

Abstract

Based on a facile three-step preparation method, Cu2O/Au/TiO2-NAs ternary heterojunction nanocomposites have been successfully synthesized by electrodepositing a Cu2O layer on the surface of Au nanoparticles (NPs) decorated highly ordered TiO2 nanotube arrays (NAs). The structure, surface morphology, chemical composition, and optical and intrinsic defects properties of the as-prepared samples are characterized by transmission and scanning electron microscopy (TEM and SEM), X-ray diffraction (XRD), UV–vis light absorbance spectra, Raman scattering, and X-ray photoelectron spectroscopy (XPS). Simultaneously, the Cu2O/Au/TiO2-NAs ternary nanohybrids exhibited progressively improved photoelectrocatalytic (PEC) performance compared with the dual Cu2O/TiO2-NAs type-II nanoheterojunctions, confirming by the photocurrent density versus testing time curve (amperometric I–t curve), open-circuit potential versus testing time curve (Voc–t curve), and electrochemical impedance spectroscopy (EIS) measurements, which were mainly ascribed to the synergistic effect of reduced interfacial charge transfer resistance and boosted energetic charge carriers generation associated with embedding Au NPs. Furthermore, the self-consistent charge transfer mechanism of Z-scheme and interband transitions mediated with Au NPs for Cu2O/Au/TiO2-NAs triple nanocomposites is proposed, which was evaluated by nanosecond time-resolved transient photoluminescence (NTRT-PL) spectra excited by 266 and 400 nm, respectively. Following this scheme, UV–vis light photocatalytic activities of Cu2O/Au/TiO2-NAs ternary nanohybrids were elaborated toward photodegradation of methyl orange (MO) in aqueous solution, and the photodegradation rate of optimum triple nanocomplex was found to be 90%.
Introduction
In the past few decades, the increasing environmental pollution with increasing development of economy threatens the fragile balance of the ecological environment, which is caused by inadequate combustion of traditional fossil energy. Fujishima and Honda1 stated that hydrogen was produced during photoelectrochemical (PEC) water splitting by using TiO2 electrode under UV irradiation in the year 1972. The PEC technique of semiconductor-based materials has received appreciable interests for the decomposition of dyes and volatile organic compounds, owing to its convenient operation, gentle reactive conditions, and no second pollutants, which utilize solar energy to directly convert organic pollutants into inorganic minerals in the form of H2O and CO2.2 Remarkably, TiO2 has been confirmed to be an excellent candidate for photo-oxidation decomposing harmful organic compounds because of its low cost, nontoxicity, physicochemical properties stability, and photocorrosion resistance characteristics.3 Yet, the intrinsic disadvantages for pure TiO2 are ineluctable, including low quantum yield and fast recombination of photoinduced electron–hole (e––h+) pairs.4 Specifically, the PEC activities of self-organized TiO2 nanotube arrays (NAs) were proved superior to those of their bulk materials, which ascribed to high specific surface area and vertical electron transportation stemmed from the nanotubular architecture.5 Nevertheless, the wide band gap (BG) for anatase TiO2-NAs corresponds to the utilization of less than 5% in the solar spectrum, while the visible light accounts for 43–46% of the total energy.6 Evidently, the embarrassment for single-component PEC photoelectrode materials is the mismatch between the powerful redox capability and sufficient photoresponse. Logically, considerable efforts have been dedicated for expanding the visible-light absorption and enhancing the photoinduced e––h+ pairs redox ability of the TiO2-NAs, for example, sensitizing with narrow band-gap (NBG) semiconductors to construct type-II heterogeneous nanocomposites, doping with transition metal ions, and fabricating nanostructures with defective characteristics. Among aforementioned various strategies, the development of heterojunction nanohybrids by coupling TiO2-NAs with NBG semiconductors, such as CdS, CdSe, CdTe, and Cu2O,7−10 have received considerable attention because of the merit in improving the charge separation efficiency and integrating respective advantages of each constituents. Moreover, conventional type-II heterojunction nanocompounds may have different photoresponse ranges, which are beneficial to capture a greater portion of the solar spectrum.11,12
It should be noted that the cadmium-based compounds are highly toxic and might cause harsh environmental impact if not recycled properly. Among the NBG semiconductors, cuprous oxide (Cu2O), a native p-type direct BG semiconductor with a cubic crystal structure and a BG energy around 1.9–2.2 eV at room temperature,13 becomes the promising PEC candidate to sensitize TiO2 due to several intrinsic advantages, such as environmental friendliness, abundance in the earth, superior visible light harvesting ability,14 and its appropriate BG alignment with TiO2. As regards these virtues, Cu2O is widely applied in the fields of electrochromic, gas sensing, hydrogen production from water splitting, and PEC organic pollutants degradation.15−17 Therefore, many studies have demonstrated that the photocatalytic activity of Cu2O/TiO2 type-II nanoheterostructures could be enhanced due to the following reasons: (i) the decorated Cu2O expanded absorption spectrum of the TiO2-NAs from the ultraviolet to visible-light range and (ii) the built-in electric field at the interface of Cu2O/TiO2 nanoheterojunctions than that of pure materials, which minimizes the recombination rate of photo-excited e––h+ pairs. Moreover, it is well established that the high aspect ratio of tubular nanostructures is favorable for adsorbing more oxygen molecules (O2), which can scavenge photogenerated conduction band (CB) electrons (e–cb) to produce more superoxide anion radical (O2–), and increasing the photocatalytic degradation ability of Cu2O/TiO2 nanocompounds. (iii) Due to the matching of the band structure between Cu2O and TiO2-NAs, both the CB and valence band (VB) of Cu2O lie higher than that of TiO2-NAs, which are facilitated to rapid transfer of photoinduced electrons from the CB of activated Cu2O into the CB of TiO2-NAs. Meanwhile, the photogenerated holes in these two semiconductors follow a reverse migration, i.e., the photoinduced holes are accumulated on the VB of Cu2O. However, these advantages are at the expense of redox abilities of Cu2O/TiO2 nanohybrids, originating from the Cu+ that was oxidized to Cu2+ by holes, which indicate a weakened built-in electric field driving force that may be caused by photocorrosion of Cu2O.18−20 Thus, the excogitation and effectuation of innovative nanoarchitecture PEC materials for high visible light harvesting and charge separation with low recombination is extremely essential and urgent.
