

SUMMARY
Understanding the fundamental mechanisms of arbovirus transmission and pathogenesis is essential to develop strategies for treatment and prevention. We previously took an in vivo evolution-based approach and identified the chikungunya virus E1 glycoprotein residue 80 to play a critical role in viral transmission and pathogenesis. In this study, we address the genetic conservation and function of position 80 and demonstrate that this residue is a key determinant in alphavirus infectivity and dissemination through modulation of viral fusion and cholesterol dependence. In addition, in studying the evolution of position 80, we identified a network of glycoprotein residues, including epidemic determinants, that regulate virus dissemination and infectivity. These studies underscore the importance of taking evolution-based approaches to not only identify key viral determinants driving arbovirus transmission and pathogenesis, but also to uncover fundamental aspects of arbovirus biology.
Keywords: alphaviruses, Chikungunya virus, E1 glycoprotein, cholesterol dependence, fusion, viral dissemination
INTRODUCTION
Viral evolution is a key driving force for arbovirus emergence and in turn can be used to study how arboviruses are transmitted and cause disease. Recent outbreaks of Venezuelan equine encephalitis virus (VEEV), West Nile virus, and chikungunya virus (CHIKV) have been retrospectively mapped to adaptive mutations in the viral glycoproteins, which have led to increases in transmission and pathogenesis as well as the adaptation to new vectors (Ebel et al., 2004; Greene et al., 2005; Moudy et al., 2007; Pesko and Ebel, 2012). Although the viral glycoproteins are not the only factors modulating viral transmission, extensive studies of natural and laboratory-derived glycoprotein variants have demonstrated that glycoproteins are critical determinants of emergence, transmission and pathogenesis of arboviruses (Brault et al., 2004; Greene et al., 2005; Stapleford et al., 2014). However, the underlying molecular mechanisms of how the viral glycoproteins regulate such processes remain elusive.
To study arbovirus transmission, we use CHIKV, a member of the Togaviridae family (genus alphavirus) transmitted by the Aedes species of mosquitoes (Higgs and Vanlandingham, 2015). CHIKV contains a single-stranded positive-sense RNA genome that encodes for four non-structural proteins (nsP1–4) and six structural proteins (CP, E3, E2, 6K, TF, and E1). The E1 fusion glycoprotein is a class-II fusion protein (Kielian and Rey, 2006), anchored to the membrane by the C-terminal end, and with an ectodomain sub-divided into domains I-III, with domain II containing the fusion loop (Voss et al., 2010). In the mature virion, E1 and the attachment protein E2 are arranged to form 80 trimeric spikes constituted of trimers of E1–E2 heterodimers (Kielian and Rey, 2006). These protein complexes mediate CHIKV internalization by receptor-mediated endocytosis and fusion within the early endosome (Hoornweg et al., 2016), where low endosomal pH triggers E1–E2 dissociation, E1 fusion loop insertion into the target membrane, and membrane fusion (Kielian and Rey, 2006).
The CHIKV glycoproteins play a significant role in CHIKV transmission, emergence, and spread. In particular, an adaptive mutation in the E1 glycoprotein (A226V) gave rise to the Indian Ocean Lineage (IOL) of CHIKV (Schuffenecker et al., 2006). The emergence of the E1-A226V variant increased infectivity and transmission by Ae. albopictus mosquitoes conferring a selective advantage over transmission by Ae. aegypti (Tsetsarkin et al., 2007; Vazeille et al., 2007). Since then, CHIKV has continued to evolve, incorporating mutations in the viral attachment glycoprotein E2 (e.g., L210Q, K252Q), which further increased CHIKV fitness in Ae. albopictus (Tsetsarkin et al., 2014; Tsetsarkin and Weaver, 2011). Collectively, the continuous step-wise evolution of CHIKV glycoproteins highlights their role as key determinants of arbovirus transmission and infectivity.
In a previous study, we took an in vivo evolution-based approach to study emerging viral determinants of CHIKV transmission and pathogenesis (Stapleford et al., 2014). Using this model, we identified two mutations in the E1 glycoprotein (V80I and A129V) that are important for CHIKV transmission and pathogenesis. Position 80 in the E1 glycoprotein is fully conserved within the alphavirus genus and is located at the tip of domain II (Rey and Lok, 2018; Voss et al., 2010) next to the fusion loop and in proximity to the residue E1–226. Given the selection of this variant in vivo and its evolutionary conservation, we hypothesize that E1-V80 plays a central role in CHIKV biology. Therefore, by studying this position we can get insights into the specific mechanisms of how the E1 glycoprotein regulates CHIKV infectivity and dissemination.
In this study, we define the specific role of the E1 glycoprotein position 80 in CHIKV biology. We combine a mutational tolerance approach where we mutate the valine at position 80 to each possible canonical amino acid, with an in vitro evolution approach in mammal and mosquito cell lines. By coupling these approaches with functional analyses in both mammal and mosquito models, we found that E1 position 80 plays a central role in alphavirus infectivity and dissemination through the modulation of viral fusion and cholesterol dependence. In addition, we demonstrate a functional link between E1 position 80 and 226 in mediating viral cholesterol dependence in vitro and viral infectivity and dissemination in mammals. Finally, we identified a network of genetically linked residues in the CHIKV structural proteins with the potential to regulate viral infectivity and dissemination. Taken together, we show that the E1 residue 80 is a key determinant for CHIKV infectivity and dissemination and that this residue functions in concert with other viral residues to orchestrate these processes.
RESULTS
Residue 80 in the chikungunya virus E1 glycoprotein tolerates aliphatic amino acids and is genetically constrained in a host-specific manner.
To investigate the role of residue E1–80 in the CHIKV replication cycle, we began by addressing the mutational tolerance of this position (Figure 1A). To do this, we took an unbiased mutagenesis approach and changed position 80 to every possible canonical amino acid. Because CHIKV alternates between a mammal host and mosquito vector, we generated each variant independently in either baby hamster kidney (BHK-21) or Ae. albopictus (C6/36) cells, passaged the viruses three times over each cell type, and addressed the genetic stability by Sanger sequencing (Figure S1A).
Figure 1. Position 80 of the E1 glycoprotein is a main determinant of alphavirus infectious particle production in vitro.
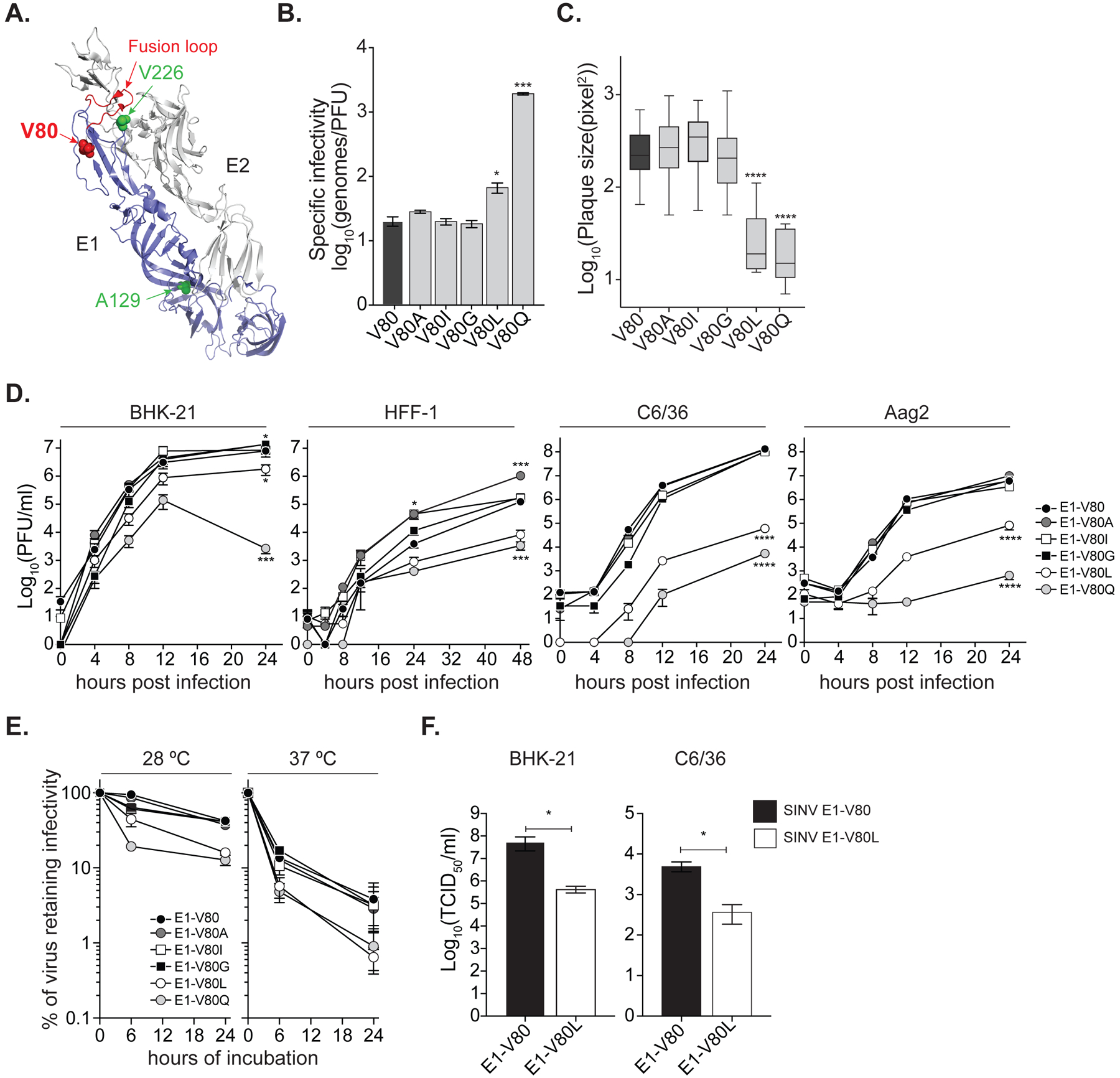
(A) Ribbon representation of the CHIKV E1/E2 heterodimer (PDBID: 3N42) highlighting the E1 monomer (blue) and the E2 monomer (gray). Residue at position 80 is shown as a red sphere. Positions E1–226 and E1–129 are highlighted in green. The E1-fusion loop is indicated in red. (B) Specific infectivity expressed as number of genomes over infectious particles (genomes/PFU). Data represent three independent repetitions, * p < 0.05, *** p < 0.001; Kruskal-Wallis test. (C) Plaque size in Vero cells, **** p < 0.0001, N > 40; Kruskal-Wallis test. (D) Multi-step viral replication curves performed in mammal BHK-21 and HFF-1, and mosquito C6/36 and Aag2 cell lines. Data represent two independent experiments, each with internal triplicates, * p<0.05, *** p<0.001 and **** p<0.0001. P values were determined by two-way ANOVA with Bonferroni post-hoc test. (E) Virion thermostability in cell-free environment. Data represent two independent experiments, 6 h at 28 °C: E1-V80Q, E1-V80L, E1-V80I and E1-V80G p< 0.0001; 24 h at 28 °C: E1-V80Q p<0.0001 and E1-V80L p<0.001; 6 h at 37 °C: E1-V80Q and E1-V80L p = 0.001. P values were determined by two-way ANOVA with Bonferroni post-hoc test. (F) Production of SINV infectious particles determined by TCID50. Data represent two independent experiments, each with internal duplicates, * p<0.05. P values were determined by Mann-Whitney U test. (B-F) The data represent the mean and the standard error of the mean (SEM). See also Figure S4 and S5.
