

Abstract
The 60-year high Bordetella pertussis (B. pertussis) infection rate in the world suggests the urgent need for new anti-pertussis vaccines. Lipopolysaccharide (LPS) of B. pertussis is an attractive antigen for vaccine development. With the presence of multiple rare sugars and unusual glycosyl linkages, B. pertussis LPS is a highly challenging synthetic target. In this work, aided by molecular dynamics simulation and modeling, the pertussis LPS like pentasaccharide was chemically synthesized for the first time. The pentasaccharide was conjugated with a powerful carrier, bacteriophage Qβ as a vaccine candidate. Immunization of mice with the conjugate induced robust anti-glycan IgG responses with IgG titers reaching several million enzyme-linked immunosorbent assay (ELISA) units. The antibodies generated were long lasting and boostable, and could recognize multiple clinical strains of B. pertussis, highlighting the potential of Qβ-glycan as a new anti-pertussis vaccine.
Keywords: bacteriophage Qβ, lipo-oligosaccharides, pertussis, synthesis, vaccines
Graphical Abstract
For the first time, the pertussis LPS like pentasaccharide bearing multiple rare sugar subunits was synthesized with high stereoselectivities and overall yield. Conjugation of the pentasaccharide with the carrier protein Qβ yielded a vaccine candidate, which induced anti-LPS antibodies in mice with broad-spectrum recognitions for B. pertussis strains.
Introduction
Pertussis is a highly contagious respiratory disease caused primarily by the infection of a Gram-negative bacterium Bordetella pertussis (B. pertussis).[1–2] However, despite the availability of vaccines such as the acellular vaccines (aPVs), 24.1 million infections and 160,700 deaths have been estimated worldwide in children younger than 5 years alone each year due to pertussis.[3] Even in developed countries such as US and Australia where the vaccination rates are high, the number of reported cases of pertussis has markedly risen during recent decades, reaching a 60 year high.[4–5] Similar trends have also been observed in Europe, UK and Canada.[6–7] Several major factor contribute to the rise of the pertussis infections. These include that some current circulating B. pertussis strains are deficient in the production of certain vaccine targets resulting in probable escape from vaccine-induced immunity.[8–9] Furthermore, the current acellular vaccine does not prevent infections and the immunity induced can only last a short time.[10–12] Therefore, there are urgent needs for a new generation of anti-pertussis vaccines.
B. pertussis expresses lipopolysaccharide (LPS) endotoxins, which are appealing targets for vaccines.[13–14] LPS can induce nitric oxide production and contribute to disease pathology by damaging respiratory ciliated cells.[15] Unlike antibodies against the exotoxins such as pertussis toxin, anti-LPS IgG antibodies can confer complement-dependent cytotoxicity against B. pertussis.[16] The antibodies could also overcome pertussis’s resistance to bactericidal activities.[16] In addition, studies have shown that the LPS structure is highly conserved across various B. pertussis strains and does not change in clinical isolates from pre- and post-vaccine era,[17] rendering it attractive for broad spectrum anti-pertussis vaccine development.
LPS of B. pertussis are comprised of lipid A linked with a branched dodeca-saccharide.[13] The pentasaccharide sequence at the non-reducing end of LPS can bind with an anti-LPS monoclonal antibody, suggesting it may be a suitable epitope target for vaccines.[18] The pertussis pentasaccharide has multiple unique structural features, which include three rare monosaccharides, i.e., 2,3-diacetamido-2,3-dideoxy D-mannuronic acid, 2-acetamido-2,4,6-trideoxy-4-methylamino L-galactose, and L-glycero-D-manno-heptose. In addition, it contains multiple 1,2-cis glycosyl linkages, which are generally difficult to form with high selectivity. With its complex structure, its synthesis has not been reported. Herein, we describe the first total synthesis of pertussis-like pentasaccharide 1 and the immunological evaluation as a potential anti-pertussis vaccine.
Results and Discussion
The synthetic design of the target pentasaccharide 1 was based on several considerations (Scheme 1). For the CDE branching trisaccharide, we envision the E unit should be installed onto D first before C as the 4-OH of D may be too hindered if the 6-OH of D is glycosylated first. To facilitate the formation of 1,2-trans linkage between C and D, Troc, which is regarded as a participating neighboring group,[19–20] was used as the N-protective group for 2-amine of C unit fucosamine (building block 4). The glycosyl linkage between amino-mannuronic acid B and fucosamine C was challenging to form directly due to the generally low reactivities of uronic acid donors and the 1,2-cis stereochemistry of the linkage. We opted for an indirect route using the 3-amino glucose derivative 5 as surrogates of amino-mannuronic acid. The 2-O-Bn and 2-N3 bearing building blocks 2 and 6 were designed to facilitate the formation of the α-glycosyl linkages between E-D and A-B units.
Scheme 1.
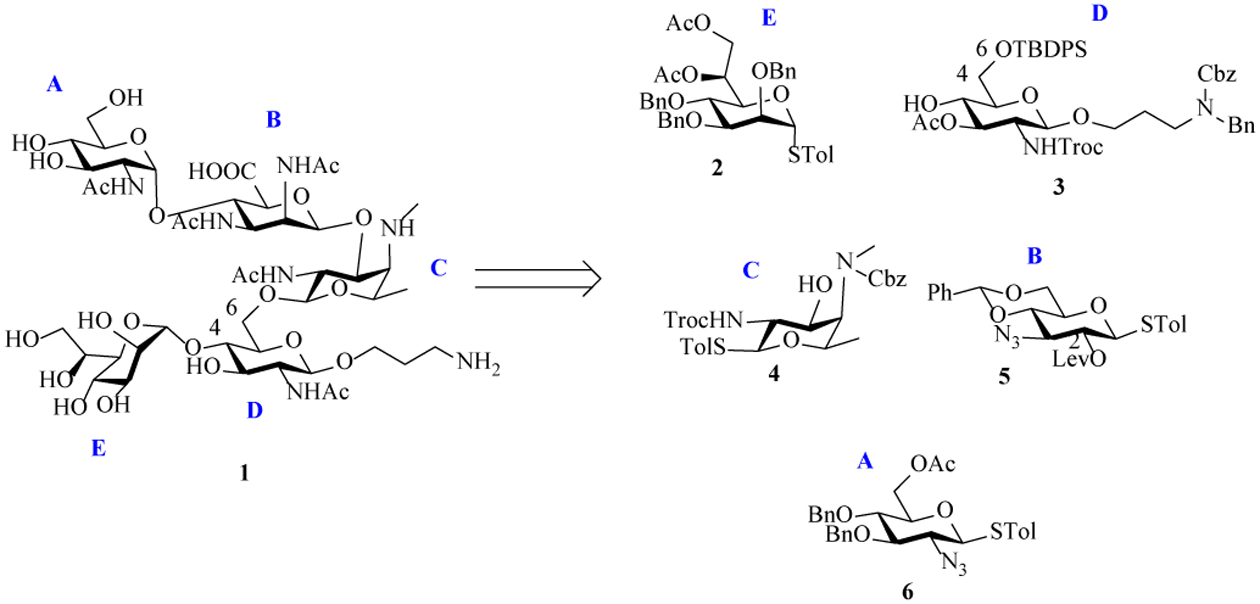
Retrosynthetic analysis of target pentasaccharide 1.
Our synthesis commenced with the preparation of building blocks for the rare sugars (2, 4, 5) in the pentasaccharide. Acid-catalyzed ketal hydrolysis of the known 3-azido glucose derivative 7[21] followed by global acetylation and treatment with p-toluenethiol (p-TolSH) and boron trifluoride etherate (BF3 Et2O) gave the thioglycoside 8 in 54% yield over 3 steps (Scheme 2). All O-acetyl groups of 8 were removed with NaOMe and 4, 6-benzylidene was installed to afford 9 in 81% yield. Protection of the free 2-OH with levulinic acid (LevOH) aided by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC HCl) and 4-dimethylaminopyridine (DMAP) yielded the first rare sugar building block 5.
Scheme 2.
