

Abstract
We have developed a rapid and sensitive single-well dual-parametric method introduced in linked RAS nucleotide exchange and RAS/RAF-RBD interaction assays. RAS mutations are frequent drivers of multiple different human cancers, but the development of therapeutic strategies has been challenging. Traditionally, efforts to disrupt the RAS function have focused on nucleotide exchange inhibitors, GTP-RAS interaction inhibitors, and activators increasing GTPase activity of mutant RAS proteins. As the amount of biological knowledge grows, targeted biochemical assays enabling high-throughput screening have become increasingly interesting. We have previously introduced a homogeneous quenching resonance energy transfer (QRET) assay for nucleotide binding studies with RAS and heterotrimeric G proteins. Here, we introduce a novel homogeneous signaling technique called QTR-FRET, which combine QRET technology and time-resolved Förster resonance energy transfer (TR-FRET). The dual-parametric QTR-FRET technique enables the linking of guanine nucleotide exchange factor-induced Eu3+-GTP association to RAS, monitored at 615 nm, and subsequent Eu3+-GTP-loaded RAS interaction with RAF-RBD-Alexa680 monitored at 730 nm. Both reactions were monitored in a single-well assay applicable for inhibitor screening and real-time reaction monitoring. This homogeneous assay enables separable detection of both nucleotide exchange and RAS/RAF interaction inhibitors using low nanomolar protein concentrations. To demonstrate a wider applicability as a screening and real-time reaction monitoring method, the QTR-FRET technique was also applied for G(i)α GTP-loading and pertussis toxin-catalyzed ADP-ribosylation of G(i)α, for which we synthesized a novel γ-GTP-Eu3+ molecule. The study indicates that the QTR-FRET detection technique presented here can be readily applied to dual-parametric assays for various targets.
Multiple cellular signaling pathways act through GTP-binding proteins. Small guanosine triphosphatases (GTPases), including RAS and heterotrimeric G proteins, play an important role by regulating correct cellular functions.1,2 The small GTPase superfamily comprise over 150 human members, which are divided into five major branches.3 RAS superfamily members act as membrane bound switches by cycling between GDP-bound inactive and GTP-bound active forms.1,4−6 This switch function is controlled by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs).6 Usually, GTPases are functional when in the active GTP-bound form, enabling interaction with their downstream effectors.7−9 Apart from small GTPases, for example, RAS, GTP is a critical cofactor for multiple disease-related signaling pathways, including G protein-coupled receptors (GPCRs). Heterotrimeric G proteins serve similar switch functions as RAS and mediate signal transmission via seven transmembrane domain GPCRs to intracellular effectors.2,10,11 G proteins are composed of three subunits, α, β, and γ, from which the α subunit (Gα) works as a GTP-binding switch. The heterotrimeric G protein cycles between an inactive GDP-bound conformation, that is primed for interaction with an activated receptor, and an active GTP-bound conformation, which can modulate the activity downstream.2,10,11 Extracellular stimulus of GPCR, having an exchange factor activity, induces the GDP dissociation from Gα and subsequent activation of Gα through GTP binding. Gα forms the constitutive heterotrimer together with Gβ and Gγ subunits, and the activation of the Gα subunit results in conformational changes leading to dissociation from the receptor and from Gβγ subunits. After activation, both Gα subunits and Gβγ subunits interact downstream with varying specificity.11,12 G protein GAPs act allosterically on Gα subunits by increasing intrinsic GTP hydrolysis, but their deactivation is also regulated by RGS (regulator of G-protein signaling) proteins, which can accelerate the intrinsic GTPase activity and enable the reformation of heterotrimers.12
GPCRs are the largest human membrane protein family and one of the most common drug targets comprising one-third of all drug sales.13,14 G proteins themselves are not as widely targeted, but interest against GPCR downstream targets is increasing.15 Also, small GTPases and especially KRAS have gained renewed interest in recent years as a drug target, but so far, their direct targeting has proved to be challenging.16−20 Due to the high interest, novel assay methods to study and target these proteins are constantly being developed. Traditional methods to study RAS and G proteins have been nucleotide binding assays utilizing labeled GDP and GTP. In the case of RAS and other small GTPases, radiolabeled nucleotides have been extensively replaced by fluorophore-conjugated nucleotides, which are mostly suitable for separation-free detection based on the properties of the nucleotide conjugated fluorophore, for example, environment sensitivity or anisotropy changes.21−25 The search for improved methods is still active, as a panel of methods has recently been introduced.26 In the case of G proteins, the number of suitable methodologies is even more limited because of the membrane environment. Thus, radiolabels and heterogeneous assay techniques have not been totally displaced.27 However, fluorophore-based techniques enabling homogeneous separation-free detection for G proteins have already started to replace the conventional heterogeneous techniques.28−32 Despite the large number of potential methods, advances can be achieved by improving assay performance or the amount of reliable data produced within a single assay.
Time-resolved fluorescence (TRF) in its different forms can potentially answer the need of improved assays. Lanthanide chelates were originally applied for heterogeneous assays, but newer methods based on chelate and cryptate structures are mainly homogeneous and often utilized in time-resolved Förster resonance energy transfer (TR-FRET).28,33−35 The advantage of TRF signal detection is the increased signal-to-background (S/B) ratio due to the low background signal resulting from long-lived fluorescence and time-gated measurements. In addition, the apparent large Stokes shift simplifies the assay development, especially for TR-FRET applications. GPCR second messengers and kinases are nowadays the most popular TR-FRET targets, but as the assays have been developed for many other targets, TR-FRET has become the primary technology for many laboratories. The search for improvements related to TR-FRET detection has been targeted mainly for improved fluorophore structures. However, especially when multiplexed assays are developed, multiple labeling steps and spectral overlap optimization may complicate the assay development and increase the unwanted background signal.36 To avoid multiple labeling steps, native tryptophan moieties have been used as one signaling moiety to study, for example, GTPases.37 However, not all proteins contain suitable tryptophan residues, and thus, monitoring of linked protein–ligand (PLIs) and protein–protein interactions (PPIs) still rely on label-conjugated assay components.
