

Abstract
Type I and III IFNs play diverse roles in bacterial infections, being protective for some but deleterious for others. Using RNA-sequencing transcriptomics we investigated lung gene expression responses to Bordetella pertussis infection in adult mice, revealing that type I and III IFN pathways may play an important role in promoting inflammatory responses. In B. pertussis–infected mice, lung type I/III IFN responses correlated with increased proinflammatory cytokine expression and with lung inflammatory pathology. In mutant mice with increased type I IFN receptor (IFNAR) signaling, B. pertussis infection exacerbated lung inflammatory pathology, whereas knockout mice with defects in type I IFN signaling had lower levels of lung inflammation than wild-type mice. Curiously, B. pertussis–infected IFNAR1 knockout mice had wild-type levels of lung inflammatory pathology. However, in response to infection these mice had increased levels of type III IFN expression, neutralization of which reduced lung inflammation. In support of this finding, B. pertussis–infected mice with a knockout mutation in the type III IFN receptor (IFNLR1) and double IFNAR1/IFNLR1 knockout mutant mice had reduced lung inflammatory pathology compared with that in wild-type mice, indicating that type III IFN exacerbates lung inflammation. In marked contrast, infant mice did not upregulate type I or III IFNs in response to B. pertussis infection and were protected from lethal infection by increased type I IFN signaling. These results indicate age-dependent effects of type I/III IFN signaling during B. pertussis infection and suggest that these pathways represent targets for therapeutic intervention in pertussis.
Pertussis, also referred to as whooping cough, is a highly contagious respiratory disease caused by infection with the bacterial pathogen Bordetella pertussis. There has been a recent resurgence in pertussis, with 48,277 reported cases in the United States in 2012, the most since 1955 (1). In older children and adults, symptoms build up to periods of severe paroxysmal coughing, often for several weeks after onset, but other complications are relatively rare (2, 3). However, for infants, pertussis can be a fatal disease because of complications, including pulmonary hypertension, high-level circulating leukocytosis, and pneumonia (4–6). A major difficulty in the treatment and prevention of pertussis is our relatively poor understanding of specific mechanisms of disease pathogenesis. In addition, the limited effectiveness of the currently used acellular vaccine that confers rapidly waning immunity as well as a lack of effective treatments contribute to these challenges (7). Antibiotics are administered to infected individuals to prevent transmission but do not typically change the clinical course of disease for the infected person (8, 9). Host-directed therapeutics that treat pertussis disease could benefit individuals with severe cough and save the lives of infected infants. Our current work has identified several host targets that are involved in inflammatory and immune responses to B. pertussis infection and are potential candidates for therapeutic intervention (10–12).
Type I IFNs (including IFN-α and IFN-β) signal through the IFN-αR (IFNAR)1/IFNAR2 heterodimeric receptor (13) and JAK/STAT pathway (14). They are major cytokines in immune responses and antiviral defense, but they are also involved in pathogenesis and inflammation in several disease models (15–18). Type I IFNs have diverse roles in bacterial infections in animal and cell culture models, providing protective effects against some of these infections and deleterious effects for others (15, 19, 20). For example, they are protective against infections by Legionella pneumophila (21), Streptococcus pneumoniae (20) and Helicobacter pylori (22), but detrimental to Mycobacterium tuberculosis (23), Listeria monocytogenes (24) and Staphylococcus aureus (25) infections. Although the mechanisms are unclear in most of these cases, the diverse activities of type I IFNs combined with the specific context of each host–pathogen interaction likely determine this dichotomy of effects. The role of type I IFNs in B. pertussis infection and disease is an understudied area. In one recent report, it was found that IFN-α–producing lung plasmacytoid dendritic cells subdue immune cell Th17 responses in B. pertussis–infected mice (26).
Type II IFN or IFN-γ signals through the IFN-γR (IFNGR) and JAK/STAT pathway (27). IFN-γ is induced in adult mouse lungs in response to B. pertussis infection (28), and IFN-γ signaling is important for host defense in mouse models of pertussis because IFNGR1 knockout (KO) adult mice fail to maintain localization of the infection to the respiratory tract and succumb to a lethal disseminating infection (29). However, these mice do not exhibit altered lung inflammatory pathology compared with wild-type (WT) mice (30).
Type III IFNs, also known as IFN-λ, are more recently discovered cytokines that can induce expression of the same downstream IFN-stimulated genes (ISGs) as type I IFN (31). Type III IFNs also signal through the JAK/STAT pathway but use a different heterodimeric receptor, type III IFN receptor (IFNLR1)/IL-10Rβ, expressed predominantly by epithelial cells as well as by several immune cells (32, 33). Type III IFNs have not yet been investigated with regards to their involvement in B. pertussis infection but have been described as critical in immune responses to many viral as well as some bacterial infections (34–36). In this study, we report transcriptomic analysis of lung gene expression changes in response to B. pertussis infection in adult mice. Our analysis identified IFNAR1 and IFNLR1 as two of the most significant upstream regulators of transcriptional responses to infection, and we hypothesized that type I and III IFNs exacerbate lung inflammatory pathology during B. pertussis infection. Because mice are incapable of coughing, lung inflammatory pathology represents the best indicator of pertussis disease in the murine model. Lung inflammatory pathology is also seen in B. pertussis–infected baboons, which exhibit the typical paroxysmal pertussis cough, as well as in fatal human infant pertussis autopsies (4, 37, 38). Our studies in the adult mice described in this report support the hypothesis that type I and III IFNs exacerbate pertussis disease. In marked contrast, we found that type I and III IFN genes were not upregulated in response to B. pertussis infection in infant mice and that increased type I IFN signaling was protective in infant mice against lethal infection, indicating age dependence in the effects of these cytokines in the pathogenesis of this infection. Mouse models of B. pertussis infection reflect the different pathogenesis of pertussis in infant and adult humans. In adult mice, disease is characterized by significant and prolonged lung inflammatory pathology from which the mice recover, but in infant mice, infection progresses to a lethal disseminating disease with no significant lung inflammation until just prior to the time of death (39). Overall, our findings indicate that type I and III IFN signaling contribute to pertussis pathogenesis and may represent important host targets for therapeutic intervention in pertussis, although the specific direction of therapy would be different between adults and infants.
Materials and Methods
Bacterial strains
In these studies, our strain of B. pertussis was a streptomycin-resistant derivative of Tohama I (40). B. pertussis was grown on Bordet–Gengou agar supplemented with 10% defibrinated sheep’s blood (LAMPIRE Biological Laboratories) and 200 μg/ml streptomycin at 37°C for 48 h.
Mouse infections
All mouse strains were on the C57BL/6 genetic background. WT, Ifnar1−/− (The Jackson Laboratory), Ifnar1S526A (IFNAR1-SA) (41), Ifnlr1−/−, Ifnar1/Ifnlr1−/− (kindly provided by S. Kotenko), Ifnb−/− (kindly provided by S. Vogel), Stat1−/− and Stat2−/− (The Jackson Laboratory) animals were all used in accordance with an Institutional Animal Care and Use Committee protocol (University of Maryland, Baltimore). Bacteria were grown for 48 h on Bordet–Gengou agar, and inocula were prepared in PBS suspension. For adult mice, 2 × 106 CFU (unless otherwise stated) were administered intranasally in a volume of 50 μl. For infant mice, bacteria were administered by aerosol, as described previously (39). The sphingosine-1–phosphate (S1P) receptor agonist AAL-R (kindly provided by H. Rosen) was dissolved in sterile water and administered intranasally at a dose of 0.5 mg/kg. Anti-mouse IFN-λ (IL-28A/B) and isotype control Abs (R&D Systems) were administered i.p. at a dose of 25 μg on the indicated days postinoculation. Y136 protein (R&D Systems) and BSA were administered s.c. at a dose of 2 μg on the indicated days postinoculation. Following infection, lungs (and other organs) were removed for analysis of bacterial burden, transcript levels, protein levels, and inflammatory histopathology. In general, groups of adult mice consisted of four to six animals of both sexes, and groups of infant mice were litters of five to nine animals of both sexes.
Protein isolation and processing
After harvest, tissues were flash-frozen using an isopropanol/dry-ice bath. The tissue was then homogenized in 2 ml of PBS with protease inhibitor (Roche) added to prevent protein degradation. ELISA was performed on dilutions of the tissue samples as per the manufacturer’s protocol (PBL Assay Science) for IFN-α, -β, and -λ kits.
RNA isolation and quantitative real-time PCR
After harvest, tissues were flash-frozen using an isopropanol/dry-ice bath. RNA extraction was achieved using the RNeasy Microarray Tissue Kit (QIAGEN) following the manufacturer’s instructions, and they were DNase treated to remove DNA contamination. Quality control on RNA was performed on a 2100 Bioanalyzer machine (Agilent Technologies). cDNA was synthesized from mRNA using a reverse transcription system (Promega) as per the manufacturer’s instructions. Quantitative real-time PCR was performed with SYBR Green/ROX Master Mix for quantitative PCR in the 7500 Fast Real-Time PCR system (Applied Biosystems). Hypoxanthine phosphoribosyltransferase (hprt) was used as a house-keeping control gene. Primers used for PCR are listed in Supplemental Table I. Expression was determined by calculating fold changes compared with PBS “sham”-infected animals using the 2−ΔΔCT method.
RNA sequencing
RNA-sequencing (RNA-seq) analysis was performed by the Informatics Resource Center, Institute for Genome Sciences, University of Maryland School of Medicine. Paired end Illumina libraries were mapped to the mouse reference, Ensembl GRCm38.74, using TopHat v1.4.0 with the default mismatch parameters. Read counts for annotated genes were calculated using HTSeq. DESeq Bioconductor package v1.5.24 was used to normalize read counts by library size to generate gene counts per million, estimate dispersion, and determine differentially expressed genes between two groups. Downstream analysis was done with differentially expressed transcripts with a false discovery rate of ≤0.05 and log2 fold change. Ingenuity Pathway Analysis (IPA) was used to compute enrichment of biological pathways using the list of differentially expressed genes. IPA was also used to analyze upstream regulators by predicting the regulators of each data set and their activation state. Each predicted upstream regulator is assigned a z-score for their activation state based on previous experimentally observed transcriptional events from the literature. A z-score of <2 predicts a regulator to be inhibitory, whereas a z-score of >2 indicates the regulator is activating. Additionally, each upstream regulator is assigned an overlap p value, a measure of statistically significant overlap between genes in the dataset and genes known to be regulated by this regulator. Reads were deposited in the National Center for Biotechnology Information Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA493118) under BioProject PRJNA493118,.