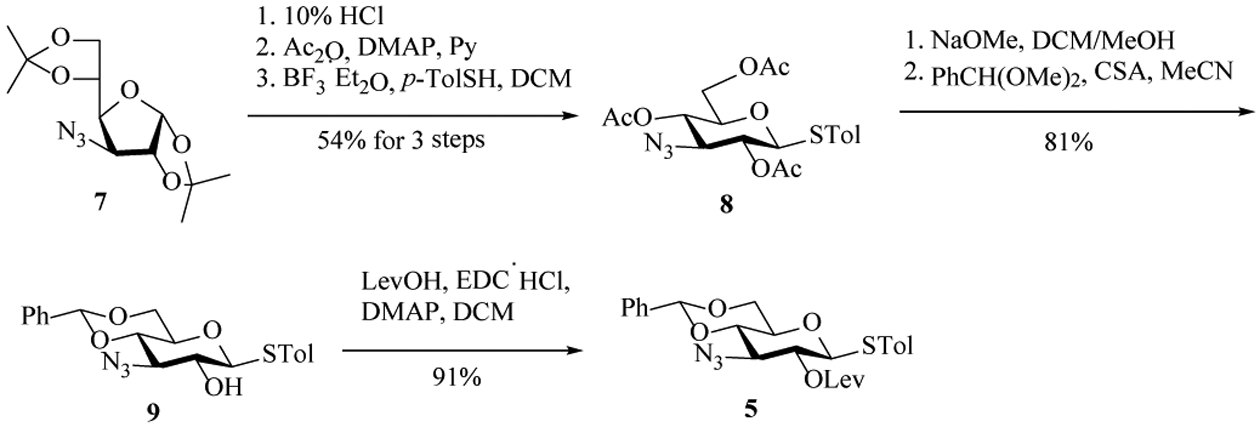
Preparation of building block 5.
Preparation of the 2-acetamido-4-amino-2,4,6-trideoxygalactose (AAT) presented significant difficulties. The first route we undertook started with bis-triflation of α-rhamnoside 10[22] to be followed by SN2 displacement of O-triflates with sodium azide. However, triflation resulted in a low yield of the desired bis-triflate with a major side product isolated containing the STol migrated to C-2 presumably due to the 1,2-trans arrangement of the α-STol and the presumed intermediate bearing 2-OTf. To overcome this, the β-rhamnoside 11[23] was prepared, which was selectively protected at the 3-OH with benzoyl (Bz) and triflated at the 2- and 4-OH (Scheme 3). Sequential displacement of the two triflates by sodium azide and potassium phthalimide gave the fucosamine analog 13 in a moderate 30–50% yield. Deprotection of the phthalimide and Bz groups with hydrazine produced the free amine 14, albeit with a relatively low 30% yield.
Scheme 3.
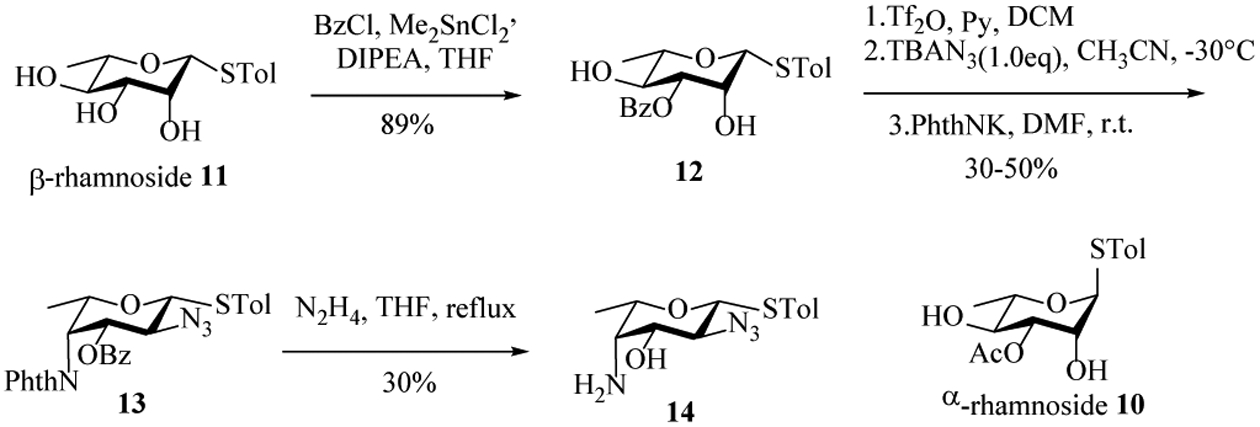
Preparation of fucosamine building block 14.
To improve the yield, we explored trichloroacetimidate as the protecting group[24] of 3-OH of β-rhamnoside 11 rather than Bz so that C4 inversion could be achieved in situ upon triflation (Scheme 4). The thioglycoside 11 was treated with trichloroacetonitrile and 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) giving 15 in a yield of 90%. The imidate 15 was subjected to trifluoromethanesulfonic anhydride (Tf2O) and pyridine at −30 °C, which formed 3,4-oxazoline ring spontaneously following nucleophilic displacement of 4-OTf by the neighboring imidate. Displacement of the 2-OTf with sodium azide in DMF at 50 °C afforded 16 in an excellent yield of 85% over 2 steps. Refluxing 16 in 4M hydrochloric acid opened the oxazoline ring and gave 14 in 89% yield with the two amine moieties on the ring differentiated. Among multiple methods investigated, monomethylation of 4-NH2 of 14 was best performed by refluxing 14 in ethyl formate and trimethylamine followed by lithium aluminum hydride reduction, which was accompanied by simultaneous reduction of the 2-N3 to amine. Selective protection of equatorial primary 2-NH2 with trichloroethyl chloroformate (TrocCl) yielded 18, which was then treated with benzyl chloroformate giving the fucosamine building block 4 in 75% yield.
Scheme 4.
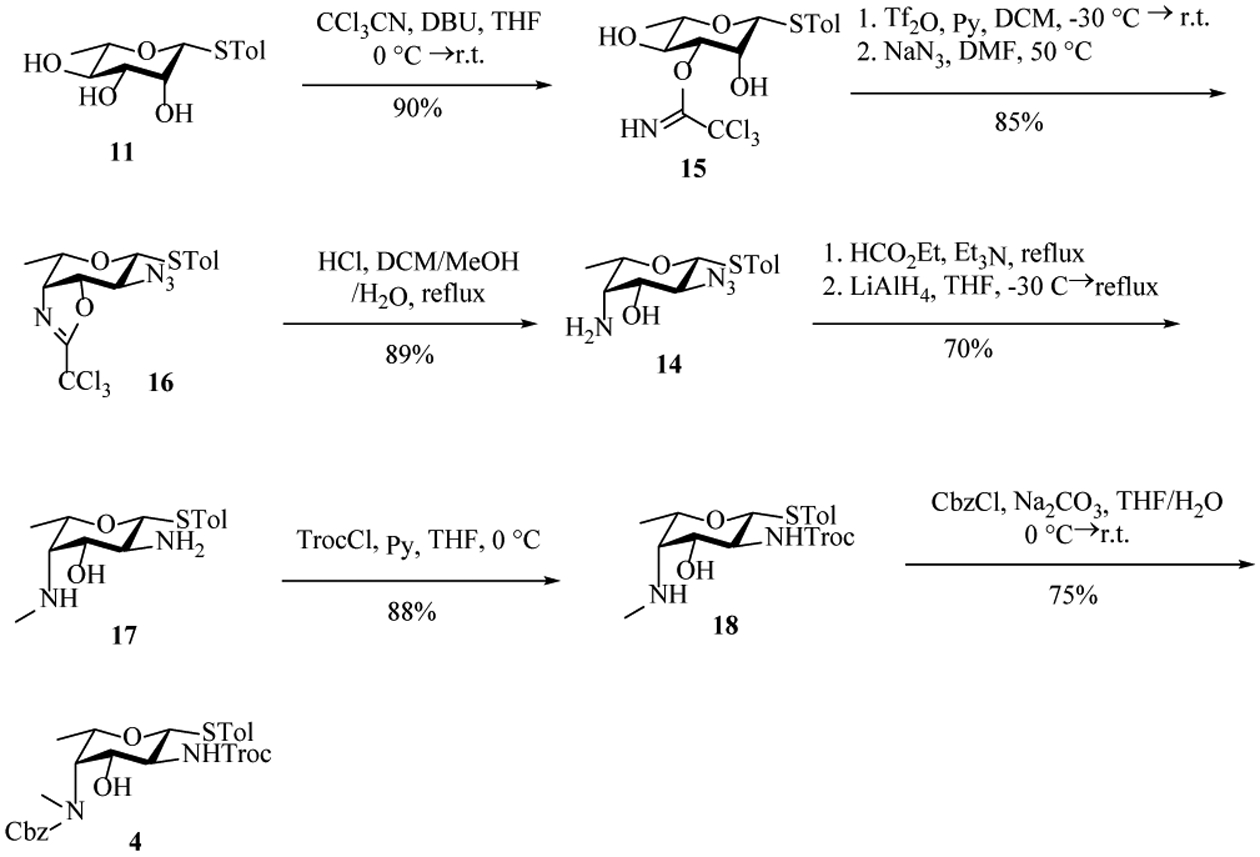
Preparation of fucosamine building block 4.
Preparation of the mannoheptose building block with the desired (L, D) configuration[25–26] started from Swern oxidation of the mannosyl thioglycoside 19. The resulting aldehyde was not stable, which was directly subjected to Wittig olefination to give the product 20 in 83% yield. Treatment of the olefin 20 with osmium tetraoxide (OsO4) and 4-methylmorpholine N-oxide (NMO) at 0 ℃ furnished diols 21 and 22 in 2.4:1 ratio (21: H-6 3.93 ppm, C-7 63.1 ppm; 22: H-6 3.98 ppm, C-7 65.0 ppm[25]), which were separated by silica gel column chromatography. The D,D- diastereoisomer 21 was then epimerized to the desired L,D-isomer 22. Acetylation of the diol afforded the desired mannoheptose building block 2 in 91% yield.