In the case of noble metal nanoparticles (NM-NPs) embedded nanostructures, i.e., semiconductor-metal-semiconductor (S-M-S) ternary nanohybrids, have attracted considerable attention in PEC application due to its excellent charge separation ability and powerful oxidation/reduction potential, which attributed to introduce Z-scheme21 and localized surface plasmon resonance (LSPR) enhancement charge-transfer (CT) mechanism into the heterostructure design.22 Simultaneously, the LSPR of plasmonic metal can improve optical absorption and scattering properties originated from the plasmon exciton coupling and local dielectric environment variation, respectively.23,24 As in representative reports, Aguirre et al.25 have demonstrated that Cu2O/TiO2 type-II binary heterostructures exhibited improved photoreduction performance of CO2 by direct Z-scheme CT process. However, Li et al.26 have synthesized TiO2-Au-Cu2O ternary heterostructure nanocomposites as a photocatalyst, which showed higher PEC performance for H2 production and CO2 photoreduction than pure Cu2O and binary TiO2/Cu2O under simulated solar-light irradiation. Apparently, the capability of charge separation and redox of Au NPs-embedded three-component nanostructures were superior to that of type-II binary nanoheterojunctions, which originated from the Z-scheme electron transportation and LSPR effect. It is also to be observed that NM-NPs could significantly enhance the photoluminescence (PL) spectra intensity of TiO2 UV and defect states emissions either via direct plasmon-induced electron transfer or radiative resonant energy transfer by the time-resolved PL, respectively.27,28
In this work, we propose a straightforward electrodepositing approach to synthesize Cu2O/Au/TiO2-NAs ternary heterostructure nanocomposites, and the characterization for its optical and chemical components was examined by UV–vis light absorbance spectra, X-ray diffraction (XRD), Raman spectra, and X-ray photoelectron spectroscopy (XPS). The as-fabricated Cu2O/Au/TiO2-NAs triple nanohybrids show an enhanced photocatalytic activities for photodegradation of methyl orange (MO) under UV–vis light irradiation in comparison to the relevant samples, including pristine TiO2-NAs, and binary Cu2O/TiO2-NAs type-II nanoheterojunctions, while their PEC performances were characterized by photocurrent density versus time response (amperometric I–t curve), the open-circuit potential versus time curve (Voc–t curve), and the electrochemical impedance spectroscopy (EIS) measurements, respectively. To the best of our knowledge, the validness of the Z-scheme CT mechanism has been controversial due to the lack of direct evidence. Yet, our results have provided further experimental evidence in favor of Z-scheme and interband transitions (i.e., Au-mediated) photoinduced interfacial CT dynamics for ternary Cu2O/Au/TiO2-NAs heterojunction nanocomposites, associating with radiative recombination PL of near band edge (NBE) and intrinsic defect states emissions, which are approved by the nanosecond time-resolved transient photoluminescence (NTRT-PL) spectra excited by 266 and 400 nm femtosecond (fs) laser source, respectively. The purpose of our study is to design and construct a well-defined Z-scheme heterostructure system with a verified, reliable CT mechanism for pushing forward the practice of a Z-scheme photocatalyst.
Results and Discussion
The surface morphology and cross-sectional features of the pristine TiO2-NAs, Au/TiO2-NAs, dual Cu2O/TiO2-NAs type-II nanoheterojunctions, and ternary Cu2O/Au/TiO2-NAs nanoheterojunctions with different Cu2O-NPs sputtering times (20, 40, and 80 s) were characterized by SEM and TEM as shown in Figure 1. Figure 1a shows the top-view SEM picture, exhibiting the open top of the nanotubes with a thin sidewall thickness of about 10 nm and a pore diameter of less than 60 nm. One of the insets in Figure 1a is the SEM image for cross-sectional morphologies of TiO2-NAs, explicitly displaying highly ordered and vertically oriented TiO2-NAs with a length of about 1 μm, and the other inset is the TEM image of individual nanotube, with an outer diameter is about 60 nm, which is in accord with the top-view SEM observation. Figure 1b illustrates the morphologies of TiO2-NAs film after Au coating and annealing at 450 °C, expositing well-isolated gold NPs with a size of less than 50 nm, which is formed by thermal migration and re-condensation of the original continuous gold granular. As will be discussed latter, the surface decoration of Au NPs on TiO2-NAs film plays a key role for the enhanced interfacial CT process. Figure 1c exhibits the top-view SEM picture of the Cu2O/TiO2-NAs nanocomposites with a Cu2O NPs deposited time of 40 s after annealing at 450 °C. The surface of Cu2O/TiO2-NAs samples became rougher as a result of Cu2O deposition. In some areas, clusters of Cu2O NPs were formed in the entrances of the TiO2 nanotube openings. The inset in Figure 1c shows the cross-sectional morphologies of Cu2O/TiO2-NAs, exhibiting a hierarchical structure for polyhedral Cu2O and TiO2-NAs, which witness the successful synthesis of dual type-II Cu2O/TiO2-NAs nanoheterojunctions. Figure 1d–f present the top-view SEM images for the Cu2O/Au/TiO2-NAs nanocomplex with increasing Cu2O electrodeposition time from 20 to 80 s after annealing at 450 °C, respectively. It could be distinctly observed that the deposited Cu2O NPs infilled the interstices of the nanorod array and the thickness of the Cu2O film increased with increasing electrodeposition time.
