM obtained from unprimed BM-MSCs (unprimed BM-MSC CM) (Fig. 1C).
Subsequently, we conducted both tube formation and cell migration assays using CM collected from BM-MSCs primed via HGF-eMSCs or unprimed BM-MSCs. The results from the Matrigel tube formation assays, which were carried out to evaluate vessel-forming capability, demonstrated that the tube length and branches were significantly higher in the human umbilical cord endothelial cells (HUVECs) treated with 30% primed BM-MSC CM compared to unprimed BM-MSC CM–treated HUVECs (Fig. 1D). In the endothelial cell (EC) migration assays, as shown in Fig. 1E, the addition of 10% primed BM-MSC CM significantly enhanced the migration of HUVECs compared to the CM from unprimed BM-MSCs or the negative control group, suggesting that cytokines released from the primed BM-MSCs augmented the mobility of the ECs (Fig. 1E).
We also found that BM-MSCs primed via HGF-eMSCs were more resistant against ischemic injury simulated by serum deprivation. Primed BM-MSCs in a serum-starved condition showed substantially higher cell viability compared to unprimed BM-MSCs determined by cholecystokinin-8 (CCK-8) and annexin V assays (Fig. 1F).
Paracrine factors secreted from cultured HGF-eMSCs or HGF cytokine directly protect cardiomyocytes from ischemic insults
Since previous studies reported direct cardioprotective effects of the HGF cytokine, we sought to examine whether HGF-eMSCs could confer direct cytoprotective effects to cardiomyocytes. Ischemic injury was induced by exposing rat neonatal cardiomyocytes to H2O2 (500 μM). The results showed that administration of CM from HGF-eMSCs (HGF-eMSC CM) or HGF cytokine significantly improved the cell viability against ischemic insults (Fig. 2). Together, the results obtained from various in vitro experiments clearly demonstrate that HGF-eMSCs not only are capable of priming BM-MSCs to promote their angiogenic properties by enhancing paracrine activity but also directly protected the cardiomyocytes through the consistent secretion of cardioprotective HGF cytokine.
Fig. 2. Direct cytoprotective effects of HGF-eMSC CM on cardiomyocytes undergoing simulated ischemic injury.
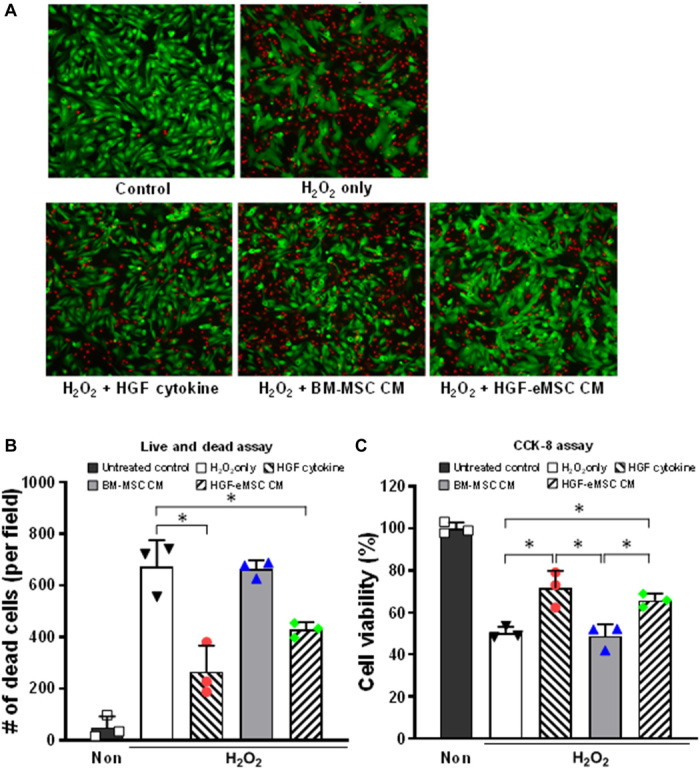
Treatment with either HGF-eMSC CM or HGF cytokine significantly promoted cell survival after serum starvation for 5 days determined by the live and dead assay (A to B) and CCK-8 assay (C). *P < 0.05 compared to each group; n = 3 per group.
In vivo priming by HGF-eMSCs improves cardiac function and reduces scar formation following MI
To examine the therapeutic effects of BM-MSCs primed with HGF-eMSCs in vivo against MI, we generated the following experimental groups: (i) sham control, (ii) untreated MI control, (iii) BM-MSC only, (iv) HGF-eMSCs only, and (v) BM-MSC/HGF-eMSC mixture. Then, the effects were verified in a rat MI model. BM-MSC only (total: 1 × 106), HGF-eMSCs only (total: 1 × 106), or BM-MSC/HGF-eMSCs (a mixture of 5 × 105 of each cell type) were loaded into hdECM patches and implanted in MI-induced rat hearts generated by left anterior descending (LAD) artery ligation to compare their therapeutic efficacy (Fig. 3). To exclude the potential risk of genetically modified HGF-eMSCs and to prevent their overgrowth in the hdECM patches, we irradiated the HGF-eMSCs at 100 Gy before loading onto the patches. No tumor was detected from in vivo tumorigenicity testing of irradiated HGF-eMSCs in nude mice (fig. S5).
Fig. 3. Schematic illustration for manufacturing BM-MSC/HGF-eMSC patch using hdECM.
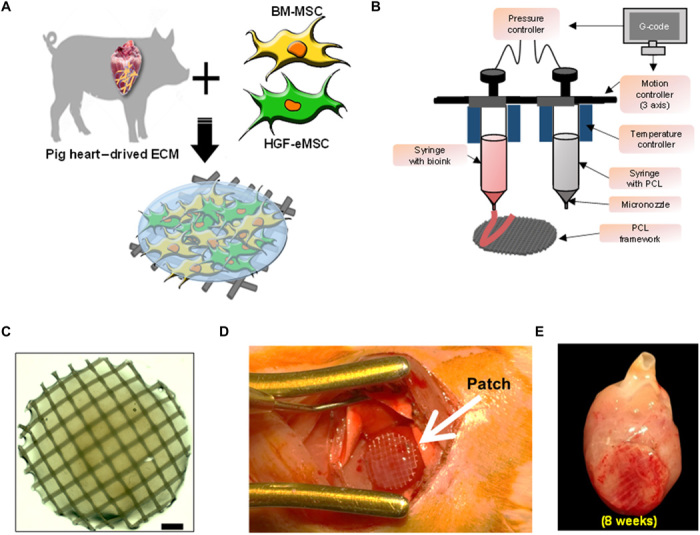
(A) Procedures for preparing hdECM bioink. (B) Macroscopic view and illustration of 3D printing system used to produce the polycaprolactone (PCL) platform. (C) Shape of hMSC patch (hMSC-PA). Scale bar, 1 mm. (D) Epicardially implanted BM-MSC/HGF-eMSC patch in MI-induced hearts (E) and at 8 weeks after implantation. Photo credit: Bong-Woo Park, The Catholic University of Korea.