Pathology
Lungs were perfused with PBS, then transferred to 10% (w/v) buffered formalin. H&E staining was performed at the Pathology and Histology Laboratory at the University of Maryland School of Medicine. Histopathological findings were scored on a scale of 0–9. Every slide was scored 0–3 in the following three categories by multiple blinded investigators: the degree of tissue consolidation, severity of bronchovascular bundle inflammation, and the percentage of bronchovascular bundles inflamed.
Statistical analysis
Data were analyzed and graphs were made using GraphPad Prism 8.0 statistics software. Each plot represents mean values with SD. In the RNA-seq data, IPA software was used to calculate z-scores for upstream regulators, and the Fisher exact test was used to determine the probability that associations between genes in our data sets and canonical pathways were significant. The Student t test was used for determining significance between two means, and two-way ANOVA was used for determining significance between multiple groups.
Results
Lung transcriptional response to B. pertussis infection in adult mice
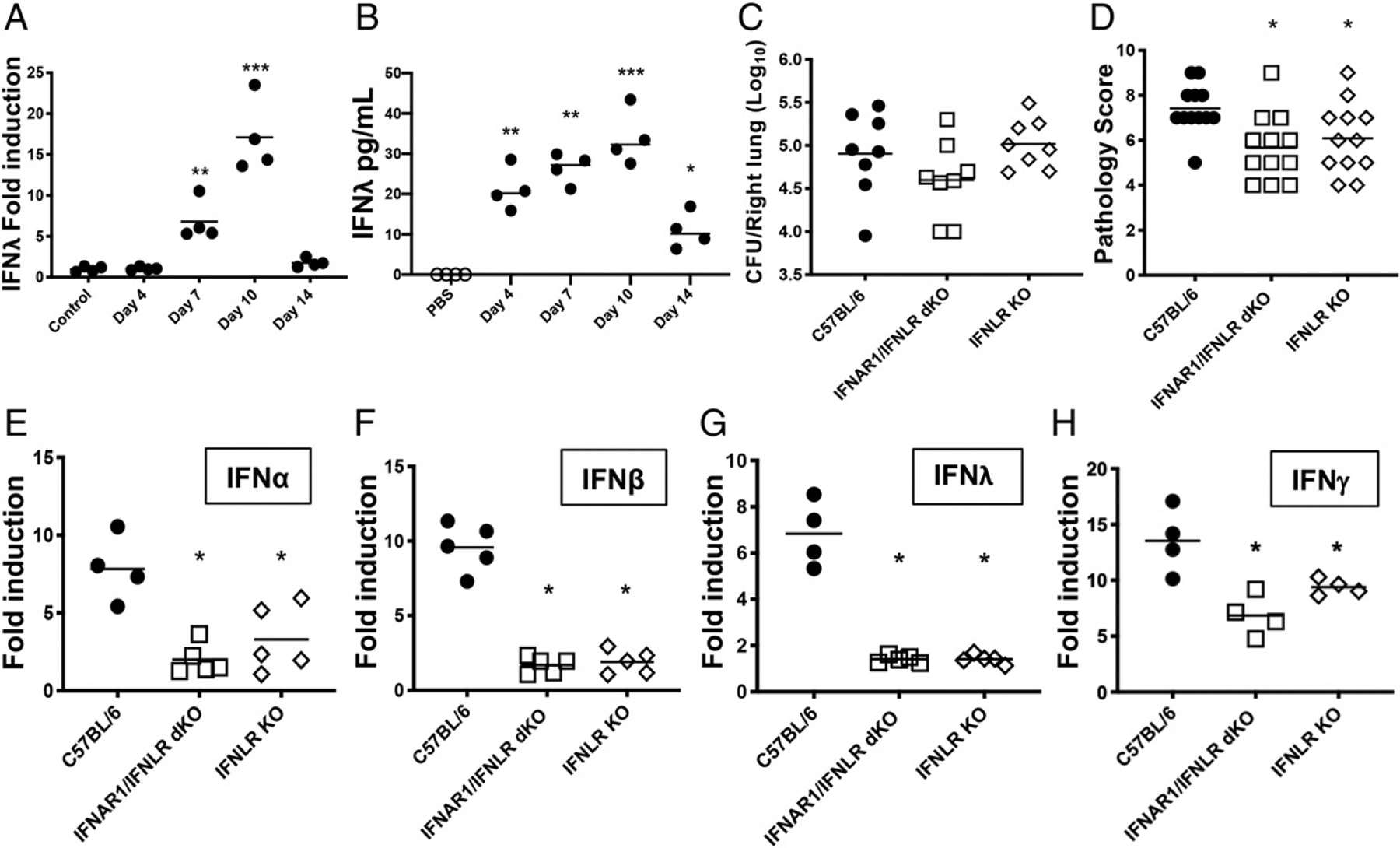
To investigate the mechanisms through which B. pertussis induces lung inflammatory pathology, we used RNA-seq to differentiate lung transcriptional responses in B. pertussis–infected adult WT mice compared with sham-infected mice at 4 d postinoculation (dpi). In total, 1537 genes were >2-fold differentially regulated in infected versus uninfected mice. Within these 1537 genes, 451 were downregulated and 1086 were upregulated in B. pertussis–infected lungs (Fig. 1A). The top 10 most differentially regulated genes (all upregulated) are shown in Fig. 1B, including 1) proinflammatory genes (Ifng, Csf3, Cxcl3), 2) genes coding for acute phase proteins associated with inflammation (Il6) (42), and 3) antimicrobial response genes (Acod1, Defb4) (43). These data demonstrate an overall antibacterial response and increase in proinflammatory gene expression in the lungs following B. pertussis infection. Furthermore, IPA was used to identify the most significant canonical pathways represented by the differentially expressed genes (top 10 shown in Fig. 1C). These pathways also reflect an inflammatory response, including the following: pattern recognition receptor recognition of bacteria (such as TLR signaling), agranulocyte and granulocyte adhesion and diapedesis (cell mobilization as part of the inflammatory response), increased inflammatory acute phase response signaling, and a reduction of anti-inflammatory IL-10 signaling. Using IPA, upstream regulators of the differentially expressed genes were predicted (Fig. 1D). This analysis predicts that the most significant upstream regulator is IFNAR1, which is essential for all type I IFN signaling (13). In addition to IFNAR1, IFNLR1 (part of the heterodimeric IFN-λ receptor) was the fifth most significant upstream regulator of transcriptional responses. Consistent with this finding, a large number of ISGs were upregulated in response to B. pertussis infection (Supplemental Fig. 1A), including genes in the top 10 most significantly upregulated (Cxcl11, Oas1h) (44, 45). These findings indicate that type I and III IFN signaling play a significant role in host responses to B. pertussis infection. Based on these data and published findings on the role of type I IFN signaling in exacerbation of other bacterial infections (17, 18), IFNAR1 was chosen as a candidate pathway for investigation of its contribution to lung inflammatory pathology during B. pertussis infection.
FIGURE 1.
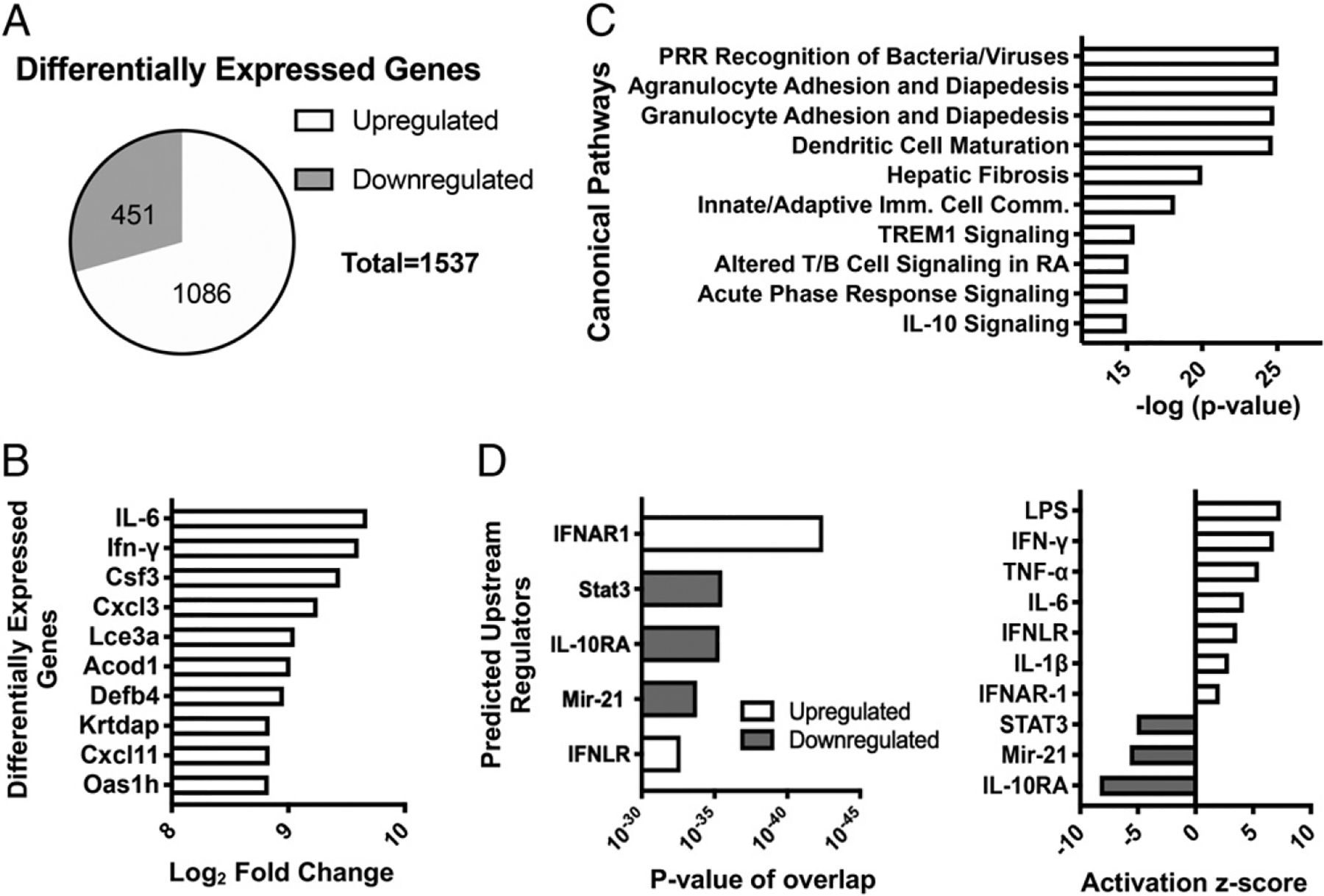
Lung transcriptional response to B. pertussis infection in adult C57BL/6 mice. Mice (n = 4 per group) were euthanized on day 4 postinoculation with B. pertussis or PBS sham inoculum, and RNA was isolated from the lungs. RNA-seq and IPA revealed (A) total number and direction of differentially expressed genes, (B) top 10 most upregulated genes, (C) top canonical pathways represented by the differentially expressed genes, and (D) predicted upstream regulators of responses, ranked by significance (left) or z-score (right).