Figure 1.

Morphological characterization of pristine TiO2 nanotube array (TiO2-NAs) films, Au/TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs nanoheterojunction complex, respectively. (a) SEM pictures of as-formed pristine TiO2-NAs with cross-sectional microstructures (inset) and TEM picture for single TiO2 nanotube (inset). (b) Top-view SEM image of Au/TiO2-NAs. (c) SEM pictures of as-fabricated Cu2O/TiO2-NAs with cross-sectional microstructures (inset). (d–f) Top-view SEM images of Cu2O/Au/TiO2-NAs with Cu2O deposited times of 20, 40, and 80 s, respectively.
The HRTEM image was performed to investigate the interface morphology of the resulting Cu2O/Au/TiO2-NAs with Cu2O deposition time 40 s ternary heterojunction nanocomposites, as illustrated in Figure S1 (Supporting Information). The HRTEM image exhibits characteristic lattice spacings of 0.23, 0.25, and 0.35 nm, corresponding to the (111) lattice plane of the Au,29 the (111) lattice plane of the Cu2O,30 and the (101) lattice plane of TiO2,31 respectively, which provides the direct evidence for the good contact between ternary Cu2O and Au/TiO2-NAs nanocomplex. According to the size distribution, it is evidently observed that the average thickness for Cu2O NPs and average diameter for Au NPs are 10 and 17 nm, respectively. Simultaneously, there seems to be no other elemental existing at the interface of nanoheterostructure.
To calculate the true content or molar ratios of Au NPs in different Cu2O/Au/TiO2-NAs ternary nanocomposites, the weight percentages (wt %) of Ti, Au, and Cu were determined by EDXRF, which is a surface-sensitive, non-destructive, and standard analytical method. Figure 2 illustrates the EDXRF spectrum recorded for Au/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs nanohybrids with different amounts of the Cu2O catalyst. It is clearly observed that the two peaks located at 2.12 and 9.71 keV in all specimens, which originated from the Mα and Lα emission lines, were successfully excited by Au.32 Simultaneously, the two peaks sited at 4.51 and 4.93 keV appeared in all samples, corresponding to the Kα and Kβ emission lines of Ti, respectively,32 while the two peaks were excited at a relatively higher photon energy, i.e., centered at 8.04 and 8.91 keV, which ascribed to the the Kα and Kβ emission lines of Cu, respectively.31 No other elements were identified in the spectra, and results indicated that Au and Cu were successfully loaded on the TiO2-NAs.
Figure 2.

Energy-dispersive X-ray fluorescence spectrum (EDXRF) of Au/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs nanoheterojunctions with different Cu2O electrochemical deposition times (20, 40, and 80 s).
According to the directly proportional relationship between the measured intensities and the concentration of elements, UniQuant software based on the fundamental parameters approach has been applied for quantitative analysis of Au/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs nanohybrids with different amounts of Cu2O, as listed in Table S1 (Supporting Information). It follows from Figure 2 that the characteristic K-lines for Cu element increasing with the wt % of Cu depositing content increased. Consequently, the characteristic peak intensities and wt % of Ti and Au decreasing as the Cu deposited amount increased. The different ratios of Cu2O/Au/TiO2-NAs ternary nanocomposites affect the PEC performance, which will be discussed in the next sections.
The crystal structures and composition of as-fabricated dual Cu2O/TiO2-NAs, and ternary Cu2O/Au/TiO2-NAs with Cu2O deposition time 20 s nanocomplex were examined by XRD before and after the photodegradation reaction, as shown in Figure S2 (Supporting Information). Figure S2a presents the anatase TiO2 (JCPDS 197921-1272) diffraction peaks of Cu2O/TiO2-NAs before photodegradation at 25.67°, 37.89°, 53.97°, and 62.82°, indexing to (1,0,1), (0,0,4), (1,0,5), and (2,0,4) lattice planes, respectively, labeled by the symbol “▼”. Meanwhile, a cubic phase Cu2O was identified by the diffraction peaks at scattering angles (2θ) of 42.41° and 70.03° that were indexed to (2,0,0) and (3,1,0) planes of Cu2O (JCPDS 65-3288), respectively, marked by the symbol “■”. In contrast to the specimen of Cu2O/TiO2-NAs before the photodegradation reaction, as illustrated as Figure S2c, no other diffraction peaks were detected in that of after the photodegradation reaction except the diffraction signals from the CuO (JCPDS 80-1916), located at 46.23° and 48.68° and indexing to (−1,1,2) and (−2,0,2) crystal planes, labeled as “◆”. It is assumed to believe that the formation of CuO was derived from the photocorrosion of Cu2O associated with VB hole, i.e., Cu+ + h+ → Cu2+.33Figure S2b,d exhibits the XRD patterns of Cu2O/Au/TiO2-NAs heterojunction nanocomposites with a loading time of 20 s of Cu2O before and after the photodegradation reaction, respectively. Both of the diffraction peak positions for (b) and (d) are the same, located at 25.67°, 37.89°, 44.33o° 52.56°, 62.13°, and 65.76°. Evidently, the anatase phase TiO2 (2θ = 25.67°, 37.89°, and 62.13°) and cubic phase Cu2O (2θ = 52.56° and 65.76°) were identified by the JCPDS 197921-1272 and JCPDS 65-3288. A supplementary peak marked with “●”, which matched the expected position of the deposited noble metals, can be explicitly observed at 44.33°, which was readily assigned to the (2,0,0) plane of Au NPs (JCPDS 04-0784) and strongly indicating that ternary Cu2O/Au/TiO2-NAs heterojunction nanohybrids have successfully been synthesized. The diffraction peaks of Cu2+ were not obviously distinguished by an XRD pattern before and after the photodegradation reaction, which indicated that the crystal structure of Cu2O was stable during the Cu2O/Au/TiO2-NAs PEC reaction process.