In mammalian cells, we found five variants bearing isoleucine (I), alanine (A), leucine (L), glycine (G), and glutamine (Q) substitutions (E1-V80I, A, L, G and Q) to be genetically stable at position 80 with no additional second-site mutations across the subgenomic region (Table 1 and Figure S1B). However, in one out of five independent repetitions, we found that E1-V80G acquired two second-site mutations, one in the E1 glycoprotein (E1-V344L) and a second in the capsid protein (CP-V56L), and E1-V80Q acquired a second-site mutation in the E1 glycoprotein (E1-V226A) (Table 1 and Figure S1B). Of the unstable variants, not all reverted to wild-type, but in some cases changed to a stable variant (E1-V80I/A/L or wild-type) or acquired second-site mutations including E1-A129V which we have previously identified in vivo (Stapleford et al., 2014) (Table 1 and Figure S2 and S3). The aliphatic nature of the residues tolerated at position E1-V80X, regardless the bulkiness of the lateral group, suggests that the hydrophobicity of position E1-V80 is crucial for CHIKV.
Table 1.
Sequence stability of the E1-V80X variants in mammalian and mosquito cell lines. See alsoFigure S1 to S3.
| Mammalian cells (BHK-21) | Mosquito cells (C6/36) | |||
|---|---|---|---|---|
| Amino acid Substitution | Position 80 Stability | Second site mutations | Position 80 Stability | Second site mutations |
| E1-V80 | Stable | Stable | ||
| E1-V80G | Stable | E1-V344L and CP-V56L | Stable | E1-T207T and CP-G199G |
| E1-V80A | Stable | Stable | ||
| E1-V80L | Stable | Stable | E1-V226A or E1-A194V | |
| E1-V80I | Stable | Stable | E1-T65A or E1-A66A:E1-D70N | |
| E1-V80Q | Stable | E1-V226A | Stable or reverted | E1-G139E or E1-V226A |
| E1-V80N | Stable, reverted or mutated to D, S or T | E1-T82P or 1-A129M | Stable or reverted | E1-T82I or E1-A129V |
| E1-V80C | Stable or reverted | E1-F95F and/or E1-A129V | Reverted | E1-A129V |
| E1-V80M | Stable, reverted or mutated to I or A | Reverted or mutated to I or A | ||
| E1-V80F | Reverted or mutated to A | E1-A129V or E1-A129M | Reverted | E1-T65T and/or E1-A129V |
| E1-V80Y | Reverted or mutated to I or G | Reverted or mutated to S | ||
| E1-V80W | Reverted or mutated to I | E1-A129M | Reverted or mutated to S | E1-A129E and CP-Q166L:E2-T2T:E2-V32I |
| E1-V80P | Stable or mutated to S | E1-T65S or E1-A239A:E1-A359N | Mutated to S | |
| E1-V80S | Stable or mutated to A | CP-P57L | Stable | E1-T41A |
| E1-V80T | Stable or mutated to N or I | E1-T82I | Stable or mutated to A | E1-V226A or CP-P137L:E2-V32I |
| E1-V80H | Reverted or mutated to T or S | E1-A129M | Not rescued | |
| E1-V80R | Reverted or mutated to L or D | Reverted | E1-A129V/M/I | |
| E1-V80K | Reverted | E1-A129V | Reverted | E1-A129V |
| E1-V80D | Stable or mutated to S | E1-H107Y | Stable or reverted | E1-V226A or E1-A129V |
| E1-V80E | Stable or mutated to A | E2-E24K | Stable or reverted | E1-V226A or E1-A342D and CP-Q166L: E2-T2T:E2-V32I |
In mosquito cells, we found E1-V80G and V80A to be genetically stable (Table 1 and Figure S1 to S3), as other variants underwent reversion, mutation to a stable variant, or accumulation of second-site mutations throughout the subgenomic region. Interestingly, E1-V80L, Q, D, E and T remained genetically stable yet acquired the mutation E1-V226A, providing further evidence for a genetic link between these two positions. In addition, we found with the E1-V80I variant, two replicates with second-site mutations: a double mutation E1-A66A:E1-D70N, or a E1-T65A substitution (Table 1 and Figure S1C). Interestingly, position 66, 70 and 226 of E1 glycoprotein have been previously implicated with CHIKV fusion and cholesterol dependence suggesting a potential functional link between position 80 and these residues in these processes (Lu et al., 1999; Tsetsarkin et al., 2011; Vashishtha et al., 1998). Importantly, given that we observed more genetic variability (Figure S1 to S3) in mosquito cells, we hypothesized that this could be because of a faster viral replication rate compared to mammalian cells, and therefore an increased probability to acquire mutations. However, we found that the replication rate of CHIKV in C6/36 cells is slower than in BHK-21 cells (Figure S4A) (Jose et al., 2017; Roberts et al., 2017). This coupled with the narrower mutational spectrum tolerated at position E1-V80 in insect compared to mammalian cells, suggests that this position is constrained in a host-specific manner. Therefore, we chose to focus on the five genetically stable E1-V80 variants generated in mammalian cells (E1-V80I, A, G, L, and Q) for further in vitro and in vivo studies.
Chikungunya virus E1-V80 affects the production of infectious virus in vitro.
Upon generation the E1-V80 variants in BHK-21 cells, we observed that two variants, E1-V80L and E1-V80Q, led to an increase in viral specific infectivity (genomes/PFU) (Figure 1B) and small plaque sizes on Vero cells compared to wild-type (Figure 1C), suggesting that these variants may be impairing the production, stability, or infectivity of CHIKV particles. To first rule out the effect of E1-V80X variants on viral RNA synthesis, we used a reporter CHIKV expressing luciferase under the control of the subgenomic promoter, such that luciferase will only be produced during active CHIKV genome replication (Figure S4A, top panel). We transfected BHK-21 cells with each viral RNA and measured luciferase expression at 4, 6 and 8 h post-transfection (hpt) (Figure S4B, left panel). We confirmed that the luciferase levels measured in the early time points (4 and 6 hpt) are strictly associated to viral replication, as infectious viral progeny cannot be detected until 8 hpt (Figure S4B, right panel). We found no differences in the luciferase levels between the variants (Figure S4B, left panel), indicating that mutations E1-V80L and E1-V80Q are not involved in the regulation of viral RNA replication in BHK-21 cells.
We have shown that the cell line used for in vitro studies can have a significant effect on the growth of CHIKV mutants (Stapleford et al., 2014). Therefore, we performed multi-step replication curves on multiple mammalian (BHK-21 and human skin fibroblasts (HFF-1)) and insect (C6/36 and Ae. aegypti (Aag2)) cell lines susceptible to CHIKV infections (Roberts et al., 2017; Zhang et al., 2018). We found that the E1-V80L and E1-V80Q significantly attenuated infectious virus production in both mammalian and mosquito cell lines (Figure 1D). In contrast, E1-V80I and E1-V80A show increased infectious virus progeny in HFF-1 cells, which is in line with previous studies of E1-V80I in mouse embryonic fibroblasts and in vivo (Stapleford et al., 2014). Interestingly, we observed that the infectious titers for E1-V80Q decrease 1.5 logs from 12 to 24 h in BHK-21 cells (Figure 1D), suggesting this variant may cause destabilization of the virion. To address virion thermostability in E1-V80Q and E1-V80L attenuation, we incubated each variant in a cell-free suspension at 28 °C (mosquito) or 37 °C (mammalian) temperatures. We found both E1-V80Q and E1-V80L showed reduced stability at both 28 °C and 37 °C (Figure 1E), suggesting that these mutations are affecting particle thermostability and therefore contributing to the reduction in infectious CHIKV particles. All together, these results show that small changes in hydrophobicity at position E1–80 can affect CHIKV virion stability and infectious virus production in vitro.