Type I IFN production is induced in response to B. pertussis infection in adult mice
Because we identified IFNAR1 as the most significant upstream regulator of transcriptional responses to B. pertussis infection, we investigated the expression of type I IFN in the lungs throughout the time course of infection. To achieve this, we infected WT mice with B. pertussis and harvested lungs at timepoints of 4, 7, 10, and 14 dpi. Bacterial loads showed a typical early peak (4 dpi) then decline at later timepoints (Fig. 2A). Results from quantitative RT-PCR showed the significant transcriptional upregulation of both IFN-α (Fig. 2B) and IFN-β (Fig. 2C) starting after 4 dpi and peaking at 10 dpi around 15-fold higher than PBS sham-inoculated controls. Protein levels for both IFN-α and IFN-β were similarly upregulated but starting at 4 dpi (prior to the start of transcriptional upregulation) (Fig. 2D, 2E), suggesting that translational regulation may account for this earlier increase in type I IFN levels. Additionally, the temporal profile of type I IFN production correlated with that of the proinflammatory cytokines IL-6, IL-1β, IFN-γ, and TNF-α in response to B. pertussis infection (Supplemental Fig. 1B), suggesting a possible link between type I IFNs and lung inflammation.
FIGURE 2.
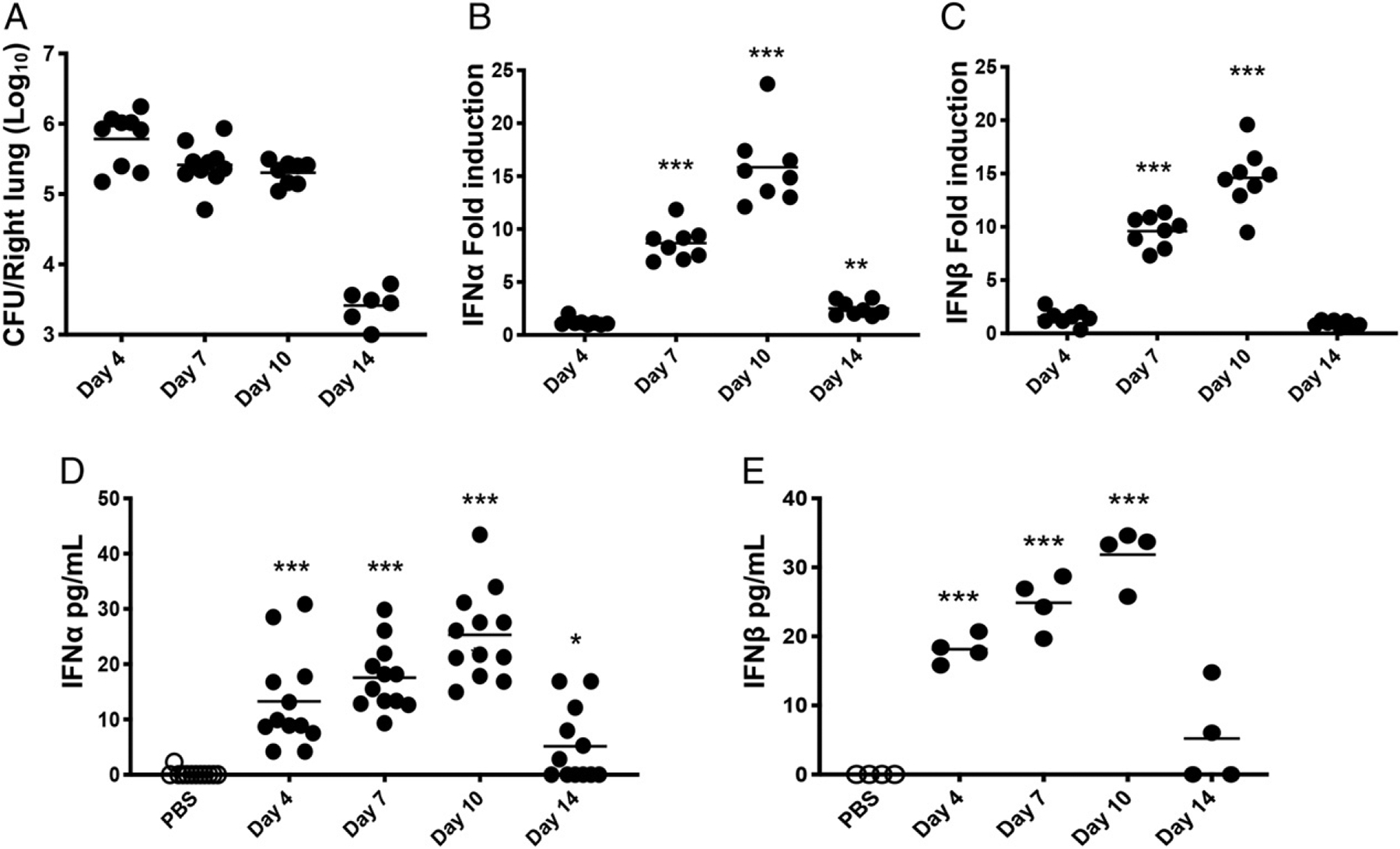
Type I IFN production induced in response to B. pertussis infection in adult mice. Mice (n ≥ 4 per group) were euthanized on the indicated dpi (x-axis) with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of (A) bacterial burdens by plating for viable counts, (B and C) mRNA levels of IFN-α and IFN-β by quantitative RT-PCR, or (D and E) IFN-α and IFN-β protein levels by ELISA. Data are representative of at least two independent experiments. *p < 0.05, ***p < 0.001 by the Student t test and two-way ANOVA.
Increased IFNAR signaling leads to exacerbation of lung inflammatory pathology in B. pertussis–infected adult mice
Having confirmed type I IFN induction by B. pertussis infection, we sought to determine the contribution of type I IFN signaling to host lung inflammation during infection. Inflammatory and stress signals act to rapidly downregulate IFNAR1 by stimulating the phosphorylation-driven ubiquitination, internalization, and degradation of this protein (46, 47). Thus, we used IFNAR1-SA mutant mice that harbor a mutation in the cytoplasmic domain of IFNAR1 (41). This mutation acts to interfere with inflammation-induced proteolytic loss of the receptor, leading to increased type I IFN signaling under inflammatory conditions (41, 48), but otherwise these mice do not display any difference in development, function, or activity of immune cells (or any other phenotypes) compared with WT mice (41, 49–51). To test the effect of B. pertussis infection in IFNAR1-SA mice, we inoculated IFNAR1-SA and WT mice with a lower dose (1 × 106 CFU) of B. pertussis because our standard dose induces near maximal levels of lung inflammatory pathology at 7 dpi in WT mice (and therefore we would not be able to observe any potential increase in inflammation in the IFNAR1-SA mice). Bacterial burdens in B. pertussis–infected IFNAR1-SA mice showed no significant difference from those in infected WT mice (Fig. 3A). Despite this lack of difference in bacterial loads, IFNAR1-SA mice had significantly higher levels of lung inflammatory pathology than WT mice at 7 dpi (Fig. 3B, 3C). Infected IFNAR1-SA mice also displayed higher gene expression of the proinflammatory cytokines IL-1β, TNF-α, IL-6, and IFN-γ than WT mice at 7 dpi (Fig. 3D). Type I and type III IFN gene expression levels were not significantly different between the two groups (data not shown), although IFNAR1-SA mice had higher expression of the ISGs MX1 (Fig. 3D) and ISG15 (data not shown), reflecting the increased level of IFNAR signaling. Together, these data demonstrate a clear role for enhanced type I IFN signaling in exacerbation of lung inflammatory pathology during B. pertussis infection.
FIGURE 3.