In order to examine the absorbance spectra and also to determine the band gaps for as-prepared samples, the UV–vis absorbance spectra were carried out, as in Figure S3a,b (Supporting Information). Figure S3a shows the optical absorption properties and corresponding band-gap energies (inset) of pristine TiO2-NAs and pure Cu2O NPs (Cu2O electrochemical deposition time is 40 s on a quartz glass substrate) after annealing 450 °C. It is obvious that the absorption edge of pristine TiO2-NAs appears at 400 nm, while the absorption edge for pure Cu2O appears at 558 nm. The derived Tauc plots are used to calculate the band-gap energy for TiO2 and Cu2O semiconductors in the inset of Figure S3a. The (αhυ)1/n versus the photon energy hυ curves plotted according to the following equation:34 (αhυ)1/n = A(hυ – Eg), where α is the absorption coefficient, h is the Planck’s constant, υ is the photon frequency, hυ is the incident light energy, Eg is the band-gap energy, and A is a proportionality constant. Simultaneously, the value of n depends on the characteristics of the transition in a semiconductor, where n = 2 for an indirect transition semiconductor, and n = 1/2 for a direct transition semiconductor. As the latter type of transition, n should be taken as 1/2 for both TiO2-NAs and Cu2O.35,36 Thus, the intercept of the tangent to the photon energy axis would give a good approximation of the Eg of the samples. The calculated band-gap values (Eg) of TiO2-NAs (black curve) and Cu2O (red curve) are evaluated to be 3.1 and 2.2 eV, which are consistent with the absorption edge, respectively.
To explore the variation in optical properties and energetic electrons origin of the as-fabricated samples in the presence of Au NPs, the UV–vis absorbance spectra of the corresponding heterojunction films are also provided in Figure S3b. It can be observed that there is an extended visible absorption region for triple Cu2O/Au/TiO2-NAs nanoheterojunctions compared with a single semiconductor. Meanwhile, it is found that the spectra shapes for all of the ternary Cu2O/Au/TiO2-NAs nanocomposites consisted of two parts, 400–500 nm and 500–600 nm, which originated from the interband transition and the LSPR of Au NPs with a size approaching 50 nm.37,38 The absorbance spectra of Au NPs produced shows a LSPR maxima at wavelength of 541 nm, indicative of well-separated Au NPs as illustrated in the inset of Figure S3b. The absorbance intensity increased with the content of Cu2O in the Cu2O/Au/TiO2-NAs nanocomplex increasing from 20 to 40 s, indicating that forming the ternary nanocomposites is beneficial to light harvesting. However, the absorbance intensity decreased with the content of Cu2O in the ternary nanocomplex increasing from 40 to 80s, which may be ascribed to the excess Cu2O deposition that blocks the excited absorption of Au NPs.
It is well known that XPS analysis is an efficient tool to examine the chemical and electronic structure. Evidence of oxygen-related intrinsic defects and compositions in Cu2O/Au/TiO2-NAs heterojunction nanocomposites is further conducted by the high-resolution XPS spectra, as shown as Figure S4 (Supporting Information). Figure S4a presents the survey spectrum of Cu2O/Au/TiO2-NAs nanohybrids, distinctly revealing the presence of Ti, Au, Cu, and O elements. Some C element can be observed since the introduction of extraneous carbon in the instrument operation or sample preparation. Figure S4b exhibits the fitting result of Ti 2p XPS spectra for as-prepared Cu2O/Au/TiO2-NAs; the black hollow circles are experimental data points, and the red solid line is the result of curve fitting. After subtracting the inelastic background, two doublets were necessary to fit the Ti 2p signal. The Ti 2p spectra were decomposed into two components originating from Ti4+ and Ti3+, including Ti4+ 2p3/2, Ti3+ 2p3/2, Ti4+ 2p1/2, and Ti3+ 2p1/2. The different binding energy (BE) values corresponding to different oxidation states of Ti atoms, the BE peaks at 458.1, 458.8, 463.8, and 464.4 eV are attributed to the Ti3+ 2p3/2, Ti4+ 2p3/2, Ti3+ 2p1/2, and Ti4+ 2p1/2 of Ti2+, respectively, which well coincide with the previous literature.39 It obviously demonstrates the presence of oxygen vacancies (Vo) in the as-formed TiO2-NAs substrate due to the experimental detection of the Ti3+ species. Simultaneously, the BE peaks located at 932.5 and 952.5 eV in Figure S4c can be ascribed to those of Cu 2p3/2 and Cu 2p1/2 from Cu2O, respectively.40 Furthermore, O 1s XPS spectra, which are illustrated in Figure S4d, were wide and approximately symmetric in the range of 526–534 eV, indicating that there were multiple oxygen states.41 The black hollow circulars are experimental data points, and the red solid line is the result of curve fitting. After fitting, we can observe that there were three kinds of chemical states of oxygen, Ti–O (530 eV), Cu–O (530.5 eV), and −OH (531.5 eV) with the increasing of BE.41,42 No satellite peaks of Cu2+ (CuO) was identified in the Cu2O/Au/TiO2-NAs films, which is consistent with the XRD analysis. Thus, we can reasonably conclude that cubic Cu2O species were indeed deposited onto the surface of Au/TiO2-NAs nanoheterojunctions, which could greatly increase the transfer and separation efficiency of charge carriers as well as the PEC performance.