With all monosaccharide building blocks in hand, we began the oligosaccharide assembly. Reaction of donor 2 with acceptor 3 proceeded smoothly promoted by p-TolSCl/AgOTf[29] affording the α-linked DE disaccharide 23 in 80% yield (JH1-C1 = 171.0 Hz[27] for the newly formed glycosyl linkage) (Scheme 6a). Removal of the t-butyldiphenylsilyl (TBDPS) of 23 exposed 6-OH on glucosamine, which was to be glycosylated by a fucosamine donor.
Scheme 6.
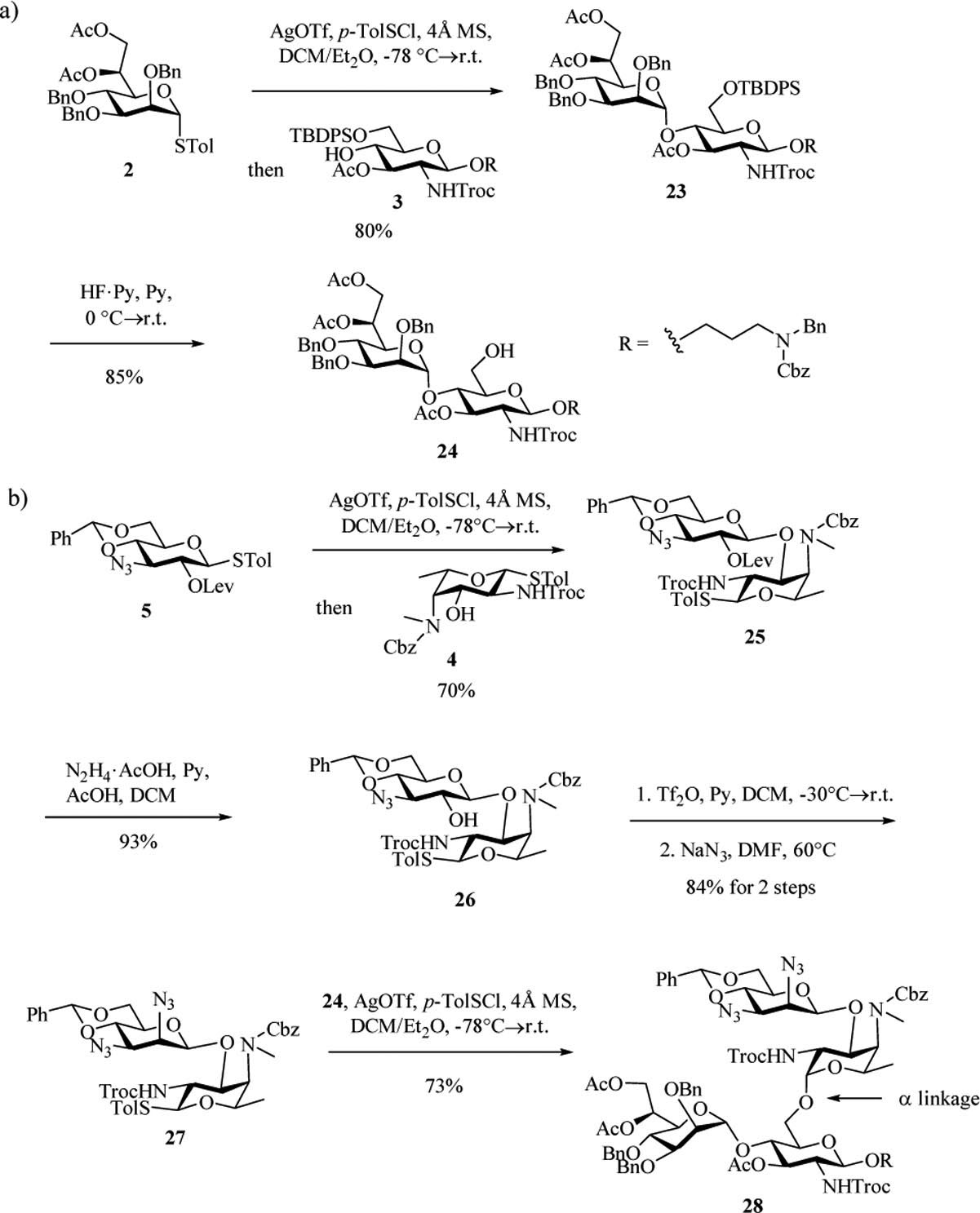
a) Formation of the ED disaccharide unit 24. b) Undesired α-anomer trisaccharide 28 was formed from glycosylation between 27 and 24.
To test the formation of the BC linkage, our original attempts used the uronic acid donors such as 29. While uronic acid building blocks have been used in the literature,[28] in our hands, these reactions gave low yields with fucosamine acceptors such as 4, presumably due to the low reactivities of these uronic acid based glycosyl donors and/or fucosamine acceptor. As an alternative, we explored the utility of 3-amino glucoside donor 5. Glycosylation of acceptor 4 by donor 5 with the promoter p-TolSCl/AgOTf at −78 ℃ successfully produced disaccharide 25 in 70% yield (Scheme 6b). Removal of 2-O-levulinoyl group from 25 exposed free hydroxyl in 26, which was then triflated and substituted by azide to furnish the mannose configuration in BC disaccharide 27. The glycosylation of 27 and disaccharide acceptor 24 was explored next. While tetrasaccharide 28 was formed in good yield (73%), surprisingly, the newly formed glycosyl linkage was α only (JH1-C1 = 171.0 Hz) despite the presence of the 2-N-Troc capable of neighboring group participation.
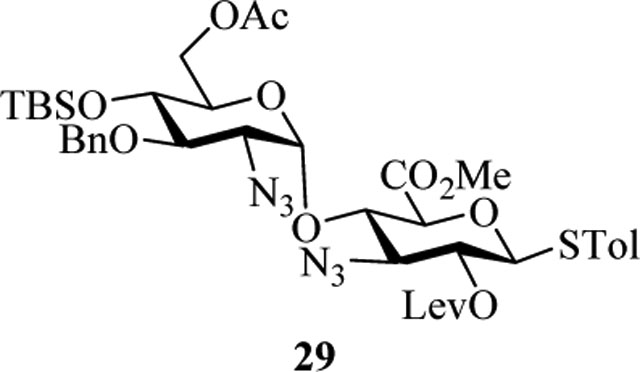
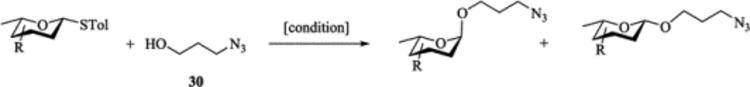
To form the desired β-isomer for CD linkage, we tested a series of reactions by varying protecting groups on the fucosamine donor with 3-azido propanol 30 as the model acceptor (Table 1). Glycosylation of donor 31 with acceptor 30 with p-TolSCl/AgOTf promoter gave a 3:2 α:β ratio using dichloromethane (DCM) as the reaction solvent (entry 1). Switching the promoter to NIS/TfOH gave a small improvement of the β selectivity (entry 2). Acetonitrile is well known to favor the formation of equatorial glycosides.[30] However, 10% acetonitrile as the co-solvent did not impact stereoselectivity much (entry 3). Increasing the amount of acetonitrile to 50% gave a low yield of the glycoside product (entry 4).
Table 1.
Investigation of the impact of protective groups on β-selective di-aminofucosylation.
