

Abstract
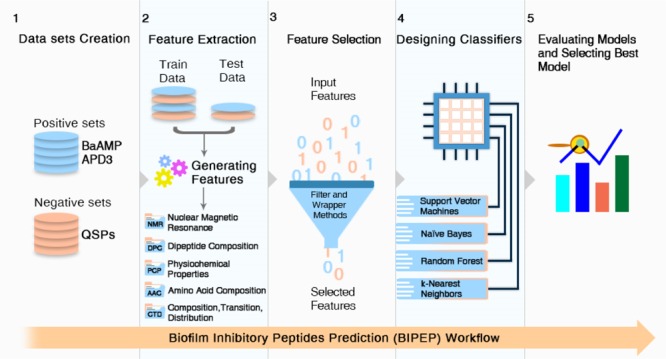
Biofilms are biological systems that are formed by a community of microorganisms in which microbial cells are connected on a surface within a self-produced matrix of an extracellular polymeric substance. On some occasions, microorganisms use biofilms to protect themselves against the harmful effects of the host body immune system and the surrounding environment, hence increasing their chances of survival against the various anti-microbial agents. Biofilms play a crucial role in medicine and industry because of the problems they cause. Designing agents that inhibit bacterial biofilm formation is very costly and takes too much time in the laboratory to be discovered and validated. Therefore, developing computational tools for the prediction of biofilm inhibitor peptides is inevitable and important. Here, we present a computational prediction tool to screen the vast number of peptide sequences and select potential candidate peptides for further lab experiments and validation. In this learning model, different feature vectors, extracted from the peptide primary structure, are exploited to learn patterns from the sequence of biofilm inhibitory peptides. Various classification algorithms including SVM, random forest, and k-nearest neighbor have been examined to evaluate their performance. Overall, our approach showed better prediction in comparison with other prediction methods. In this study, for the first time, we applied features extracted from NMR spectra of amino acids along with physicochemical features. Although each group of features showed good discrimination potential alone, we used a combination of features to enhance the performance of our method. Our prediction tool is freely available.
Introduction
According to the National Institutes of Health (NIH), about 80% of bacterial pathogen form biofilms.1 These highly adhesive populations are associated with a great number of health problems because of two major reasons: first, about 65% of all human microbial infections and near 80% of chronic infection are caused by these biofilms and second, their strong, protective, and multicellular structure make them up to 10–1000 fold more resistant to the hosts’ defense systems and traditional antimicrobials than planktonic bacteria.2
Formation of these adhesive population of different microorganisms including bacteria, archaea, fungi and protists is at the root of many serious chronic infections.3 This sessile community of different microorganisms is able to grow on a diverse range of biotic and abiotic surfaces. This ability helps them to be created in a variety of environments.
The formation of a biofilm is a complex process and consists of five steps. In the first stage of formation, planktonic bacteria using van der Waals powers or flagella which is a weak, reversible adhesion are absorbed into a surface (reversible connection). This phase is mainly related to the capacity of microbes to interact and cooperate through quorum sensing (QS) with other cells, which microbes do by releasing and responding to small diffusible signal molecules. In the second stage, hydrophobic forces between the bacterium and extracellular matrices increase, when bacterial appendages overcome physically repulsive forces, resulting in the decreased repulsion between them (irreversible attachment). This irreversible attachment is due to the bacteria being able to secrete a complicated range of extracellular polymeric substances, including proteins, polysaccharides, and DNA. In the third phase, through cell division, the biofilm rises when colonization has started the biofilm (proliferation). The matrix of previous species in the colony can then be adhered to by a few microbial species that do not have the capacity of attachment. Maturation is the final stage of biofilm development. During this stage, through certain physiological modifications in form, size, efflux pumps, oxygen gradient, division of labor, and so forth, the biofilm becomes more specialized. Microbial colonies are able to become antibiotic-resistant when the biofilm is fully developed. Bacteria acquire the capacity to spread and colonize new substratum in a critical phase of biofilm formation (dispersal). Different variables control this significant step, including shear stress or enzymes that degrade the extracellular matrix, such as dispersion B and deoxyribonuclease. Some anti-biofilm agents are needed to stop the impact of microbes’ multicellular mode lifestyle (biofilm).3
In recent years, a large number of infections caused by organisms that are resistant to drugs appeared as a direct consequence of widespread use of antibiotics. These problems motivate studies in the field of antimicrobial peptides (AMPs) to produce viable alternatives for current antibiotics.4 AMPs, also called host defense peptides, attracted researchers’ attention, and scientists have developed various AMPs. The interest in AMPs for the treatment of the biofilm has increased in recent decades. To be more exact, the types of AMPs that can prevent the formation of the biofilm around bacteria through the aforementioned processes are called biofilm inhibiting peptides (BIPs).