It is well known that the EIS technique is a powerful method to evaluate the interfacial CT and separation process between the electrode and electrolyte solution, which is significantly influenced by the PEC performance of the photocatalyst. To further explore the interfacial CT resistance occurring at the binary type-II and ternary nanoheterojunctions, the characteristic Nyquist plots of the specimens recorded at an open-circuit potential under AM 1.5 simulated solar light irradiation are depicted in Figure S5 (Supporting Information). Figure S5a presents the Nyquist plots of pristine TiO2-NAs, binary Cu2O/TiO2-NAs, and ternary Cu2O/Au/TiO2-NAs heterojunction nanohybrids, and the equivalent circuit is shown in Figure S5b. The Nyquist plots are generated between the imaginary component of the impedance (Z″) and the real component (Z′). In the equivalent circuit, Rs, Rct, Ws, and CPE represented the solution resistance, CT resistance, Warburg impedance, and constant phase element of the non-ideal frequency-dependent capacitance, respectively.43 All curves exhibited the characteristic arc spectra, in which the smaller arc represents the lower CT resistance at the interface between the electrode and electrolyte. It has been found that the arc radius on the plot of the Cu2O/TiO2-NAs curve is smaller than that of the pure TiO2-NAs film without being decorated by Cu2O, which demonstrates that the type-II nanoheterojunctions formed between Cu2O and TiO2-NAs promote the separation of electron–hole (e––h+) pairs and the CT of interfacial electron. Furthermore, with increasing deposition time of Cu2O for the Cu2O/Au/TiO2-NAs nanocomplex from 20 to 80s, the arc radii of EIS curves decrease with more Cu2O amount (from 20 to 40s) and then increase when the Cu2O deposition time is 80 s. The Cu2O/Au/TiO2-NAs with a Cu2O decorated time of 40 s exhibit the minimum interfacial electron CT resistance and optimum conductivity. This result indicates that the interactions between Au NPs and Cu2O/TiO2 heterojunction could effective reduce electron transport resistance and accelerate the electron mobility by exciting the Au-mediated synergistic effect. A proper deposition amount of Cu2O can improve the conductivity and enhance interfacial CT, but excessive Cu2O may suppress the CT.
In order to further clarify the separation efficiency of photogenerated charge carriers of as-prepared specimens, amperometric I–t curves were recorded and are exhibited in Figure S6 (Supporting Information). The photo-generated current of the samples was tested for 280 s, and the time interval between the light-on and the light-off was 35 s in 0.5 M Na2SO4 aqueous solution under intermittent simulated solar light irradiation. The photocurrent increased significantly, reached a relatively steady state, and dramatically decreased back to approximately zero when the lamp was turned on–off cycle, demonstrating that the well-ordered TiO2-NAs photoelectrodes represented good reproducibility and stability under several on–off cycles of intermittent light illumination. The pristine TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs films deposited Cu2O at different times (20, 40, and 80 s) exhibited photocurrent density values of 0.023, 0.11, 0.18, 0.33, 0.26 mA/cm2, respectively. The bare TiO2-NAs films exhibited the least photocurrent response due to its wide band gap, which is limited to absorb only ∼5% or less of solar spectrum. Under the same condition, Cu2O/TiO2-NAs showed a higher photocurrent density, manifesting the type-II heterojunction formed between Cu2O and TiO2-NAs, which facilitates the separation of photogenerated e––h+ pairs than bare TiO2-NAs. Simultaneously, for the reason of photocorrosion, the photocurrent density of Cu2O/TiO2-NAs gradually decreases during the PEC measurement, which coincided with the XRD analysis. The photocurrent density explicitly increases for coupling Cu2O/TiO2-NAs with Au NPs, which acted as an electron storage center. The Cu2O/Au/TiO2-NAs with a deposited time of 40 s displayed the best photocurrent response, which is three times higher than that of the Cu2O/TiO2-NAs electrode. When the Cu2O deposition time was 80 s, the Au/TiO2-NAs was completely covered by an excess amount of Cu2O, which may block the energetic electrons photoexcitation triggered by the interband-transition effect, resulting in the reduced photocurrent. Therefore, the enhanced amperometric I–t properties for Cu2O/Au/TiO2-NAs heterojunction nanohybrids can be attributed to the synergistic effect of the favorable energy level between Cu2O and TiO2-NAs and facilitated charge separation mediated by Au metal.
The open-circuit potential (Voc) is a key parameter used to pinpoint photogenerated e––h+ pairs accumulation under simulated solar light irradiation. Voc–t curves for the specimens were tested for 350 s, and the light-on and light-off time interval was 50 s, as shown in Figure S7 (Supporting Information). It can be clearly observed that the Voc instantaneously increased when the light irradiation was turned on and remained stable because of the competition between photogenerated charge accumulation and recombination. When the light irradiation was turned off, the Voc decayed gradually in virtue of accumulated electrons successively captured by electron acceptors and recombined with holes. The Voc for pristine TiO2-NAs was the least, which can be ascribed to limited optical absorption caused by the wide band gap. The Voc for Cu2O/TiO2-NAs exhibited higher than that of pure TiO2-NAs owing to the enhanced electron accumulation, which is caused by the injection of electrons to the CB of TiO2 due to the formation of the Cu2O/TiO2-NAs p–n heterojunctions. It can be seen that the Voc values improved with the Cu2O deposition time increasing from 20 to 40 s and then decreased with a more deposition time was 80 s, which is consistent with the change in trend of EIS and amperometric I–t curves measurements. The deposition of Au NPs increased the separation rate of the photogenerated e––h+ pairs and enhanced the accumulation of electrons within the Cu2O/TiO2-NAs.