Previously, we have introduced a single-label homogeneous quenching resonance energy transfer (QRET) technique, for example, for small GTPases and heterotrimeric G proteins, using a Eu3+-chelate conjugated GTP.38−40 We have also shown that QRET enables similar analytical performance as the dual-label TR-FRET.40 Additionally, we recently introduced quencher modulated time-resolved Förster resonance energy transfer (QTR-FRET) as a method to reduce TR-FRET background fluorescence by introducing a soluble quencher molecule.41 Now, we introduce the QTR-FRET method for dual-parametric kinetic studies with KRAS and G(i)α. QTR-FRET combines the single-label QRET, used now for binding studies with Eu3+-GTP and protein, with a dual-label TR-FRET method enabling simultaneous monitoring of two reactions, PLI and PPI. To demonstrate the analytical performance of the QTR-FRET platform, we monitored Eu3+-GTP association and followed RAF-RBD-Alexa680 binding to Eu3+-GTP-loaded KRAS. Additionally, we studied γ-GTP-Eu3+ association to G(i)α, which was linked to monitor pertussis toxin-catalyzed G(i)α ADP-ribosylation. These assays clearly demonstrated that the QTR-FRET approach can improve the functionality of the TR-FRET measurements by increasing the S/B ratio and that two reactions can be monitored simultaneously by using only two labeled molecules and a soluble quencher.
Experimental Section
Kinetic Monitoring of KRAS Nucleotide Exchange and KRAS/RAF-RBD Interaction
Detailed lists of materials and instrumentation, cloning, production, and purification of KRAS and related proteins (Figures S1 and S2), Alexa680 conjugations, and γ-GTP-Eu3+ synthesis (Figures S3–S5) are presented in the Supporting Information (SI). In addition, detailed protocols for KRAS/RAF-RBD interaction concentration optimization, KRAS enzymatic glycosylation, and data analysis are presented in the SI. All presented assays were performed in triplicate unless otherwise indicated.
We selected 50 and 200 nM KRAS for further nucleotide association and protein–protein interaction monitoring. Assays were performed using protocol 1 in a 15 μL final volume. First, we assayed two known KRAS-inhibiting designed ankyrin repeat proteins (DARPins) to monitor their functional concentrations.42 DARPins (K27 and K55) were titrated up to 20 μM in a reaction with KRAS (200 nM), Eu3+-GTP (25 nM), MG (24 μM), and SOScat (10 nM). Signals were monitored 15 min after SOScat addition, following RBD-Alexa680 (25 nM) addition and a second measurement after 15 min. DARPins K27 and K55 were thereafter used in single selected concentrations. KRAS was incubated with K27 (500 nM) and K55 (5 μM) for 10 min in a 7 μL volume. Detection solution (6 μL) containing Eu3+-GTP (25 nM), MG (24 μM), and RBD-Alexa680 (25 nM) was added, and signals were monitored after 5 min at 615 and 730 nm. Nucleotide association and KRAS/RAF-RBD interaction were launched by 10 nM SOScat (2 μL), and signals were monitored during a 60 min incubation at RT. Thereafter, nucleotide association and KRAS/RAF-RBD interaction were separated. Again, KRAS was incubated with K27 and K55 for 10 min. A QRET detection solution (4 μL) containing Eu3+-GTP (25 nM) and MG (24 μM) was added, and signals were monitored. After 10 nM SOScat (2 μL) addition, signals were kinetically monitored for 1000 s, before 25 nM of RBD-Alexa680 (2 μL) was added, and signal monitoring was continued for 20 min.
G(i)α ADP-Ribosylation
Protocols for synthesis, characterization, and functionality testing of γ-GTP-Eu3+ are presented in the SI (Figures S3–S6). In the enzymatic assays with G(i)α (Figure S7), 150 nM G(i)α and various Pertussis toxin (PTX, recombinant form of the PTX S1-subunit)43,44 concentrations (0–600 nM) were used. An ADP-ribosylation reaction (60 min, RT) was performed at a 4.5 μL volume, using an EDTA-containing assay buffer and bio-NAD+ (25 nM). γ-GTP-Eu3+ (25 or 50 nM) was added together with 0 or 10 μM GTP in a MgCl2-containing assay buffer, enabling γ-GTP-Eu3+ loading monitored at 615 and 730 nm (30 min). Thereafter, 25 nM SA-Alexa680 was added, and signals were monitored multiple times during a 90 min incubation at a 15 μL volume. Finally, MG (20 or 35 μM) was added, and signals were further monitored at a 17 μL volume after 30 min of incubation.
Results and Discussion
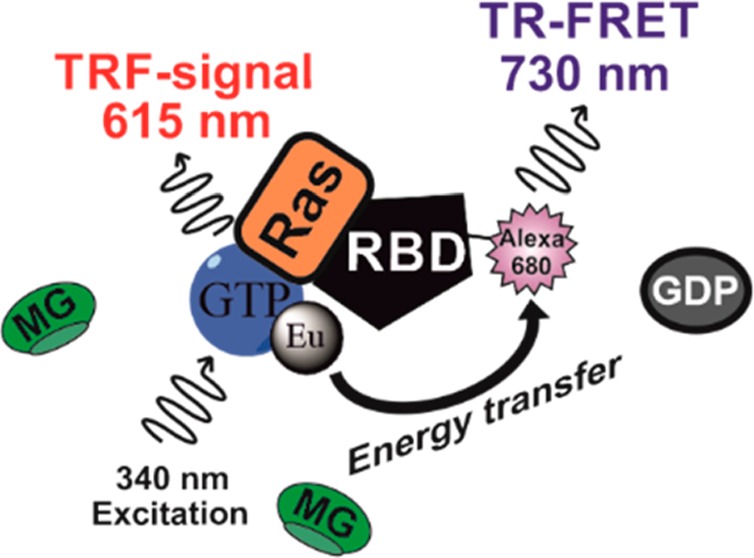
In nature, many enzymatic reactions are coupled with other reactions occurring simultaneously or in a specific order. Currently, the methods enabling simultaneous detection of multiple reactions are scarce. Previously, we introduced a method named as QTR-FRET, which was demonstrated to reduce background signals in TR-FRET assays.41 QTR-FRET combines the TR-FRET-type energy transfer between lanthanide chelate (donor) and acceptor fluorophore and the use of soluble quencher, here malachite green (MG), to reduce the signal from the nonbound Ln3+ ligand following the QRET principle. When lanthanide chelate, for example, Eu3+-chelate, is conjugated to a small molecule, it enables the detection of PLI in the presence of a soluble quencher as shown earlier with multiple different targets monitored using the QRET technique.45 Energy transfer processes are not self-excluding, and thus, the second reaction between Eu3+-chelate and an acceptor-conjugated molecule can be monitored from a single well (Figure 1). In this work, we studied Eu3+-GTP binding to KRAS in the coupled reaction to monitor active KRAS interaction with RAF-RBD-Alexa680. In the assay, the reaction can be inhibited in two steps: (1) SOScat-induced KRAS activation and (2) KRAS-GTP interaction with RAF-RBD. These reactions can be monitored from a single well in real time (Figure 1). To demonstrate the wider applicability of the QTR-FRET technique, we also monitored G(i)α ADP-ribosylation and simultaneous loading with γ-GTP-Eu3+ (Figure S7).