Subsequently, to monitor cardiac remodeling and the recovery of left ventricular function, we regularly performed echocardiography. Baseline echocardiography 1 week after MI (PRE-IN) showed that the EF and fractional shortening (FS) did not differ significantly among the groups (Fig. 4, A to D). However, subsequent echocardiography results clearly demonstrated that while the EF and FS were constantly preserved in the BM-MSC/HGF-eMSC mixed groups until week 8, these functional parameters were significantly lower in other experimental groups, such as the MI control, BM-MSC only, and HGF-eMSC only groups (Fig. 4, A to D, and fig. S6). Although the HGF-eMSC only group exhibited cardiac function similar to the BM-MSC/HGF-eMSC mixed group by week 4, ventricular function was substantially deteriorated at week 8, probably because of the eventual death of the irradiated HGF-eMSCs (Fig. 4, A to D). The results from additional experiments testing nonirradiated HGF-eMSC demonstrated that, regardless of the irradiation, therapeutic efficacy of the HGF-eMSCs/BM-MSC mixture group was comparable or higher than that of the HGF-eMSC alone group (fig. S7). The results of several parameters for cardiac remodeling—such as left ventricular internal diastolic dimension (LVIDd), left ventricular internal systolic dimension (LVISd), septal wall thickness (SWT), and posterior wall thickness (PWT)—suggested that the overall cardiac remodeling of the hearts in the BM-MSC/HGF-eMSC mixed group was significantly reduced compared to other experimental groups (Fig. 4, E to H). Similarly, results from Masson’s trichrome staining using cardiac tissue harvested at 8 weeks exhibited an area of fibrosis (%) that was substantially smaller in the BM-MSC/HGF-eMSC group than in the other experimental groups (Fig. 4, I to K).
Fig. 4. Implantation of the BM-MSC/HGF-eMSC patch improved cardiac function and reduced adverse cardiac remodeling.

(A to B) LVEF and EF change at 8 weeks after intervention. CON, control. (C to D) FS and FS change at 8 weeks after intervention. (E) LVIDd. (F) LVISd. (G) SWT. (H) PWT. *P < 0.05 compared to sham group; †P < 0.05 compared to control group; ‡P < 0.05 compared to BM-MSC group; §P < 0.05 compared to HGF-eMSC group. n = 5 to 9 for each group. N.S., not significant. (I) Representative images from the four experimental groups showing cardiac fibrosis after staining with Masson’s trichrome in the hearts harvested 8 weeks after MI and (J) their quantification results such as percentage of viable myocardium and (K) percent fibrosis. *P < 0.05 compared to control group; †P < 0.05 compared to BM-MSC group; ‡P < 0.05 compared to HGF-eMSC group. n = 5 to 6 for each group. Scale bar, 5 mm.
Primed BM-MSCs increase capillary density and salvage myocardium
Consistent with our previous observation from in vitro experiments showing greater survival against simulated ischemic insults, primed BM-MSCs remained relatively healthy and within the patch until 8 weeks after implantation (Fig. 5, A to C). Through the TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) assay, we observed positive TUNEL signals in less than 5% of the primed BM-MSCs, indicating that most of the primed BM-MSCs encapsulated within the hdECM patch, which were attached to the ischemic hearts, remained viable even after 8 weeks (Fig. 5, A to C).
Fig. 5. Survival of in vivo primed BM-MSC in MI hearts and their effects on vascular regeneration and cardioprotection.

(A to C) Survival of hMSCs and HGF-eMSC within the cardiac patch after 8 weeks from implantation into the MI-induced rat hearts and their quantification summary. (A) Representative images of hMSCs [DiI (1.1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocynanine perchlorate), red], HGF-eMSC [DiO (3,3ʹ-Dioctadecyloxacarbocyanine perchlorate), green], TUNEL signal (gray), and 4′,6-diamidino-2-phenylindole (DAPI) for nucleus (blue) within the patch made by using hdECM. (B) Number of either DiI- or DiO-positive MSCs. (C) Number of TUNEL signal–positive MSCs. Scale bars, 50 μm. n = 5 per group. (D) Representative images of capillaries stained with fluorescein isothiocyanate (FITC)–IB4 (green) on the infarct zone (IZ), border zone (BZ), and remote zone at 8 weeks after MI and (E to G) their quantification summary in three areas. For quantification, the number of capillaries on five randomly selected fields in each heart was counted. Scale bars, 50 μm. *P < 0.05 compared to control group; †P < 0.05 compared to BM-MSC group; ‡P < 0.05 compared to HGF-eMSC group. n = 5 per group. (H) Representative immunostaining images with cardiac-specific TNNT2 (red), TUNEL signal (gray), and DAPI for nucleus (blue) at 8 weeks after MI and (I and J) their quantification summary. Scale bars, 50 μm. *P < 0.05 compared to control group; †P < 0.05 compared to BM-MSC group; ‡P < 0.05 compared to HGF-eMSC group. n = 5 to 9 for each group.
Since paracrine factors secreted from the BM-MSCs are involved in vascular regeneration that could substantially contribute to cardiac regeneration following MI, we examined the effects of primed BM-MSCs on vascular regeneration in the MI-induced hearts. To visualize the functional cardiac vessels, we perfused isolectin B4 (IB4) conjugated to a green fluorescent protein (GFP) into the heart before tissue harvesting 8 weeks after the patch implantations. The fluorescent image assessments demonstrated that the number of capillaries (in mm2) in both the border zone and infarct zone of the hearts from the BM-MSC/HGF-eMSC mixed group were substantially higher than the control and the BM-MSCs or HGF-MSC only groups (Fig. 5, D to G).