In order to interpret the generation, migration, and trap ultrafast dynamics process of the photogenerated charge carriers and also to clarify the radiative recombination electrons transitions of the interfacial energy band structures, we have adopted the method of transient PL spectrum to evaluate the CT process for the as-prepared binary and ternary heterojunction nanocomposites. Figure 3a presents the three-dimensional (3D) NTRT-PL spectra for the Cu2O/TiO2-NAs nanocomplex with every 1.5 ns time interval evolution under 266 nm irradiation. To better demonstrate the variation of the PL peak position and intensity versus time evolution, two-dimensional (2D) NTRT-PL spectroscopy is used, as illustrated in Figure 3b. It is explicitly observed that the Cu2O/TiO2-NAs sample exhibits 430 nm (ca. 2.8 eV) and 560 nm (ca. 2.2 eV) transient PL emission peaks with 6 ns time evolution, which are ascribed to the conduction band electrons (e–CB) radiative recombination associated with Vo defect states levels in TiO2 and near-band-edge (NBE) photoinduced electrons direct transition recombination of Cu2O,44 respectively.
Figure 3.
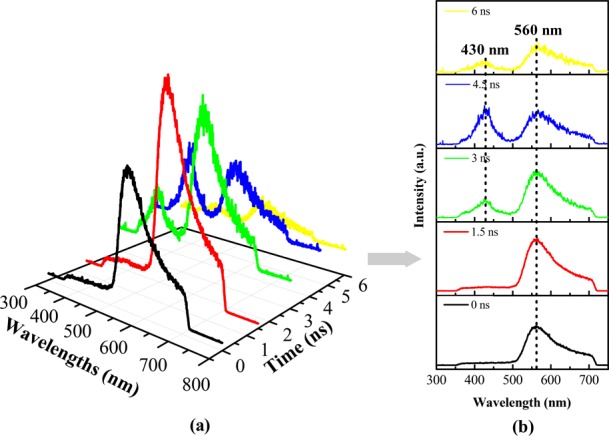
(a) Three-dimensional and (b) two-dimensional nanosecond time-resolved transient photoluminescence (NTRT-PL) spectra of the binary Cu2O/TiO2-NAs nanocomposites, respectively.
As we can see, Figure 4 exhibits the NTRT-PL spectra of ternary Cu2O/Au/TiO2-NAs heterojunction nanohybrids with different Cu2O NPs deposition times of 20, 40, and 80 s, excited at 266 nm at room temperature, respectively. For the Cu2O/Au/TiO2-NAs nanocomplex with depositing times of 20 and 40 s, the NTRT-PL spectra show a distinct blue shift of transient PL emission peaks, centered from 2.4 to 2.6 eV with time evolution, which originated from photogenerated e–CB radiative recombination associated with T3+ shallow trap states in TiO2.45 In addition, the NTRT-PL spectra for the Cu2O/Au/TiO2-NAs nanocomplex with a deposition time of 80 s situated at 385, 431, and 558 nm of transient PL emission peaks with time evolution, the peak at 385 nm is assigned to the NBE photogenerated e––h+ pairs direct recombination of TiO2.46 As stated above, the peaks at 431 and 558 nm are pertained to the e–CB radiative PL related with Vo defect states in TiO2 and free exciton recombination for Cu2O, respectively.
Figure 4.

NTRT-PL spectra of the ternary nanosized Cu2O/Au/TiO2-NAs heterojunction films deposited Cu2O with different times of (a) 20 s, (b) 40 s, and (c) 80 s excited at 266 nm, respectively.
Based on the above experimentally observed blue-shifted phenomenon of NTRT-PL emission, we present a conceivable mechanism for the interfacial CT process in the binary Cu2O/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs heterojunction nanohybrids under 266 nm irradiation, as schematically illustrated in Figure 5. It is comprehensible that there is no CT process before individual Cu2O and pristine TiO2-NAs contact, accompanying with atmospheric molecular oxygen (O2) that was adsorbed on the surface of the Cu2O and the TiO2-NAs, respectively.47 Consequently, the surface energy bands of the Cu2O and TiO2-NAs are rather flat. The band-gap energy (Eg) for Cu2O and TiO2 semiconductors are 2.2 and 3.1 eV, and their Fermi levels (EF) are −4.7 eV and −4.1 eV (relative to the energy of the vacuum), respectively,25 as the schematic illustration of the band gap structure presents in Figure 5a.
Figure 5.

Schematic illustration of (a) band gap structure for individual Cu2O and TiO2 semiconductors before contact, (b) band gap structure for binary Cu2O/TiO2-NAs nanoheterojunctions, (c) photogenerated charge carriers excited and traditional type-II transient transfer pathway for Cu2O/TiO2-NAs nanocomposites under UVC light irradiation, and (d) photogenerated charge carriers Z-scheme transient transfer for ternary Cu2O/Au/TiO2-NAs nanohybrids under UVC light irradiation.