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Reaction Condition | Yield | α : β |
| 1 | 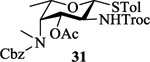 |
[p-TolSCl + AgOTf]a, DCM | 89% | 3 : 2 |
| 2 | 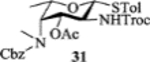 |
[NIS + TfOH]b, DCM | 92% | 2 : 3 |
| 3 | 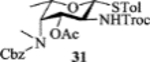 |
[NIS + TfOH], DCM/MeCN = 9/1 | 90% | 2 : 3 |
| 4 | 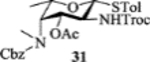 |
[NIS + TfOH], DCM/MeCN = 1/1 | 25% | α only |
| 5 |  |
[NIS], DCM | 60% | 5 : 1 |
| 6 | 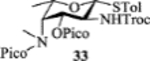 |
[NIS], DCM | <5% | N/A |
| 7 | 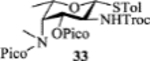 |
[p-TolSCl + AgOTf], DCM | 30% | α only |
| 8 | 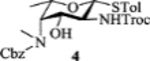 |
[NIS + TfOH], DCM | 80% | 3 : 1 |
| 9 |  |
[NIS + TfOH], DCM | 91% | 1 : 2 |
| 10 |  |
[NIS + TfOH], DCM | <5% | N/A |
| 11 | 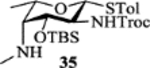 |
[p-TolSCl + AgOTf], DCM | 60% | 1 : 2.5c |
[NIS + TfOH]: NIS, TfOH, 4Å MS, −78°C
[p-TolSCl + AgOTf]: p-TolSCl, AgOTf, 4Å MS, −78°C
Ratios determined from NMR spectra of the crude reaction mixtures
The 2-picoloyl was pioneered by the Demchenko group as a remote participating group, which can form a hydrogen bond with the acceptor and direct the addition of the acceptor to the activated glycosyl donor with high syn selectivity.[31] However, in our system, introduction of picoloyl onto the 3-OH of the fucosamine donor gave worse β selectivity (entry 5). Switching the 4-NCbz to 4-NPico or leaving the 3-OH unprotected in the fucosamine donor did not change the stereoselectivity much either (entries 6, 7 and 8). The 3-Lev bearing donor 34 favored the formation of the β-anomer with acceptor 30 (α:β = 1:2, entry 9). Unfortunately, the α-glycoside became the major product (α:β = 2:1) when 34 glycosylated disaccharide acceptor 24.
One possible reason for the difficulty in forming β-glycoside is the epimerization of β-glycoside during reaction as the α-glycoside is expected to be more stable due to the anomeric effect. To test this, the β-glycoside product was subjected to the glycosylation condition with additional 1 eq TfOH. No appreciable amounts of the α-glycoside were found suggesting the β-glycoside once formed was stable under the reaction condition.
To better understand the preference for α-glycoside formation using donors 31–34, molecular modeling of the postulated oxocarbenium intermediate of donor 31 was performed. To obtain conformers covering wide configuration space, molecular dynamics (MD) simulations were performed at 900 K. From the resulting MD frames, 100 conformers with equal time intervals were extracted, and further optimized using the B3LYP/6–31G* method. The optimized 100 conformers were then sorted by their energies and the 20 most stable conformers were selected for geometrical comparison. Only 4 out of those 20 conformers exhibited the anticipated neighboring group participation by the carbonyl oxygen of 2-NHTroc with a distance of 1.58–1.59 Å between the carbonyl oxygen and C1 (d2 in Table S1). Intriguingly, most of the conformers revealed a potential remote group participation by the carbonyl oxygen of 4-NMeCbz (d3 = 1.51–1.55 Å in Table S1, Figure S1). It is possible that the Cbz moiety hindered the approach of a nucleophile to the anomeric center from the β-face of the oxacarbenium ion, leading to the formation of a large amount of the α anomer. The possible remote participation by Cbz led to the design of donor 35, where the 4-methyl amino group was unprotected. Donor 35 failed to be activated by NIS/TfOH to glycosylate 3-azidopropanol presumably due to neutralization of TfOH by the free secondary amine in the donor. Changing the promoter to p-TolSCl/AgOTf gave the β anomer (β:α = 2:1) as the major product (entry 11, Table 1). Interestingly, the reaction of 35 with acceptor 24 gave excellent β-selectivity (Scheme 7). Following Cbz protection, the two anomers were separated providing 36α (10%) and 36β (73%) after the 2 steps. While synthesis of α-fucosamine glycosides bearing 2,4-diamino groups has been reported,[22] to the best of our knowledge, this is the first time that such a β-linked fucosamine with 4-methylated amine has been produced.
Scheme 7.
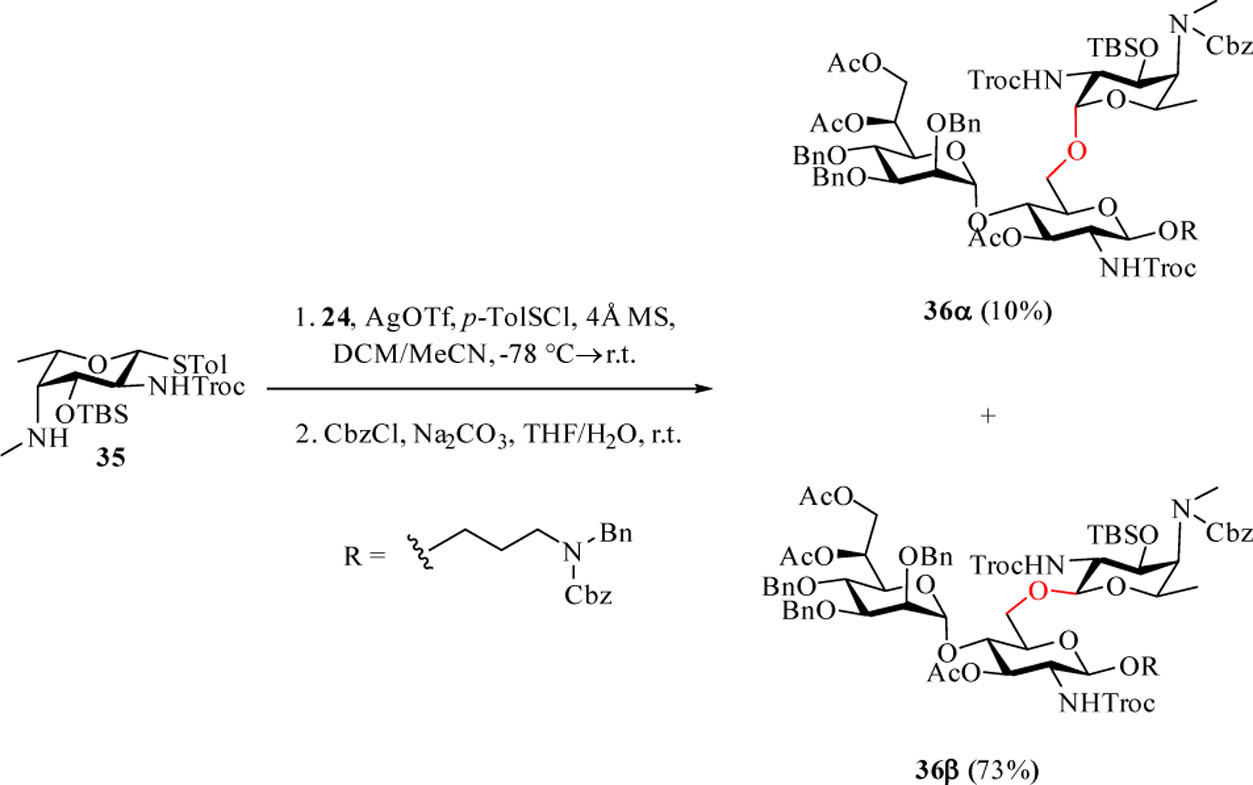
Stereoselective formation of β-amino fucoside 36β.
With the challenge of β-amino fucoside formation overcome, we next focused on assembling the full pentasaccharide. After TBS removal from 36β, the acceptor 37 was glycosylated with donor 5 yielding tetrasaccharide 38 (Scheme 8). The 2-O-Lev group was removed with hydrazine hydrate in 84% yield. The newly liberated 2-OH on 39 was then subjected to triflation, whereupon nucleophilic displacement of 2-O-triflate by NaN3[32] afforded mannose configuration on 40 in 86% yield over 2 steps. The benzylidene group of 40 was cleaved with trifluoroacetic acid (TFA) in presence of H2O to give the diol 41 in 86% yield. Selective oxidation of the 6-OH[33] followed by methylation with methyl iodide produced the tetrasaccharide acceptor 42 in 66% yield over the 2 steps. Subsequent glycosylation of 42 by donor 6 completed the backbone construction and gave the fully protected pentasaccharide 43 in 63% yield.
Scheme 8.