To investigate whether primed BM-MSCs protected the myocardium from ischemic insults, we quantified the viable myocardium 8 weeks after MI by immunostaining for cardiac troponin T (TNNT2) antibodies in all experimental groups (Fig. 5, H to J). There was a substantially higher number of TNNT2-positive cardiomyocytes in the BM-MSC/HGF-eMSC group than in the other groups, including the BM-MSC and HGF-eMSC groups (Fig. 5, H to I, and fig. S8). The TUNEL staining results further demonstrated that less than 1% of the TNNT2-positive cardiomyocytes in all the experimental groups were TUNEL positive (Fig. 5J). These results clearly and consistently support the notion that the primed BM-MSCs conferred substantial salutary effects to the ischemic myocardium.
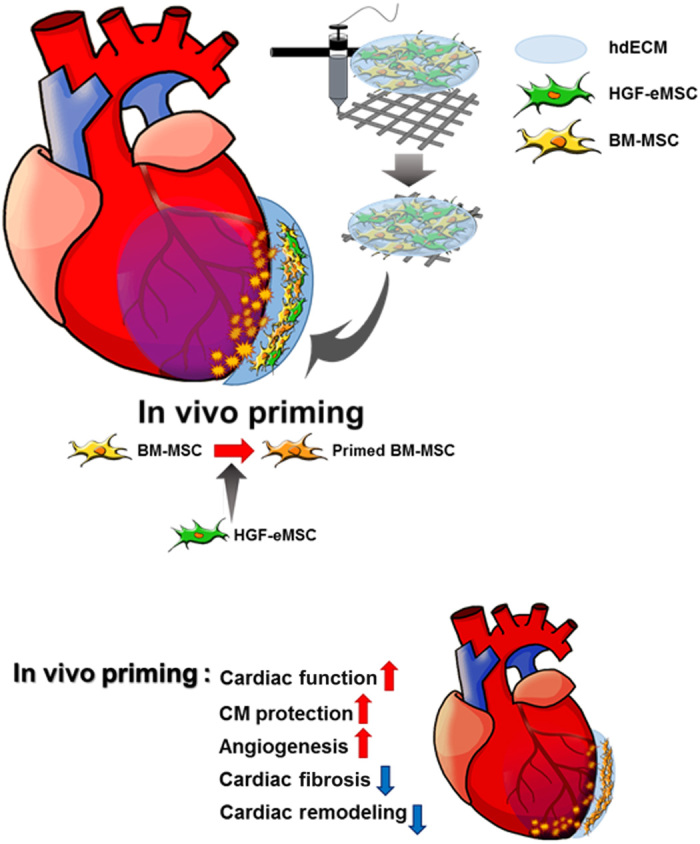
Collectively, these in vivo results indicate that BM-MSCs primed via HGF-eMSCs survived better within the 3D patch and enhanced vascular regeneration through the prolonged secretion of beneficial paracrine factors, which ultimately augmented heart function and restored the injured myocardium (Fig. 6).
Fig. 6. Schematic diagram of the underlying mechanism of in vivo priming of BM-MSCs with HGF-eMSC.
DISCUSSION
Despite varied outcomes from several clinical trials evaluating the therapeutic efficacy of MSCs in treating heart failure, because of several inherent advantages, MSCs are still considered one of the most promising candidates for cell-based cardiac regeneration therapies to date (3). Thus, particularly in the field of adult stem cells, the focus has been on developing effective methods to augment the therapeutic potential of MSCs (14). In the present study, we sought to develop a strategy to enhance the therapeutic efficacy of MSCs in vivo by coencapsulating BM-MSCs with engineered HGF-eMSCs within 3D cardiac patches, which were then implanted in the epicardium of MI hearts.
Our results demonstrated that the BM-MSCs were primed within the epicardially attached cardiac patch through the sustained secretion of paracrine factors, specifically HGF, from the HGF-MSCs. Subsequently, those empowered BM-MSCs survived for longer periods and released greater amounts of beneficial paracrine factors capable of salvaging damaged myocardium and regenerating vasculatures. Although the concept of priming cells to bolster therapeutic capacity is not entirely novel and it has been previously tested in various other cell types, such as several types of immune cells (3, 13) and hepatocytes (20), the fact that this study was the very first to achieve priming in intact hearts in vivo after transplantation of BM-MSCs onto MI hearts should be appreciated.
To prime BM-MSCs in vivo, we encapsulated BM-MSCs together with HGF-eMSCs genetically engineered to continuously release HGF. We chose HGF as a major inducer for in vivo priming since HGF is a renowned pleiotropic factor that is involved in multiple biological activities, such as cell proliferation and survival, angiogenesis, and antifibrotic and antiapoptotic activities (21–24). Previous studies showed that overexpression of the HGF gene in the myocardium or the transplantation of HGF-overexpressing MSCs promoted cardiac function and vascular regeneration and prevented cellular apoptosis. MSCs pretreated with HGF cytokines displayed greatly enhanced therapeutic potential to repair damaged organs, such as livers (20). In regard to the use of genetically engineered HGF-eMSCs generated by lentiviral vector encoding human telomerase reverse transcriptase (hTERT) and c-Myc reprogramming factors, to exclude the risk for tumorigenicity, particularly since c-Myc is an oncogene, we irradiated (100 Gy) the cultured HGF-eMSCs before patch generation to prevent their overgrowth. Subsequently, we observed that the proliferation rate of HGF-eMSCs was markedly decreased in in vitro culture, and no tumor was detected from in vivo tumorigenicity testing using nude mice. Notably, microscopic observations of rat heart tissues harvested at weeks 4 and 8 after implantation revealed that most of the hMSCs were located within the patch and they did not migrate to the neighboring myocardium until 8 weeks following implantation. Most of DiI (1.1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)–positive hMSCs, which we prelabeled with CM-DiI (red fluorescence) before patch generation to monitor their migration, were detected within the patch, not in the myocardium. These results further verify that our strategy of encapsulating genetically engineered cells in patches located outside the heart is a suitable strategy that can prevent undesirable outcomes from genetically modified cells in the heart.