In the case of the Cu2O NPs that were deposited onto the surface of TiO2-NAs without light irradiation, a Cu2O/TiO2-NAs type-II integrated heterojunction barrier is formed at the interface between the Cu2O and TiO2-NAs, owing to the alignment of their different EF as stated above, which caused the surface energy band of the Cu2O/TiO2-NAs nanocomplex to bend, as schematic displayed in Figure 5b. According to the literature reported previously,47 the energy difference value (EDv) of affinity (χ) between Cu2O and TiO2-NAs, that is, the calculated CB offset (ΔEc), is 0.81 eV, and the EDv of the VB (i.e., the VB offset, ΔEv) between them is 1.8 eV. The quantities previously determined are referred to the vacuum level. Simultaneously, based on the relationship between the potential of the normal hydrogen electrode (NHE), ENHE, and the energy of the vacuum (Evac), Evac = −ENHE – 4.44 (eV),48 associating with the average Eg for Cu2O (2.2 eV) and TiO2-NAs (3.1 eV) obtained from the Tauc plots, ΔEc, ΔEv, and the energy level of CB (ECB) for TiO2 (−0.26 eV vs NHE),49 the ECB for Cu2O was determined to be −1.07 eV. Similarly, following this procedure for the energy level of VB (EVB) in Cu2O/TiO2-NAs were calculated to be 1.13 eV/2.94 eV (vs NHE), respectively. Thus, the formation of Cu2O/TiO2-NAs staggered type-II heterojunction can greatly facilitate the electron transfer, and the surface energy band of both in the heterojunction is bent resultantly, as schematic illustrated in Figure 5c.
In the circumstances of the type-II Cu2O/TiO2-NAs heterojunction nanocomposites irradiated by 266 nm light, it is distinctly observed that the TiO2-NAs films surface was incompletely covered by Cu2O NPs as exhibited in Figure 1c, i.e., TiO2-NAs are partly exposed, which represented that both Cu2O NPs and TiO2-NAs have the opportunity to absorb irradiated light. As plotted in 2D NTRT-PL spectra in Figure 3b, in the initial UVC photoexcitation (t = 0 ns), plenty of electrons in the VB are promoted across the band gap to the CB to produce e–CB, remaining holes in the VB to create h+VB, owing to the incident photon energy (ca. 4.7 eV) that is greater than the band-gap thresholds of Cu2O and TiO2-NAs, respectively. Immediately after the UVC light irradiation, the Cu2O/TiO2-NAs system no longer generated photoinduced e––h+ pairs. Atmosphere O2 can trap the e–CB from the CB of Cu2O to yield superoxide radical anions (·O2–), i.e., O2 + e–CB → ·O2–,47 ascribing to the ECB of Cu2O (−1.07 eV vs NHE) that is more negative than the redox potential of O2/·O2– (−0.33 eV vs NHE),50 while h+VB diffuse to the TiO2-NAs surfaces to combine with the adsorbed water (OH–) to generate hydroxyl radicals (·OH), that is, OH– + h+VB → ·OH, benefiting from the EVB of TiO2-NAs (2.94 eV vs NHE) that is more positive than that of OH–/·OH (2.72 eV vs NHE).51 Conceivably, the concentration of adsorbed O2– for Cu2O NPs is much less than that of TiO2-NAs, leading to the concentration of e–CB for Cu2O that is far more than that of TiO2-NAs in the prime of irradiation (t = 0–1.5 ns), which attributes to the specific surface area of the former that is much smaller than that of the latter. With the concentration of e–CB for Cu2O increasing when the recording time was increased (t = 1.5 ns), the intensity of the transient PL peak sited at 560 nm yet becomes more stronger compared with t = 0 ns, which is associated with the e–CB–h+VB pairs direct radiative recombination (e–CB + h+VB → hν560 nm). With the increased spectral recording time (t = 3 ns), photogenerated carriers driven by the built-in electric field, the ΔEc can supply the intrinsic driving force for photogenerated e–CB migrating from the CB of Cu2O to the CB of TiO2, while the ΔEv should promote the photoexcited h+VB transfer from the VB of TiO2 to the VB of Cu2O in the opposite direction,52 which directly witnessed the CT route of the conventional Cu2O/TiO2 type-II heterojunction,53 just as schematized in Figure 5c. So, we observe that two transient PL emission peaks located at 430 and 560 nm in the NTRT-PL spectrum, which attributed to OV trapping states of TiO2 radiative transition with the h+VB (OV + h+VB → F+ + hν430 nm) and photogenerated e–CB–h+VB pairs direct radiative recombination in Cu2O, respectively. With the continuous traditional type-II interfacial CT of photogenerated e–CB between Cu2O and TiO2-NAs (t = 4.5 ns), the intensity of transient PL sited at 430 and 560 nm appeared as one falls and another rises. At this moment, the e–CB of TiO2 cannot react with the dissolved O2 to produce ·O2– as the active species because the ECB of TiO2 is −0.26 eV versus NHE, which is more positive than that of O2/·O2– (−0.33 eV vs NHE); besides, the h+VB of Cu2O is unable to oxidize with H2O to generate ·OH owing to the EVB of Cu2O that is 1.13 eV versus NHE, which is more negative than that of OH–/·OH (2.72 eV vs NHE), shown in Figure 5c. Thus, the amount of active species (·O2– and ·OH) is decreasing gradually. The unceasing consumption of photoexcited e–CB resulted from radiative recombination between OV and h+VB in the last stage of the spectral recording time (t = 6 ns), giving rise to the attenuated transient PL intensity of both.