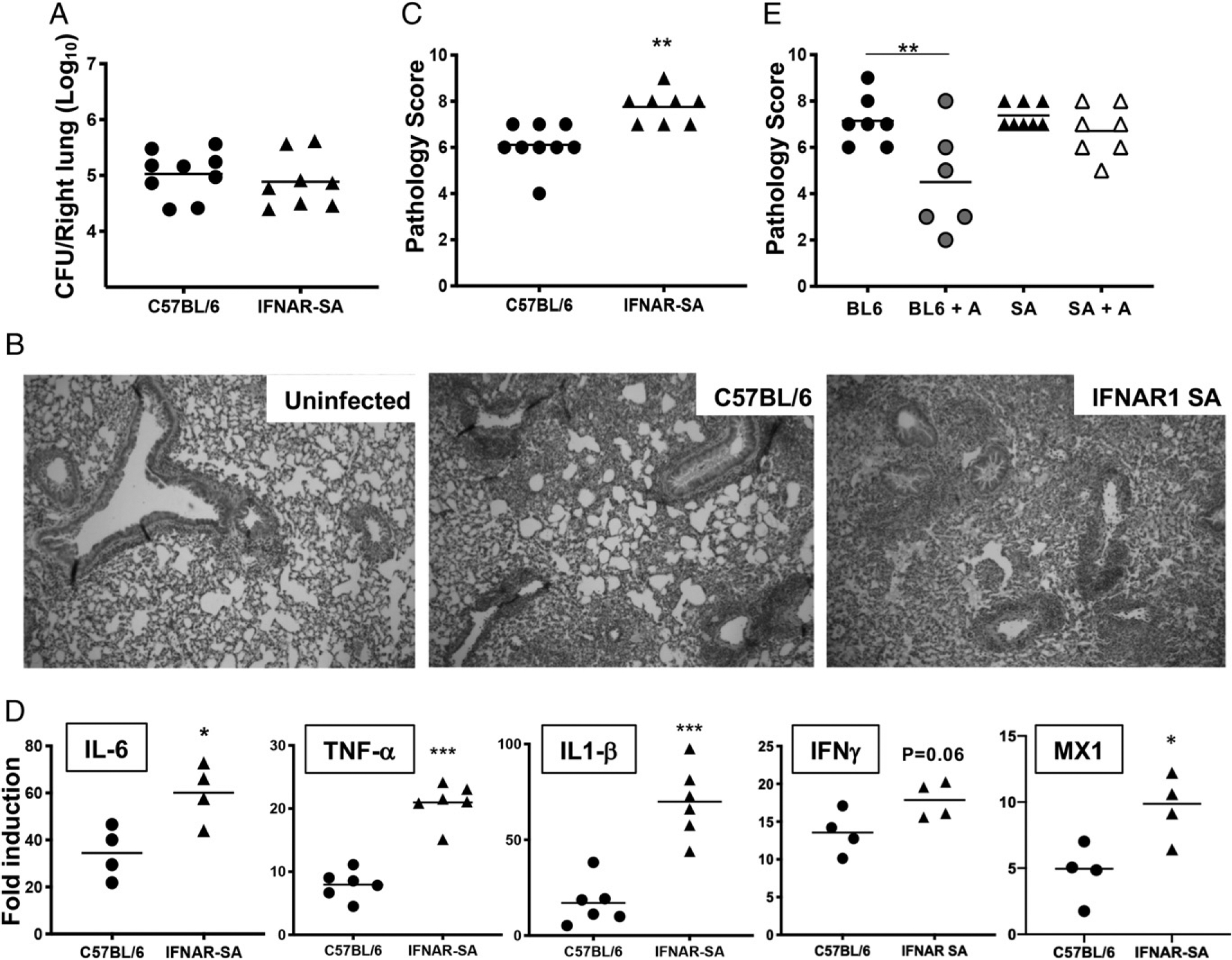
Increased IFNAR1 signaling leads to exacerbated lung inflammation in B. pertussis–infected adult mice. C57BL/6 or IFNAR-SA mice (n ≥ 4 per group) were euthanized on day 7 postinoculation with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of outcomes. (A) Bacterial burdens. (B) Representative H&E-stained inflammatory pathology images (original magnification 340) of uninfected and infected C57BL/6 and infected IFNAR1-SA mouse lung sections. (C) Inflammatory pathology scores assessed from lung histology sections. (D) Fold induction (infected versus sham inoculated) of IL-6, TNF-α, IL-1β, IFN-γ, and MX1 mRNA levels. (E) Infected C57BL/6 (BL6) or IFNAR1-SA (SA) mice (n ≥ 6 per group) were treated with AAL-R (+A) or PBS intranasally 24 h postinoculation and euthanized on day 7 postinoculation for assessment of lung inflammatory pathology scores. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by the Student t test and two-way ANOVA.
Previously, we showed that treating B. pertussis–infected mice with the S1P receptor agonist AAL-R resulted in a reduction of pulmonary inflammatory pathology (11, 12). A recent report suggested that AAL-R reduces inflammation by promoting the internalization and degradation of IFNAR1 (52). We found that AAL-R administration reduced lung inflammatory pathology at 7 dpi in WT mice but not in IFNAR1-SA mice inoculated with our standard dose of B. pertussis (Fig. 3E). This indicates that AAL-R has no effect in mice that cannot downregulate IFNAR signaling because of defective internalization, and therefore, the therapeutic activity of this drug is dependent upon elimination of IFNAR1 and inhibition of type I IFN signaling in B. pertussis–infected mice. In addition, this finding further supports an important role for type I IFN signaling in exacerbation of pulmonary inflammation induced by B. pertussis infection.
Impaired type I IFN signaling leads to a reduction in lung inflammatory pathology during B. pertussis infection in adult mice
Because enhanced IFNAR signaling led to an increase in lung histopathology and inflammation, we predicted that reduction in type I IFN signaling would reduce inflammatory pathology in B. pertussis–infected mice. We investigated this by examining outcomes in mutant mouse strains deficient in some aspect of type I IFN signaling, including mice with KO mutations in IFN-β (a type I IFN), STAT1, and STAT2 [transcription factors that mediate type I and type III IFN signaling (14)]. B. pertussis infection of these mutant mice showed no significant difference in bacterial burden from that in WT mice (Fig. 4A). However, lung inflammatory pathology scores at 7 dpi were significantly lower in each of the mutant mice compared with those in WT mice (Fig. 4B). Of these, STAT1 KO mice showed the lowest levels of lung inflammation, which may indicate a role for other STAT1-dependent factors (such as IFN-γ) in promoting lung inflammation. Overall these data are again consistent with a role for the type I IFN pathway in promoting lung inflammatory pathology during B. pertussis infection.
FIGURE 4.
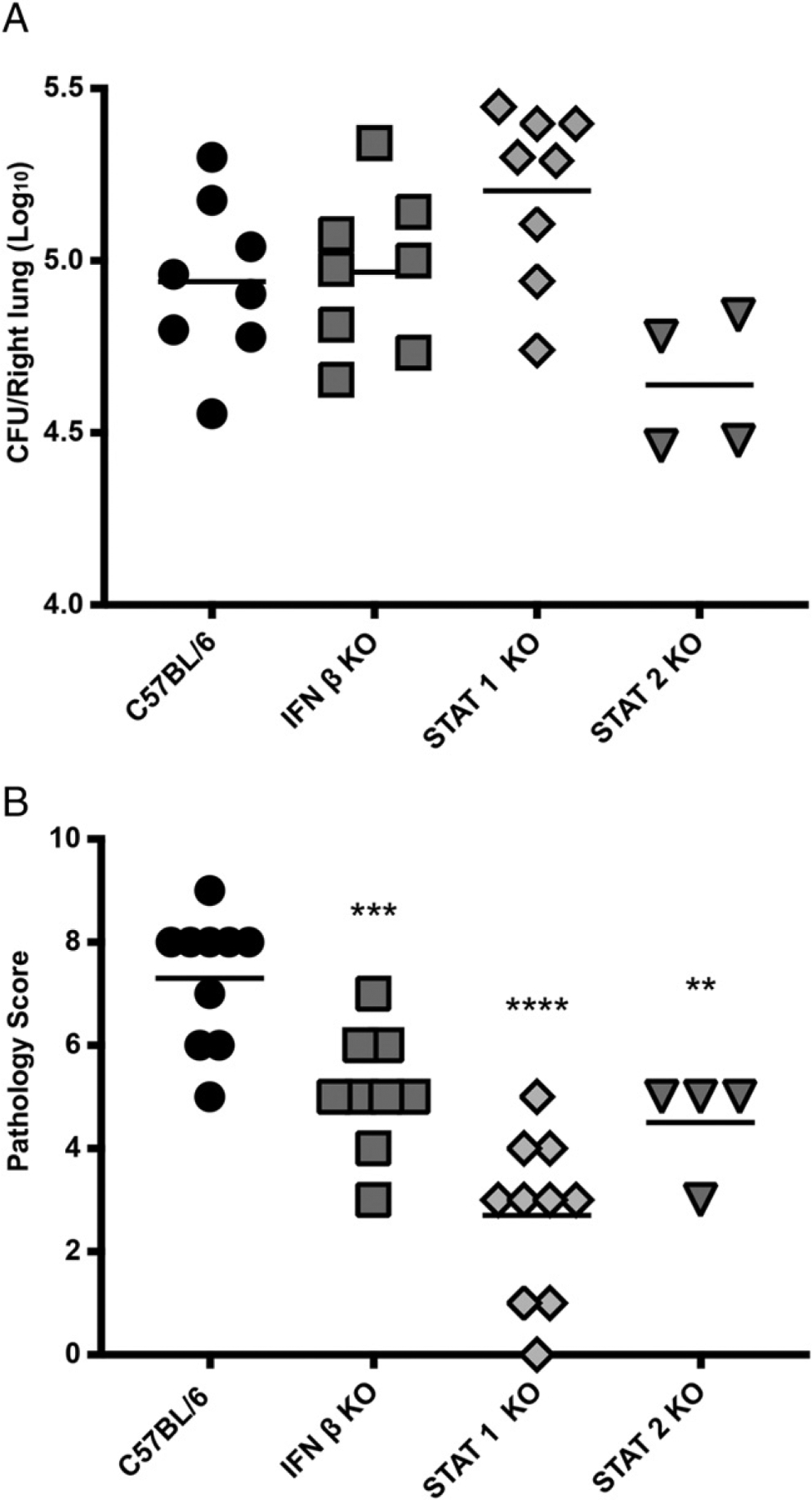
Impaired type I IFN signaling leads to a reduction in lung inflammation in adult mice. C57BL/6, IFN-β KO, STAT1 KO, and STAT2 KO mice (n ≥ 4 per group) were euthanized on day 7 postinoculation with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of outcomes. (A) Bacterial burdens. (B) Inflammatory pathology scores assessed from lung histology sections. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA.