One of the critical hurdles for the clinical application of BM-MSCs for cardiac cell therapy is their low survival and poor engraftment in the heart. Given the inherent characteristics of the heart as a contractile organ and owing to the undesirable microenvironments in the ischemic myocardium, the implanted BM-MSCs easily die or leak out of the injection sites and washout through venous shunts. Therefore, to improve the survival of transplanted BM-MSCs and thereby ensure prolonged secretion of favorable paracrine factors toward injured myocardium, we aimed to generate hdECM-derived cardiac patches to serve as carriers of a cell reservoir, as well as to provide structural and potential vasculature support to the infarcted area. Our results indicated that both primed and unprimed BM-MSCs appeared to survive better within the patch until 8 weeks after implantation, as evidenced by the minimal number of detectable TUNEL-positive MSCs in most of the experimental groups. We presume that the hdECM-derived cardiac patches provided a complimentary microenvironment that enabled more resistance against hostile conditions in ischemic tissues, particularly during the early stage of implantation. Surviving MSCs continuously released beneficial paracrine factors and, consequently, restored the injured myocardium and promoted the vascular regeneration. In terms of the techniques we used to engineer the cardiac patches, in direct contrast to several previous studies that simply used natural ECMs or synthetic materials to produce cardiac patches, we took an innovative approach that used a mixture of lyophilized porcine hdECM with MSCs as bioink. By using lyophilized porcine hdECMs that allow for convenient storage and easy transport of cell-free ECMs, our study demonstrated the potential clinical benefit of BM-MSCs in translational research in the field of cardiac repair.
In summary, in the current study, we suggested a proof-of-concept platform coined as in vivo priming that maximizes the therapeutic potential of BM-MSCs to treat heart disease. When BM-MSCs were placed together with HGF-eMSCs engineered to continuously secrete HGF in a cardiac patch that was epicardially implanted in the hearts, paracrine secretion by the BM-MSCs was remarkably bolstered and the paracrine factors were stably secreted toward the diseased hearts. Despite literature movement away from BM-MSCs, our study indicates the remarkable potential that BM-MSCs still have as a source of cell-based cardiac therapy if used effectively—a potential that, if studied under scrutiny, can open a new doorway to the future of cardiac stem cell therapy.
MATERIALS AND METHODS
Isolation of MSCs derived from human bone marrow
BM-MSCs were obtained from the Catholic Institute of Cell Therapy (CIC) (Catholic MASTER Cells, CIC, Korea). Human bone marrow aspirates were obtained from the iliac crest of healthy donors aged 20 to 55 years with approval from the Institutional Review Board of Seoul St. Mary’s Hospital (approval numbers KIRB-00344-009 and KIRB-00362-006). The bone marrow aspirate from each donor who consented was collected and sent to the Good Manufacturing Practice (GMP)–compliant facility of the CIC (Seoul, Korea; www.cic.re.kr) for the isolation, expansion, and quality control of human BM-MSCs. The marrow mixtures were centrifuged at 793g and 4°C for 7 min to obtain marrow pellets. After removal of the supernatant, red blood cells (RBCs) were removed by adding and suspending the pellets in a 10-fold volume of sterile distilled water. RBC-deprived samples were then suspended in an MSC growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM)–low glucose (PAA) and 20% fetal bovine serum (FBS; Gibco). They were added to 100-mm tissue culture dishes (TPP) and incubated at 37°C with 5% CO2. MSC growth medium was used for all cell expansion procedures unless stated otherwise. The media were replaced twice a week. The cells were detached when they reached 70 to 90% confluence and were replated at a density of 5 × 103 to 8 × 103 cells/cm2. The cells were expanded two to four passages in the GMP-compliant facility.
Generation of engineered HGF-eMSCs
BM-MSCs (Catholic MASTER Cells) were obtained from the CIC (Seoul, Korea). To generate engineered immortalized MSCs, we first produced replication-incompetent lentiviral vectors each containing c-Myc and hTERT. At this time, a gene construct expressing the tetracycline transactivator protein was inserted together to use the Tet-off system. These lentiviral vectors were extracted and quantified from DH5α Escherichia coli cells using the EndoFree Plasmid Maxi Kit (QIAGEN). To amplify the lentiviruses, Lenti-X cell (Clontech Laboratories) was transduced using Lipofectamine PLUS reagent (Thermo Fisher Scientific). The amplified lentiviruses were transduced to BM-MSCs at 100 multiplicity of infection using RetroNectin (Clontech Laboratories). After infection, Zeocin (500 μg/ml) and puromycin (1 μg/ml) were sequentially added to the culture media of the stabilized cells to select the cells infected with lentiviruses. These selected immortalized MSCs were highly proliferative, and the engineered MSCs were expanded and banked for further therapeutic gene transfection.
To generate HGF-expressing immortalized MSCs, the human HGF gene containing a replication-incompetent lentiviral vector was also prepared and amplified. After the transfection of the human HGF gene containing lentiviral vector to the immortalized MSCs, hygromycin (25 μg/ml) was added for the selection of lentiviral-infected cells. Next, HGF-eMSCs were isolated as a monoclonal cell population by the limiting dilution method. The final monoclonal cells were selected by HGF protein secretion, proliferation rate, and other MSC phenotypes. The HGF-eMSCs were cultured in low glucose–containing DMEM (Gibco) supplemented with 10% FBS (Gibco), basic FGF (5 ng/ml; PeproTech), and doxycycline (2 μg/ml; Clontech Laboratories) at 37°C and 5% CO2.
Enzyme-linked immunosorbent assay
ELISA was performed to confirm the expression levels of HGF. Three different passages of HGF-eMSCs were used for the ELISA. The HGF-eMSCs were seeded at a density of 1 × 105 cells per well in 12-well plates (Corning) with 1 ml of CM. After 48 hours at 37°C in a 5% CO2 incubator, the supernatants were harvested and centrifuged to remove cell debris. The HGF concentrations were determined using a Human HGF ELISA kit (R&D Systems) according to the manufacturer’s instructions. The absorbance of the samples at 450 nm was measured using a SpectraMax 190 microplate reader (Molecular Devices).