The effects of BIPs on different types of microorganisms have already been investigated in several kinds of studies. Accordingly, the anti-biofilm agents can carry out various actions such as targeting the QS signaling molecules, extracellular polymeric substance matrix, genes involved in biofilm formation, adhesion, and so forth.5,6 In a study by Batoni et al., it has been verified that BIPs can be proper alternatives to conventional antibiotic treatments.7
The current focus is on implementing methods that are able to synthetize more potent biofilm inhibitory peptides. Metagenomics is a rapidly growing field of research that studies uncultured organisms to know the real variety and functions of microbes.8 Because of advances in next-generation sequencing technology, we are now able to reconstitute complete or draft genomes from metagenomic sequences.9,10 Because sequences extracted from different microbial environments such as soil, water, ancient remains of animals, or the digestive systems of animals and humans are good candidates for the extraction of different bioactive peptides, such as BIPs, an approach that can reduce the number of candidate sequences for experimental validation is in high demand in metagenomics.11−13
Advances in computational tools based on amino acid sequences have greatly influenced the peptide research field. Currently, there are a variety of sequence-based predictors for different classification tasks in a wide range from AMP prediction14 up to the prediction of anti-cancer15,16 and QS peptides.17 These sequence-based techniques can be useful in identifying the best candidate BIPs in a wet-lab prior to synthesis and testing against pathogens. Computational tools like BIOFIN18 and dPABBs19 have been developed for the prediction of BIPs. Both of them use BaAMPs, which is a well-known database containing experimentally validated BIPs (http://www.baamps.it/), to create a positive data set.20 In BIOFIN, random forest (RF) and SVM learning techniques were employed for the prediction of BIPs, based on the different composition features of peptides. dPABBs employs amino acid compositions (AACs) for learning the SVM and RF-based classifier models.
Although, the aforementioned methods have played an important role in gaining knowledge to predict BIPs, the prediction accuracy needs to be improved in order to minimize the number of false positives and access better BIP candidates for experimental validation. In addition, for machine learning tasks, feature representation is fundamentally important for improved performance. Using a good feature combination will enable capture of the characteristics of data and support effective machine learning. The present study aims to develop a computational approach to more accurately predict anti-biofilm peptides. To the best of our knowledge, in the current work for the first time, combinations of different descriptors, such as the composition, transition, and distribution (CTD) of different features besides the newly introduced NMR-based features were used to map peptide sequences to numeric feature vectors as the input for prediction methods. The proposed approach was found to perform better than several existing approaches for predicting BIPs.
Results and Discussion
Compositional Analysis
We used a histogram to display the average AAC of the two groups (BIPs and non-BIPs) (Figure 1). As one can see in the figure, BIPs had more numerous positively charged residues (K, H, R), while the non-BIPs had more negatively charged residues (D, E). Additionally, the number of large amino acids (F, R, W, Y) in the BIP group was significant, whereas the tiny amino acids (A, C, D, G, S, T) were mainly found in the non-BIPs. These results indicate the importance of using the physiochemical features in the training process of our machine.
Figure 1.

Comparison of the average AAC between two classes.
We conducted dipeptide compositional analyzes between BIPs and non-BIPs to understand dipeptide residue preference. Analysis of DPC revealed that 54 out of 400 possible dipeptides differed considerably between BIPs and non-BIPs (Welch’s t-test; P-value 0.01). Of these, RI, IR, KK, RV, RR, WR, RW, VR, KR, RK and LF, FF, SL, LS, FT, SF, TQ, FN, SG, TF were the top 10 most abundant dipeptides in BIPs and non-BIPs. Figure 2 reveals, significantly, DPC between BIPs and non-BIPs.
Figure 2.

Comparison of the average dipeptide AAC between two classes.
Feature Extraction and Feature Selection
A total of 1024 features were obtained for the peptides of both groups from the PyDPI package and IAMPE tool. Table 1 presents all the feature vectors used in this study, along with the number of features in each group. For detailed information about each of the feature vectors, see Supporting Information.
Table 1. Presentation of Different Feature Vectors Used in This Study with the Number of Generated Features in Each Set.
| feature sets | AAC | DPC | CTD | PCP | NMR | total |
| number of generated features | 20 | 400 | 504 | 15 | 40 | 979 |
Only 980 features were associated with the separation of our classes after t-test analysis. These 980 features were considered the inputs for the REF algorithm and SelectKBest from the scikit-learn package of Python. Finally, 150 best features were obtained for our data. We moved across a different number of features, from 100 to 200 features, and the most optimal results for our method were reached, when we choose 150 features. Most of the features were from the CTD group (108 features). Twelve features were from the NMR set. Nine features were selected from the AAC group. Thirteen features were chosen from DPC. Finally, only eight features were selected from the PCP group. These 150 features are listed in Table S5.