IFNAR1 KO mice have similar levels of lung inflammation to WT mice when infected with B. pertussis
To obtain further support for a role of IFNAR signaling in lung inflammation during B. pertussis infection, we investigated outcomes of infection in IFNAR1 KO mice. B. pertussis–infected IFNAR1 KO adult mice displayed no significant difference in bacterial burden compared with WT mice (Fig. 5A). Surprisingly, IFNAR1 KO mice had equivalent levels of lung inflammatory pathology to WT mice at 7 and 10 dpi (Fig. 5B) and at any other time points examined (data not shown). Further investigation of host responses in infected IFNAR1 KO mice revealed significantly increased IFN-λ gene expression (~7-fold) compared with infected WT mice (Fig. 5C). This relative increase in IFN-λ expression was not observed in infected IFN-β, STAT1, and STAT2 KO mice (Fig. 5C), in which we had observed reduced levels of lung inflammatory pathology (in fact, in infected STAT1 and STAT2 KO mice, IFN-γ expression was significantly lower than WT levels, perhaps because IFN-λ amplifies expression of its own gene). In contrast, type I IFN gene expression was not significantly upregulated in infected IFNAR1 KO mice, although IFN-γ was expressed at WT levels (Supplemental Fig. 2A–C). We hypothesized that the increased expression of type III IFN could explain the WT levels of lung inflammation seen in the infected IFNAR1 KO mice, in effect “compensating” for the loss of type I IFN signaling. To test this hypothesis, IFNAR1 KO or WT mice were inoculated with B. pertussis and then treated 0, 2, and 4 dpi with a neutralizing anti–IFN-λ Ab and with a viral protein (Y136) previously shown to inhibit IFN-λ signaling (53). Control-infected mice were treated with an equivalent isotype Ab and BSA. Results from 7 dpi showed that, despite no effect on bacterial loads (Fig. 5D), infected IFNAR1 KO mice treated to reduce IFN-λ signaling had significantly reduced levels of lung inflammatory pathology compared with control-infected mice (Fig. 5E). Treated IFNAR1 KO mice also had reduced levels of IFN-λ gene upregulation (Fig. 5F), again indicating that IFN-λ amplified expression of its own gene. Treatment did not affect gene expression levels of the other IFN genes (Supplemental Fig. 2D–F). Because our RNA-seq analysis had shown that IFNLR1 (IFN-λR component) was one of the most significant upstream regulators of host transcriptional responses to infection (Fig. 1D), these findings together implicate type III IFN as an additional factor contributing to lung inflammatory pathology during B. pertussis infection.
FIGURE 5.
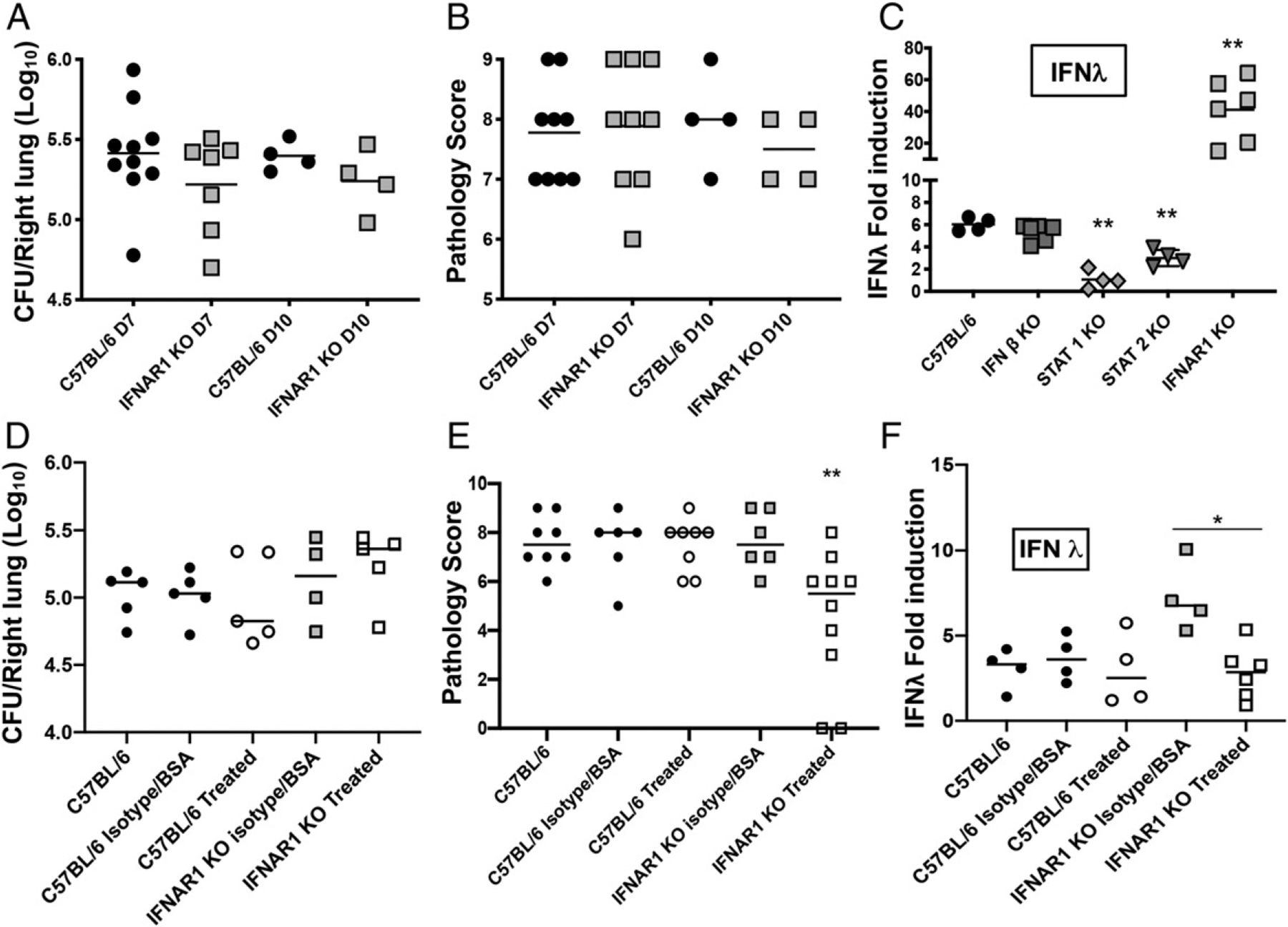
IFNAR1 KO mice show no significant difference in lung inflammatory pathology from WT mice, a role for type III IFNs. C57BL/6 and IFNAR1 KO mice (n ≥ 4 per group) were euthanized on day 7 or day 10 postinoculation with B. pertussis and lungs were dissected for assessment of (A) bacterial burdens, and (B) inflammatory pathology scores assessed from lung histology sections. (C) C57BL/6, IFN-β KO, STAT1 KO, STAT2 KO, and IFNAR1 KO mice (n ≥ 4 per group) were euthanized on day 7 postinoculation with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of fold induction (infected versus sham inoculated) of IFN-λ mRNA levels. (D–F) B. pertussis–infected C57BL/6 and IFNAR1 KO mice (n ≥ 4 per group) were treated on 0, 2, and 4 dpi with a neutralizing anti–IFN-λ Ab and a viral protein, Y136, previously shown to inhibit IFN-λ signaling (treated). Control-infected mice were treated with an equivalent isotype Ab and BSA. Lung bacterial burdens (D), inflammatory pathology (E), and IFN-λ mRNA levels (F) were assayed at 7 dpi. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA.
Type III IFNs contribute to lung inflammatory pathology in B. pertussis–infected adult mice
No previous information has been published on IFN-λ in B. pertussis infection, so our first step was to analyze the profile of expression throughout infection. We found that in infected adult WT mice, IFN-λ gene expression followed a similar pattern to that of type I IFNs, with upregulation starting after 4 dpi and peaking at 10 dpi (Fig. 6A). IFN-λ protein levels followed a similar pattern to those of type I IFN as well, with measurable levels seen from 4 dpi to 14 dpi, peaking at 10 dpi (Fig. 6B).
FIGURE 6.
Type III IFN is upregulated in response to B. pertussis infection and contributes to lung inflammation in adult mice. (A and B) C57BL/6 mice (n ≥ 4 per group) were euthanized on the indicated dpi (x-axis) with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of (A) fold induction (infected versus sham inoculated) of IFN-λ mRNA levels and (B) IFN-λ protein levels. (C and D) C57BL/6, IFNLR1 KO, or IFNAR1/IFNLR1 double KO mice were euthanized on day 7 postinoculation with B. pertussis, and lungs were dissected for assessment of (C) bacterial burdens and (D) inflammatory pathology. (E–H) Fold induction (infected versus sham inoculated) of mRNA levels of the indicated IFN genes at day 7 postinoculation with B. pertussis or PBS sham inoculum in adult C57BL/6, IFNLR1 KO, or IFNAR1/IFNLR1 double KO mice (n ≥ 4 per group). Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by Student t test and two-way ANOVA.
We reasoned that if the WT levels of lung inflammatory pathology observed in infected IFNAR1 KO mice (Fig. 5B) were due to the increased level of IFN-λ expression in the lungs of these mice (Fig. 5C), then mice deficient in both IFNAR1 and IFNLR1 should have reduced levels of lung inflammatory pathology because neither type I nor type III IFN signaling would occur. Upon infection of IFNAR1/IFNLR1 double KO mice, there were no significant differences in bacterial burden compared with those in WT mice (Fig. 6C). However, lung inflammatory pathology scores were significantly lower in the double KO mice than in WT mice (Fig. 6D). Interestingly, the same was true for IFNLR1 KO mice (in which type I IFN signaling would be functional), indicating that loss of type III IFN signaling alone is sufficient to reduce lung inflammatory pathology during B. pertussis infection. Analysis of IFN gene expression in the lungs of IFNLR1 KO and IFNAR1/IFNLR1 double KO mice revealed that neither type I nor type III IFN was significantly upregulated in response to infection at 7 dpi, in contrast to their robust upregulation in WT mice (Fig. 6E–G). Upregulation of IFN-γ gene expression in these mice was blunted compared with WT mice but was still significantly above background levels (Fig. 6H). These data indicate that type III IFNs contribute to lung inflammatory pathology in conjunction with type I IFNs but also that type III IFN signaling is necessary for upregulation of type I (and III) IFN gene expression in response to B. pertussis infection.