Cell proliferation analysis
To determine the proliferation rate of the HGF-eMSCs, 4 × 105 cells were seeded into a T75 flask. After 3 to 4 days, the cells were harvested, and the proliferation rate was measured by the trypan blue (Gibco) exclusion method using a single-use hemocytometer (INCYTO). The cumulative PDL of the HGF-eMSCs was determined by the summation of PD. The PDL was calculated using (logN − logN0)/log2, where N is the number of harvested cells and N0 is the number of seeded cells.
MSC marker analysis
The immunophenotype of the MSCs was analyzed using flow cytometry. The cells were detached from the flask using trypsin, then centrifuged at 1500 rpm for 5 min, and resuspended in staining/wash buffer [phosphate-buffered saline (PBS) with 2% FBS)]. For staining, the BM-MSCs and HGF-eMSCs were washed with staining/wash buffer and incubated for 30 min in the dark at 4°C with Stemflow hMSC Analysis Kit (BD) according to the manufacturer’s instructions. The MSCs were characterized using the following conjugated monoclonal antibodies: CD90 fluorescein isothiocyanate (FITC) (Clone: 5E10), CD105 PerCP-Cy5.5 (Clone: 266), CD73 allophycocyanin (APC) (Clone: AD2), and phycoerythrin (PE) negative cocktail (CD34, CD11b, CD19, CD45, and HLA-DR). Flow cytometry was conducted using an LSRFortessa cell analyzer (BD Biosciences) and analyzed using FlowJo version 10 or BD FACSDiva software.
Karyotyping
To karyotype the HGF-eMSCs, we used the karyotyping service of EONE laboratories.
Irradiation and in vitro analysis
Before in vivo administration, cryopreserved HGF-eMSCs were irradiated with 100-Gy radiation using a high-level gamma irradiation device (MDS Nordion) at Korea Atomic Energy Research Institute, Advanced Radiation Technology Institute. The temperature of the HGF-eMSCs was kept below −50°C during irradiation using specially fabricated devices. The irradiated HGF-eMSCs were thawed, and 5 × 105 cells were plated in six-well plates (Corning) with 5 ml of CM. For the in vitro analysis, the samples were prepared as triplicate time points. Supernatant was harvested from the sample wells, centrifuged at 1500 rpm for 5 min, aliquoted without cell debris, cryopreserved at −80°C until the experimental end point, and then subjected to ELISA analysis. The cell viability was measured in the sample wells by trypan blue exclusion.
Coculture
BM-MSCs and HGF-eMSCs were indirectly cocultured using a Transwell culture system (0.4-μm pore polyethylene terephthalate membrane; Corning) for 3 days. The BM-MSCs (1 × 105) were preplated onto a 12-well plate for 3 hours, and then, the inserts containing HGF-eMSCs (1 × 106) with 2% ECM patch were placed into the wells. The cells were cultured in BM-MSC growth medium (DMEM; Gibco) containing 10% FBS (Gibco) with 1% penicillin-streptomycin (Gibco) for 3 days at 37°C with 5% CO2. To produce CM, the Transwell insert was removed and then washed with PBS. Culture media was changed to DMEM without FBS or growth factor. After 3 days of culture, the supernatants were collected and kept at −80°C.
RNA preparation and RT-PCR
Total RNA was extracted using TRIzol reagent (Life Technologies) following the manufacturer’s instructions. cDNA synthesis was carried out with 500 ng of total RNA using Takara PrimeScript RT Master Mix (Takara Bio, RR036A). Gene expression was measured via the TB Green Premix Ex Taq (Takara, RR420A) using a LightCycler 480 II (Roche). qRT-PCR was performed in triplicate in three independent experiments. Relative quantification was calculated with the 2–ΔΔCt method using the housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase) as reference for normalization.
Tube formation assay
Undiluted basement membrane matrix (250 μl; Matrigel, BD Biosciences) was added to each well of a 24-well plate and solidified by incubation at 37°C for 30 min. HUVECs (1 × 105) were plated onto each Matrigel-coated well containing different types of media (EGM2, EBM, BM-MSCs, and HGF-eMSC–primed BM-MSC CM) and then incubated at 37°C with 5% CO2 for 12 hours. After removing the media, 4% paraformaldehyde (PFA) was added for fixation, the cells were visualized using a light microscope, and formation of tube structures was calculated using ImageJ software.
EC migration assay
HUVECs (5 × 104) were seeded onto the upper layer of Transwell inserts (8-μm pore) with EBM basal medium and then placed onto a 24-well plate containing the test media (EGM2, EBM, BM-MSCs, and HGF-eMSC–primed BM-MSC CM). The cells were then allowed to migrate for 6 hours at 37°C with 5% CO2. The migrated cells were fixed in 4% PFA for 10 min, and staining was performed using 0.1% crystal violet (Sigma-Aldrich) for 10 min. After rinsing the Transwell membrane with distilled water, the upper side of the membrane was gently swiped with a cotton swab to remove nonmigrated cells. The migrated cells were counted using a light microscope, and the stained area of the membrane was calculated using ImageJ software.
Neonatal cardiomyocyte isolation
Neonatal rat cardiomyocytes were isolated from 1-day-old Sprague-Dawley rat hearts, following the guidelines and under the approval of the Animal Care and Use Committee at the Catholic University of Korea. Ventricular tissue was incubated with 0.1% trypsin solution (Welgene) in Hanks’ balanced salt solution (HBSS; Welgene) with gentle agitation at 4°C overnight. The next day, the digested tissue was transferred to a petri dish on ice, minced, and collected into a conical tube. Digestion solution [5 ml of collagenase B (1 mg/ml) in HBSS; Roche] was added to the heart tissue and triturated using a 5-ml pipette 10 times and incubated in a 37°C water bath for 5 min with gentle agitation. Supernatant containing the cardiomyocytes was transferred to growth media [DMEM containing 10% FBS (Gibco) with 1× antibiotic-antimycotic (Gibco)], and the undigested heart tissue was resuspended in 5 ml of digestion solution. An additional digestion step was performed, and the cell-containing supernatant was collected. The digestion step was repeated five times, followed by centrifugation for 5 min at 1500 rpm and resuspension of the cell pellet in growth media. Contaminating fibroblasts and ECs were removed by preplating for 2 hours at 37°C with 5% CO2. After the preplating step, nonadherent cardiomyocytes were transferred to a new conical tube, resuspended in growth media, then plated on 0.1% gelatin (Welgene)–coated culture plates at a density of 1 × 105 cells/cm2, and cultured at 37°C and 5% CO2. The culture medium was changed every other day.