An interesting analysis was conducted to demonstrate that a combination of different features along with NMR feature set leads to better results. In this analysis, the SVM machine was trained separately for each attribute group on first independent data set (including 20 positive and negative samples). The predicted class for all samples in the independent data set was extracted. Because the actual class of this test data set was available, we were able to examine the strength of various features. The diagram in Figure 3 presents the number of samples found correctly by each feature.
Figure 3.
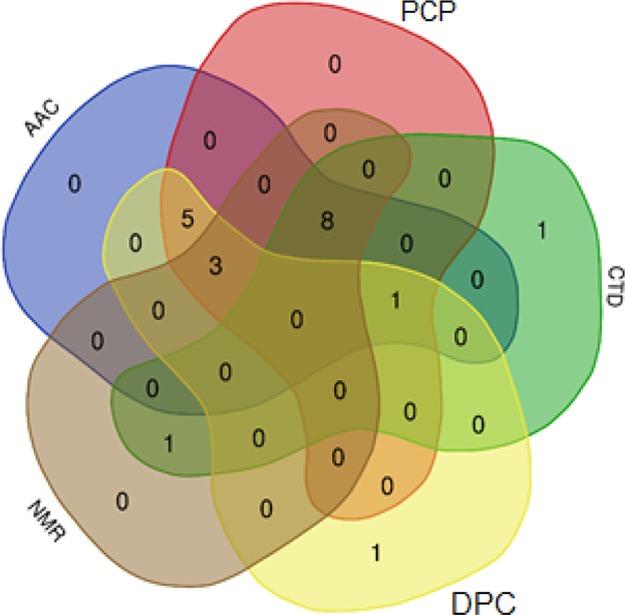
Power of various features in the right prediction of samples in the first independent data set.
Model Evaluation with Different Classifiers
Table 2 shows the performance of different classification models for our data. For evaluation of the models in the sense of TP rate (sensitivity) and FP rate (specificity), receiver operating characteristic (ROC) curves were plotted using an Orange Version 3.11.0.A ROC curve is a graphical plot that demonstrates the ability of a binary classifier system when its discrimination threshold is diverse. Figure 4 represents ROC curves for models used in BIPEP.
Table 2. Performance Evaluation Metrics with Different Classification Models Based on the Best 150 Features Selected.
| methods | accuracy | sensitivity | specificity | AUC |
|---|---|---|---|---|
| RF | 0.87 | 0.87 | 0.87 | 0.90 |
| kNN | 0.88 | 0.88 | 0.88 | 0.90 |
| Naïve Bayes | 0.85 | 0.87 | 0.85 | 0.91 |
Figure 4.
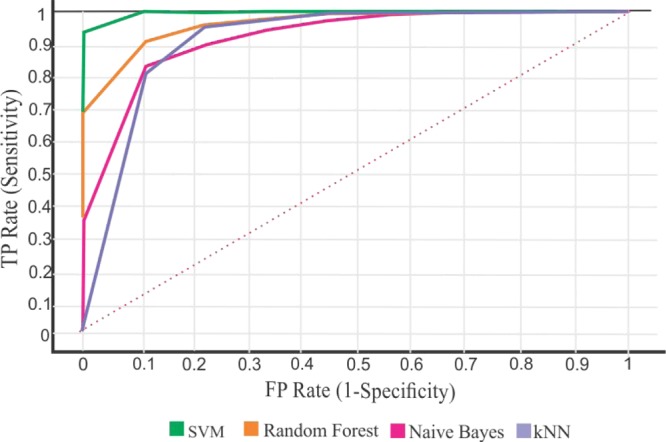
Ability of various binary classifier systems in term of ROC curves.
SVM Model Based on the Selected Feature
The most important features were selected for the SVM model (rbf kernel with cost = 0.1). We assessed the performance of SVM using five-fold cross validation. In fact, we ran our models with 90% data as a train set and 10% data as a test set, and ran this step 150 times. As illustrated in Figure 5, different evaluation parameters were calculated in all runs. The average values of the different performance metrics are listed in Table 3.
Figure 5.
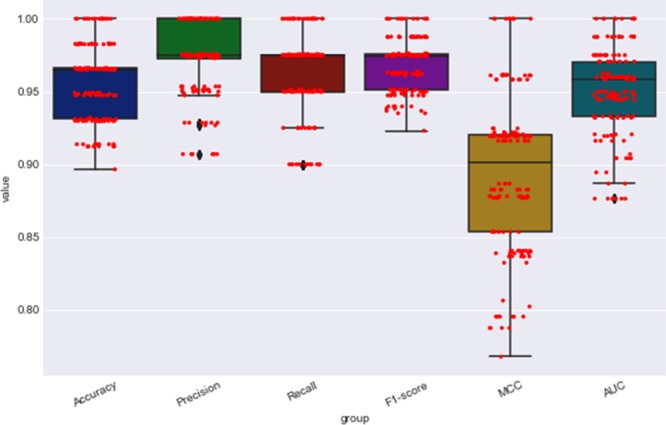
Comparison of various evaluation parameters was calculated in all runs for the five-fold cross-validation method. The red points in the figure illustrate the distribution of the different performance measures in the 150 runs.