Increased IFNAR signaling protects infant mice from B. pertussis–induced lethality
The pathogenesis of B. pertussis infection in infant (7 d old) mice is markedly different from that in adult mice, as exemplified by higher bacterial loads in the lung, dissemination of infection to other organs, leukocytosis, and death (39). In addition, significant lung inflammation in B. pertussis–infected infant mice is not seen until just prior to death (around 2 wk postinoculation in our model), in contrast to the pronounced and prolonged lung inflammation in adult mice (39). To investigate whether differences in type I and III IFN expression and signaling between adult and infant mice might contribute to the contrasting outcomes of infection, we first analyzed the profile of type I and III IFN gene expression in the lungs of infant WT mice during B. pertussis infection. As shown in Fig. 7A and 7B, in contrast to the robust upregulation in adult mice, no significant transcriptional upregulation of IFN-α or IFN-λ was seen through 12 d of infection in infant mice, and gene expression was downregulated compared with sham-infected mice at some time points. There was a similar lack of transcriptional upregulation of IFN-β in infected infant mice (data not shown). However, type I IFN responses were activated in B. pertussis–infected IFNAR1-SA infant mice, with increased levels of expression of the ISGs, ISG15, and MX1 compared with infected WT mice (Fig. 7C, 7D). Type I and III IFN gene expression levels were equivalent to those in WT mice (data not shown), whereas upregulation of IFN-γ gene expression was blunted (Fig. 7E). Remarkably, infected IFNAR1-SA infant mice suffered a significantly lower level of lethality than infected WT infant mice (Fig. 8A), indicating that increased type I IFN signaling is protective for infant mice, in contrast to its deleterious effect in adult mice. B. pertussis–infected IFNAR1 KO infant mice died at the same time as WT mice (data not shown). At 7 dpi IFNAR1-SA infant mice had significantly lower levels of leukocytosis than WT infant mice (Fig. 8B), correlating with the reduced lethality in this group. IFNAR1-SA infant mice had similar bacterial loads in the lungs, liver, and spleen as WT mice, although the majority of IFNAR1-SA infant mice had no detectable bacteria in the blood, in contrast to the high levels in WT mice (Supplemental Fig. 3A). Inflammatory cytokine gene expression was generally lower in the lungs of infected IFNAR1-SA infant mice than in those of WT infant mice, although there was no difference in IL-6 expression (Supplemental Fig. 3B–D). Together, these data indicate that type I IFN signaling mediates overall protective effects against B. pertussis infection in infant mice and that a possible contributor to the susceptibility of infant mice to lethal pertussis is the lack of type I and III IFN gene upregulation in response to infection in these mice.
FIGURE 7.
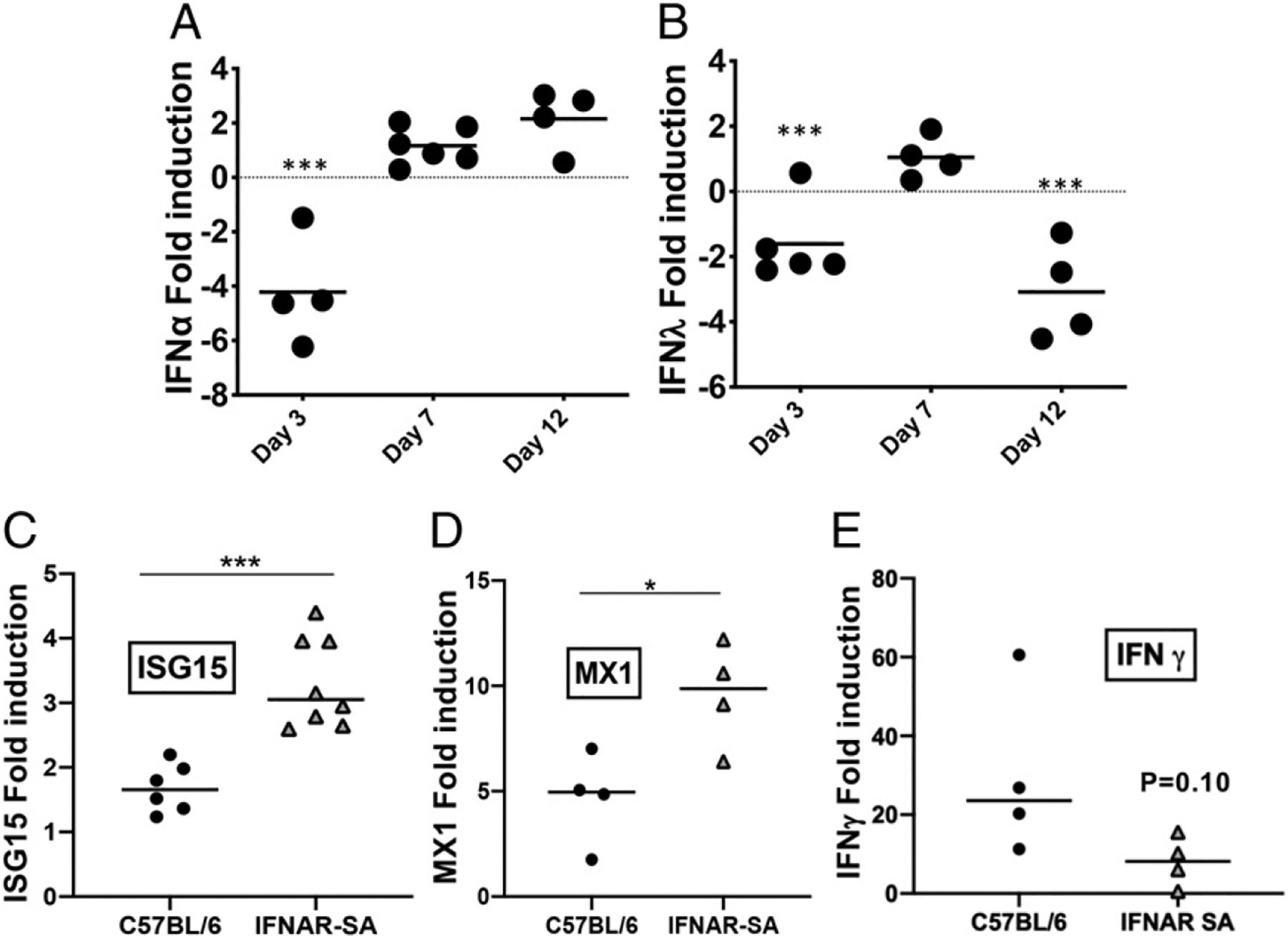
Type I and III IFNs are not upregulated by B. pertussis infection in infant WT mice. (A and B) Infant C57BL/6 mice (n ≥ 4 per group) were euthanized on the indicated dpi (x-axis) with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of fold induction (infected versus sham inoculated) of (A) IFN-α and (B) IFN-λ mRNA levels. (C–E) Infant C57BL/6 and IFNAR1-SA mice (n ≥ 6 per group) were euthanized on day 7 postinoculation with B. pertussis or PBS sham inoculum, and lungs were dissected for assessment of fold induction (infected versus sham inoculated) of the ISGs (C) ISG15 and (D) MX1 or (E) IFN-g mRNA levels. Data are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by Student t test.
FIGURE 8.
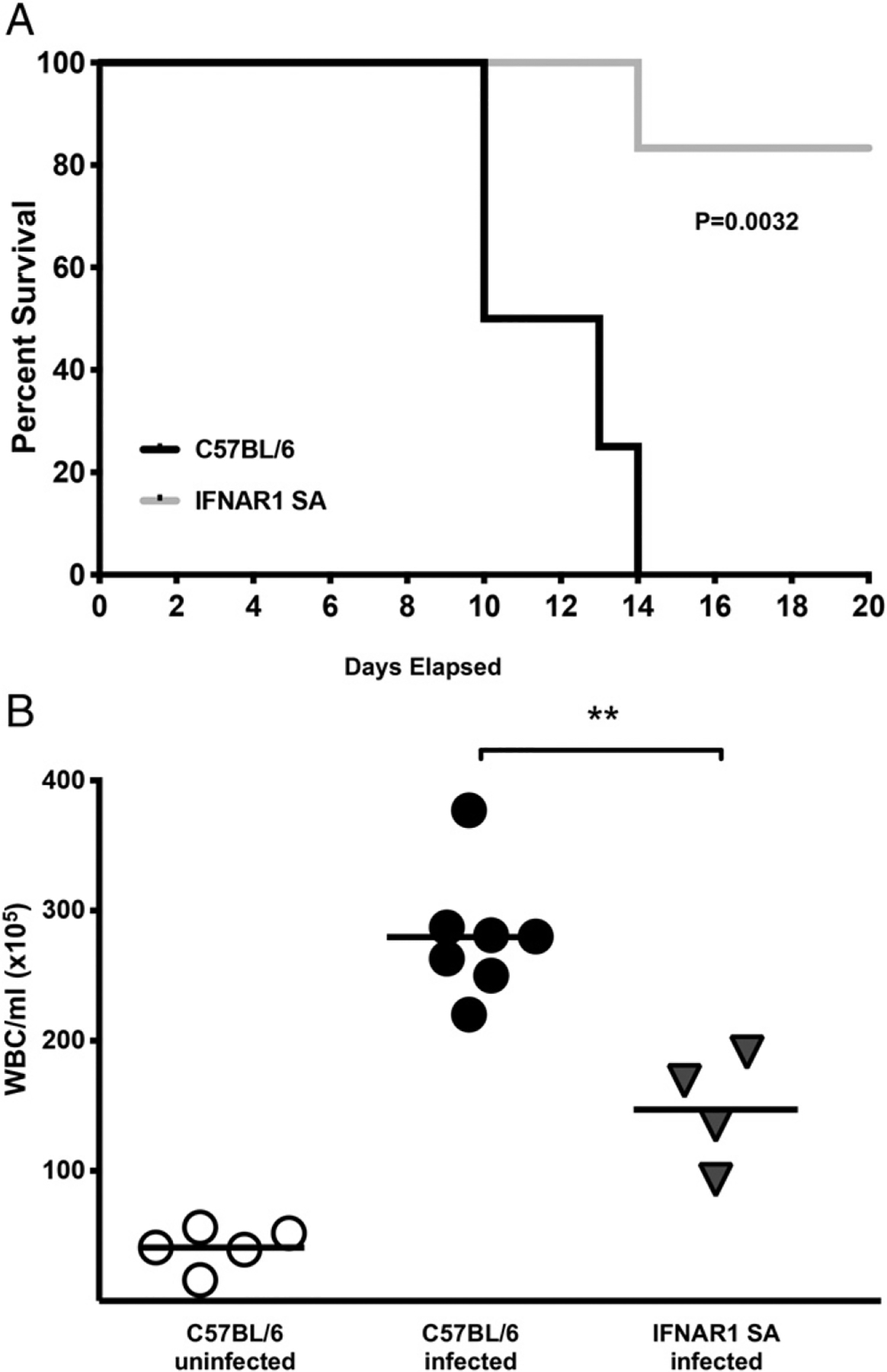
Increased IFNAR signaling is protective against lethal B. pertussis infection in infant mice. (A) Time course of survival of infant C57BL/6 and IFNAR1-SA mice after inoculation with B. pertussis. (B) Increase in WBC counts in blood of infant C57BL/6 and IFNAR1-SA mice 7 dpi with B. pertussis. Data are representative of at least two independent experiments. **p < 0.01, Student t test or log-rank (Mantel–Cox) test (for survival curves).