Finally, the valine residue at position 80 of the E1 glycoprotein is fully conserved within the alphavirus genus (Figure S5). Thus, we addressed the effect of E1-V80L substitution on the in vitro replication cycle of the distantly related alphavirus Sindbis virus (SINV). We infected BHK-21 and C6/36 at a MOI of 0.01, and quantified infectious particles at 24 hpi. We observed a reduction of infectious virus progeny of the SINV E1-V80L variant compared to the wild-type counterpart in both mammal and mosquito cell lines (Figure 1F). These data show that E1 position 80 impacts infectious virus production not only in CHIKV but in other alphaviruses as well.
CHIKV E1-V80L and E1-V80Q reduce viral infection and dissemination in Aedes aegypti mosquitoes.
Given their attenuation in mosquito cell lines, we hypothesized that these variants may affect CHIKV infection and dissemination in mosquitoes. We first infected Ae. aegypti mosquitoes with a high-dose artificial blood meal (106 PFU/ml) of wild-type E1-V80, E1-V80A as an unbiased control, and E1-V80L and allowed infection to progress for 7 days. We found that although the E1-V80L variant can infect Ae. aegypti mosquitoes at similar levels as wild-type (Figure 2A, upper panel), there was a significant reduction in the amount of infectious virus in the mosquito bodies (Figure 2A, lower panel). In addition, viruses bearing the E1-V80L substitution showed impaired dissemination to legs and wings, with infectious particles detected in only 19 of the 44 infected mosquitoes (Figure 2A, upper panel). Interestingly, we found that although E1-V80A had no impact on infection or dissemination in mosquitoes (Figure 2A, upper panel), there was a significant reduction in infectious virus in the legs and wings (Figure 2A, lower panel).
Figure 2. Chikungunya virus E1-V80L and E1-V80Q variants are attenuated and showed impaired dissemination in Ae. aegypti mosquitoes.
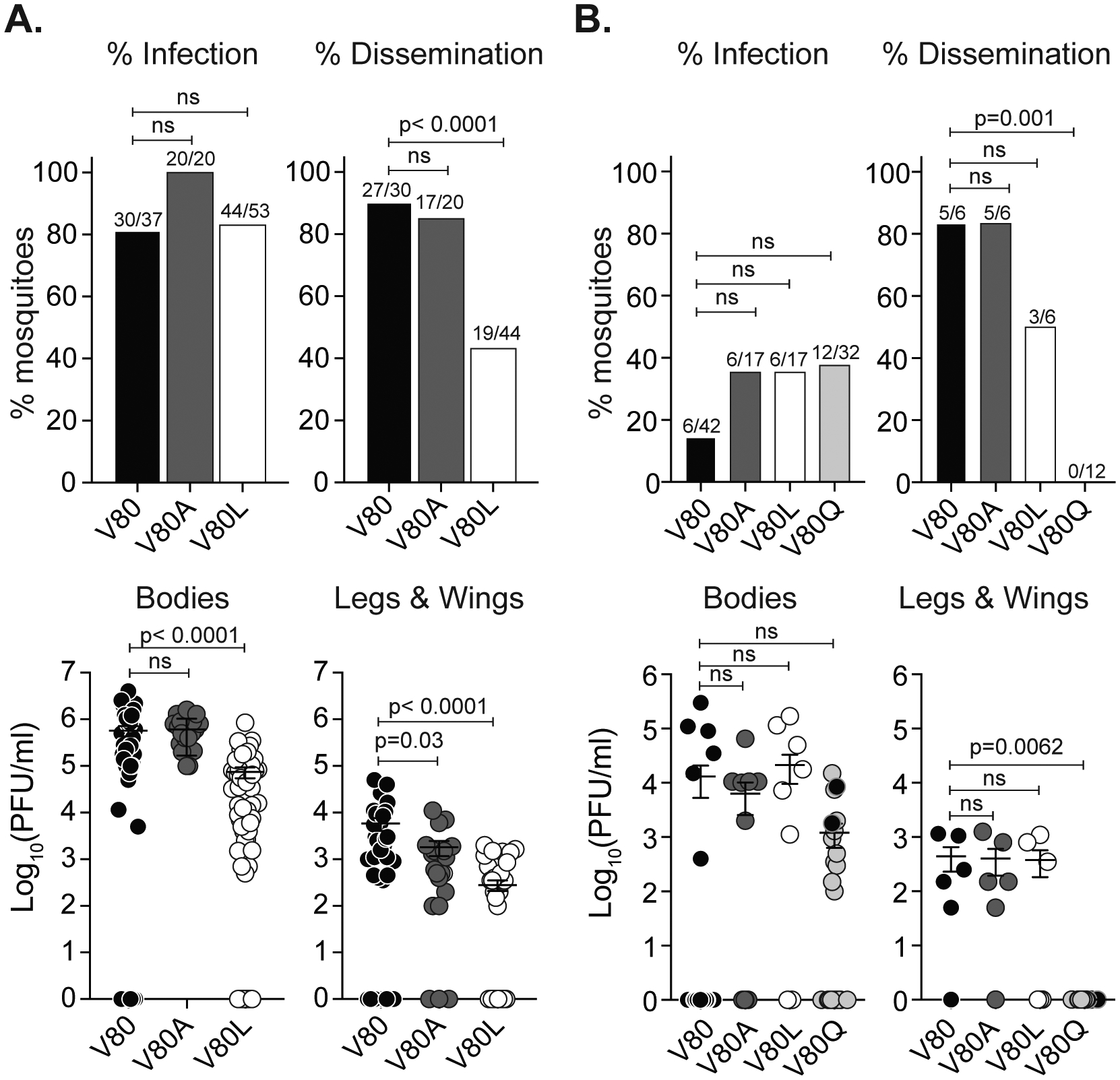
7 day old Ae. aegypti mosquitoes were infected with 106 PFU/ml (A) or 104 PFU/ml (B) of CHIKV E1-V80, V80A, V80L (A and B) and E1-V80Q (B) via artificial blood-meals. Viral titers of bodies, and legs and wings were determined after 7 (A) or 14 (B) days post infection by plaque assay on Vero cells. Error bars represent mean and SEM, (N=17–53 mosquitoes). Data represent two independent experiments. P values were determined by Kruskal-Wallis test. Percentage of infection or dissemination were determined as the number of mosquitoes with positive bodies or legs and wings over the total of engorged mosquitoes or positive bodies, respectively. P values were determined by Fisher’s exact test with Bonferroni correction. E1-V80Q with second-site mutation and reverted plaque size phenotype are represented as black circles. See also Figure S6.
To address the effect of the variant E1-V80Q, for which we were unable to obtain high titer stocks, we infected mosquitoes with a low-dose of each virus (104 PFU/ml) and allowed the infection to progress for 14 days. We found that at low doses, the variants had a trend towards higher infection rates compare to wild-type (Figure 2B, upper panel) and E1-V80Q yielded lower, although not statistically significant, viral loads in the bodies of mosquitoes (Figure 2B, lower panel). Interestingly, based on the distinctive plaque size exhibited by E1-V80Q (Figure 1C) we observed that viruses isolated from two mosquitoes reverted in plaque size phenotype (Figure S6A). We Sanger sequenced the full viral subgenomic region from these two mosquitoes and found that in addition to the E1-V80Q mutation, each individual virus acquired an additional second-site mutation in the E1 glycoprotein: E1-N20Y or E1-M88L (Figure S6A). Both residues E1–20 and E1–88 are located in regions implicated in modulation of viral fusion (Voss et al., 2010; Zheng et al., 2011), suggesting that the attenuation observed for these mutants could be associated to a defect in fusion (Figure S6B and S6C). Importantly, of all the mosquitoes infected with E1-V80Q, including the ones with reversion in their plaque phenotype, none were able to disseminate into the legs and wings and this effect was also observed for E1-V80L, although less pronounced (Figure 2B). Taken together, these results indicate that position E1–80 is important for CHIKV infection and dissemination in Ae. aegypti mosquitoes and may affect viral fusion.
CHIKV E1-V80L and E1-V80Q are attenuated in mice and impair dissemination to secondary organs.
In addition to reduced infection and dissemination in mosquitoes, we observed that E1-V80L and E1-V80Q were impaired in mammalian cells and thus we asked whether these mutations attenuate CHIKV in mice. We infected adult C57BL/6J mice with 1000 PFU of E1-V80, V80A, V80L, and V80Q in the left footpad and addressed infectious viral loads and dissemination two days post infection (Figure 3A), which has been described as the peak of viremia of CHIKV for this model (Gardner et al., 2010). Interestingly, we found reduced levels of E1-V80L in the ipsilateral quadricep and calf muscles (Figure 3B), primary sites of infection, as well as in the spleen and liver (Figure 3C), secondary sites of infection. Strikingly, we were unable to recover infectious E1-V80L particles from the heart and brain of infected mice indicating that this virus has impaired dissemination to specific organs (Figure 3C, lower panel). The E1-V80Q variant had reduced titers in the footpad (Figure 3B) and we did not detect infectious virus in the serum or other secondary organs, suggesting a restriction of this virus to the site of injection (Figure 3C).
Figure 3. Chikungunya virus E1-V80L and E1-V80Q variants are attenuated and showed impaired dissemination in mice.
(A) Schematic representation of the workflow. C57BL/6J mice were inoculated with E1-V80 (N=10), E1-V80A (N=8), E1-V80L (N=11) and E1-V80Q (N=6). Viral infectious particles were determined in the primary targeted organs (B) or secondary targeted organs (C). Data represents three independent experiments. P values were determined by Kruskal-Wallis test. (D) Kaplan-Meier survival curves of C57BL/6J Ifnar −/− mice inoculated with E1-V80 (black dots), E1-V80L (open dots), E1-V80Q (gray dots) or DMEM control (dashed dots) and monitored during 5 days (E1-V80L: p= 0.0009, E1-V80Q: p<0.0001, N=4; log-rank Mantel-Cox test). (E) Number of viral infectious particles was determined at the endpoint of the experiment. All results are expressed as PFU/ml of tissue homogenate. The plaque assay detection limit is indicated as a dashed line. Error bars indicate SEM. See also Figure S7.