CCK-8 assay
The viability of the BM-MSCs and the HGF-eMSC–primed BM-MSCs cultured under serum starvation conditions (72 hours) was assessed using the CCK-8 assay. The CCK-8 reagent (Dojindo Laboratories) was added to a well containing 10% of the total culture medium volume. After a 2-hour incubation, the supernatant was transferred to a 96-well plate and the optical density was measured at a wavelength of 450 nm.
Annexin V assay
The cells were dissociated with 0.25% trypsin-EDTA (Thermo Fisher Scientific) and then stained using the Alexa Fluor 488 annexin V/Dead Cell Apoptosis Kit with Alexa Fluor 488 annexin V and propidium iodide (Invitrogen, V13241) according to the manufacturer’s instructions. The stained cells were then analyzed by flow cytometry (BD LSRFortessa). Fluorescence emission was measured at 530 nm and 575 nm using 488-nm excitation.
Live and dead assay
The live and dead assay was performed using the LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen, L3224) according to the manufacturer’s instructions. The medium was changed to phenol red–free DMEM with a calcein and ethidium homodimer-1 dye mixture and incubated for 30 min; then, the labeled cells were analyzed under a fluorescence microscope (Nikon).
Manufacturing cardiac patches with hdECM
The hdECM was prepared as described previously (25). Briefly, heart tissue from a 6-month-old Korean domestic pig was purchased from a livestock product market with supplier approval (fig. S9). We dissected the left ventricle from the complete porcine heart, which was cut into smaller pieces. Then, the small pieces of heart tissues were soaked in 1% SDS (Affymetrix) solution for 48 hours, followed by treatment with 1% Triton X-100 solution in PBS (Biosesang) for 1 hour. Next, the decellularized tissues were placed in PBS for 3 days to remove the residual detergent (fig. S9). Subsequently, the hdECM were lyophilized, pulverized in liquid nitrogen, and digested in 10 ml of 0.5 M acetic acid solution (Merck Millipore) at a final concentration of 3.3 w/v% (330 mg of hdECM powder) supplemented with 33 mg of pepsin powder. The digested hdECM solution was filtered through a 40-μm pore mesh, aliquoted in 1 ml, and stored at −20°C for further experiments. Before manufacturing the cardiac patches, the hdECM solution was adjusted to a neutral pH of 7.4 by adding 10 M NaOH solution on ice to avoid gelation of the hdECM (fig. S9).
To generate the hdECM patches, we first produced disk-shaped polycaprolactone (PCL) that is 8 mm in diameter and 0.5 mm high as a supporting framework by using a 3D printer according to the generated code. Then, we built up the hdECM patches by mixing the MSCs (total: 1 × 106 per ml) and hdECM (20 mg/ml) on the PCL framework. Subsequently, the hdECM patches were exposed to ultraviolet light for about 60 s to initiate a vitamin B2–induced postcrosslinking process (light intensity, 30 mW/cm2) and kept them at 37°C for 24 hours for additional thermal cross-linking. After all these procedures, the final thickness and diameter of the hdECM patches averaged 3 and 8 mm, respectively.
MI model and patch delivery
All of the surgical interventions and the pre- and postsurgical animal care were provided in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Survival Surgery provided by the Institutional Animal Care and Use Committee in the School of Medicine at the Catholic University of Korea (approval number: CUMS-2015-0048-04). Male Fischer 344 rats (8 weeks old and 160 to 180 g; KOATECH, Korea) were anesthetized with 2% inhaled isoflurane and intubated via the trachea with an 18-gauge intravenous catheter. The rats were then mechanically ventilated with medical-grade oxygen. The animals were placed on a 37°C heating pad to prevent cooling during the procedure. After shaving the chest, a left thoracotomy was performed. MI was achieved by tying a suture with sterile polyethylene glycol tubing (22 gauge) placed into the LAD artery for 1 min, and then, the knot was permanently ligated using a 7-0 Prolene suture. To establish baseline left ventricular function, the EF and regional wall motion abnormalities were examined after operation day 7 (inclusion criterion: EF < 45% by echocardiographic evaluation). On the same day, the rats were anesthetized again using isoflurane inhalation, intubated, and mechanically ventilated. The animal chest was reopened, and the pericardium was partially removed from the infarcted heart. Then, BM-MSC, HGF-eMSC, and BM-MSC/HGF-eMSC patches were implanted directly onto the epicardium using sutures. The chest was closed aseptically, and antibiotics and 0.9% normal saline solution were administered. All rats received immunosuppressants (azathioprine, 2 mg/kg; cyclosporine A, 5 mg/kg; methylprednisolone, 5 mg/kg) daily, as described previously.
Echocardiography
The assessment of cardiac function was performed by echocardiography. The rats were lightly anesthetized with inhaled isoflurane, and physiological data were recorded by using a transthoracic echocardiography system equipped with a 15-MHz L15-7io linear transducer (Affiniti 50G, Philips). Echocardiograms were performed before the patch attachment and at 2, 4, and 8 weeks after patch attachment. The echocardiography operator was blinded to the group allocation during the experiment. SWT, PWT, left ventricular internal diastolic dimension (LVIDd), and left ventricular internal systolic dimension (LVISd), which are indexes of left ventricular adverse remodeling, and EF and FS, which are indexes of left ventricular systolic function, were calculated with the following equations, respectively
Capillary density measurement
At the time of euthanasia, the hearts were perfused with GFP-conjugated IB4 from Griffonia simplicifolia for 15 min at room temperature. The hearts were fixed in 4% paraformaldehyde overnight, and then, blocks were made. After storage at −20°C, the heart was cross-sectioned into 4-μm sections starting at the top of the apex using a microtome (Leica, RM2255, Germany). The number of capillaries were counted in five random microscopic fields using a fluorescence microscope (Nikon) and expressed as the number of capillaries per square millimeter of tissue area.