Figure 1.

Dual-parametric nucleotide exchange and RAS/RAF-RBD interaction assay based on the QTR-FRET principle. In the nucleotide exchange, RAS-bound GDP is dissociated in the presence of GEF (SOScat), enabling Eu3+-GTP association. Bound Eu3+-GTP is protected from the soluble quencher (MG), and an increase in TRF signal is monitored at 615 nm. Formation of Eu3+-GTP-RAS enables RBD-Alexa680 binding, and an increased TR-FRET signal is monitored at 730 nm. Inhibitors can block either Eu3+-GTP association, resulting low signals in TRF- and TR-FRET measurements, or the RAS/RAF-RBD interaction, rendering an unaffected TRF signal but low TR-FRET signal. Additionally, MG lowers the Eu3+-GTP ensuing background signal in TR-FRET measurements, thus providing an improved S/B ratio. The QTR-FRET principle enables simultaneous and discriminating protein–ligand (PLI) and protein–protein interaction (PPI) inhibitor selections in a coupled real-time assay.
QTR-FRET Enables Dual-Parametric Eu3+-GTP Association and KRAS/RAF-RBD Interaction Monitoring
The RAF protein is a serine/threonine kinase and downstream interactor of RAS, which occurs through RAF-RBD, binding exclusively to active GTP-RAS with a reported Kd of approximately 20 nM.7−9,46 In addition to biological importance, this GTP-RAS-specific binding has been utilized in many assays to study RAS.42 We selected the GTP-RAS-dependent binding of RAF-RBD as a model to demonstrate the QTR-FRET principle in a dual-parametric assay, monitoring nucleotide exchange and GTP-RAS interaction with RAF-RBD in a single well. Simultaneous monitoring is especially beneficial for inhibitor screening, as effects on a nucleotide binding state can be monitored through Eu3+-GTP (615 nm), and inhibition of either nucleotide exchange or RAS/RAF-RBD interaction is measured from the Alexa680 channel at 730 nm (Figure 1).
To prove the QTR-FRET assay functionality, we first performed KRAS titration (0–600 nM) using fixed SOScat (10 nM) and RBD-Alexa680 (25 nM) concentrations. The assay was performed using two protocols with varying detection: (1) Eu3+-GTP (25 nM) and MG (24 μM) were premixed and added simultaneously, or (2) Eu3+-GTP was added first, followed by addition of MG. Using the first protocol, the TRF signal at 615 nm could be monitored in real time, and only a minor increase in the signal was observed after RBD-Alexa680 addition (Figure S8A). Using the second protocol, S/B ratios without MG were low, but the introduction of MG dramatically increased the S/B ratio to 12 (Figure S8B).
We also monitored TR-FRET signals at 730 nm to measure the RAS/RAF-RBD interaction using the two given protocols. Using protocol 1, a clear TR-FRET signal increase was detected in KRAS titration after Alexa680-RBD addition (Figure S8C). Without RBD-Alexa680, the S/B ratio compared to the GDP-blocked reaction was 2.3, which stems from the Eu3+-GTP signal protection upon binding to KRAS. This protection could also be seen at 730 nm, although the signal levels were very low. However, with RBD-Alexa680, the monitored S/B ratio was 10, clearly demonstrating the interaction between Eu3+-GTP loaded KRAS and RBD-Alexa680. The TR-FRET signal was also monitored without a quencher, as shown with protocol 2 (Figure S8D). Without MG and RBD-Alexa680, the S/B ratio was minimal, and the addition of RBD-Alexa680 did not dramatically improve the monitored S/B ratio. Even though the KRAS-dependent TR-FRET signal between Eu3+-GTP and RBD-Alexa680 is clear, a high background signal, also in the presence of GDP, was detected with the used concentrations. By adding MG, this high background level was reduced approximately 5-fold, whereas the specific TR-FRET signal was reduced by less than 2-fold. This improved the S/B ratio from 2.2 to 5.9, respectively. Given the higher S/B ratios and utility of real-time monitoring when following protocol 1 (Eu3+-GTP added in complex with MG), it was selected for all further assays. On the basis of these results, we also kept the KRAS concentration at 200 nM or lower in subsequent assays. The selected low nM concentrations are in accordance to the ones used previously in RAS/RAF interaction assays utilizing TR-FRET.42,47
Eu3+-GTP Association and KRAS/RAF-RBD Interaction Can Be Monitored in Real Time
Next, we tested the functionality of the assay in a real-time measurement by monitoring Eu3+-GTP association and KRAS/RAF-RBD interaction. To demonstrate the biologically relevant functionality of the assay we selected two recently demonstrated DARPins as a tool compounds for specific KRAS inhibition.42 First, saturating concentrations for GDP-RAS-specific DARPin K27 and GTP-RAS-specific DARPin K55 were determined (Figure S9).42 On the basis of the DARPin titration, we selected 0.5 μM and 5 μM as saturating concentrations for K27 and K55, respectively. Using these concentrations, we assayed the association and KRAS/RAF-RBD interaction separately by adding RBD-Alexa680 after near maximal Eu3+-GTP loading (Figure 2). We observed increased TRF signals (615 nm) without DARPin and with K55, compared to the reactions without SOScat or with K27. This reflects complete blocking of a nucleotide exchange by K27 but not with K55. In all cases, the RBD-Alexa680 addition did not dramatically change the signal observed at 615 nm (Figure 2A). On the other hand, TR-FRET signals monitored at 730 nm, which were low in all cases before RBD-Alexa680 addition, increased after RBD-Alexa680 addition in reactions with SOScat but without DARPins (Figure 2B). When TR-FRET signals were compared before and after RBD-Alexa680 addition, the S/B ratio of 7.4 was achieved after 10 min of incubation. When the reaction was compared to the reactions without SOScat or with SOScat and in the presence of K27 or K55, the observed S/B ratios were 1.7, 1.5, and 1.8, respectively. This reflects no RBD-Alexa680 interaction with KRAS. These results are consistent with the notion that K27 inhibits the Eu3+-GTP association to KRAS, and thereafter, the RBD-Alexa680 interaction cannot occur. Additionally, K55 allows Eu3+-GTP association, but blocks the KRAS interaction with RBD-Alexa680 (Figure 2).42
Figure 2.