One possible explanation for the restriction in dissemination, especially for the E1-V80Q variant is that the virus was not able to replicate in the injection site and therefore was not able to establish a systemic infection. To verify that E1-V80Q and E1-V80L viruses were infectious albeit attenuated, we took advantage of a susceptible mouse model lacking the interferon α/β receptor (Ifnar−/−) (Gardner et al., 2012). We infected Ifnar−/− mice with 1000 PFU of E1-V80, V80L, V80Q and a carrier control via footpad injection and monitored mice daily for signs of disease (Figure 3D). All mice succumbed to the infection compared to the uninfected control (Figure 3D); however, we observed an attenuation of E1-V80L and E1-V80Q through the delay in symptoms and death compared to wild-type (Figure 3D and Figure S7). To examine the dissemination to the different organs, we analyzed the infectious viral loads in the primary and secondary target organs at the humane endpoints (Figure 3E). Notably, both E1-V80L and E1-V80Q were able to disseminate to all the tested organs, including heart and brain (Figure 3E). However, it is important to note that mice infected with the different variants succumbed to the infection at different days, therefore viral titers between variants cannot be compared. Taken together, these data show that E1-V80L and E1-V80Q variants are indeed infectious in vivo and that residue E1–80 plays a critical role in viral dissemination in both insects and mice.
CHIKV residue E1-V80 impacts virion fusion dynamics and pH sensitivity.
Given the proximity of residue E1–80 to the E1 glycoprotein fusion loop (Figure 1A) as well as the identification of second-site mutations (E1-V226A, E1-N20Y, and E1-M88L) in regions implicated viral fusion (Voss et al., 2010; Zheng et al., 2011), we hypothesized that the viral attenuation and the impaired dissemination observed for the E1-V80 variants are associated with defects in viral entry.
To test this hypothesis, we generated ZsGreen reporter viruses of E1-V80, V80L, V80Q and V80A to visualize infected cells and performed cellular binding and fusion assays (Figure S4C). E1-V80Q variant was excluded from this study due to the inability to reach concentrated titers. First, we addressed virion binding by incubating BHK-21 cells with gradient purified variants and measured the amount of membrane-bound virus after 5 and 60 min by RT-qPCR. We found that after 5 min E1-V80L showed reduced binding compared to E1-V80 which was restored after 60 min, suggesting that this variant may have a defect early in cell binding (Figure 4A). Next, we performed fusion-from-without experiments on BHK-21 cells at a MOI of 10 to overcome potential binding defects. We found that E1-V80 and E1-V80A showed a threshold pH for fusion of ~5.9, similar to what has been previously described for CHIKV (Hoornweg et al., 2016; Sanchez-San Martin et al., 2013; Stapleford et al., 2014). Remarkably, the fusion of E1-V80L with host cells was significantly reduced at multiple pH treatments compared to controls (Figure 4B), shifting the threshold pH for fusion to ~5.5. As an alternative approach to look at pH dependence, we took advantage of the lysosomotropic agents ammonium chloride and bafilomycin A1, which deacidify the endosomal compartments and block viral fusion (Kielian et al., 1984; Sourisseau et al., 2007). We found that E1-V80L was more sensitive to each of these compounds compared to wild-type confirming that this residue can sense and respond to pH changes (Figure 4C). Interestingly, we found that E1-V80A was sensitive to ammonium chloride treatment yet this may be through a different mechanism than the one underlying the fusion defect of E1-V80L or through off target effects of the compound. Finally, to address differences in the fusion kinetics of the E1-V80 variants, we performed an ammonium chloride by-pass assay, in which viruses that are able to fuse more efficiently display a reduced sensitivity to ammonium chloride blockade at earlier time points than those that take longer to enter and fuse. We found that E1-V80L and E1-V80A displayed a reduced infectivity at all the time points tested compared to wild-type (Figure 4D), indicating significant impairments of E1-V80L and E1-V80A in rate of viral fusion and infectivity. All together these data indicate that position E1–80 is a determinant of viral fusion and that altering this residue can regulate viral fusion dynamics.
Figure 4. CHIKV residue E1–80 impacts virion fusion dynamics, pH sensitivity and cholesterol dependence.
(A) CHIKV binding assay. BHK-21 cells were incubated with an equivalent of 100 particles per cell of purified ZsGreen E1-V80, A, and L variants (based on viral genomes) at 4 °C and in the presence of 20 mM ammonium chloride for 5 or 60 min. Then, cells were washed extensively with PBS, and RNA genomes were quantified by RT-qPCR. Data represent three independent experiments, each with internal duplicates, *p<0.05. (B) CHIKV fusion-from-without. Purified ZsGreen viruses were bound to BHK-21 cells at a MOI of 10 for 1 h, at 4 °C. After binding, cells were treated at the indicated pHs for 2 min at 37 °C. pH was neutralized, and the amount of ZsGreen expressing cells was determined 16 hpi. Data represent two independent experiments, each with internal triplicates, * p<0.05, ** p<0.01, *** p < 0.001. (C) Effect of lysosomotropic drugs on CHIKV mutant variants. BHK-21 cells were pre-treated for 3 h with increasing concentrations of ammonium chloride (left panel) or Bafilomycin A1 (right panel), and then infected with purified ZsGreen viruses at a MOI of 1. Data represent two independent experiments, each with internal triplicates, * p<0.05, ** p<0.01, *** p < 0.001. (D) Ammonium chloride by-pass assay. BHK-21 cells were pre-incubated for 1 h, at 4 °C. Purified ZsGreen E1-V80, V80A, and V80L variants (MOI= 0.1) were added to the cells and incubated for 1 h, at 4 °C. Then, cells were incubated at 37 °C, and 20 mM ammonium chloride was added at the indicated time points. Data represent two independent experiments, each with internal duplicates, ** p<0.01, *** p < 0.001, **** p < 0.0001. (E) Cholesterol depletion. BHK-21 cells were treated with increased concentrations of Methyl-β-cyclodextrin (MβCD) for 1 h at 37 °C. Infections with ZsGreen-viruses were performed at a MOI of 1 for 1 h, and subsequently complete media with 20 mM ammonium chloride was added to impair viral spreading. Data represent three independent experiments, each with internal triplicates, ***p < 0.001. (F) Cholesterol repletion. Cholesterol depleted BHK-21 cells were treated with media containing 200 μg/ml soluble cholesterol or media alone. Infections with ZsGreen viruses were performed at a MOI of 1 for 1 h, and subsequently complete media with 20 mM ammonium chloride was added. Data represent two independent experiments, each with internal triplicates, ** p<0.01, *** p<0.001, **** p < 0.0001. All data represents the mean and SEM. P values were determined by one or two-way ANOVA with Bonferroni post-hoc test. See also Figure S4.
Residue E1–80 is involved in CHIKV E1 cholesterol-dependent entry.
It has been described for alphaviruses that the fusion loop insertion and fusion is promoted by the presence of cholesterol in the target membrane (Guardado-Calvo et al., 2017; Kielian et al., 2000). Because we found that residue E1–80 regulates fusion dynamics, we asked whether the E1-V80 variants may be affected by the levels of cholesterol in the plasma membrane. To address this, we treated BHK-21 cells with methyl beta-cyclodextrin (MβCD) (Hoornweg et al., 2016; Mahammad and Parmryd, 2015; Zidovetzki and Levitan, 2007), and measured the effect of cholesterol only during entry. We found that while cholesterol depletion had no effect on the E1-V80A infectivity compared to wild-type, E1-V80L infectivity was significantly impaired by this treatment (Figure 4E), indicating that the E1-V80L variant has an increased sensitivity to cholesterol depletion. To confirm that the reduced infectivity observed for E1-V80L in the presence of MβCD was due to the depletion of cholesterol from the plasma membrane, we replenished BHK-21 depleted cells with water-soluble cholesterol. We found that upon cholesterol replenishment, the infectivity of wild-type and E1-V80A were completely restored to untreated levels. Interesting, we found that cholesterol replenishment only partially restored the infectivity of E1-V80L (Figure 4F), suggesting that this variant has an increased dependence on cellular cholesterol compared to wild-type. Taken together, these studies show that position E1-V80 is involved in CHIKV cholesterol-dependent entry in mammalian cells.
E1 residues 80 and 226 function together in CHIKV infectivity and dissemination.
Position E1–226 has been shown to influence the cholesterol requirement for CHIKV, SFV and SINV viral entry (Lu et al., 1999; Tsetsarkin et al., 2011; Vashishtha et al., 1998). Given the selection of the E1-V226A second-site variant during the E1-V80L adaptation in C6/36 cells (Table 1, and Figure S1 to S3), and that E1-V226A substitution in CHIKV IOL results in decreased cholesterol dependence for fusion (Hoornweg et al., 2016), we hypothesized that the acquisition of the E1-V226A mutation will restore the E1-V80L cholesterol dependence. To test this hypothesis, we introduced the E1-V226A mutation in our E1-V80L infectious clone, generated the double mutant virus (E1-V80L:V226A) and confirmed stability by sequencing the subgenomic region after three passages on BHK-21 cells. We found that the E1-V80L:V226A variant partially restored the E1-V80L plaque size phenotype (Figure 5A), suggesting that this mutation rescues the impaired viral spreading observed for E1-V80L. We next addressed the capacity of E1-V80L:V226A to restore E1-V80L reduced infectivity in cholesterol-depleted cells. Indeed, we found that the E1-V80L:V226A variant reduced the cholesterol dependence observed for E1-V80L (Figure 5B), confirming that residues E1–80 and E1–226 may contribute to similar functions. Finally, we asked whether the E1-V226A substitution is able to rescue the E1-V80L in vivo attenuation and dissemination phenotype observed in mice. We infected C57BL/6J mice via footpad injection with ~1000 PFU of E1-V80, V80L or V80L:V226A variants. We found that E1-V80L:V226A double mutant rescued the attenuated phenotype observed for E1-V80L in primary organs (Figure 5C). In addition, in secondary organs the E1-V80L:V226A variant rescued the dissemination phenotype observed in the heart for E1-V80L (Figure 5D), suggesting a potential functional link between these two residues in at least dissemination to this organ. Taken together, these results demonstrate that the highly conserved position 80 in the E1 glycoprotein is a key determinant in alphavirus infectivity, fusion, and dissemination functioning in concert with E1–226 to regulate CHIKV cholesterol dependence.