Table 3. Comparison of Different Evaluation Metrics in a 5-Fold Cross Validation Method.
| evaluation parameters | accuracy | sensitivity | specificity | F1-score | MCC | AUC |
| five-fold cross validation | 0.95 | 0.97 | 0.96 | 0.97 | 0.89 | 0.96 |
Comparative Analysis
The performance of our SVM model, with a combination of different compositional feature sets and NMR features, was compared with the performance of the latest BIP prediction tools. Two independent data sets were used to judge the prediction ability of our method compared to existing approaches. Table 4 presents the evaluation parameters for various predictive tools. The results demonstrate that our model is more efficient in predicting biofilm inhibitory peptides. Moreover, to avoid a biased comparison toward selecting specific training data sets, the training sets of BioFIN and dPABBs have been used to compare the prediction power of BIPEP with two other models. As illustrated in Table S9, BIPEP was found to perform better predictions than the dPABBs and the BioFIN in terms of all performance metrics. This finding supports the hypothesis that a combination of different feature sets to makeup a feature space consisting of optimal relevant features, will lead to better results.
Table 4. Evaluation Parameters Value Among Various BIP Predictive Tools on Our First Independent Data set.
| independent data set | predictive tools | sensitivity (%) | specificity (%) | accuracy (%) | MCC (%) |
|---|---|---|---|---|---|
| first independent data set | dPABBs | 70 | 100 | 85 | 33.33 |
| BioFIN | 0 | 100 | 10 | 0 | |
| our model | 90 | 90 | 90 | 80 | |
| second independent data set | dPABBs | 94.44 | 81.25 | 88.82 | 77.19 |
| BioFIN | 0 | 100 | 42.55 | 0 | |
| our model | 98.14 | 85 | 92.55 | 85.03 |
Online Prediction Server
The developed user-friendly web-server is accessible at http://cbb1.ut.ac.ir/BIPClassifier/Index. This service will be able to obtain single or multiple peptides to calculate a wide variety of features that were mentioned in this paper. Moreover, the classifier service can exploit these features to predict whether each peptide has a biofilm inhibition property. All results that are represented in this section have been extracted using this web server.
Discussion
BIPs are natural peptides that have been attracting a great deal of attention in recent years because of their ability to eliminate biofilms.3 These peptides naturally code in many microorganisms. On the other hand, metagenomic samples contain a diverse community of bacteria, protozoa, fungi, and archaea, which are potentially a great source of BIPs. However, the main obstacle in analyzing these samples is the amount of data and difficulty of handling this data without a powerful computational tool to reduce the number of candidates for experimental validation.
In this paper, we have described a computational tool that can be used to predict biofilm inhibitory peptides. As was mentioned before, identifying and designing BIPs using experimental approaches is time consuming and costly. The identification of BIPs with computational tools is important in order to reduce appropriate candidates for experimental validation and helpful to biological labs. The current research describes a computing technique based on SVM that can predict BIPs more accurately than previously reported methods.
In this investigation, all available sequences of BIPs have been used to create our positive data set20,21 and Quorum sensing peptide (QSP) peptides were used to construct the negative data set.19 When we compared two data sets with respect to their AAC, it became apparent that positively charged amino acids like K, H, and R are more abundant in BIPs. A previous study by Segev-Zarko et al.35 showed that peptides rich in the number of Leu (K) and Lys (L) exhibit potential inhibitory activity against biofilms. Cationic residues assist BIPs to act against their target correctly. For instance, peptide’s biofilm inhibitory activity against its target will decrease by replacing cationic residues with an acidic residue.36
Preparing a computational approach to predict biofilm inhibitory peptides was a great challenge for two reasons. First, the number of experimentally validated BIPs is limited. Second, these peptide sequences have a variety of lengths and primary structures. Thus, we could not make proper predictions simply according to features based on the composition of different amino acids. In this work, a combination of features was used to map sequences onto feature vectors, which were subsequently used as inputs in SVM for the prediction of BIPs. For the first time, the CTD of different features and features based on NMR was used for prediction. These feature sets, along with AAC, DPC, and physiochemical features, led to the creation of our final feature space, which consists of 108 CTD, 12 NMR, 9 AAC, 13 DPC, and 8 PCP. The absence of each of them would change our final result.
As illustrated in Figure 3, all features have the ability to separate two data sets, but merging these feature sets helped us achieve the most optimal results. For example, the machine learned with DPC, has the ability to predict all BIPs correctly but is unable to predict non-BIPs. On the other hand, the SVM-based model, which trained with the NMR feature set, was effective in predicting BIPs and non-BIPs, but had true negatives and false positives, which were corrected with other feature sets. These results support our hypothesis that using multiple sets of attributes leads to better results. With the exception of AAC and DPC, the rest of the selected feature groups in this study were used for the first time in BIP prediction. On the other hand, different classifiers were used in this study, but the SVM-based model displays good performance as compared to kNN-, Naïve Bayes-, and RF-based models.