Real-time monitoring of the coupled nucleotide association and KRAS/RAF-RBD interaction with DARPins K27 and K55. Eu3+-GTP (25 nM) association and RBD-Alexa680 interaction were monitored at 340/615 nm (A) and 340/730 nm (B) in the absence (red) or presence of 10 nM SOScat (black) or reactions with SOScat and selected DARPin, K27 (blue) or K55 (magenta). Eu3+-GTP association kinetics were first monitored after SOScat addition, and monitoring was continued similarly after RBD-Alexa680 (25 nM) addition. Data represent mean ± SD (n = 3).
Next we performed combined real-time measurements using a reaction where both Eu3+-GTP and RBD-Alexa680 were present before the SOScat addition. These assays confirmed the observation that K27 blocks both nucleotide exchange and subsequent KRAS/RAF-RBD interaction, but K55 blocks only the interaction between Eu3+-GTP-loaded KRAS and RBD-Alexa680 (Figures 2 and 3). As the Eu3+-GTP association is slower than the subsequent interaction with RBD-Alexa680, kinetics in both 615 nm (Figure 3A) and 730 nm (Figure 3B) channels primarily report the slower reaction, explaining the short lag phase seen at 730 nm. The assay performed with 50 nM KRAS showed nearly identical results as the assay with 200 nM KRAS but only with a decreased S/B ratio (data not shown). With 50 nM KRAS, especially the TR-FRET signal level monitored at 730 nm was compromised. These results clearly indicate that the single-well QTR-FRET assay can be used, instead of two separate assays, to monitor nucleotide exchange and KRAS/RAF-RBD interactions at the same time in real time.
Figure 3.

Single-well real-time monitoring of coupled nucleotide association and KRAS/RAF-RBD interaction reactions with DARPins K27 and K55. Eu3+-GTP (25 nM) association at 340/615 nm (A) and KRAS/RAF-RBD interaction at 340/730 nm (B) were kinetically monitored in a single reaction with KRAS (200 nM) and RBD-Alexa680 (25 nM) after SOScat (10 nM) addition. Reaction without SOScat (black) showed no signal increase over time, while a fast TRF signal increase is observed with SOScat (red) followed by a slightly delayed TR-FRET increase. K27 (blue) blocks totally the nucleotide exchange and subsequent RBD-Alexa680 interaction, showing no TR-FRET or TRF signal increase, as K55 (magenta) blocked only the RBD-Alexa680 interaction, with only a minor effect to the TRF signal observed. All measurements were performed in the presence of 24 μM MG. Data represent mean ± SD (n = 3).
HTS Compatible QTR-FRET Technique Enables Monitoring of Reactions Influencing KRAS Biology
KRAS activity is regulated on many levels, including through post-translational modifications.4,5 In addition to prenylation of the CAAX-motif, KRAS can also be, for example, glycosylated.48 Bacterial toxin TpeL is one of the enzymes which monoglycosylates small GTPases from the switch I domain (Thr35), and it is reported to prevent the KRAS interaction with RAF-RBD.48 TpeL was selected for QTR-FRET glycosylation assays, which were performed with GDP- or GMPPNP-loaded KRAS and two UDP-sugar donors, UDP-GlcNAc and UDP-Glu. TpeL has reported to not prefer a KRAS activity state but prefers UDP-GlcNAc over UDP-Glu.48 The QTR-FRET assay confirmed the TpeL UDP-GlcNAc preference, which was 30-fold over UDP-Glu, but it also showed a 10-fold preference to GDP-KRAS over GMPPNP-KRAS when monitored at 615 nm (Figure 4A, Table S1). In the assay with GMPPNP-KRAS, TpeL could not completely inhibit the nucleotide exchange with either one of the used UDP-sugars, but the level of glycosylation with UDP-GlcNAc was again higher. This observed GDP-KRAS-preferring behavior of TpeL is similar as reported with TpeL-related glycosylating toxin, the lethal toxin of Clostridium sordellii.49,50 As TpeL KRAS glycosylation is reported as a KRAS/RAF-RBD interaction blocker and not for nucleotide exchange, QTR-FRET results from the nucleotide exchange were further confirmed with a conventional QRET nucleotide exchange assay. QRET assay results were in line with the ones from QTR-FRET and confirmed the enzyme preference to UDP-GlcNAc and GDP-KRAS (Figure S10, Table S1). As TpeL blocked the nucleotide exchange, leading to a prevented KRAS/RAF-RBD interaction, no further information using QTR-FRET was observed compared to traditional QRET, when the KRAS/RAF-RBD interaction was monitored at 730 nm (Figure S11).