Figure 5. E1 residues 80 and 226 function together in CHIKV infectivity and dissemination.
(A) Plaque size in Vero cells was quantified using ImageJ, **** p < 0.0001, ** p< 0.01, N > 40; Kruskal-Wallis test. (B) BHK-21 cells were treated with 5 mM MβCD for 1 h at 37 °C. Infections were performed at a MOI of 1 for 1 h, and complete media with 20 mM ammonium chloride was added. Data represent two independent experiments, each with internal triplicates, *** p <0.001, ** p <0.01; One-way ANOVA with Bonferroni post-hoc test. (C-D) Mice were inoculated with 1000 PFU of E1-V80, V80L or V80L:V226A variants. Viral infectious particles were determined in the primary targeted organs (C) or secondary targeted organs (D). Results are expressed as PFU/ml of tissue homogenate. V80 (N=5), V80L (N=8), V80L:V226A (N=6). Data represent two independent experiments. Error bars indicate SEM. P values were determined by Kruskal-Wallis test.
DISCUSSION
Arboviral diseases are undergoing a global expansion (Vignuzzi and Higgs, 2017; Wilder-Smith et al., 2017), pointing to the urgent need to study fundamental aspects of arboviral biology to identify new therapeutic targets. We have previously identified position 80 in the E1 glycoprotein as a hotspot of viral transmission and pathogenesis (Stapleford et al., 2014). In the present work, we investigated the specific role of this conserved position in CHIKV biology and found that residue E1–80 is involved in CHIKV infectivity and dissemination through the modulation of viral fusion and cholesterol dependence.
Position 80 in the E1 glycoprotein is located within β-strand c at the tip of domain II, adjacent to the fusion loop, and near a structurally conserved glycerophospholipid binding pocket (GPL) (Figure 6A) (Guardado-Calvo et al., 2017; Voss et al., 2010). Our studies show that by changing this single amino acid we can regulate the CHIKV viral replication cycle, providing powerful tools to study arbovirus transmission and pathogenesis. In particular, the increased in vitro replication observed for the E1-V80A and V80I in HFF-1 cells, which have a competent innate immunity signaling pathway (Chen et al., 2017) is intriguing and suggests that this position can potentially be involved in other steps of the CHIKV replication cycle. Notably, the impact of position 80 in viral attenuation, is not exclusive to CHIKV, as we observed similar levels of attenuation in SINV, another member of the alphavirus genus. Such observations point to a generalized function of the E1–80 residue as an arboviral modulator of pathogenesis and dissemination. Further research aimed towards defining analog residues in other arboviral type-II fusion proteins would help to expand this concept and provide clues to develop broad-spectrum antivirals.
Figure 6. CHIKV E1-V80 mutational network.
(A) Ribbon representation of the CHIKV E1 glycoprotein (PDBID: 3N42). Second-site mutations found only in mammalian, mosquitoes or both mammalian and mosquito experiments are represented as orange, purple and green spheres, respectively. Detail of the tip of domain II (residues 53–108 and 218–235) is shown in the inset. Residue E1–80 is highlighted in red. Residues E1–226, E1–66 and E1–70, previously described as cholesterol-dependent mutants are underlined. Fusion loop, ij loop, bc-loop, β-strand c, β-strand d, and the GPL binding pocket are indicated. (B) Representation of a single trimeric spike showing position E2–E24 (black spheres) and E2-V32 (green spheres) are indicated (PDBID: 3J2W). E1 (blue) and E2 (orange) glycoproteins from different heterodimers are depicted in different color intensities for clarity. (C) Schematic representation of a single trimeric spike with the transmembrane domains and the capsid protein (PDBID: 3J2W). In the inset is shown the globular domains of the capsid protein (residues 113 to 261). The red dashed-line indicates the intrinsically disordered domain from the capsid protein absent in the crystal structure (residues 1 to 113). For clarity the mutations are depicted in only one monomer.
Our mutagenesis analysis showed host-specific constraints acting on position E1–80 with second-site mutations arising within the tip of domain II (E1-T65, A66, D70, T82, M88, H107 and V226) or proximal to the linker between domain I and III (E1-N20, G139, V344, A342 and A359) (Figure 6A), suggesting that each group of residues could function along with E1-V80. Notably, both the tip of domain II as well as the linker region, are associated to membrane fusion (Kielian and Rey, 2006; Zheng et al., 2011), albeit through different mechanisms. While the tip of domain II is implicated with insertion of the fusion loop into the target membrane (Gibbons et al., 2003; Gibbons and Kielian, 2002) and interaction with lipids (Guardado-Calvo et al., 2017), the linker region has been associated to the stabilization of the E1 post-fusion hairpin during the low-pH-dependent refolding (Zheng et al., 2011). In addition, we observed the emergence of synonymous mutations such as CP-G199G, E2-T2T, E1-A66A, E1-F95F, E1-T207T or E1-A239A. This raises an alternative explanation for where mutations in response to E1-V80X variants could arise to compensate RNA elements within this region of the viral genome. Further work should be done in this regard, but we can hypothesize that both protein and RNA elements around E1-V80 shapes CHIKV evolution.
We found intriguing that although the mutations E1-V80I and E1-A129V arose together in the saliva of Aedes mosquitoes (Stapleford et al., 2014), in our in vitro evolution experiments, position E1–129 remains stable in the five replicates of E1-V80I in both mammalian or mosquito cells. This could be because of positive effects on fitness conferred by the E1-V80I mutation alone and suggests that the emergence of E1-V80I and E1-A129V in mosquitoes could be associated to a different process or evolutionary trajectory. Nonetheless, although we did not observe linkage between V80I and A129V, we did find position E1–129 linked to the reversion of position E1–80 in 57% of the E1-V80X unstable variants (Table 1 and Figures S1 to S3). In addition, Sanger sequencing of E1-V80F passaging shows the presence of both the original and mutated codons at positions 80 and 129 (not shown). This observation highlights a direct link between E1-V80 and E1-A129 and provides further evidence of an epistatic interaction between these two positions.
Finally, we found nonsynonymous mutations in the E2 glycoprotein (E2-E24K, and E2-V32I) and in the capsid protein (CP-V56L, CP-P57, CP-P137L and CP-Q166L) (Figure 6B and 6C). Despite that the overall alphavirus conservation of these positions is low, the proline residue at position 137 in the capsid protein is fully conserved (Sharma et al., 2018), and it will be an interesting target to study potential compensatory mutations between these two proteins. In addition, the glutamic acid at position 24 has been described as part of E2-E2 Van der Walls contacts (Voss et al., 2010), suggesting that the non-conservative E2-E24K mutation might be affecting the stability of this interaction.
Nonetheless, our data demonstrates that E1–80 is an essential determinant of viral fusion, and altering this residue regulates viral fusion dynamics. Thus, position E1–80 and the compensatory mutations that emerged during the in vitro passages can potentially be involved in a common mechanism regulating viral fusion. We speculate that compensatory mutations within the fusion pocket are associated with the fusion loop insertion and/or lipid binding, where the second-site mutations in the linker region could be associated to a different mechanism, possibly favoring fusion by stabilizing the pre-fusion to post-fusion conformational transition. In addition, compensatory mutations in other CHIKV structural proteins may function with E1-V80 to facilitate CHIKV entry and fusion.
It has been widely described that the fusion loop insertion and fusion are promoted by the presence of cholesterol in the target membrane (Kielian et al., 2000). In this study, we demonstrated that position 80 of E1 glycoprotein regulates CHIKV cholesterol dependence in mammalian cell lines. In addition, passaging of E1-V80 variants on C6/36 drives the emergence of residues E1–226, E1–70 and E1–66 as second-site mutations (Figure 6A and Figure S1), which were previously associated to cholesterol dependence of CHIKV infectivity (Hoornweg et al., 2016; Tsetsarkin et al., 2011). In particular, selection experiments on the attenuated E1-V80L variant in C6/36 cells favored the emergence of the E1-V226A variant. One potential explanation is that C6/36 cells, because of their reduced cholesterol levels (Hafer et al., 2009), impose a selective pressure on E1-V80L that results in the acquisition of the mutation E1-V226A, a mutation described to reduce the cholesterol requirements for CHIKV entry (Hoornweg et al., 2016). Indeed, our genetic studies support this hypothesis, given that the incorporation of E1-V226A into the E1-V80L infectious clone compensates for the viral spreading, cholesterol dependence and infectivity defects observed for E1-V80L (Figure 5). In this study, we have found a region implicated in modulation of cholesterol dependence at the tip of domain II, which involves position 80. We speculate, that the ij loop (containing residue 226), the β-strand c (containing residue 80) and the bc loop (containing residues 66 and 70) (Figure 6A) work concerted to accommodate cholesterol within the tip of domain II during the fusion process. A recent study identified a conserved GPL binding pocket involving residues from the bc loop, fusion loop and ij loop in E1 glycoprotein essential for fusion loop insertion (Guardado-Calvo et al., 2017). In addition, the authors propose that additional small head group lipids (such as cholesterol) are required to create space between the bulky head group to allow fusion loop insertion. Structural studies will be needed to address the role of position 80 in cholesterol dependence which could be implicated with direct E1–80 mediated interactions, or through modulation of the loops dynamic during the conformational transition and/or insertion in the target membrane.