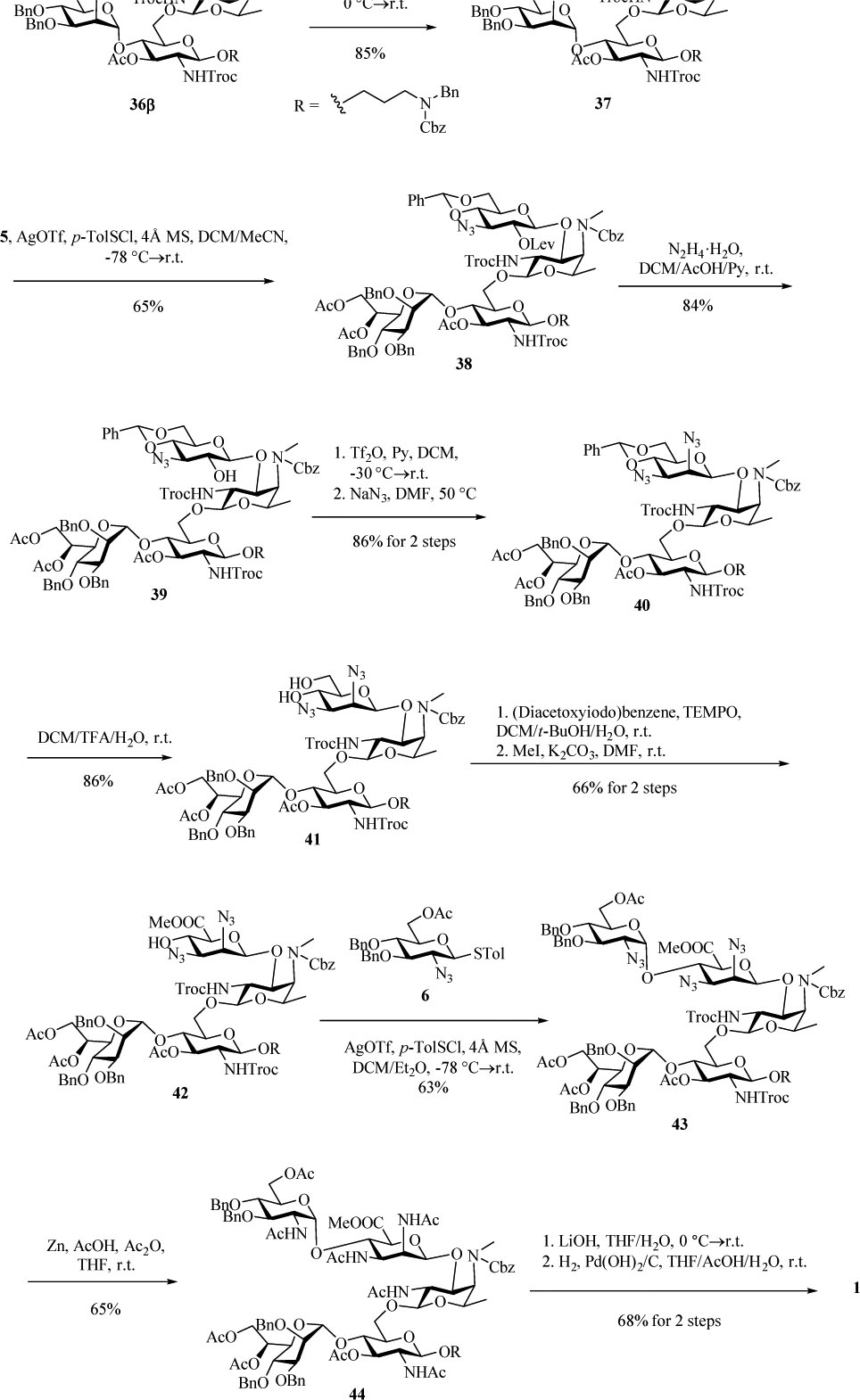
Completion of the synthesis of the target pentasaccharide 1.
The deprotection of 43 was carried out by first converting all azido and NHTroc groups to acetamido with zinc powder, acetic acid and acetic anhydride in THF to produce 44 in 65% yield (Scheme 8). A major side product isolated was found to carry one dichloroethyloxycarbonyl group due to partial reduction of Troc, which was converted to the desired product by base promoted hydrolysis and subsequent acetylation. Saponification of 44 with lithium hydroxide followed by hydrogenolysis over Pd(OH)2 gave the deprotected pentasaccharide 1, completing the first total synthesis of pertussis like pentasaccharide.
As carbohydrates are typically T cell independent B cell antigens, to generate high titers of anti-glycan IgG antibodies, it is important to link the carbohydrates to an immunogenic carrier capable of activating helper T cells.[34–35] We have previously demonstrated that bacteriophage Qβ is a powerful carrier to deliver tumor associated carbohydrate antigens.[36] To elicit anti-pertussis antibody responses, we investigated the conjugation of pertussis pentasaccharide 1 with Qβ.
For bioconjugation, pertussis pentasaccharide 1 was first treated with CSCl2[37] to convert the free amine to a thiocyanate moiety. However, the highly reactive CSCl2 also reacted with secondary amine on Unit C based on NMR and mass spectrometry (MS) analysis. We next investigated N-hydroxysuccinimide (NHS) active ester chemistry by treating 1 with adipic acid di-NHS ester 45 (Scheme 9a) forming NHS ester 46. 46 was then incubated with bacteriophage Qβ in PBS buffer at pH 7.4, which successfully introduced the pertussis pentasaccharide onto Qβ. On average, there were 235 copies of the glycan per Qβ particle from MS analysis of the conjugate (Figure S2).
Scheme 9.
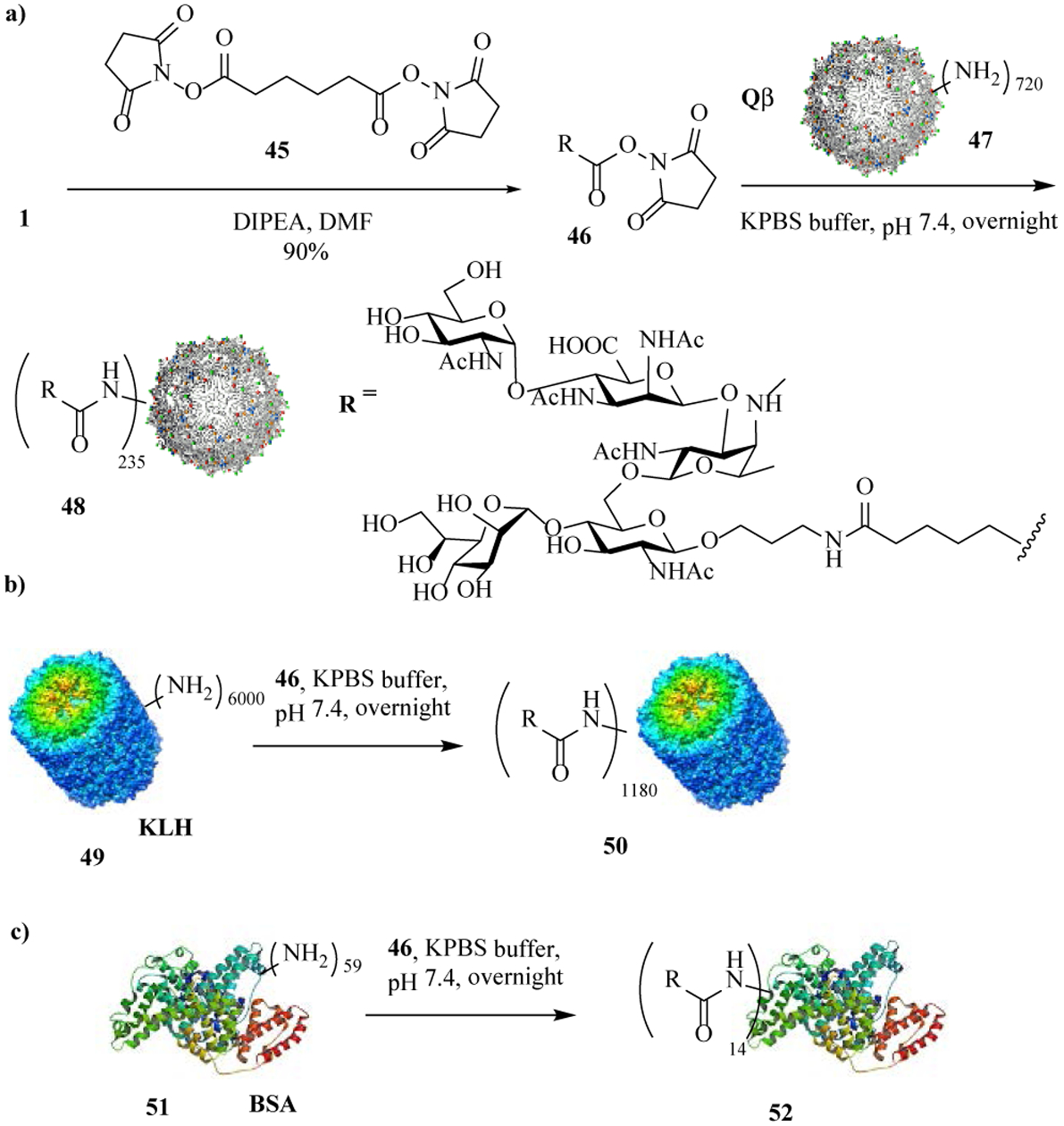
Preparation of protein-glycan conjugates.
To compare effect of carrier protein in eliciting anti-glycan antibodies, a gold standard carrier, i.e., keyhole limpet hemocyanin (KLH), was also investigated. Conjugation of 46 and KLH resulted in 1180 copies of the glycan per KLH (Scheme 9b), as determined by the anthrone-sulfuric acid assay.[38] For the ELISA analysis of serum antibodies, a bovine serum albumin (BSA)-glycan conjugate 52 bearing 14 copies of glycan per BSA was prepared using 46 (Scheme 9c).