In the present study, we introduced features based on NMR spectra for the first time and investigated their importance in BIP prediction. Although these features alone did not provide a good separation between the two classes (BIPs and non-BIPs), the synergistic combination of this new feature set with other features improved the predictive power of the BIPEP tool. As illustrated in Figure 3, NMR features have an additive effect on other features in the data set that increase the predictive power of the tool. In fact, without using these features, we would have misidentified the number of peptides in the non-BIP class. Moreover, in our web server, the user can easily compute NMR features for other classification tasks in different scopes. This feature can be exploited in a variety of applications that need predictions based on amino acid sequences.
The performance of our BIP prediction model was also compared to two other BIP prediction servers (BioFIN and dPASBBs), two validation data sets were made and used for comparison. Our model was found to achieve higher accuracy than the two other methods. This result might be due to the use of a combination of different feature sets and the making of a feature space with the optimal relevant features from a diverse composition of measures.
Conclusions
Biofilms are known for causing different microbial diseases. These highly structured communities are resistant to various antibiotics, so designing new BIPs is extremely necessary. However, designing BIPs using laboratory methods takes a great amount of time and is a difficult task. Reducing the number of BIPs for experimental validation with computational tools effectively decreases the time and cost. Developing a prediction tool for BIPs was a challenging work because of a small number of experimentally validated BIPs. Despite this, this study used a combination of different feature groups and selected associated attributes with different feature selection, improving results. From another perspective, metagenome environments are excellent candidates for extracting different types of bioactive peptides, but screening these environments is not an easy task because of the vast volumes of data. The method presented in this study can be used and significantly reduces the number of potential candidates for laboratory work. Moreover, the webserver for this tool is available on http://cbb1.ut.ac.ir/BIPClassifier/Index and can supplement existing tools/techniques in predicting BIPs.
Materials and Methods
Data Set Preparation
Positive Sets
To construct the positive data set, sequences of anti-biofilm peptides have been gathered from databases that are publicly accessible. A total of 202 unique peptides were downloaded from the BaAMP database,20 a comprehensive database of BIPs and APD3,21 another database of BIPs. Figure 6 provides a summary of the positive data sets.
Figure 6.

BaAMP and APD3 data set used for constructing our positive data set. As shown in the figure, these two data sets had 12 peptide intersections.
Negative Sets
QSPs were selected for constructing the negative data set based on the method explained in ref (19). Quorum-sensing peptides are oligopeptides secreted by Gram-positive bacteria into the extracellular space, thus enabling bacteria to form the biofilm. The 88 non-anti-biofilm peptides that make up the negative database of the dPABBs tool were retrieved from http://ab-openlab.csir.res.in/abp/antibiofilm/ and used to form our negative data set. In order to have the same number of positive and negative peptides, the synthetic minority oversampling technique was performed to oversample the minority class.22
Data set Construction for Independent Validation
We needed to develop an approach to compare our method with the existing prediction tools, dPABBs and BioFIN. The comparison was made through the construction of two validation data sets from positive and negative peptides, which do not exist in the train data set of our models, and two other methods that we wanted to compare.
Two different independent data sets were used for comparison with existing methods. The first independent data set is the same data set used in dPABBs (contains 10 BIPs and 10 non-BIPs). CD-HIT23 (with −c 1.0, means 100% identity), confirmed that these 20 data points do not exist in the training set of BioFIN (Table S6). We conducted a literature survey to obtain more biofilm inhibitory peptides for validation. In a study done by Hancock et al.24 a QSAR model was used to predict BIPs and compare their prediction with experimentally validated BIPs. We used this experimental validation data set as a positive set for our second independent data set for validation of the presented model and comparison with other methods. To increase the number of negative data sets, QSPs were selected for this purpose. QSPs are oligopeptides used as auto-inducing molecules by Gram-positive bacteria in intraspecies QS that are known to enable the biofilm phenotype. A total of 80 unique QSPs were retrieved from the QSPpred web server to generate positive (training + independent) data sets. The CD-HIT tool confirmed (with −c 0.8, means 80% identity) that these 108 positive data points and 80 negative data points don’t have similarity values higher than 80 percent identity to the training set of our model (Table S7).
Moreover, the performances of these methods were compared in terms of their sensitivity, specificity, accuracy, and Mathew’s correlation coefficient (MCC) values for the two data sets.