Figure 4.

Dual-parametric KRAS glycosylation assay. (A) TpeL-catalyzed KRAS glycosylation was studied with two UDP-sugars (25 μM) and KRAS (200 nM) loaded with GDP or GMPPNP. After a 30 min TpeL reaction, an Eu3+-GTP (25 nM) exchange was performed with SOScat (10 nM), and signals were monitoring at 615 nm. With GDP-KRAS, UDP-GlcNAc (black) was preferred over UDP-Glu (blue) as seen from the efficient nucleotide exchange blocking. Similarly, UDP-GlcNAc (red) was preferred over UDP-Glu (magenta) when the assay was performed with GMPPNP-KRAS. (B) When the TpeL titration using UDP-GlcNAc was performed after SOScat-induced KRAS loading with Eu3+-GTP (black) or Eu3+-GDP (red), clear KRAS/RAF-RBD interaction blocking could be monitored at 730 nm. With Eu3+-GDP, no TR-FRET signal was monitored after RBD-Alexa680 (25 nM) addition as the KRAS/RAF-RBD interaction cannot occur. KRAS loading with Eu3+-GTP enabled the KRAS/RAF-RBD interaction, which was blocked in a TpeL concentration-dependent manner. Data represent mean ± SD (n = 3).
To study if glycosylation would also block the KRAS/RAF-RBD interaction, we modified the QTR-FRET assay scheme by performing the nucleotide exchange with either Eu3+-GTP or Eu3+-GDP before the TpeL reaction. The assay confirmed the expected KRAS/RAF-RBD interaction blocking in TpeL titration with UDP-GlcNAc (Figure 4B). It also proved the correct assay functionality as no TR-FRET signal was observed after RBD-Alexa680 addition if Eu3+-GDP was used in KRAS nucleotide loading. Nucleotide association with Eu3+-GTP or Eu3+-GDP was equal, and the glycosylation could not be monitored based on the TRF signal at 615 nm (data not shown). The important role of the KRAS switch I domain on PPI has already been known, and on the basis of our data, the glycosylation of Thr35 seems to block not only the KRAS interaction with RAF-RBD but also with SOScat.51
Dual Parametric QTR-FRET Technique Can Be Readily Converted for Different Targets and Reactions
Assays performed with KRAS already proved the functionality of the QTR-FRET technique for the biochemical studies and as a potential inhibitor screening tool. To further highlight the QTR-FRET technique applicability for different targets, we performed a dual-parametric assay on PTX-catalyzed ADP-ribosylation of G(i)α, a heterotrimeric GTP-binding monomer. Pertussis toxin is composed of five noncovalently bound subunits (PtxS1–S5), which are arranged in an AB5-type configuration.43 The QTR-FRET assay utilized a recombinant form of the PtxS1 subunit, which is the PTX subunit responsible for G(i)α ADP-ribosylation.43,44 As Eu3+-GTP used with KRAS (2′/3′-labeled) has too low of an affinity to Gα proteins, a novel γ-GTP-Eu3+ molecule was prepared for this purpose (Figures S3–S5).52 The new γ-GTP-Eu3+ was first tested for G(i)α binding, and the determined binding affinity was 25 ± 1 nM in the used QRET assay with 20 nM G(i)α (Figure S6). On the basis of these results, the newly introduced γ-GTP-Eu3+ binds to G(i)α at the expected affinity, and at least in the current system, the functionality of the molecule is improved compared to the previously introduced nonhydrolyzable GTP analog (Eu-GTP).40,52
Next, the QTR-FRET assay was utilized to detect PTX-catalyzed ADP-ribosylation of G(i)α. Here, G(i)α was loaded with γ-GTP-Eu3+ using the MgCl2/EDTA exchange, and biotin-NAD+ reacted with the G(i)α protein in the reaction with PTX (Figure S7). PTX titration (0–600 nM) was performed under constant conditions using 150 nM G(i)α and 25 nM biotin-NAD+. Thereafter, G(i)α was loaded with two concentrations of γ-GTP-Eu3+ (25 or 50 nM), and ADP-ribosylation was monitored in the presence of SA-Alexa680 and MG. When ADP-ribosylation was monitored at 730 nm, a clear increase in the background signal was monitored with the increased γ-GTP-Eu3+ concentration (Figure 5A). This background signal was decreased by MG, as observed from the S/B ratios calculated by comparing the signal of the GTP-blocked reaction before and after MG addition (Figure 5B). With 25 nM γ-GTP-Eu3+, monitored S/B ratios before and after MG were 4.3 and 6.0, respectively. As expected, the effect of MG was higher with 50 nM γ-GTP-Eu3+, where the S/B ratio was increased from 3.8 to 7.4, respectively (Figure 5B). Eu3+ signals (615 nm) from the same reactions were also monitored to confirm that the increased PTX concentration had no effect on TRF signal levels (Figure 5C). Thus, the TRF signal increase was solely due to γ-GTP-Eu3+ loading. The S/B ratio increased along with the increasing γ-GTP-Eu3+ concentration, as the G(i)α loading was more complete (Figure 5D). This increase in G(i)α loading also explains the improved functionality monitored with 50 nM γ-GTP-Eu3+ using the channel at 730 nm and the QTR-FRET protocol with MG (Figure 5). In all the assays, γ-GTP-Eu3+ and MG were added separately to identify the effect of each individual component. However, as seen with KRAS, the addition of preformed γ-GTP-Eu3+ and the MG mixture solution can improve the QTR-FRET functionality further (data not shown).
Figure 5.