With the Qβ-glycan conjugate 48 in hand, we explored its ability to generate anti-glycan antibodies. C57BL/6 mice were immunized subcutaneously with three biweekly injections of 48 containing 2 μg or 8 μg glycan or the KLH-glycan conjugate 50 (2 μg glycan), along with monophosphoryl lipid A (MPLA) as the adjuvant. Sera were collected from those mice one week after each injection. Unconjugated Qβ was used for immunization for the control group of mice. ELISA analysis of post-immune sera was performed using the BSA-glycan conjugate for ELISA well coating to determine the levels of anti-glycan antibodies. Sera from Qβ-glycan immunized mice showed 3000 fold enhancement in mean anti-glycan IgG antibody titers compared to those from the control group of mice immunized by unconjugated Qβ (Figure 2a). Immunization with 48 at 2 μg and 8 μg doses of glycans did not show significant differences in antibody levels suggesting the lower dose of 2 μg was sufficient. Compared to KLH conjugates, Qβ-glycan elicited significantly higher IgG antibody responses suggesting the superiority of Qβ (Figure 2a). IgG isotype analysis showed that all major subclasses of IgG antibodies were induced with a preference towards IgG2b and IgG2c, and Th1 type immune responses (Figure 2b).[39]
Figure 2.
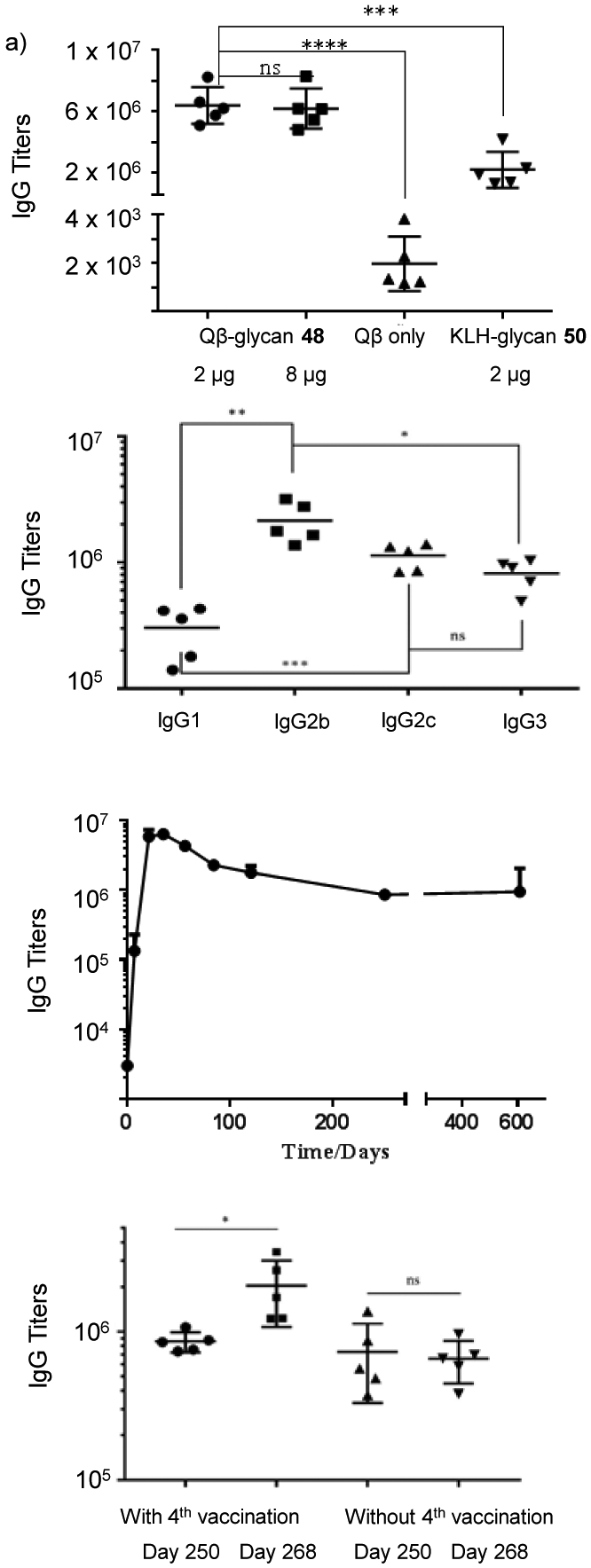
Immunological evaluation of Qβ-glycan conjugate vaccine candidate 48. a) Comparison of serum anti-glycan IgG titers from mice immunized at difference doses or with different carrier proteins. b) Anti-glycan IgG antibodies persisted over 600 days. Mice were immunized at the dose of 2 μg with 48 on days 0, 14 and 28. c) The levels of anti-glycan IgG subtypes (day 35). d) The anti-glycan IgG antibody levels could be boosted by the 4th vaccination on day 261 suggesting memory B cells were generated by immunization. In comparison, the levels of anti-glycan IgG antibodies in mice, which had received the first three injections of 48 but not the fourth vaccination on day 261, did not change significantly on day 268. ELISA titers were calculated as the highest fold of serum dilution that gave optical density of 0.1 higher than the background in ELISA assays. Each symbol represents one mouse, with the long horizontal line for each group representing the mean value of each group. Statistical analyses were performed using Graphpad Prism using student t test. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns: not significant.
For a successful vaccine, it is critical that the antibody responses can persist for a long period of time. The anti-glycan IgG responses induced by Qβ-glycan conjugate 48 were monitored over time. The IgG antibody levels reached the maximum on day 35 and maintained at a high level even on 607 days following the initial immunization (Figure 2c). In contrast, no anti-IgG antibodies were detectable in mice immunized with KLH- glycan conjugate 52 on day 607 further supporting the advantage of Qβ carrier. In order to test whether the antibody responses are boostable, some mice received an additional vaccination on day 261 after the first immunization, which resulted in doubling of the anti-glycan IgG antibody titers on day 268 indicating memory B cells were successfully induced by vaccination (Figure 2d).
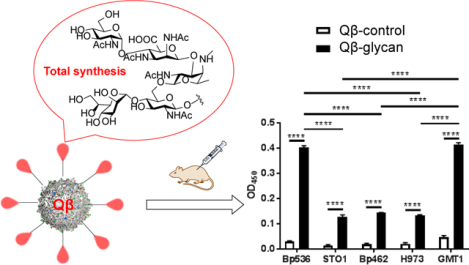
To establish the potential of Qβ-glycan as anti-pertussis vaccines, we analyzed the binding of post-immune sera with the native LPS isolated from B. pertussis bacteria. Sera from Qβ-glycan immunized mice could bind with the native LPS (Figure S4). Next, the abilities of the post-immune sera to recognize LPS on the bacterial surface were evaluated. The prototype laboratory strain Bp536 and circulating clinical strains from the US (STO1, H973 and GMT1) and Argentina (Bp462) isolated in the last two decades (Table S1) were utilized. The binding of these strains with the post-immune sera was analyzed by ELISA. Control sera from Qβ-immunized mice did not recognize all five strains strongly (Figure 3). In comparison, sera from Qβ-glycan 48 immunized mice (Qβ-glycan) resulted in high optical densities, demonstrating strong binding to B. pertussis strains. The bindings of two of the B. pertussis strains (Bp536 and GMT1) were higher than those to STO1, Bp462 and H973 suggesting better recognition and possibly higher production of LPS by Bp536 and GMT1 strains.
Figure 3.