Feature Extraction
Peptide sequences must be mapped to numeric feature vectors before being used as inputs in supervised learning classifiers. In this study, many different categories of features, including AAC, dipeptide composition-DPC and others, were used. These feature sets have been used for binary classification in different studies.14,15,25−27
In addition, based on the CTD of different features (polarity, polarizability, molecular weight, etc.),28,29 physicochemical and nuclear magnetic resonance (NMR) features, the corresponding feature vectors were extracted for the prediction of BIPs. All of the features, except NMR, such as the physiochemical features (PCP) were computed with PyDPI, a free python package for extracting variety of features from protein and peptide sequences.30 The physicochemical and NMR features were computed using the IAMPE web tools available at http://cbb1.ut.ac.ir/FeatureCalc/Index.
Feature Selection
Feature selection or variable selection, as a preprocessing stage, was the method used to select a subset of relevant features (variables, predictors) for model construction. Because of the importance of this step, we used several methods. We performed both the filter base method and the wrapper method for this step. Finally, the intersection of features that were reported by both methods was used for further analysis.
First, we trained our machine with all features separately. For example, the AAC feature was calculated for our data and then the SVM was used to investigate the performance of the machine for this feature set. These steps were repeated for all five group of features (AAC–DPC–PCP–CTD–and NMR features) (Table S8), which were extracted in the previous step. Then, we used a t-test-based selection method to determine which features separated the two class with a p-value smaller than 0.05. Among 1024 features, only 980 features aligned with these data. We then used recursive feature elimination (RFE) and SelectKBest from the scikit learn package in python to draw out the features that contributed the most out of the BIPs that correlate strongly to the classification variable. RFE is a feature selection method that fits a model and removes the weakest features until the defined number of features is achieved. In the SelectKBest method, features are chosen according to the highest k scores. To overcome any bias toward a specific split of training and testing data sets, this step was conducted to evaluate different data set splitting (100 splits). As a result, the intersection of features selected with two various methods comprise our feature space.
Construction of Machine-Learning-based Prediction Models
Support Vector Machine-based Prediction
A support vector machine is a supervised model which is used for classification tasks. Because of the small size of our data set, it was better to use a nonparametric method. Thus, we have therefore used SVM to predict BIPs because it is a nonparametric method and the most commonly used supervised learning technique.14,18,19,27,31 This kernel based classification algorithm forms a decision surface between positive and negative data to classify samples. According to the kernel structure, a linear or nonlinear hypersurface may discriminate between classes. Polynomial and radially basis functions are among the powerful nonlinear kernels. SVM classifier prediction capacity relies primarily on the type of kernel function selected to map the input information to the feature space. Because of the importance of choosing the optimal kernel and other appropriate parameters for the SVM base model, we applied a Grid search for the different amounts of SVM parameters. Grid-searching refers to the process of scanning data to configure optimal parameters for a given model.
RF-based Prediction
RF or random decision forest is an ensemble learning method that employ hundreds or thousands of independent decision trees to perform classification and regression.32,33 A detailed description of the RF algorithm has been reported elsewhere.34
The prediction ability of RF mainly depends on the three most important parameters of this classifier (i.e., the number of trees, number of attributes considered at each step, and the minimum number of samples required to split an internal node). By using a grid search upon these important variables, we are able to optimize their values.
Moreover, we examined other nonparametric classifiers, k-nearest neighbor (kNN) and Naïve Bayes for BIP prediction. However, the SVM still performed better than the others, so it was chosen as a classifier for our machine.
Cross-Validation Measure
In statistical prediction, cross-validation methods are often used to examine a predictor for its effectiveness in practical application. We used five-fold cross-validation to evaluate the performance of the predictive model used in this study. In the five-fold cross-validation, the whole data set is randomly split into five sets, out of which one set (test) is tested by a model developed on the remaining four sets (training). This process is then iterated 150 times.
Performance Evaluation
We evaluated the performance of our model using several metrics. We used sensitivity, specificity, accuracy, and MCC to evaluate the performance of the proposed model as calculated by the following formulae
where TP and TN are correctly predicted positive and negative examples, respectively. Similarly, FP and FN are wrongly predicted positive and negative examples, respectively, and MCC is the Mathew’s correlation coefficient.
Data Availability
An open source for both positive and negative data sets that related to this study hosted at (http://cbb1.ut.ac.ir/datasets/negative_data.txt), (http://cbb1.ut.ac.ir/datasets/positive_data.txt) and (http://cbb1.ut.ac.ir/datasets/independent_data.xlsx).
Acknowledgments
This research was in part supported by a grant from the Iran National Science Foundation (INSF) (funding reference number 93026701).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04119.