Finally, we showed that in adult C57BL/6, both the E1-V80L and E1-V80Q variants had impaired dissemination to secondary organs compared to E1-V80. However, this impairment was restored in Ifnar−/− mice (Figure 3). It has been described that decreasing de novo cholesterol and fatty acid biosynthesis is a physiological response to viral infection through the IFNAR signaling pathway (York et al., 2015). This suggests, that the impaired dissemination observed in wild-type mice could be associated to reduction of available cholesterol in the membrane, which resulted in impaired E1-V80L and E1-V80Q dissemination to target organs. Although the increased infectivity and dissemination observed in Ifnar−/− mice could be associated to many other regulatory processes, such as host specific innate and adaptive antiviral immunity (Nair et al., 2017), it will be interesting to study the effect of cholesterol levels on the E1-V80L and E1-V80Q variants in Ifnar−/− mouse models. Similar associations can be modulating the impaired dissemination observed in Ae. aegypti, suggesting that cholesterol levels in the midgut could be responsible for determining the degree of dissemination of these variants.
In summary, we show that the E1 residue 80 is a key determinant in alphavirus infectivity and dissemination through modulation of viral fusion and cholesterol dependence, and that it is functionally linked with other residues to orchestrate viral fusion, cholesterol dependence, infectivity and dissemination. In addition, these data provide evidence towards the potential use of the tip of domain II as a druggable target, gearing towards the development of broad spectrum of alphavirus antivirals and opening an avenue towards the use of mutational tolerance approaches as a way to identify stable substitutions guiding the development of attenuated vaccines.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth Stapleford (Kenneth.stapleford@nyumc.org)
EXPERIMENTAL MODEL DETAILS
Cell lines
Mammalian cell lines were maintained at 37 °C in 5% CO2. Baby hamster kidney (BHK-21, ATCC CCL-10) and Human foreskin fibroblast (HFF-1, ATCC SCRC-1041) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS; Atlanta biologicals), 1% non-essential amino acids (NEAA, Gibco) and 1% penicillin-streptomycin (P/S, Gibco). Vero cells (ATCC CCL-81) were grown in DMEM with 10% newborn calf serum (NBCS; Gibco) and 1% P/S. Mosquitoes cell lines were maintained at 28 °C in 5% CO2. Ae. aegypti cells (Aag-2), obtained from Dr. Paul Turner, Yale University (Morley et al., 2018), were grown in DMEM supplemented with 10% FBS and 10% NEAA. Ae. albopictus cells (C6/36, ATCC CRL-1660) were maintained in L-15 medium (Corning) supplemented with 10% FBS, 1% tryptose phosphate broth, 1% NEAA, and 1% P/S. All the cell lines were confirmed free of mycoplasma.
Viruses
Wild-type chikungunya virus (CHIKV) was rescued from the CHIKV strain 06–049 (AM258994) infectious clone as previously described (Coffey and Vignuzzi, 2011). A CHIKV La Reunion infectious clone expressing ZsGreen and Firefly luciferase was constructed by standard molecular biology techniques found below. The Sindbis virus (SINV) infectious clone was obtained from Dr. Benjamin tenOever (Mt. Sinai Icahn School of Medicine).
Mouse models
6–9 week old male and female C57BL/6J (Grenier et al., 2017; Martin et al., 2018) and 6 week old male and female Ifnar−/− mice (Martin et al., 2018) were bred and reared in house. Animal experiments were performed in accordance with all NYU School of Medicine Institutional Animal Care and Use Committee guidelines (IACUC). All mouse studies were performed using biosafety level 3 conditions.
Mosquitoes
Aedes aegypti mosquitoes (Poza Rica, Mexico, P20) were obtained from Dr. Gregory Ebel, Colorado State University (Ruckert et al., 2017). Mosquitoes were reared and maintained in Memmert humidified chambers at 28 °C with 70% humidity and 12 h diurnal light cycle. All mosquito studies were performed using biosafety level 3 conditions.
METHOD DETAILS
Site-directed mutagenesis and reporter viruses
To generate the CHIKV E1 glycoprotein position 80 variants, each amino acid substitution was first introduced into a cloning vector by site-directed mutagenesis using Phusion DNA polymerase (Thermo-Scientific). Primer sequences are provided in Table S1. Each cloning plasmid was Sanger sequenced (Genewiz) from common 5’ XhoI and 3’ NotI restriction enzyme sites to confirm each mutation and the absence of second-site variants. The XhoI/NotI fragment of each cloning vector was sub-cloned into the CHIKV full-length infectious clone by standard molecular biology techniques and subsequently Sanger sequenced to confirm each mutation.
A CHIKV La Reunion infectious clone expressing ZsGreen and Firefly luciferase was constructed by standard molecular biology techniques. First, an AvrII restriction enzyme site was inserted 5’ of the sub-genomic promoter by site-directed mutagenesis using the primers (Forward 5’ CACTAATCAGCTACACCTAGGATGGAGTTCATCCC 3’ and Reverse 5’ GGGATGAACTCCATCCTAGGTGTAGCTGATTAGTG 3’). The CHIKV sub-genomic promoter was then amplified by PCR (Forward 5’ CCTAGGCCATGGCCACCTTTGCAAG 3’ and Reverse 5’ ACTAGTTGTAGCTGATTAGTGTTTAG 3’) and sub-cloned into the AvrII site to generate a CHIKV infectious clone containing two sub-genomic promoters. Finally, the ZsGreen and Firefly luciferase cassettes were amplified by PCR (Primers in Table S1) from CHIKV infectious clones obtained from Andres Merits (University of Tartu) and sub-cloned into the AvrII restriction enzyme site. The complete cassette and sub-genomic regions were sequenced to ensure there were no second-site mutations. ZsGreen and Firefly reporter constructs containing individual E1 mutations were constructed by sub-cloning the XhoI/NotI fragment from the unmarked infectious clones into ZsGreen and Firefly luciferase backgrounds.
Generation of virus stocks
To generate infectious virus, 10 μg of each CHIKV plasmid was linearized overnight with NotI (Thermo-Scientific), phenol:chloroform extracted, ethanol precipitated, and resuspended in nuclease-free water at 1 μg/μl. In vitro transcribed viral RNAs were produced using the SP6 mMESSAGE mMACHINE kit (Ambion) following the manufacturer’s instructions. After RNA synthesis, samples were DNase treated and RNAs were purified by phenol:chloroform extraction, ethanol precipitation, and resuspension at a concentration of 1 μg/μl. All RNAs were stored at −80 °C until electroporation. SINV infectious clone were digested with XhoI and PmeI respectively and prepared as describe above. Infectious virus was produced by electroporating BHK-21 or C6/36 cells with in vitro transcribed viral RNAs. Briefly, cells were trypsinized, washed twice with ice-cold DPBS, and resuspended at 1×107 cells/ml in PBS. 390 μl of cells were mixed with 10 μg of in vitro transcribed RNA and added to a 2 mm electroporation cuvette (BioRad). BHK-21 cells were electroporated with 1 pulse at 1.2 kV, 25 μF, with infinite resistance and C6/36 cells were electroporated with 1 pulse at 250 V, 550 μF, with a resistance of 25Ω. Cells were allowed to recover for 10 min at room temperature, transferred into 6 ml of warm DMEM, and placed in a T25 flask at 37 °C for 72 h. Virus was harvested, clarified at 1,200 x g for 5 min (Passage 0, P0), and 3 ml of the P0 were used to infect T-175 flask to generate working stocks (Passage 1, P1). All working stocks were Sanger sequenced. Viral titers were determined by plaque assay. In brief, 10-fold dilutions of each virus in DMEM were added to a monolayer of Vero cells for 1 h at 37 °C. Following incubation, cells were overlaid with 0.8% agarose in DMEM containing 2% NBCS and incubated at 37 °C for 72 h. The cells were fixed with 4% formalin, the agarose plug removed, and plaques visualized by crystal violet staining. All ZsGreen viruses were purified over a 20% sucrose cushion by ultracentrifugation at 25,000 rpm for 2 h and resuspended in virus infection media (DMEM containing 0.2% bovine serum albumin (BSA), 1 mM HEPES pH 7.4 and 1% P/S).
Experimental passaging
Each E1-V80X variant was generated by electroporation of in vitro transcribed RNA into either BHK-21 or C6/36 cells as described above. Each variant was then passaged three times in the respective cell type from which was generated (Figure S1A). To initiate each passage, each E1-V80X variant was diluted 1:2 in DMEM with no serum (approximate effective MOI ~ 0.05 to 0.1 for stable and mutated variants; and MOI <0.01 for attenuated variants, such as E1-V80Q) and added to cells for 1 h at 28 °C (insect cells) or 37 °C (mammalian). After incubation, the cells were washed with PBS, complete media was added, and the cells were incubated for 24 h at their respective temperature. Output titers from each passage ranged between 105 to 106 PFU/ml for stable and mutated variants, and from 103 to 104 PFU/ml for attenuated variants. Viral genomes from the different E1-V80X variants were sequenced at the end of passage 3. Briefly, RNA extractions were performed using TRIzol™Reagent (Invitrogen™) and viral RNA was used directly for cDNA synthesis using the Maxima H minus-strand kit (Thermo-Scientific). CHIKV cDNA was then used to generate two PCR amplicons to cover the entire CHIKV sub-genomic region, the amplicons were purified using the Nucleospin PCR and Gel Extraction kit (Macherey-Nagel), and then Sanger sequenced (Genewiz) to address variant genetic stability and second-site mutations. Each individual E1-V80X virus was generated and passaged between three to five independent times, and in all the cases the tip of the domain II was fully sequenced. The full sub-genomic region was fully sequenced in at least two independent passages (Figure S1 and S2).