Determination of fibrosis
Masson’s trichrome staining (Sigma-Aldrich) was performed to determine the fibrotic area, wall thickness scar area, and viable myocardium of the MI hearts. Briefly, five paraffin slides were preincubated in a 37°C dry oven before deparaffinization and rehydration. The paraffin sections were then refixed for 1 hour in 56°C Bouin’s solution. The sections were stained using Weigert’s iron hematoxylin solution for 15 min at room temperature and then also stained using Biebrich scarlet-acid fuchsin solution for 20 min at room temperature. Last, the sections were counterstained with aniline blue for 15 min, followed by incubation in 1% acetic acid for 2 min at room temperature. Tissue washes were performed between each step. Collagen fibers appeared blue, and viable myocardium appeared red. Imaging of the heart sections was performed with a slide scanner (Pannoramic MIDI). All other items, including the fibrotic area, were quantified using ImageJ software.
Immunohistochemistry
Immunofluorescence was performed on 4-um-thick paraffin sections. After deparaffinization and rehydration, antigen retrieval with target retrieval solution (Dako) was performed in a decloaking chamber. The sections were blocked and incubated with diluted primary antibody (Dako) at 4°C overnight. The primary antibody used in this study was mouse anti-TNNT2 (Abcam; 1:200). After washing three times with 1% Tween 20 in PBS, the samples were incubated with secondary antibody for 60 min at room temperature in the dark. The secondary antibody used in this study was anti-mouse immunoglobulin G Alexa Fluor 594 (Invitrogen; 1:500). After washing again with 1% Tween 20 in PBS, the sections were stained with 4′,6-diamidino-2-phenylindole solution (VECTASHIELD) for nuclear staining and then mounted on slides. Imaging of the heart sections was performed with an LSM 800 laser scanning microscope with Airyscan processing (Zeiss).
TUNEL assay
To assess apoptosis in vivo 8 weeks after patch attachment, the animals were euthanized and TUNEL assays (Thermo Fisher Scientific) were performed. Briefly, five paraffin sections were deparaffinized and rehydrated, and the TUNEL assay was performed on 4-um-thick sections. Following permeabilization with 0.1% sodium citrate containing 0.5% Triton X-100 for 15 min, the reaction was carried out in a dark 37°C dry oven for 1 hour. Imaging of the heart sections was performed with an LSM 800 laser scanning microscope with Airyscan processing (Zeiss).
Data analysis
All quantitative data are shown as means ± SEM unless otherwise indicated. The statistical differences between two groups were analyzed by two-tailed Student’s t tests. The statistical differences among three or more groups were also analyzed by analysis of variance (ANOVA) with Bonferroni’s post hoc analysis. Results were considered statistically significant when the P value was less than 0.05.
Supplementary Material
Acknowledgments
We would like to thank S. Oh at the City University of Hong Kong for instrumental discussions, directions, and edits for this manuscript. Funding: This study was supported by the National Research Foundation of Korea (NRF) grant (2016R1C1B2015529) and the Bio & Medical Technology Development Program grant (NRF-2017M3A9B3061954) funded by the Ministry of Science and ICT. This study was also supported by CityU Research Project (9610355 to K.B.) and the Hong Kong Research Grants Council (21100818 to K.B.). This research was also supported by Basic Science Research Program through the NRF funded by the Ministry of Education (2015R1A6A3A04059015) and Ministry of Science and ICT, Korea, under the ICT Consilience Creative program (IITP-2019-2011-1-00783) supervised by the Institute for Information & Communications Technology Planning & Evaluation (J.J.). Author contributions: Experimental conception and design, acquisition of data, analysis and interpretation of data, and manuscript drafting and revising: B.-W.P., S.-H.J., and S.D. Experimental conception and design, acquisition of data, and analysis and interpretation of data: S.M.L., J.-H.P., H.K., J.-W.H., S.L., H.-J.K., H.-Y.K., and S.J. Experimental conception and design, financial support, and administrative support: D.-W.C. Experimental conception and design, financial support, administrative support, manuscript drafting and revising, and final approval of the manuscript: J.J., K.B., and H.-J.P. Competing interests: B.-W.P., S.-H.J., S.D., S.M.L., H.-Y.K., D.-W.C., J.J., and H.-J.P. are inventors on a patent application related to this work filed by The Catholic University of Korea, SL BIGEN Inc., and POSTECH [application nos. PCT/KR2018/008903 (international) and 10-2018-0091280 (South Korea) filed on 6 August 2018]. D.-W.C. owns equity in T&R Biofab Inc. and EDmicBio Inc. J.J. owns equity in EDmicBio Inc. and holds stock options in T&R Biofab Inc. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay6994/DC1
Fig. S1. Characterization of HGF-eMSC.
Fig. S2. Cellular characteristics of irradiated HGF-eMSC.
Fig. S3. Cellular behavior of irradiated HGF-eMSC within the hdECM patch.
Fig. S4. Investigation of the duration of priming effect in BM-MSC by HGF-eMSC.
Fig. S5. Examination of the tumorigenicity of HGF-eMSCs.
Fig. S6. Representative echo images of all experimental groups at 8 weeks after interventions.
Fig. S7. Effects of nonirradiated HGF-eMSC for priming BM-MSCs and their therapeutic potential for MI.
Fig. S8. Representative immunofluorescence staining images with cardiac-specific maker proteins.
Fig. S9. Schematic illustration demonstrating the procedures for generating decellularized pig hdECM.
Table S1. Primer sequences used for qRT-PCR analysis.