Detailed information about computation of different feature vectors, name and group of 150 best features; first independent data set for validation; second independent data set for validation; performance of presented models on separate feature vectors; and performance of BIPEP with training sets of BioFIN and dPABBs (PDF)
Author Contributions
K.K. and F.F.A. conceived and designed the study; F.F.A. collected and curated the sequence data sets and analysis, developed the models, and drafted the manuscript. K.K. contributed to the initial idea. S.A. contributed to biological insights. A.B. critically revised the manuscript. S.B. designed the web-server and implemented the server. All the coauthors contributed to the discussion, read, and approved the final manuscript. K.K. and S.A. corresponding authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Jamal M.; Ahmad W.; Andleeb S.; Jalil F.; Imran M.; Nawaz M. A.; Hussain T.; Ali M.; Rafiq M.; Kamil M. A. Bacterial Biofilm and Associated Infections. J. Chin. Med. Assoc. 2018, 81, 7–11. 10.1016/j.jcma.2017.07.012. [DOI] [PubMed] [Google Scholar]
- Costerton J. W.; Stewart P. S.; Greenberg E. P. Bacterial Biofilms : A Common Cause of Persistent Infections. Science 1999, 284, 1318. 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Joo H.-S.; Otto M. Molecular Basis of In Vivo Biofilm Formation by Bacterial Pathogens. Chem. Biol. 2012, 19, 1503–1513. 10.1016/j.chembiol.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantz L. A.; Francisco S. Consequences of the Widespread Use of Antibioties. Calif. Med. 1954, 81, 1–3. [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Moser C.; Wang H.-Z.; Høiby N.; Song Z.-J. Strategies for Combating Bacterial Biofilm Infections. Int. J. Oral Sci. 2015, 7, 1–7. 10.1038/ijos.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackman G.; Coenye T. Quorum Sensing Inhibitors as Anti-Biofilm Agents. Curr. Pharm. Des. 2015, 21, 5. 10.2174/1381612820666140905114627. [DOI] [PubMed] [Google Scholar]
- Batoni G.; Maisetta G.; Brancatisano F. L.; Semih Esin M. C. Use of Antimicrobial Peptides Against Microbial Biofilms: Advantages and Limits. Curr. Med. Chem. 2011, 18, 256. 10.2174/092986711794088399. [DOI] [PubMed] [Google Scholar]
- Russell J. B.; Rychlik J. L. Factors That Alter Rumen Microbial Ecology. Science 2001, 292, 1119–1122. 10.1126/science.1058830. [DOI] [PubMed] [Google Scholar]
- Albertsen M.; Hugenholtz P.; Skarshewski A.; Nielsen K. L.; Tyson G. W.; Nielsen P. H. Genome Sequences of Rare, Uncultured Bacteria Obtained by Differential Coverage Binning of Multiple Metagenomes. Nat. Biotechnol. 2013, 31, 533. 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- Stewart R. D.; Auffret M. D.; Warr A.; Wiser A. H.; Press M. O.; Langford K. W.; Liachko I.; Snelling T. J.; Dewhurst R. J.; Walker A. W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 1–11. 10.1038/s41467-018-03317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves L. D. F.; Westmann C. A.; Lovate G. L.; de Siqueira G. M. V.; Borelli T. C.; Guazzaroni M. Metagenomic Approaches for Understanding New Concepts in Microbial Science. Int. J. Genomics 2018, 2018, 1–15. 10.1155/2018/2312987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M.; Ghazi A.; Beloqui A.; Vieites J. M.; López-Cortés N.; Marín-Navarro J.; Nechitaylo T. Y.; Guazzaroni M.-E.; Polaina J.; Waliczek A.; et al. Functional Metagenomics Unveils a Multifunctional Glycosyl Hydrolase from the Family 43 Catalysing the Breakdown of Plant Polymers in the Calf Rumen. PLoS One 2012, 7, e38134 10.1371/journal.pone.0038134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. E.; Beys-da-Silva W. O.; Santi L.; Berger M.; Vainstein M. H.; Guimarães J. A.; Vasconcelos A. T. R. A Potential Source for Cellulolytic Enzyme Discovery and Environmental Aspects Revealed through Metagenomics of Brazilian Mangroves. AMB Express 2013, 3, 65. 10.1186/2191-0855-3-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra P.; Yan J.; Li J.; Fong S.; Siu S. W. I. AmPEP: Sequence-Based Prediction of Antimicrobial Peptides Using Distribution Patterns of Amino Acid Properties and Random Forest. Sci. Rep. 2018, 8, 1697. 10.1038/s41598-018-19752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B.; Basith S.; Shin T. H.; Choi S. MLACP : Machine-Learning-Based Peptides Prediction of Anticancer. Oncotarget 2017, 8, 77121–77136. 10.18632/oncotarget.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Zhou C.; Chen H.; Song J. ACPred-FL : A Sequence-Based Predictor Using Effective Feature Representation to Improve the Prediction of Anti-Cancer Peptides. Bioinformatics 2018, 34, 4007–4016. 