Dual-parametric G(i)α assay for γ-GTP-Eu3+ association and ADP-ribosylation monitoring. (A) PTX catalyzed G(i)α ADP-ribosylation was studied with biotin-NAD+ and SA-Alexa680 as a TR-FRET acceptor. ADP-ribosylated and non-ADP-ribosylated G(i)α were loaded with 25 (red and black) or 50 nM (magenta and blue) γ-GTP-Eu3+ using MgCl2/EDTA exchange, and TR-FRET signals at 730 nm were monitored before (solid) and after (dashed) MG addition. (B) Signal-to-background (S/B) ratio was significantly improved upon MG addition (solid vs dashed) with 25 (black) and 50 nM (red) γ-GTP-Eu3+ concentrations. (C) No differences in the QRET signals, monitoring γ-GTP-Eu3+ association at 615 nm, were detected between ADP-ribosylated (dashed) and non-ADP-ribosylated (solid) G(i)α using 25 (black) or 50 nM (blue) Eu3+-GTP. γ-GTP-Eu3+ association could be blocked with 10 μM GTP in both reactions, 25 nM (red) or 50 nM (magenta) γ-GTP-Eu3+. (D) ADP-ribosylation (dashed) had no effect on γ-GTP-Eu3+ association (dashed vs solid), but only γ-GTP-Eu3+ concentration, 25 (black) or 50 nM (red), affected the observed S/B ratio compared to the reaction blocked with 10 μM GTP. Data represent mean ± SD (n = 3).
Conclusions
Here, we have demonstrated the applicability of the QTR-FRET detection method for dual-parametric HTS compatible assays by monitoring a panel of KRAS reactions and G(i)α ADP-ribosylation. The QTR-FRET technique combines the single-label QRET technique with the dual-label TR-FRET readout, enabling highly sensitive detection of two reactions from a single well in real-time. TR-FRET is widely used to study PPIs and PLIs. However, since TR-FRET relies on energy transfer between donor and acceptor, both conjugated to distinct molecules, multiplexing of these assays can be challenging. Here, we demonstrated with KRAS the functionality of QTR-FRET in coupled detection of PLIs and PPIs, using nonmodified KRAS target protein. The method can be used to separate inhibitors affecting either GDP- or GMPPNP-KRAS, as shown with specific DARPins. In addition, we monitored over a 5-fold background signal reduction with QTR-FRET compared to traditional TR-FRET, which improved the monitored S/B ratio significantly in the KRAS/RAF-RDB assay. Single-assay detection showed even improved functionality compared to individual reactions monitored using QRET or TR-FRET. Similarly in the G(i)α ADP-ribosylation assay, a clear increase in the S/B ratio was observed in the presence of MG, and the effect is even more pronounced when γ-GTP-Eu3+ loading is enhanced by using a higher concentration of γ-GTP-Eu3+. G proteins possess lower GTP binding affinity compared to small GTP binding proteins, e.g., KRAS, and thus, the preference for higher γ-GTP-Eu3+ concentrations was expected.52 On the basis of these results, the QTR-FRET assay can be readily converted to new targets, and the technique can be beneficial both for reducing the TR-FRET background and enabling dual-parametric assays. The QTR-FRET technique is expected to become a valuable and widely used research tool in the future.
Acknowledgments
This work is supported by the Academy of Finland (296225/K.K., 329012/K.K., 323433/K.K., 138584/H.H., 296093/H.H., and 295296/A.T.P.), Finnish Cultural Foundation (K.K.), Moikoisten Syöväntutkimussäätiö (K.K.), Sigrid Jusélius Foundation (A.T.P.), and University of Turku. This project is funded in part with federal funds from the National Cancer Institute, National Institutes of Health (NIH), under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, and the mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government. We thank members of the FNLCR Protein Expression Laboratory for help with cloning (Jennifer Mehalko and Vanessa Wall), E. coli expression (Troy Taylor, Nitya Ramakrishnan, and John-Paul Denson), protein purification (John-Paul Denson, Jennifer Mehalko, Peter Frank, Simon Messing, and Troy Taylor), and nucleotide exchange (Peter Frank).
Glossary
Abbreviations:
- DARPin
Designed ankyrin repeat protein
- GAP
GTPase-activating protein
- GDP
Guanosine diphosphate
- GEF
Guanine nucleotide exchange factor
- GMPPNP
5′-Guanylyl imidodiphosphate
- GPCR
G protein-coupled receptor
- GTP
Guanosine triphosphate
- MG
Malachite green
- PLI
Protein–ligand interaction
- PPI
Protein–protein interaction
- PTX
Pertussis toxin
- QTR-FRET
Quencher modulated time-resolved Förster resonance energy transfer
- QRET
Quenching resonance energy transfer
- RBD
RAS-binding domain
- RGS
Regulator of G-protein signaling
- SOScat
Son of Sevenless catalytical domain
- TpeL
Clostridium perfringens toxin
- TRF
Time-resolved fluorescence
- TR-FRET
Time-resolved Förster resonance energy transfer
- UDP-GlcNAc
Uridine diphosphate N-acetylglucosamine
- UDP-Glu
Uridine diphosphate glucose
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05126.
Experimental section, including materials and instrumentation, and the protocols for cloning, production, and purification of KRAS and related proteins; RBD- and SA-Alexa680 labeling; γ-GTP-Eu3+ synthesis and characterization; KRAS concentration optimization; KRAS enzymatic glycosylation; G(i)α loading test with γ-GTP-Eu3+; and data analysis. Supporting results, include γ-GTP-Eu3+ binding affinity to G(i)α, G(i)α ADP-ribosylation principle, KRAS concentration optimization, DARPin titrations, and KRAS glycosylation in QTR-FRET and QRET assays. (PDF)
Author Present Address
∥ M. Holderfield: Quanta Therapeutics, 1700 Fourth St., San Francisco, California 94158, United States.
The authors declare the following competing financial interest(s): Kari Kopra and Harri Harma have commercial interest through QRET Technologies, Ltd.