Discussion
In this study, we analyzed the transcriptome of adult mouse lung responses to B. pertussis infection and identified IFNAR1 and IFNLR1 as receptors involved in responses to this infection. We showed that type I and III IFNs are produced in mouse lungs in response to B. pertussis infection, but that upregulation of these responses is delayed, initiating several days into the infection. In addition, type I and III IFN levels and signaling correlate with increased proinflammatory cytokine levels and with increased lung inflammatory pathology. In contrast, in infant mice, upregulation of type I and III IFN expression in lungs was not observed in response to B. pertussis infection, and increased type I IFN signaling was protective for these mice against lethal infection.
Our results from transcriptomic analysis of lung responses to B. pertussis infection at 4 dpi identified gene expression responses that were dominated by acute phase proinflammatory and antibacterial functions as observed in other such studies (54, 55). However, we were surprised to identify IFNAR1 and IFNLR1 as highly significant upstream activators of these responses. There have been very few reports of the involvement of type I IFN (and none of type III IFN) in responses to B. pertussis infection. In one study, IFN-α–producing lung plasmacytoid dendritic cells were found to suppress Th17 cell differentiation during early B. pertussis infection in adult mice (26), indicating a possible role for type I IFNs in shaping host immune responses to this infection. We found that B. pertussis infection in adult mice induced type I and III IFN gene upregulation in the lungs but that the initiation of these responses was significantly delayed, starting after 4 dpi, in comparison with typical early type I and III IFN responses in viral infections (56) (we found no upregulation of type I and III IFN genes at 1–3 dpi; data not shown). The mechanism of induction of this delayed response is unknown, but we speculate that initiation of inflammatory pathology in the lungs may provide a source of molecules that induce these responses. One possible inducer is unmethylated CpG DNA, the detection of which by TLR9 in endosomes is a major pathway for induction of type I IFN expression (57). In contrast to CpG-rich bacterial DNA, mammalian DNA has few unmethylated CpG sequences, although they are enriched in so-called CpG islands, particularly associated with gene promoters (58). Although mammalian DNA released after tissue damage during infection does not easily gain access to endosomes, studies have shown that mammalian self-DNA can be delivered to endosomes and trigger type I IFN production by complexing with antimicrobial peptides such as LL37 or β-defensins (59, 60). Therefore, the delay in upregulation of type I and III responses may correspond with pathology-induced release of host DNA in the inflammatory environment of the lungs, although we cannot rule out a contribution from bacterial DNA. Our studies indicate the presence of significant levels of free DNA in the lungs of B. pertussis–infected adult mice and a possible contribution of TLR9 to lung inflammation in this model (J. Ardanuy, A. Migneault, and N. Carbonetti, unpublished observations). Further investigation will be needed to establish this pathway as an inducer of type I and III IFN responses during B. pertussis infection.
Type I IFNs have diverse effects on different bacterial infections in experimental models, being protective for some but detrimental for others (19). Our study shows that type I IFN responses have no effect on bacterial loads in the lung but promote lung inflammatory pathology in adult mice, putting B. pertussis infection in the category for which type I IFN responses are detrimental. This conclusion is supported by our results from both IFNAR1-SA mice, in which increased type I IFN signaling correlated with increased lung inflammatory pathology, and various mutant mouse strains deficient in one or another aspect of type I IFN signaling, in which lower levels of lung inflammatory pathology were observed. For the latter, it is interesting that STAT1-deficient mice had the lowest levels of pathology because STAT1 is a transcription factor common to type I, II, and III IFN signaling (although STAT1 also mediates signaling through other receptors). This suggests that all three IFNs may contribute to lung inflammatory pathology, including IFN-γ, which is highly upregulated in the lungs of infected WT adult mice, further upregulated in infected IFNAR1-SA adult mice, and which may account for some of the remaining pathology in the IFNR KO mice.
Surprisingly, the one KO mouse strain in which no reduction in lung inflammatory pathology was observed was the IFNAR1 KO mouse, but this is due to the increased expression of type III IFN in response to B. pertussis infection in these mice. This is supported by our findings that inhibition of type III IFN signaling in these mice significantly reduced lung inflammatory pathology and also by the IFNLR KO and IFNAR1/IFNLR1 double KO mouse experiments in which lower levels of lung pathology were observed. This indicates that there may be redundancy and overlap of type I and III IFN responses in terms of downstream signaling leading to inflammatory pathology. Consistent with this, we found that type III IFN gene upregulation in response to B. pertussis infection in adult mice had an identical temporal profile to that of type I IFN. Although type I and III IFNs signal through the same intracellular pathways, differential cellular expression of their receptors is thought to mediate different outcomes of their signaling (56). Interestingly, for influenza virus infection, type III IFN plays a predominantly anti-inflammatory role in the lungs (61), in contrast to its apparent proinflammatory effect in our model. In addition, our observation that loss of type I IFN signaling led to increased type III IFN gene expression, whereas loss of type III IFN signaling abrogated type I (and III) IFN gene upregulation in B. pertussis–infected adult mice indicates a complex regulatory interplay between the two IFNs in this infection. These data suggest that type III IFN signaling is required for type I IFN induction and that type I IFN signaling inhibits type III IFN expression. In a fungal infection model, type I IFNs were found to prime optimal expression of type III IFNs (62), but the opposite appears to be the case in our model. Further investigation of this cross-regulation and the responses downstream of type I and III IFN signaling, including roles of various ISGs that are upregulated, will be necessary to elucidate specific mechanisms leading to their upregulated expression and to their exacerbation of lung inflammatory pathology during B. pertussis infection.
Our results indicating the protective effects of type I IFN signaling in B. pertussis–infected infant mice are in stark contrast to the deleterious effects in adult mice and demonstrate age dependence in outcome of these responses. The lack of upregulation of type I and III IFN genes in response to B. pertussis infection in infant mice may contribute to their increased susceptibility to lethal infection (and likely accounts for the observation that IFNAR1 KO infant mice show no difference from WT mice in time to death). Our finding that IFNAR1-SA infant mice are relatively resistant to lethal infection and have reduced levels of pathogenic outcomes is consistent with this idea. IFN-α administration was shown to protect infant mice against respiratory syncytial virus infection and overcome age-related differences in IgA production in response to the infection (63). In another study, age-dependent effects of type I IFNs were demonstrated by the observation that type I IFNs protected infant mice treated with TLR ligands from lethal inflammatory responses, whereas they enhanced the same inflammatory response in adult mice (64). In addition, several studies with human cord blood versus adult cells indicate a deficiency in type I IFN responses to infection or TLR agonists in the infant cells (65–67), consistent with the lack of responses we observed in our infected infant mice.
The implications of these findings are very interesting because these identified pathways could potentially be targeted postexposure to B. pertussis in combination with antibiotic therapy, either to reduce symptoms and severity of disease in adults or to enhance protection from severe disease in infants. Because type I and III IFN responses to B. pertussis infection serve no apparent antibacterial role, evidenced by the lack of effects on bacterial loads in the lungs in the various mouse strains, they may be valid targets for therapeutic intervention to reduce respiratory pathology in typical pertussis. We have previously shown that treatment of B. pertussis–infected adult mice with S1P receptor–targeting drugs significantly reduces lung inflammatory pathology (11, 12) and that a proposed anti-inflammatory mechanism of these drugs involves internalization and degradation of surface expressed IFNAR1 (52). We previously found that one of these drugs, AAL-R, abrogates type I IFN gene upregulation in a human monocyte cell line exposed to B. pertussis (11), and our present study, showing a lack of anti-inflammatory effect of AAL-R treatment in B. pertussis–infected IFNAR1-SA mice, is also consistent with the proposed role of S1P receptor–targeting drugs in downregulating type I IFN responses. Therefore, these and other drugs that target type I and III IFN responses may prove beneficial in treatment of individuals suffering from pertussis. In the bigger picture, this approach can potentially be applied to other bacterial infections in which type I IFN signaling is either detrimental or beneficial to provide therapy additional to antibiotic treatment.
Supplementary Material
Acknowledgments
We thank Carbonetti laboratory members for helpful feedback on this project, the Informatics Resource Center at the Institute for Genome Sciences for help with RNA-seq, and Sergei Kotenko and Stefanie Vogel for supplying mice.
This work was supported by National Institutes of Health Grants AI101055, AI135465, and AI141372 (to N.H.C.), F31AI136377 (to J.A.), and CA092900 (to S.Y.F.).