REFERENCES AND NOTES
- 1.Pittenger M. F., Mackay A. M., Beck S. C., Jaiswal R. K., Douglas R., Mosca J. D., Moorman M. A., Simonetti D. W., Craig S., Marshak D. R., Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Nesselmann C., Ma N., Bieback K., Wagner W., Ho A., Konttinen Y. T., Zhang H., Hinescu M. E., Steinhoff G., Mesenchymal stem cells and cardiac repair. J. Cell. Mol. Med. 12, 1795–1810 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin J. Q., Zhu J., Ankrum J. A., Manufacturing of primed mesenchymal stromal cells for therapy. Nat. Biomed. Eng. 3, 90–104 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Roura S., Gálvez-Montón C., Mirabel C., Vives J., Bayes-Genis A., Mesenchymal stem cells for cardiac repair: Are the actors ready for the clinical scenario? Stem Cell Res. Ther. 8, 238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh A., Singh A., Sen D., Mesenchymal stem cells in cardiac regeneration: A detailed progress report of the last 6 years (2010-2015). Stem Cell Res. Ther. 7, 82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gnecchi M., He H., Liang O. D., Melo L. G., Morello F., Mu H., Noiseux N., Zhang L., Pratt R. E., Ingwall J. S., Dzau V. J., Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 11, 367–368 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Li W., Ma N., Ong L.-L., Nesselmann C., Klopsch C., Ladilov Y., Furlani D., Piechaczek C., Moebius J. M., Lützow K., Lendlein A., Stamm C., Li R.-K., Steinhoff G., Bcl-2 engineered MSCs inhibited apoptosis and improved heart function. Stem Cells 25, 2118–2127 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Yang J., Zhou W., Zheng W., Ma Y., Lin L., Tang T., Liu J., Yu J., Zhou X., Hu J., Effects of myocardial transplantation of marrow mesenchymal stem cells transfected with vascular endothelial growth factor for the improvement of heart function and angiogenesis after myocardial infarction. Cardiology 107, 17–29 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Haider H. K., Jiang S., Idris Niagara M., Ashraf M., IGF-1–overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1α/CXCR4 signaling to promote myocardial repair. Circ. Res. 103, 1300–1308 (2008). [DOI] [PubMed] [Google Scholar]
- 10.J. Muratsu, F. Sanada, Y. Taniyama, Y. Ikeda-Iwabu, R. Otsu, K. Shibata, M. K. Brulé, H. Rakugi, Ryuichi Morishita, HGF gene therapy for therapeutic angiogenesis in peripheral artery disease, in Therapeutic Angiogenesis, Y. Higashi, T. Murohara, Eds. (Springer Singapore, 2017), pp. 133–144. [Google Scholar]
- 11.Carvalho J. L., Braga V. B. A., Melo M. B., Campos A. C. D. A., Oliveira M. S., Gomes D. A., Ferreira A. J., Santos R. A. S., Goes A. M., Priming mesenchymal stem cells boosts stem cell therapy to treat myocardial infarction. J. Cell. Mol. Med. 17, 617–625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji S. T., Kim H., Yun J., Chung J. S., Kwon S.-M., Promising therapeutic strategies for mesenchymal stem cell-based cardiovascular regeneration: From cell priming to tissue engineering. Stem Cells Int. 2017, 3945403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei Z. Z., Zhu Y.-B., Zhang J. Y., McCrary M. R., Wang S., Zhang Y.-B., Yu S.-P., Wei L., Priming of the cells: Hypoxic preconditioning for stem cell therapy. Chin. Med. J. 130, 2361–2374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Cássia Noronha N., Mizukami A., Caliári-Oliveira C., Cominal J. G., Rocha J. L. M., Covas D. T., Swiech K., Malmegrim K. C. R., Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res. Ther. 10, 131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White I. A., Sanina C., Balkan W., Hare J. M., Mesenchymal stem cells in cardiology. Methods Mol. Biol. 1416, 55–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cashman T. J., Gouon-Evans V., Costa K. D., Mesenchymal stem cells for cardiac therapy: Practical challenges and potential mechanisms. Stem Cell Rev. Rep. 9, 254–265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X., Cohen M. V., Downey J. M., Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 24, 225–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ardehali H., Signaling mechanisms in ischemic preconditioning. Circ. Res. 99, 798–800 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Qiu Y., Tang X.-L., Park S.-W., Sun J.-Z., Kalya A., Bolli R., The early and late phases of ischemic preconditioning. Circ. Res. 80, 730–742 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Li R., Rong W., Han M., Cui C., Feng Z., Sun X., Jin S., Therapeutic effect of hepatocyte growth factor-overexpressing bone marrow-derived mesenchymal stem cells on CCl4-induced hepatocirrhosis. Cell Death Dis. 9, 1186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitta K., Day R. M., Ikeda T., Suzuki Y. J., Hepatocyte growth factor protects cardiac myocytes against oxidative stress-induced apoptosis. Free Radic. Biol. Med. 31, 902–910 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Ueda H., Nakamura T., Matsumoto K., Sawa Y., Matsuda H., Nakamura T., A potential cardioprotective role of hepatocyte growth factor in myocardial infarction in rats. Cardiovasc. Res. 51, 41–50 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Sonnenberg S. B., Rane A. A., Liu C. J., Rao N., Agmon G., Suarez S., Wang R., Munoz A., Bajaj V., Zhang S., Braden R., Schup-Magoffin P. J., Kwan O. L., DeMaria A. N., Cochran J. R., Christman K. L., Delivery of an engineered HGF fragment in an extracellular matrix-derived hydrogel prevents negative LV remodeling post-myocardial infarction. Biomaterials 45, 56–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura T., Mizuno S., Matsumoto K., Sawa Y., Matsuda H., Nakamura T., Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J. Clin. Invest. 106, 1511–1519 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pati F., Jang J., Ha D.-H., Kim S. W., Rhie J.-W., Shim J.-H., Kim D.-H., Cho D.-W., Printing three-dimensional tissue analogues with decellularized extracellular matrix bioink. Nat. Commun. 5, 3935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay6994/DC1
Fig. S1. Characterization of HGF-eMSC.
Fig. S2. Cellular characteristics of irradiated HGF-eMSC.
Fig. S3. Cellular behavior of irradiated HGF-eMSC within the hdECM patch.
Fig. S4. Investigation of the duration of priming effect in BM-MSC by HGF-eMSC.
Fig. S5. Examination of the tumorigenicity of HGF-eMSCs.
Fig. S6. Representative echo images of all experimental groups at 8 weeks after interventions.
Fig. S7. Effects of nonirradiated HGF-eMSC for priming BM-MSCs and their therapeutic potential for MI.
Fig. S8. Representative immunofluorescence staining images with cardiac-specific maker proteins.
Fig. S9. Schematic illustration demonstrating the procedures for generating decellularized pig hdECM.
Table S1. Primer sequences used for qRT-PCR analysis.