Supplementary Material
References
- Goitre L.; Trapani E.; Trabalzini L.; Retta S. F. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol. Biol. 2014, 1120, 1–18. 10.1007/978-1-62703-791-4_1. [DOI] [PubMed] [Google Scholar]
- Oldham W. M.; Hamm H. E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Wennerberg K.; Rossman K. L.; Der C. J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- Konstantinopoulos P. A.; Karamouzis M. V.; Papavassiliou A. G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discovery 2007, 6, 541–555. 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- Ahearn I. M.; Haigis K.; Bar-Sagi D.; Philips M. R. Regulating the regulator: post-translational modification of Ras. Nat. Rev. Mol. Cell Biol. 2012, 13, 39–51. 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J.; Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- Rajalingam K.; Schreck R.; Rapp U. R.; Albert S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta, Mol. Cell Res. 2007, 1773, 1177–1195. 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Leicht D. T.; Balan V.; Kaplun A.; Singh-Gupta V.; Kaplun L.; Dobson M.; Tzivion G. Raf kinases: function, regulation and role in human cancer. Biochim. Biophys. Acta, Mol. Cell Res. 2007, 1773, 1196–1212. 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J. RAS and downstream RAF-MEK and PI3K-AKT signaling in neuronal development, function and dysfunction. Biol. Chem. 2016, 397, 215–222. 10.1515/hsz-2015-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera T. M.; Vanhauwe J.; Thomas T. O.; Medkova M.; Preininger A.; Mazzoni M. R.; Hamm H. E. Insights into G protein structure, function, and regulation. Endocr. Rev. 2003, 24, 765–781. 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- Syrovatkina V.; Alegre K. O.; Dey R.; Huang X. Y. Regulation, signaling and physiological functions of G-proteins. J. Mol. Biol. 2016, 428, 3850–3868. 10.1016/j.jmb.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderovski D. P.; Willard F. S. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int. J. Biol. Sci. 2005, 1, 51–66. 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M.; Masuram S.; Schiöth H. B. The druggable genome: evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A. P.; Smrcka A. V. Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discovery 2018, 17, 789–803. 10.1038/nrd.2018.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysin S.; Salt M.; Young A.; McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus K.; Mattos C. Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin. Cancer Res. 2015, 21, 1810–1818. 10.1158/1078-0432.CCR-14-2148. [DOI] [PubMed] [Google Scholar]
- Lim S. M.; Westover K. D.; Ficarro S. B.; Harrison R. A.; Choi H. G.; Pacold M. E.; Carrasco M.; Hunter J.; Kim N. D.; Xie T.; Sim T.; Janne P. A.; Meyerson M.; Marto J. A.; Engen J. R.; Gray N. S. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem., Int. Ed. 2014, 53, 199–204. 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel J.; Cromm P. M.; Zimmermann G.; Grossmann T. N.; Waldmann H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- Ryan M. B.; Corcoran R. B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- Ahmadian M. R.; Wittinghofer A.; Herrmann C. Fluorescence methods in the study of small GTP-binding proteins. Methods Mol. Biol. 2002, 189, 045–063. 10.1385/1-59259-281-3:045. [DOI] [PubMed] [Google Scholar]
- Rojas R. J.; Kimple R. J.; Rossman K. L.; Siderovski D. P.; Sondek J. Established and emerging fluorescence-based assays for G-protein function: Ras-superfamily GTPases. Comb. Chem. High Throughput Screening 2003, 6, 409–418. 10.2174/138620703106298509. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Zhang Y.; Shacter E.; Zheng Y. Mechanism of the guanine nucleotide exchange reaction of Ras GTPase - Evidence for a GTP/GDP displacement model. Biochemistry 2005, 44, 2566–2576. 10.1021/bi048755w. [DOI] [PubMed] [Google Scholar]
- Evelyn C. R.; Ferng T.; Rojas R. J.; Larsen M. J.; Sondek J.; Neubig R. R. High-throughput screening for small-molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J. Biomol. Screening 2009, 14, 161–172. 10.1177/1087057108328761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze Z.; Waller A.; Wu Y.; Romero E.; Edwards B. S.; Wandinger-Ness A.; Sklar L. A. Identification of a small GTPase inhibitor using a high-throughput flow cytometry bead-based multiplex assay. J. Biomol. Screening 2010, 15, 10–20. 10.1177/1087057109352240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D.; Stephen A. G.; Turbyville T. J.; Holderfield M. New weapons to penetrate the armor: Novel reagents and assays developed at the NCI RAS Initiative to enable discovery of RAS therapeutics. Semin. Cancer Biol. 2019, 54, 174–182. 10.1016/j.semcancer.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlack J. M.; Parkhill A. L. Assay of G protein-coupled receptor activation of G proteins in native cell membranes using [35S]GTP gamma S binding. Methods Mol. Biol. 2003, 237, 135–143. 10.1385/1-59259-430-1:135. [DOI] [PubMed] [Google Scholar]
- Frang H.; Mukkala V. M.; Syystö R.; Ollikka P.; Hurskainen P.; Scheinin M.; Hemmilä I. Nonradioactive GTP binding assay to monitor activation of g protein-coupled receptors. Assay Drug Dev. Technol. 2003, 1, 275–280. 10.1089/15406580360545080. [DOI] [PubMed] [Google Scholar]
- Miyano K.; Sudo Y.; Yokoyama A.; Hisaoka-Nakashima K.; Morioka N.; Takebayashi M.; Nakata Y.; Higami Y.; Uezono Y. History of the G protein-coupled receptor (GPCR) assays from traditional to a state-of-the-art biosensor assay. J. Pharmacol. Sci. 2014, 126, 302–309. 10.1254/jphs.14R13CP. [DOI] [PubMed] [Google Scholar]