Abbreviations used in this article:
- dpi
day postinoculation
- IFNAR
IFN-αR
- IFNAR1-SA
Ifnar1S526A
- IFNLR1
type III IFN receptor
- IPA
Ingenuity Pathway Analysis
- ISG
IFN-stimulated gene
- KO
knockout
- RNA-seq
RNA-sequencing
- S1P
sphingosine-1–phosphate
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Clark TA 2014. Changing pertussis epidemiology: everything old is new again. J. Infect. Dis 209: 978–981. [DOI] [PubMed] [Google Scholar]
- 2.Heininger U, Klich K, Stehr K, and Cherry JD. 1997. Clinical findings in Bordetella pertussis infections: results of a prospective multicenter surveillance study. Pediatrics 100: E10. [DOI] [PubMed] [Google Scholar]
- 3.Mattoo S, and Cherry JD. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev 18: 326–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paddock CD, Sanden GN, Cherry JD, Gal AA, Langston C, Tatti KM, Wu KH, Goldsmith CS, Greer PW, Montague JL, et al. 2008. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin. Infect. Dis 47: 328–338. [DOI] [PubMed] [Google Scholar]
- 5.Somerville RL, Grant CC, Grimwood K, Murdoch D, Graham D, Jackson P, Meates-Dennis M, Nicholson R, and Purvis D. 2007. Infants hospitalised with pertussis: estimating the true disease burden. J. Paediatr. Child Health 43: 617–622. [DOI] [PubMed] [Google Scholar]
- 6.Winter K, Zipprich J, Harriman K, Murray EL, Gornbein J, Hammer SJ, Yeganeh N, Adachi K, and Cherry JD. 2015. Risk factors associated with infant deaths from pertussis: a case-control study. Clin. Infect. Dis 61: 1099–1106. [DOI] [PubMed] [Google Scholar]
- 7.Klein NP, Bartlett J, Rowhani-Rahbar A, Fireman B, and Baxter R. 2012. Waning protection after fifth dose of acellular pertussis vaccine in children. N. Engl. J. Med 367: 1012–1019. [DOI] [PubMed] [Google Scholar]
- 8.Altunaiji S, Kukuruzovic R, Curtis N, and Massie J. 2007. Antibiotics for whooping cough (pertussis). Cochrane Database Syst. Rev (3): CD004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Bettiol S, Thompson MJ, Roberts NW, Perera R, Heneghan CJ, and Harnden A. 2014. Symptomatic treatment of the cough in whooping cough. Cochrane Database Syst. Rev (9): CD003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scanlon KM, Gau Y, Zhu J, Skerry C, Wall SM, Soleimani M, and Carbonetti NH. 2014. Epithelial anion transporter pendrin contributes to inflammatory lung pathology in mouse models of Bordetella pertussis infection. Infect. Immun 82: 4212–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skerry C, Scanlon K, Ardanuy J, Roberts D, Zhang L, Rosen H, and Carbonetti NH. 2017. Reduction of pertussis inflammatory pathology by therapeutic treatment with sphingosine-1-phosphate receptor ligands by a pertussis toxin-insensitive mechanism. J. Infect. Dis 215: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skerry C, Scanlon K, Rosen H, and Carbonetti NH. 2015. Sphingosine-1-phosphate receptor agonism reduces Bordetella pertussis-mediated lung pathology. J. Infect. Dis 211: 1883–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SH, Cohen B, Novick D, and Rubinstein M. 1997. Mammalian type I interferon receptors consists of two subunits: IFNaR1 and IFNaR2. Gene 196: 279–286. [DOI] [PubMed] [Google Scholar]
- 14.Rauch I, Müller M, and Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAK-STAT 2: e23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carrero JA 2013. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int. Immunol 25: 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crow YJ 2011. Type I interferonopathies: a novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci 1238: 91–98. [DOI] [PubMed] [Google Scholar]
- 17.Davidson S, Maini MK, and Wack A. 2015. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J. Interferon Cytokine Res 35: 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McNab F, Mayer-Barber K, Sher A, Wack A, and O’Garra A. 2015. Type I interferons in infectious disease. Nat. Rev. Immunol 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovarik P, Castiglia V, Ivin M, and Ebner F. 2016. Type I interferons in bacterial infections: a balancing act. Front. Immunol 7: 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macrì G, Ruggeri A, et al. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol 178: 3126–3133. [DOI] [PubMed] [Google Scholar]
- 21.Schiavoni G, Mauri C, Carlei D, Belardelli F, Pastoris MC, and Proietti E. 2004. Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J. Immunol 173: 1266–1275. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, Chiba T, Fuss IJ, Kitani A, and Strober W. 2010. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest 120: 1645–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE III, Freedman VH, and Kaplan G. 2001. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. USA 98: 5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, et al. 2004. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med 200: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin FJ, Gomez MI, Wetzel DM, Memmi G, O’Seaghdha M, Soong G, Schindler C, and Prince A. 2009. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest 119: 1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu V, Smith AA, You H, Nguyen TA, Ferguson R, Taylor M, Park JE, Llontop P, Youngman KR, and Abramson T. 2016. Plasmacytoid dendritic cell-derived IFNa modulates Th17 differentiation during early Bordetella pertussis infection in mice. Mucosal Immunol. 9: 777–786. [DOI] [PubMed] [Google Scholar]
- 27.Schroder K, Hertzog PJ, Ravasi T, and Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 28.Andreasen C, Powell DA, and Carbonetti NH. 2009. Pertussis toxin stimulates IL-17 production in response to Bordetella pertussis infection in mice. PLoS One 4: e7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahon BP, Sheahan BJ, Griffin F, Murphy G, and Mills KH. 1997. Atypical disease after Bordetella pertussis respiratory infection of mice with targeted disruptions of interferon-gamma receptor or immunoglobulin mu chain genes. J. Exp. Med 186: 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skerry CM, Cassidy JP, English K, Feunou-Feunou P, Locht C, and Mahon BP. 2009. A live attenuated Bordetella pertussis candidate vaccine does not cause disseminating infection in gamma interferon receptor knockout mice. Clin. Vaccine Immunol 16: 1344–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donnelly RP, and Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J. Interferon Cytokine Res 30: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, Mendoza JL, Garcia KC, McLauchlan J, Höfer T, Stanifer ML, and Boulant S. 2018. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 14: e1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanoni I, Granucci F, and Broggi A. 2017. Interferon (IFN)-λ takes the helm: immunomodulatory roles of type III IFNs. Front. Immunol 8: 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen TS, and Parker D. 2016. Microbial pathogenesis and type III interferons. Cytokine Growth Factor Rev. 29: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotenko SV, and Durbin JE. 2017. Contribution of type III interferons to antiviral immunity: location, location, location. J. Biol. Chem 292: 7295–7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Syedbasha M, and Egli A. 2017. Interferon lambda: modulating immunity in infectious diseases. Front. Immunol 8: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawal M, Cohen M, Irazuzta JE, Kumar R, Kirton C, Brundler MA, Evans CA, Wilson JA, Raffeeq P, Azaz A, et al. 2009. Fulminant pertussis: a multi-center study with new insights into the clinico-pathological mechanisms. Pediatr. Pulmonol 44: 970–980. [DOI] [PubMed] [Google Scholar]
- 38.Zimmerman LI, Papin JF, Warfel J, Wolf RF, Kosanke SD, and Merkel TJ. 2018. Histopathology of Bordetella pertussis in the baboon model. Infect. Immun DOI: 10.1128/IAI.00511-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scanlon KM, Snyder YG, Skerry C, and Carbonetti NH. 2017. Fatal pertussis in the neonatal mouse model is associated with pertussis toxin-mediated pathology beyond the airways. Infect. Immun DOI: 10.1128/IAI.00355-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carbonetti NH, Artamonova GV, Mays RM, and Worthington ZE. 2003. Pertussis toxin plays an early role in respiratory tract colonization by Bordetella pertussis. Infect. Immun 71: 6358–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, Yu Q, Zheng H, Carbone CJ, Katlinskaya YV, et al. 2014. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol. Med 6: 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruys E, Toussaint MJ, Niewold TA, and Koopmans SJ. 2005. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. B 6: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, et al. 2013. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 110: 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu SY, Sanchez DJ, Aliyari R, Lu S, and Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 109: 4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang CH, Wei L, Pfeffer SR, Du Z, Murti A, Valentine WJ, Zheng Y, and Pfeffer LM. 2007. Identification of CXCL11 as a STAT3-dependent gene induced by IFN. J. Immunol 178: 986–992. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs SY 2012. Ubiquitination-mediated regulation of interferon responses. Growth Factors 30: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar KG, Krolewski JJ, and Fuchs SY. 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J. Biol. Chem 279: 46614–46620. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, Grigoriadou C, Aldabe R, Diehl JA, and Fuchs SY. 2009. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5: 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, Brice A, Davar D, Sanders C, Kirkwood JM, Rui H, et al. 2016. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 15: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. 2017. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, Carbone CJ, Yu Q, Katlinski KV, Katlinskaya YV, et al. 2019. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell 35: 33–45.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teijaro JR, Studer S, Leaf N, Kiosses WB, Nguyen N, Matsuki K, Negishi H, Taniguchi T, Oldstone MB, and Rosen H. 2016. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-a autoamplification. Proc. Natl. Acad. Sci. USA 113: 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J, Smirnov SV, Lewis-Antes A, Balan M, Li W, Tang S, Silke GV, Pütz MM, Smith GL, and Kotenko SV. 2007. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc. Natl. Acad. Sci. USA 104: 9822–9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banus S, Pennings J, Vandebriel R, Wester P, Breit T, Mooi F, Hoebee B, and Kimman T. 2007. Lung response to Bordetella pertussis infection in mice identified by gene-expression profiling. Immunogenetics 59: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Connelly CE, Sun Y, and Carbonetti NH. 2012. Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection. Infect. Immun 80: 4317–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazear HM, Schoggins JW, and Diamond MS. 2019. Shared and distinct functions of type I and type III interferons. Immunity 50: 907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bao M, and Liu YJ. 2013. Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. Protein Cell 4: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deaton AM, and Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev. 25: 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, Chamilos G, Feldmeyer L, Marinari B, Chon S, et al. 2014. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. [Published erratum appears in 2015 Nat. Commun. 6: 6595] Nat. Commun. 5: 5621. [DOI] [PubMed] [Google Scholar]
- 60.Tewary P, de la Rosa G, Sharma N, Rodriguez LG, Tarasov SG, Howard OM, Shirota H, Steinhagen F, Klinman DM, Yang D, and Oppenheim JJ. 2013. β-Defensin 2 and 3 promote the uptake of self or CpG DNA, enhance IFN-α production by human plasmacytoid dendritic cells, and promote inflammation. J. Immunol 191: 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, and Andreakos E. 2017. Interferon-l mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46: 875–890.e6. [DOI] [PubMed] [Google Scholar]
- 62.Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, Pitler A, Whitehead I, Obar JJ, Durbin JE, et al. 2017. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol DOI: 10.1126/sciimmunol.aan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hijano DR, Siefker DT, Shrestha B, Jaligama S, Vu LD, Tillman H, Finkelstein D, Saravia J, You D, and Cormier SA. 2018. Type I interferon potentiates IgA immunity to respiratory syncytial virus infection during infancy. Sci. Rep 8: 11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Deriaud E, Jiao X, Braun D, Leclerc C, and Lo-Man R. 2007. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J. Exp. Med 204: 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Danis B, George TC, Goriely S, Dutta B, Renneson J, Gatto L, Fitzgerald-Bocarsly P, Marchant A, Goldman M, Willems F, and De Wit D. 2008. Interferon regulatory factor 7-mediated responses are defective in cord blood plasmacytoid dendritic cells. Eur. J. Immunol 38: 507–517. [DOI] [PubMed] [Google Scholar]
- 66.Lissner MM, Thomas BJ, Wee K, Tong AJ, Kollmann TR, and Smale ST. 2015. Age-related gene expression differences in monocytes from human neonates, young adults, and older adults. PLoS One 10: e0132061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang B, Kang W, Zuo J, Kang W, and Sun Y. 2017. The significance of type-I interferons in the pathogenesis and therapy of human immunodeficiency virus 1 infection. Front. Immunol 8: 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.