10.1093/bioinformatics/bty451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Hu J.; Li F.; Song J. Comparative Analysis and Prediction of Quorum-Sensing Peptides Using Feature Representation Learning and Machine Learning Algorithms. Briefings Bioinf. 2018, 21, 106–119. 10.1093/bib/bby107. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Sharma A. K.; Jaiswal S. K.; Sharma V. K. Prediction of Biofilm Inhibiting Peptides : An In Silico Approach Preparation of Dataset. Front. Microbiol. 2016, 7, 949. 10.3389/fmicb.2016.00949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Gupta P.; Kumar R.; Bhardwaj A. DPABBs : A Novel in Silico Approach for Predicting and Designing. Sci. Rep. 2016, 6, 21839. 10.1038/srep21839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca M.; Maccari G.; Maisetta G.; Batoni G. BaAMPs: The Database of Biofilm-Active Antimicrobial Peptides. Biofouling 2015, 31, 193. 10.1080/08927014.2015.1021340. [DOI] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. APD3 : The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, 1087–1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla N. V.; Bowyer K. W.; Hall L. O.; Kegelmeyer W. P. SMOTE : Synthetic Minority Over-Sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. 10.1613/jair.953. [DOI] [Google Scholar]
- Li W.; Godzik A. Cd-Hit : A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Haney E. F.; Brito-sánchez Y.; Trimble M. J.; Mansour S. C.; Hancock R. E. W. Computer-Aided Discovery of Peptides That Specifically Attack Bacterial Biofilms. Sci. Rep. 2018, 8, 1871. 10.1038/s41598-018-19669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Mittal P.; Madhu M. K.; Sharma V. K. IL17eScan: A Tool for the Identification of Peptides Inducing IL-17 Response. Front. Immunol. 2017, 8, 1430. 10.3389/fimmu.2017.01430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Kapoor P.; Gautam A.; Chaudhary K.; Kumar R.; Chauhan J. S.; Tyagi A.; Raghava G. P. S. Computational Approach for Designing Tumor Homing Peptides. Sci. Rep. 2013, 3, 1607. 10.1038/srep01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B.; Basith S.; Shin T. H.; Lee G. MAHTPred : A Sequence-Based Meta-Predictor for Improving the Prediction of Anti-Hypertensive Peptides Using Effective Feature Representation. Bioinformatics 2018, 35, 2757. 10.1093/bioinformatics/bty1047. [DOI] [PubMed] [Google Scholar]
- Govindan G.; Nair A. S.. Composition, Transition and Distribution (CTD)—A Dynamic Feature For Predictions Based on Hierarchical Structure Of Cellular Sorting. 2011 Annual IEEE India Conference; IEEE, 2011.
- Manavalan B.; Subramaniyam S.; Shin T. H.; Kim M. O.; Lee G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715. 10.1021/acs.jproteome.8b00148. [DOI] [PubMed] [Google Scholar]
- Cao D.-S.; Liang Y.-Z.; Yan J.; Tan G.-S.; Xu Q.-S.; Liu S. PyDPI: Freely Available Python Package for Chemoinformatics, Bioinformatics, and Chemogenomics Studies. J. Chem. Inf. Model. 2013, 53, 3086–3096. 10.1021/ci400127q. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Kapil R.; Dhakan D. B.; Sharma V. K. MP3 : A Software Tool for the Prediction of Pathogenic Proteins in Genomic and Metagenomic Data. PLoS One 2014, 9, e93907 10.1371/journal.pone.0093907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B.; Lee J.; Lee J. Random Forest-Based Protein Model Quality Assessment ( RFMQA ) Using Structural Features and Potential Energy Terms. PLoS One 2014, 9, e106542 10.1371/journal.pone.0106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Lee K.; Joung I.; Joo K.; Brooks B. R.; Lee J. Sigma-RF: Prediction of the Variability of Spatial Restraints in Template-Based Modeling by Random Forest. BMC Bioinf. 2015, 16, 94. 10.1186/s12859-015-0526-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random Forests. Mach. Learn. 2001, 45, 5–32. 10.1023/a:1010933404324. [DOI] [Google Scholar]
- Segev-Zarko L.; Saar-Dover R.; Brumfeld V.; Maria Luisa Mangoni Y. S. Mechanisms of Biofilm Inhibition and Degradation by Antimicrobial Peptides. Biochem. J. 2015, 468, 259. 10.1042/bj20141251. [DOI] [PubMed] [Google Scholar]
- Daep C. A.; Lamont R. J.; Demuth D. R. Interaction of Porphyromonas Gingivalis with Oral Streptococci Requires a Motif That Resembles the Eukaryotic Nuclear Receptor Box Protein-Protein Interaction Domain. Infect. Immun. 2008, 76, 3273–3280. 10.1128/iai.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An open source for both positive and negative data sets that related to this study hosted at (http://cbb1.ut.ac.ir/datasets/negative_data.txt), (http://cbb1.ut.ac.ir/datasets/positive_data.txt) and (http://cbb1.ut.ac.ir/datasets/independent_data.xlsx).