- Sridharan R.; Zuber J.; Connelly S. M.; Mathew E.; Dumont M. E. Fluorescent approaches for understanding interactions of ligands with G protein coupled receptors. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 15–33. 10.1016/j.bbamem.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goricanec D.; Stehle R.; Egloff P.; Grigoriu S.; Pluckthun A.; Wagner G.; Hagn F. Conformational dynamics of a G-protein α subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E3629–E3638. 10.1073/pnas.1604125113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange P. G. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br. J. Pharmacol. 2010, 161, 1238–1249. 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siitari H.; Hemmilä I.; Soini E.; Lövgren T.; Koistinen V. Detection of hepatitis B surface antigen using time-resolved fluoroimmunoassay. Nature 1983, 301, 258–60. 10.1038/301258a0. [DOI] [PubMed] [Google Scholar]
- Degorce F.; Card A.; Soh S.; Trinquet E.; Knapik G. P.; Xie B. HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr. Chem. Genomics 2009, 3, 22–32. 10.2174/1875397300903010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergin E.; Dogan A.; Parmaksiz M.; Elçin A. E.; Elçin Y. M. Time-resolved fluorescence resonance energy transfer [TR-FRET] assays for biochemical processes. Curr. Pharm. Biotechnol. 2016, 17, 1222–1230. 10.2174/1389201017666160809164527. [DOI] [PubMed] [Google Scholar]
- Faklaris O.; Cottet M.; Falco A.; Villier B.; Laget M.; Zwier J. M.; Trinquet E.; Mouillac B.; Pin J.-P.; Durroux T. Multicolor time-resolved Förster resonance energy transfer microscopy reveals the impact of GPCR oligomerization on internalization processes. FASEB J. 2015, 29, 2235–2246. 10.1096/fj.14-260059. [DOI] [PubMed] [Google Scholar]
- Bill A.; Blockus H.; Stumpfe D.; Bajorath J.; Schmitz A.; Famulok M. A homogeneous fluorescence resonance energy transfer system for monitoring the activation of a protein switch in real time. J. Am. Chem. Soc. 2011, 133, 8372–8379. 10.1021/ja202513s. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Ligabue A.; Wang Q.; Syrjänpää M.; Blaževitš O.; Veltel S.; van Adrichem A. J.; Hänninen P.; Abankwa D.; Härmä H. A homogeneous quenching resonance energy transfer assay for the kinetic analysis of the GTPase nucleotide exchange reaction. Anal. Bioanal. Chem. 2014, 406, 4147–4156. 10.1007/s00216-014-7795-7. [DOI] [PubMed] [Google Scholar]
- Kopra K.; van Adrichem A. J.; Salo-Ahen O. M. H.; Peltonen J.; Wennerberg K.; Härmä H. High-throughput dual screening method for Ras activities and inhibitors. Anal. Chem. 2017, 89, 4508–4516. 10.1021/acs.analchem.6b04904. [DOI] [PubMed] [Google Scholar]
- Rozwandowicz-Jansen A.; Laurila J.; Martikkala E.; Frang H.; Hemmilä I.; Scheinin M.; Hänninen P.; Härmä H. Homogeneous GTP binding assay employing QRET technology. J. Biomol. Screening 2010, 15, 261–267. 10.1177/1087057109358921. [DOI] [PubMed] [Google Scholar]
- Syrjänpää M.; Vuorinen E.; Kulmala S.; Wang Q.; Härmä H.; Kopra K. QTR-FRET: Efficient background reduction technology in time-resolved förster resonance energy transfer assays. Anal. Chim. Acta 2019, 1092, 93. 10.1016/j.aca.2019.09.045. [DOI] [PubMed] [Google Scholar]
- Guillard S.; Kolasinska-Zwierz P.; Debreczeni J.; Breed J.; Zhang J.; Bery N.; Marwood R.; Tart J.; Overman R.; Stocki P.; Mistry B.; Phillips C.; Rabbitts T.; Jackson R.; Minter R. Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat. Commun. 2017, 8, 16111. 10.1038/ncomms16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein P. E.; Boodhoo A.; Armstrong G. D.; Cockle S. A.; Klein M. H.; Read R. J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. 10.1016/S0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- Ashok Y.; Miettinen M.; de Oliveira D. K. H.; Tamirat M. Z.; Näreoja K.; Tiwari A.; Hottiger M. O.; Johnson M. S.; Lehtiö L.; Pulliainen A. T. Discovery of compounds inhibiting the ADP-ribosyltransferase activity of pertussis toxin. ACS Infect. Dis. 2020, na. 10.1021/acsinfecdis.9b00412. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Härmä H. Quenching resonance energy transfer (QRET): a single-label technique for inhibitor screening and interaction studies. New Biotechnol. 2015, 32, 575–580. 10.1016/j.nbt.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Herrmann C.; Horn G.; Spaargaren M.; Wittinghofer A. Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J. Biol. Chem. 1996, 271, 6794–6800. 10.1074/jbc.271.12.6794. [DOI] [PubMed] [Google Scholar]
- Winter J. J.; Anderson M.; Blades K.; Brassington C.; Breeze A. L.; Chresta C.; Embrey K.; Fairley G.; Faulder P.; Finlay M. R.; Kettle J. G.; Nowak T.; Overman R.; Patel S. J.; Perkins P.; Spadola L.; Tart J.; Tucker J. A.; Wrigley G. Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. J. Med. Chem. 2015, 58, 2265–2274. 10.1021/jm501660t. [DOI] [PubMed] [Google Scholar]
- Guttenberg G.; Hornei S.; Jank T.; Schwan C.; Lü W.; Einsle O.; Papatheodorou P.; Aktories K. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J. Biol. Chem. 2012, 287, 24929–24940. 10.1074/jbc.M112.347773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C.; Ahmadian M. R.; Hofmann F.; Just I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J. Biol. Chem. 1998, 273, 16134–16139. 10.1074/jbc.273.26.16134. [DOI] [PubMed] [Google Scholar]
- Schorch B.; Heni H.; Zahaf N. I.; Brummer T.; Mione M.; Schmidt G.; Papatheodorou P.; Aktories K. Targeting oncogenic Ras by the Clostridium perfringens toxin TpeL. Oncotarget. 2018, 9, 16489–16500. 10.18632/oncotarget.24740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhaeizadeh H.; Amin E.; Nakhaei-Rad S.; Dvorsky R.; Ahmadian M. R. The RAS-effector interface: isoform-specific differences in the effector binding regions. PLoS One 2016, 11, e0167145 10.1371/journal.pone.0167145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinski J. A.; Zera E. M.; Angleson J. K.; Wensel T. G. High affinity interactions of GTPgammaS with the heterotrimeric G protein, transducin. Evidence at high and low protein concentrations. J. Biol. Chem. 1996, 271, 12919–12924. 10.1074/jbc.271.22.12919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.