Virus genome replication and luciferase assay
BHK-21 or C6/36 cells (20,000 cells/well) were transfected with 90 ng of each reporter viral RNA via Lipofectamine 2000 reagent (Invitrogen™) following the manufacturer’s instructions. At each time point, the supernatant was removed, and 100 μl of Steady-Glo firefly luciferase reagent (Thermo-Scientific) was added directly to the wells. Luminescence was quantified using the Perkin Elmer EnVision™ plate reader under the US LUM 96 settings. In addition, the supernatant collected at the different time points was used to determine the presence of progeny infectious particles. Briefly, collected supernatants were used to infect naïve BHK-21 cells for 1 h. After incubation, supernatant was removed, cells washed two times with PBS, and 100 μl of complete media was added to each well. Luminescence was quantified after 24 h as described above.
Plaque assay
Viral replication cycles were determined in BHK-21, C6/36, and Aag2 cells by infecting each cell type at a MOI of 0.01 in serum-free media at 37 °C for 1 h. Cells were washed twice with PBS, and complete media was added. At the indicated time points 10% of the supernatant was collected and replaced with fresh complete media. Viral titers were quantified by plaque assay as described below. For SINV, viral titers at 24 h post infection were quantified by TCID50. In brief, Vero cells were seeded into 96-well plates (10,000 cells/well). Ten-fold dilutions of each virus were made in DMEM and 100 μl of virus was added to each well. Virus-cell mixtures were incubated at 37 °C with 5% CO2 for 7 days. Following incubation, cells were fixed with 4% formalin, and visualized using crystal violet.
Thermostability assay
Virus stocks were diluted to 5 × 104 PFU/ml in complete media and 60 μl aliquots were made. Aliquots were incubated in BSL-3 tissue culture incubators at 28°C or 37°C. Aliquots were taken for infectious particles quantification by plaque assay at 0, 6 and 24 h. Percentage of virus retaining infectivity was estimated as the percentage of infectious particles at the indicated timepoint relative to time point 0.
Plaque size quantification
Plaque assays were performed in 6 well plates and plaques were photographed using Chemidoc touch Imaging System (Biorad) at a fixed distance. Plaque sizes were measured in pixels using ImageJ software (Schneider et al., 2012).
Mosquito infections
Aedes aegypti mosquitoes (Poza Rica, Mexico, P20, obtained from Dr. Gregory Ebel, Colorado State University) were infected via artificial blood meals with high or low viral loads (106 or 104 PFU/ml, respectively). Briefly, viruses were mixed 1:2 with pre-washed rabbit whole blood (BioIVT) supplemented with 5 mM ATP. Female mosquitoes were allowed to feed on 37 °C blood meals through an artificial membrane for 60 to 90 min. Engorged females were identified, sorted and incubated at 28 °C with 10% sucrose ad libitum for 7 day or 14 days for high and low viral loads infections, respectively. After incubations, legs and wings were removed, and placed in 2 ml round bottom tubes with 500 μl of PBS containing one 5 mm stainless steel bead (QIAGEN), homogenized with the Tissue-Lyser II (QIAGEN), and debris was pulled down at 1200 rpm for 10 min. Viral titers in sample homogenates were determined by plaque assay using Vero cells.
Mouse infections
C57BL/6J and Ifnar−/− mice were anesthetized by inhalation of isoflurane (Henry Schein Animal Health), and infected via footpad injection with 103 PFU of each mutant variant in 50 μl of DMEM. Mock-infected animals received DMEM alone. C57BL/6J mice were euthanized 2 days post infection by CO2 inhalation. Ifnar−/− were monitored for clinical signs of diseases twice a day and weighed at 24 h intervals. After reaching the humane endpoint all the mice were euthanized by CO2 inhalation. Blood was collected by cardiac puncture. Organs including footpad, calf and quadriceps muscles, liver, heart, brain and spleen were collected in 500 μl of PBS containing one or two 5 mm stainless steel beads (QIAGEN), homogenized with the Tissue-Lyser II (QIAGEN), and debris was pulled down at 8,000 rpm for 10 min. Viral titers in tissue homogenates were determined by plaque assay using Vero cells.
Viral RT-qPCR
RNA extractions were performed as described above, and the number of viral genomes/ml was quantified by RT-qPCR using the Taqman® RNA-to-CT One-Step RT-PCR kit (Applied Biosystems™) and CHIKV primers and probe targeting an amplicon in nonstructural protein 4 (nsP4) (Forward: 5’ TCACTCCCTGCTGGACTTGATAGA 3’; reverse: 5’ TTGACGAACAGAGTTAGGAACATACC 3’) and the probe (5’-(6-carboxyfluorescein)-AGGTACGCGCTTCAAGTTCGGCG-(black-holequencher)]-3) as described previously (Stapleford, 2014). A standard curve was generated for each data set using in vitro transcribed CHIKV RNAs.
Virus Binding assays
For binding assays, BHK-21 cells were preincubated with media containing 20 mM ammonium chloride for 1 h, at 4 °C. Purified ZsGreen viruses were diluted in binding buffer (RPMI, 0.2% BSA, 10 mM HEPEs, 20 mM ammonium chloride) and an equivalent of 100 particles per cell (based on viral genomes) were incubated with BHK-21 cells for 5 or 60 min at 4 °C. After incubation, the cells were washed three times with ice-cold PBS, and RNA was extracted with TRIzol™ Reagent (Invitrogen™) as described previously. CHIKV RNA was quantified by RT-qPCR as described above.
Virus fusion-from-without assays
BHK-21 cells (35,000 cells/well) were incubated with binding buffer (RPMI, 0.2% BSA, 10 mM HEPES, 20 mM ammonium chloride) for 1 h, at 4°C. Then, purified ZsGreen viruses were added at a MOI of 10 for 1 h, at 4°C. Unbound virus was removed and viral fusion was induced by adding fusion buffer for 2 min at 37°C (RPMI, 0.2% BSA, 30 mM succinic acid, 10 mM HEPES, adjusted to each pH described and pre-warmed at 37°C). After incubation, the pH curve was removed and replaced with complete media with 20 mM ammonium chloride. After incubation at 37°C for 18 h, cells were fixed with 1% paraformaldehyde (PFA). After fixation, cells were permeabilized with 0.25% triton X-100, stained with 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, Invitrogen), and quantified on a CellInsight CX7 High-content microscope (Thermo-Scientific) using the HCS Navigator Version 6.6.1 (Thermo-Scientific). Infected cells were scored based on a cut-off for three standard deviations from the negative uninfected control.
Lysosomotropic drug sensitivity
BHK-21 cells (30,000 cells/well) were seeded in 96 well plates, 24 h before treatment. Cells were pretreated for 3 h in serum free media containing increasing concentrations of ammonium chloride or bafilomycin A1 (EMD Millipore). Following incubation, cells were infected with each ZsGreen virus at a MOI of 1 in the presence of each compound for 1 h at 37 °C. Cells were washed extensively, complete media containing each compound was added, and cells were incubated at 37 °C for 16 h. In addition, control cells were treated with equivalent dilutions of the drug with no virus, and virus with no drug. After incubation, cells were fixed with 4% PFA and ZsGreen positive cells were quantified as described above.
Ammonium chloride by-pass assay
BHK-21 cells (35,000 cells/well) were seeded in 96 well plates 24 h before treatment. Cells were preincubated for 1 h in infection media (DMEM, 0.2% BSA, 1 mM HEPES). Then, cells were infected with each ZsGreen virus at a MOI of 0.1 for 1 h, at 4 °C. After incubation, the plate was placed at 37 °C and complete media containing 20 mM final concentration of ammonium chloride was added into the cells at the indicated timepoints. Cells were incubated at 37 °C for 16 h. After incubation, cells were fixed with 4% PFA and ZsGreen positive cells were quantified as described above.
Cholesterol depletion and repletion
BHK-21 cells (30,000 cells/well) were seeded in 96 well plates, 24 h before treatment. Cells were pretreated for 1 h with increasing concentrations of methyl-beta-cyclodextrin (MβCD). Following incubation, cells were washed once with PBS and infected with each ZsGreen virus at a MOI of 1 for 1 h at 37 °C. The cells were then washed three times, complete media containing 20 mM ammonium chloride was added, and cells were incubated at 37 °C for 18 h. After incubation, cells were fixed, stained, and ZsGreen positive cells were quantified as described above. For cholesterol repletion experiments, cholesterol depleted cells were treated for 1 h with 200 μg/ml of water-soluble cholesterol (Sigma), then infected and treated as described above.
Viral RNA sequence alignments
All Sanger sequencing data were aligned to the CHIKV strain 06–049 (AM258994) reference sequence using SeqMan Pro (DNASTAR).