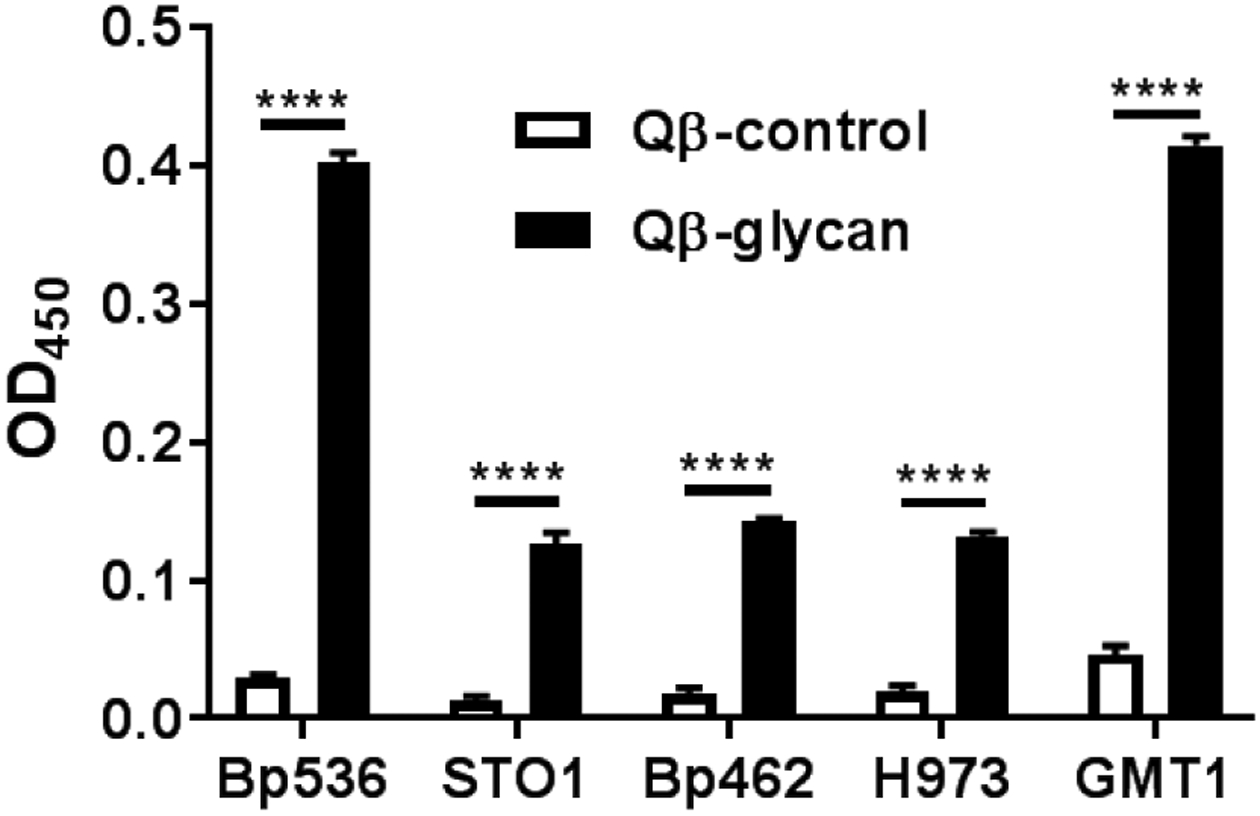
Recognition of B. pertussis strains by sera from Qβ-glycan 48 immunized mice as determined by ELISA. B. pertussis strains were used to coat 96 well microtiter plates seeded at 108 colony forming units. Strains were incubated with either Qβ- or Qβ-glycan 48 immune serum followed by incubation with anti-mouse horseradish peroxidase (HRP)-conjugated antibody. Average values with standard error means are represented. Samples were assayed in technical triplicates and a Two-Way ANOVA with Sidak’s multiple comparison post-hoc test was performed for statistical analysis. **** p < 0.0001.
Conclusion
We report the first synthesis of the unique pentasaccharide from B. pertussis LPS. As this pentasaccharide contains three unusual sugar units (2-acetamido-2,4,6-trideoxy-4-methylamino L-galactose, mannoheptose, and 2,3-diacetamido mannuronic acid), it has presented significant challenges in building block preparation as well as stereochemical control in glycosylations. The 1,2-cis linkage between 2,3-diacetamido mannuronic acid and the 2-acetamido-2,4,6-trideoxy-4-methylamino L-galactose was achieved through SN2 like inversion of 2-O stereochemistry upon glycoside formation using the corresponding 3-amino glucose donor. For the glycosylation using 2,3,6-trideoxy-4-methylamino-2-acetamido L-galactose donor, its 4-methylamino moiety was originally protected as a Cbz group. Interestingly, despite the presence of Troc group on the 2-N position, the Cbz protected donor 30 gave low 1,2-trans selectivity. Through molecular modeling, it was discovered that the Cbz moiety could conformationally hinder the approach of the acceptor to form 1,2-trans glycoside. This hypothesis led to the design of donor 34 bearing a free 4-methylamino group, which produced the desired glycoside product with high 1,2-trans selectivity. Following glycosylations, the pertussis pentasaccharide was successfully deprotected and produced in a conjugatable form for the first time. The synthetic methodologies developed and knowledge gained can be potentially applied to synthesis of other microbial glycans.
The availability of pertussis like pentasaccharide has enabled the analysis of its potential as an antigen for anti-pertussis vaccine development. Immunization of mice with the conjugate of the pentasaccharide with a protein carrier, bacteriophage Qβ, led to anti-glycan IgG titers of several million ELISA units. Compared to a gold standard glycan carrier KLH, the Qβ-glycan conjugate elicited significantly higher IgG antibody titers against the glycan. The IgG antibody levels produced by the Qβ conjugate were long lasting and could be further enhanced by a booster injection. The antibodies recognized well both the native LPS and currently circulating strains of B. pertussis. These results confirm that the synthetic pentasaccharide can be a suitable epitope, providing a promising lead towards the development of new anti-pertussis vaccines.
Supplementary Material
Figure 1.
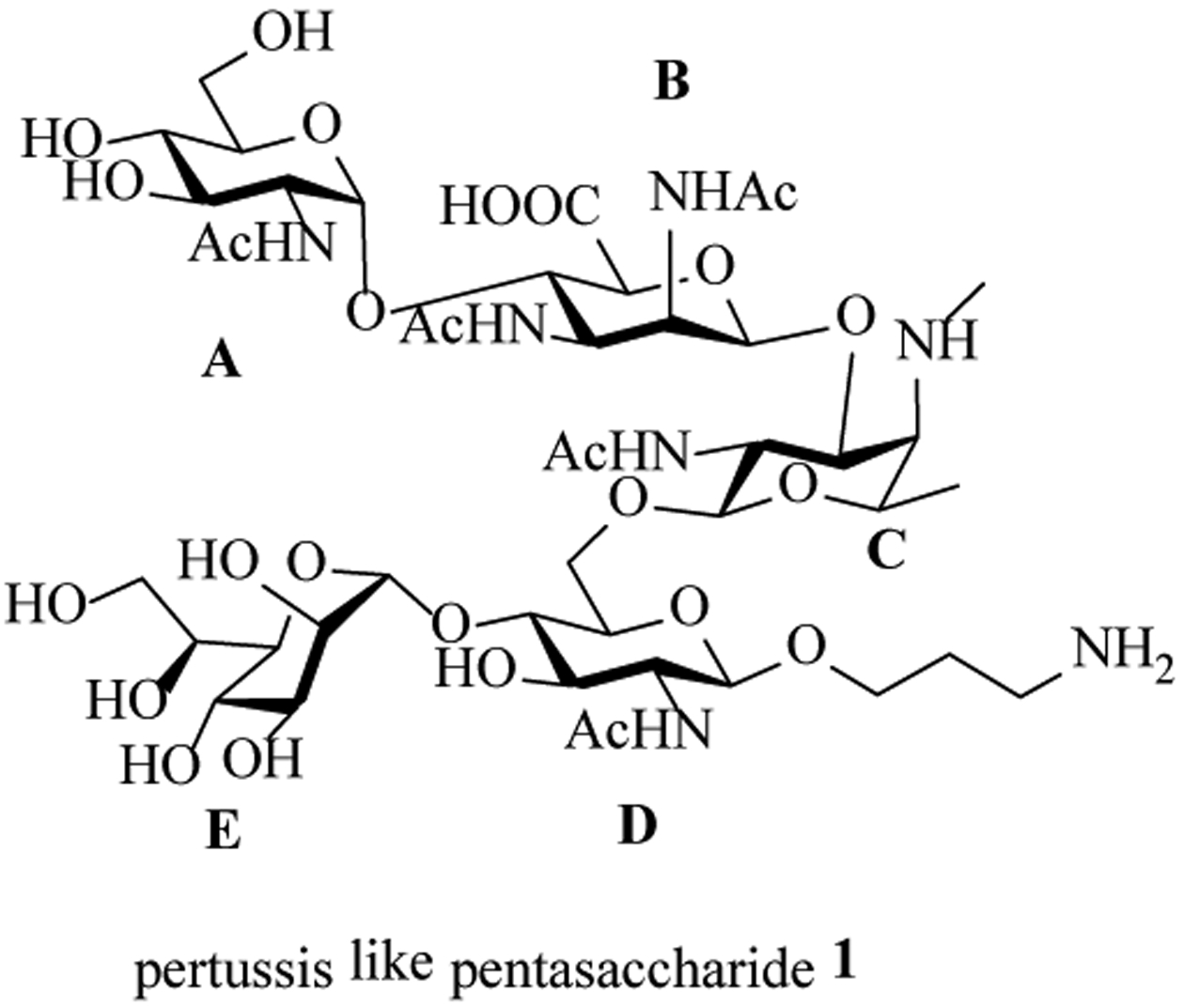
Structure of the pertussis like pentasaccharide 1.
Scheme 5.
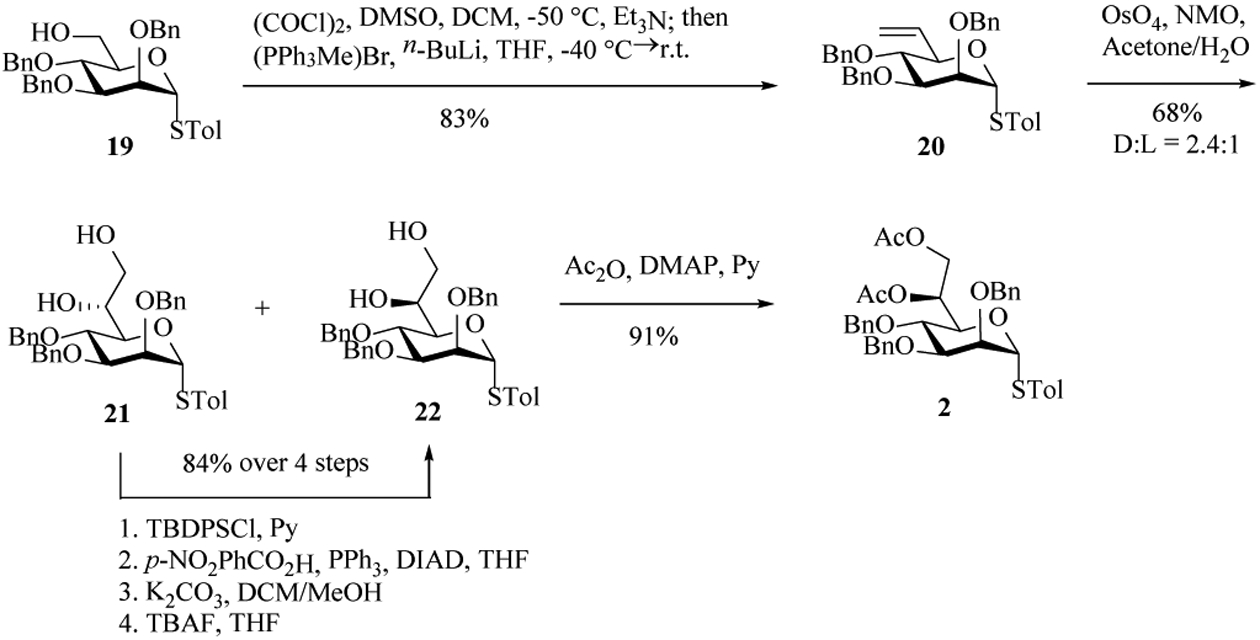
Preparation of the mannoheptose building block 2.
Acknowledgements
We are grateful for partial supports from the National Institute of Allergy and Infectious Diseases, NIH (R01AI146210, R01AI125560) and the National Cancer Institute, NIH (R01CA225101). We would like to thank Dr. Jennifer Maynard and Ms. Andrea DiVenere (Univ. of Texas) for helpful discussions.
Footnotes
Supporting information of this article can be found under:https://doi.org/10.1002/anie.%23%23%23%23.
References
- [1].Carbonetti NH, Curr. Opin. Infect. Dis 2016, 29, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cattelan N, Dubey P, Arnal L, Yantorno OM, Deora R, Pathog. Dis 2016, 74, ftv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yeung KHT, Duclos P, Nelson EAS, Hutubessy RCW, Lancet Infect. Dis 2017, 17, 974–980. [DOI] [PubMed] [Google Scholar]
- [4].Rumbo M, Hozbor D, Hum. Vaccin. Immunother 2014, 10, 2450–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].CDC, MMWR Morb. Mortal. Wkly. Rep 2018, 67, 1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sealey KL, Belcher T, Preston A, Infect. Genet. Evol 2016, 40, 136–143. [DOI] [PubMed] [Google Scholar]
- [7].Dorji D, Mooi F, Yantorno O, Deora R, Graham RM, Mukkur TK, Med. Microbiol. Immunol 2018, 207, 3–26. [DOI] [PubMed] [Google Scholar]
- [8].Bowden KE, Weigand MR, Peng YC, Sammons PK, Knipe S, Rowe K, Loparev LA,VS, Weening M, Tondella K, Williams ML, M. M, mSphere 2016, 1, e00036–00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cattelan N, Jennings-Gee J, Dubey P, Yantorno OM, Deora R, Infect. Immun 2017, 85, e00373–00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tartof SY, Lewis M, Kenyon C, White K, Osborn A, Liko J, Zell E, Martin S, Messonnier NE, Clark TA, Skoff TH, Pediatrics 2013, 131, e1047–e1052. [DOI] [PubMed] [Google Scholar]
- [11].Warfel JM, Edwards KM, Curr. Opin. Immunol 2015, 35, 48–54. [DOI] [PubMed] [Google Scholar]
- [12].Warfel JM, Zimmerman LI, Merkel TJ, Proc. Natl. Acad. Sci. USA 2014, 111, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Caroff M, Brisson J-R, Martin A, Karibian D, FEBS Lett. 2000, 477, 8–14. [DOI] [PubMed] [Google Scholar]
- [14].Caroff M, Aussel L, Zarrouk H, Martin A, Richards JC, Thérisod H, Perry MB, Karibian D, J. Endotoxin Res 2001, 7, 63–68. [PubMed] [Google Scholar]
- [15].Flak TA, Goldman WE, Cell. Microbiol 1999, 1, 51–60. [DOI] [PubMed] [Google Scholar]
- [16].Weiss AA, Mobberley PS, Fernandez RC, Mink CM, Infect. Immun 1999, 67, 1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].AlBitar-Nehme S, Basheer SM, Njamkepo E, Brisson J-R, Guiso N, Caroff M, Carbohydr. Res 2013, 378, 56–62. [DOI] [PubMed] [Google Scholar]
- [18].Niedziela T, Letowska I, Lukasiewicz J, Kaszowska M, Czarnecka A, Kenne L, Lugowski C, Infect. Immun 2005, 73, 7381–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Enugala R, Carvalho LC, Dias Pires MJ, Marques MM, Chem. Asian. J 2012, 7, 2482–2501. [DOI] [PubMed] [Google Scholar]
- [20].Ellervik U, Magnusson G, Carbohydr. Res 1996, 280, 251–260. [DOI] [PubMed] [Google Scholar]
- [21].Guo J, Frost JW, J. Am. Chem. Soc 2002, 124, 10642–10643. [DOI] [PubMed] [Google Scholar]
- [22].Emmadi M, Kulkarni SS, Nature Protoc. 2013, 8, 1870–1889. [DOI] [PubMed] [Google Scholar]
- [23].Crich D, Picione J, Org. Lett 2003, 5, 781–784. [DOI] [PubMed] [Google Scholar]
- [24].van den Bos LJ, Boltje TJ, Provoost T, Mazurek J, Overkleeft HS, van der Marel GA, Tetrahedron Lett. 2007, 48, 2697–2700. [Google Scholar]
- [25].Mulani SK, Cheng K-C, Mong K-KT, Org. Lett 2015, 17, 5536–5539 and references cited therein. [DOI] [PubMed] [Google Scholar]
- [26].Stanetty C, Baxendale IR, Eur. J. Org. Chem 2015, 2015, 2718–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bock K, Pedersen C, J. Chem. Soc. Perkin Trans 2 1974, 293–297. [Google Scholar]
- [28].Walvoort MTC, Moggré G-J, Lodder G, Overkleeft HS, Codée JDC, van der Marel GA, J. Org. Chem 2011, 76, 7301–7315. [DOI] [PubMed] [Google Scholar]
- [29].Huang X, Huang L, Wang H, Ye XS, Angew. Chem. Int. Ed 2004, 43, 5221–5224. [DOI] [PubMed] [Google Scholar]
- [30].Satoh H, Hansen HS, Manabe S, van Gunsteren WF, Hunenberger PH, Chem J. Theory Comput. 2010, 6, 1783–1797. [DOI] [PubMed] [Google Scholar]
- [31].Yasomanee JP, Demchenko AV, J. Am. Chem. Soc 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- [32].Mo K-F, Li X, Li H, Low LY, Quinn CP, Boons G-J, J. Am. Chem. Soc 2012, 134, 15556–15562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].van den Bos LJ, Codée JDC, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS, van der Marel GA, Org. Lett 2004, 6, 2165–2168. [DOI] [PubMed] [Google Scholar]
- [34].Liakatos A, Kunz H, Curr. Opin. Mol. Therap 2007, 9, 35–44. [PubMed] [Google Scholar]
- [35].Yin Z, Huang X, J. Carbohydr. Chem 2011, 31, 143–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wu X, McKay C, Pett C, Yu J, Schorlemer M, Ramadan S, Lang S, Behren S, Westerlind U, Finn MG, Huang X, ACS Chem. Biol 2019, 14, 2176–2184 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yin Z, Dulaney S, McKay C, Baniel C, Kaczanowska K, Ramadan S, Finn MG, Huang X, ChemBiochem 2016, 17, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leyva A, Quintana A, Sánchez M, Rodríguez EN, Cremata J, Sánchez JC, Biologicals 2008, 36, 134–141. [DOI] [PubMed] [Google Scholar]
- [39].Ausiello CM, Urbani F, la Sala A, Lande R, Cassone A, Infect. Immun 1997, 65, 2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.