Protein sequence alignments and LOGOS
Sequence alignments were performed using the MUSCLE software with default parameters (Edgar, 2004). We generated sequence alignments of 13 representative members of the alphavirus genus: Chikungunya virus IOL (CHIKV, ACS29297.1), O’nyong’nyong virus (ONNV, NP_740711.1), Sindbis virus (SINV, NP_740677.1), Semliki forest virus (SFV, P03315), Sagiyama virus (SAGV, Q9JGK8), Venezuelan equine encephalitis virus (strain Trinidad donkey (VEEV, P09592), Aura virus (AURAV, Q86925), Eastern equine encephalitis virus (EEEV, P08768), Mayaro virus (strain Brazil) (MAYAB, Q8QZ72), Ross river virus (RRVN, P13890), Middelburg virus (MIDDV, Q80S27), Barmah forest virus (BFV, P89946) and Western equine encephalitis virus (WEEV, P13897). Sequence logos describing the conservation between residues 62 and 99 of the E1 glycoprotein were generated with a sequence alignment of 13 representative members of the alphavirus genus using WebLogo software (Crooks et al., 2004). The x-axis indicates the position within the alignment and the y-axis indicates the information content. The height of each stack indicates conservation and the height of each letter is proportional to the frequency of that residue in that position in the alignment.
Protein representations.
All protein representations were performed using the program Pymol (https://pymol.org/).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism and R-studio software. Each experiment was completed at least two independent times with internal biological duplicates unless otherwise noted in the figure legends. The exact number of biological replicates (n) can be found in the figure legends. Data are represented as the mean ± standard error of the mean (SEM). Fisher’s exact test, Kruskal-Wallis, one-way and two-way ANOVA tests with Bonferroni post-hoc test, log-rank Mantel-Cox test, and Mann-Whitney U statistical tests were performed and specifically indicated in the figure legends. P-values > 0.05 were considered non-significant (ns), * p<0.05, ** p<0.01, *** p < 0.001, **** p < 0.0001.
Supplementary Material
Acknowledgements.
We would like to thank all members of the Stapleford lab for helpful comments on this manuscript. We also thank Meike Dittmann’s lab, Ken Cadwell, Kamal Khanna and Stephen Yeung who kindly provided essential reagents. M.G.N is supported by a Jan Vilcek/David Goldfarb Fellowship from the New York University Department of Microbiology, New York University and M.R. is supported in part by the Public Health Service Institutional Research Training Award T32 AI007180.
Footnotes
Declaration of Interests.
The authors declare there are no conflicts of interest.
REFERENCES
- Brault AC, Powers AM, Ortiz D, Estrada-Franco JG, Navarro-Lopez R, and Weaver SC (2004). Venezuelan equine encephalitis emergence: enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc Natl Acad Sci U S A 101, 11344–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Long M, Xiao B, Xiong Y, Chen H, Chen Y, Kuang Z, Li M, Wu Y, Rock DL, et al. (2017). Transcriptomic profiles of human foreskin fibroblast cells in response to orf virus. Oncotarget 8, 58668–58685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey LL, and Vignuzzi M (2011). Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J Virol 85, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel GD, Carricaburu J, Young D, Bernard KA, and Kramer LD (2004). Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am J Trop Med Hyg 71, 493–500. [PubMed] [Google Scholar]
- Gardner CL, Burke CW, Higgs ST, Klimstra WB, and Ryman KD (2012). Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology 425, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, and Suhrbier A (2010). Chikungunya virus arthritis in adult wild-type mice. J Virol 84, 8021–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons DL, Erk I, Reilly B, Navaza J, Kielian M, Rey FA, and Lepault J (2003). Visualization of the target-membrane-inserted fusion protein of Semliki Forest virus by combined electron microscopy and crystallography. Cell 114, 573–583. [DOI] [PubMed] [Google Scholar]
- Gibbons DL, and Kielian M (2002). Molecular dissection of the Semliki Forest virus homotrimer reveals two functionally distinct regions of the fusion protein. J Virol 76, 1194–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene IP, Paessler S, Austgen L, Anishchenko M, Brault AC, Bowen RA, and Weaver SC (2005). Envelope glycoprotein mutations mediate equine amplification and virulence of epizootic venezuelan equine encephalitis virus. J Virol 79, 9128–9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier JM, Yeung ST, Qiu Z, Jellison ER, and Khanna KM (2017). Combining Adoptive Cell Therapy with Cytomegalovirus-Based Vaccine Is Protective against Solid Skin Tumors. Front Immunol 8, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardado-Calvo P, Atkovska K, Jeffers SA, Grau N, Backovic M, Perez-Vargas J, de Boer SM, Tortorici MA, Pehau-Arnaudet G, Lepault J, et al. (2017). A glycerophospholipid-specific pocket in the RVFV class II fusion protein drives target membrane insertion. Science 358, 663–667. [DOI] [PubMed] [Google Scholar]
- Hafer A, Whittlesey R, Brown DT, and Hernandez R (2009). Differential incorporation of cholesterol by Sindbis virus grown in mammalian or insect cells. J Virol 83, 9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs S, and Vanlandingham D (2015). Chikungunya virus and its mosquito vectors. Vector Borne Zoonotic Dis 15, 231–240. [DOI] [PubMed] [Google Scholar]
- Hoornweg TE, van Duijl-Richter MKS, Ayala Nunez NV, Albulescu IC, van Hemert MJ, and Smit JM (2016). Dynamics of Chikungunya Virus Cell Entry Unraveled by Single-Virus Tracking in Living Cells. J Virol 90, 4745–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, Taylor AB, and Kuhn RJ (2017). Spatial and Temporal Analysis of Alphavirus Replication and Assembly in Mammalian and Mosquito Cells. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian M, Chatterjee PK, Gibbons DL, and Lu YE (2000). Specific roles for lipids in virus fusion and exit. Examples from the alphaviruses. Subcell Biochem 34, 409–455. [DOI] [PubMed] [Google Scholar]
- Kielian M, and Rey FA (2006). Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol 4, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian MC, Keranen S, Kaariainen L, and Helenius A (1984). Membrane fusion mutants of Semliki Forest virus. J Cell Biol 98, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YE, Cassese T, and Kielian M (1999). The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J Virol 73, 4272–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S, and Parmryd I (2015). Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol Biol 1232, 91–102. [DOI] [PubMed] [Google Scholar]
- Martin PK, Marchiando A, Xu R, Rudensky E, Yeung F, Schuster SL, Kernbauer E, and Cadwell K (2018). Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol 3, 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley VJ, Noval MG, Chen R, Weaver SC, Vignuzzi M, Stapleford KA, and Turner PE (2018). Chikungunya virus evolution following a large 3’UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol 4, vey012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy RM, Meola MA, Morin LL, Ebel GD, and Kramer LD (2007). A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am J Trop Med Hyg 77, 365–370. [PubMed] [Google Scholar]
- Nair S, Poddar S, Shimak RM, and Diamond MS (2017). Interferon regulatory factor-1 (IRF-1) protects against chikungunya virus induced immunopathology by restricting infection in muscle cells. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesko KN, and Ebel GD (2012). West Nile virus population genetics and evolution. Infect Genet Evol 12, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FA, and Lok SM (2018). Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 172, 1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GC, Zothner C, Remenyi R, Merits A, Stonehouse NJ, and Harris M (2017). Evaluation of a range of mammalian and mosquito cell lines for use in Chikungunya virus research. Sci Rep 7, 14641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckert C, Weger-Lucarelli J, Garcia-Luna SM, Young MC, Byas AD, Murrieta RA, Fauver JR, and Ebel GD (2017). Impact of simultaneous exposure to arboviruses on infection and transmission by Aedes aegypti mosquitoes. Nat Commun 8, 15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-San Martin C, Nanda S, Zheng Y, Fields W, and Kielian M (2013). Cross-inhibition of chikungunya virus fusion and infection by alphavirus E1 domain III proteins. J Virol 87, 7680–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, et al. (2006). Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med 3, e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Kesari P, Kumar P, and Tomar S (2018). Structure-function insights into chikungunya virus capsid protein: Small molecules targeting capsid hydrophobic pocket. Virology 515, 223–234. [DOI] [PubMed] [Google Scholar]
- Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost MC, Fsihi H, et al. (2007). Characterization of reemerging chikungunya virus. PLoS Pathog 3, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleford KA, Coffey LL, Lay S, Borderia AV, Duong V, Isakov O, Rozen-Gagnon K, Arias-Goeta C, Blanc H, Beaucourt S, et al. (2014). Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microbe 15, 706–716. [DOI] [PubMed] [Google Scholar]
- Tsetsarkin KA, Chen R, Yun R, Rossi SL, Plante KS, Guerbois M, Forrester N, Perng GC, Sreekumar E, Leal G, et al. (2014). Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat Commun 5, 4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsarkin KA, McGee CE, and Higgs S (2011). Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol J 8, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsarkin KA, Vanlandingham DL, McGee CE, and Higgs S (2007). A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsarkin KA, and Weaver SC (2011). Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog 7, e1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashishtha M, Phalen T, Marquardt MT, Ryu JS, Ng AC, and Kielian M (1998). A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J Cell Biol 140, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M, Thiria J, Dehecq JS, Fontenille D, Schuffenecker I, et al. (2007). Two Chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS One 2, e1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, and Higgs S (2017). The Bridges and Blockades to Evolutionary Convergence on the Road to Predicting Chikungunya Virus Evolution. Annu Rev Virol 4, 181–200. [DOI] [PubMed] [Google Scholar]
- Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, and Rey FA (2010). Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712. [DOI] [PubMed] [Google Scholar]
- Wilder-Smith A, Gubler DJ, Weaver SC, Monath TP, Heymann DL, and Scott TW (2017). Epidemic arboviral diseases: priorities for research and public health. Lancet Infect Dis 17, e101–e106. [DOI] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, et al. (2015). Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, et al. (2018). Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Sanchez-San Martin C, Qin ZL, and Kielian M (2011). The domain I-domain III linker plays an important role in the fusogenic conformational change of the alphavirus membrane fusion protein. J Virol 85, 6334–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, and Levitan I (2007). Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 1768, 1311–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.