As the ternary Cu2O/Au/TiO2-NAs nanoheterostructures were constructed by the electrodepositing Cu2O NPs layer on the surface of Au NPs-coated TiO2-NAs with different Cu2O deposition times of 20 and 40 s, an S-M-S Schottky barrier is formed at the junction between the metal and semiconductor due to the alignment of their different EF, which resulted in the bending of the whole surface energy band. It is noteworthy that adsorption of atmospheric O2 on the radiative exposed surface of TiO2-NAs has been replaced by the decoration of Au NPs. For the Cu2O/Au/TiO2-NAs nanohybrids was irradiated by monochromatic UVC illumination, a large number of electrons are promoted from the VB across the band gap to the CB upon excitation since the wavelength of the incident photons, 266 nm (ca. 4.7 eV), is much less than the threshold wavelengths of semiconductor Cu2O and TiO2-NAs, respectively. Until transiently terminated for the 266 nm irradiation, the ternary heterojunction no longer generated e––h+ pairs. Based on the bent energy band structure of Cu2O/Au/TiO2-NAs schematically illustrated in Figure 5d, if the Cu2O/Au/TiO2-NAs ternary nanocomposites follow the conventional type-II CT pathway under UVC irradiation, the photogenerated e–CB in the CB of Cu2O would migrate to the CB of TiO2-NAs, while the h+VB in the VB of TiO2-NAs would transfer to the VB of Cu2O in a reverse direction, an expected transient PL peak should be observed associating with e–CB radiative recombination with h+VB in Cu2O as the same as NTRT-PL for Cu2O/TiO2-NAs (as illustrated in Figure 3).
More than that, Serpone et al.28 have reported previously that the lifetime (τe) of charge carriers trapped by shallow defects is more shorter than that of deep defects trapping. Combined with the inversely proportional relation of τe between recombination probability (Pe),54 we have a sound reason to believe that the radiative Pe for photoexcited e–CB trapped by a shallow acceptor of surface defects is much greater than that of recombinative with deep-trapping defect levels, meaning that a blue shift phenomenon of transient PL peaks would be observed with the gradually decreased concentration of e–CB. Conversely, a red shift phenomenon of transient PL peaks would be detected with the gradually increased concentration of e–CB, which attributed to e–CB that would preferentially transit to the defect-state energy levels closest to the CB.55 Supposing the ternary Cu2O/Au/TiO2-NAs nanocomplex abide by conventional type-II nanoheterojunction CT routes, the gradually increasing concentration of e–CB for TiO2 will come into being, we would deservedly detect the red shift phenomenon of transient PL peaks related to e–CB recombination with discrete OV trapping states in TiO2. Yet, the NTRT-PL spectra for Cu2O/Au/TiO2-NAs with Cu2O depositing times of 20 and 40 s, as illustrated in Figure 4a,b, could not be able to monitor the transient PL peak with respect to radiative transition in Cu2O, and as shown in the result of NTRT-PL spectra measurement above, a blue shift instead of a red shift for transient PL peaks has been explicitly presented, which is contradictory to the interfacial CT process of conventional type-II nanoheterojunction, demonstrating that the efficient CT across the interfacial domain within Cu2O/Au/TiO2-NAs composites follow another transfer channel than the heterojunction.
Therefore, at the initial time of photoexcitation (t = 0 ns), the population of e–CB resides in the CB of Cu2O and TiO2-NAs, respectively. It can thus be reasonably inferred that the photoexcited e–CB transfer from the CB of TiO2-NAs to Au NPs with the driving force of the Schottky barrier until a thermodynamic equilibrium is reached anew, owing to the embedded Au-NPs, had an EF (−4.9 eV vs vacuum level, i.e., 0.45 eV vs NHE)56 substantially lower than that of TiO2-NAs (−0.36 eV vs NHE). The accumulated e–CB at Au-NPs further induced the nonradiative recombination with the photogenerated h+VB of Cu2O (1.13 eV vs NHE),57 mediating a Z-scheme interfacial CT pathway from the CB of TiO2-NAs, through Au NPs, and then migrated to the VB of Cu2O. Au NPs act as “electron conveyer” to promote the charge-carrier transfer.
Such a vectorial CT avenue led to e–CB concentration decreasing at the CB of TiO2-NAs with the increasing spectrum recording time (0–6 ns), accompanying by the competition mechanism between the photogenerated charge separation process and radiative recombination associated with OV defects.55 So, we can distinctly observed the blue-shift phenomenon of transient PL peaks from 523 nm (525 nm) to 466 nm (435 nm) for ternary Cu2O/Au/TiO2-NAs nanoheterojunctions with a Cu2O depositing time of 20 s (40s), resulting from a larger percentage of e–CB transit to the shallow OV trapping levels to radiate shorter PL wavelengths with decreased e–CB concentration in TiO2-NAs, which supported a straightforward evidence for the presence of Z-scheme CT. As exhibited in Figure 4a,b, the NTRT-PL blue shift becomes more pronounced with the increase in deposition time of Cu2O from 20 to 40 s, correlating well with the h+VB concentration as the VB increases with increasing amount of Cu2O, which indicates that the Z-scheme interfacial CT process of Cu2O/Au/TiO2-NAs with a Cu2O deposition time of 40 s is more efficient than that of 20 s. However, as shown in Figure 4c, the NTRT-PL spectrum for Cu2O/Au/TiO2-NAs with a Cu2O depositing time of 80 s presented three transient PL peaks, 558, 431, and 385 nm, which originated from the direct radiative recombination for Cu2O, shallow-trapping of OV defects, and direct radiative recombination for TiO2-NAs, respectively. In the initial stage of 266 nm irradiation (t = 0 ns), a large number of e–CB were accumulated in the CB of Cu2O owing to most of the incident photons that were absorbed by surface-covered Cu2O-NPs films compared with the substrate of TiO2-NAs, the photon energy of incident light is greater than its band-gap energy. In the next evolution of spectrum recording time (t = 1.5 ns), e–CB would tend to transfer from the CB of Cu2O to the CB of TiO2-NAs driven by built-in electric field force following the traditional double CT mechanism, accompanying with transient PL peaks emitted at 558 and 431 nm, which originated from the direct contact between Cu2O and TiO2-NAs substrate with the increased amount of Cu2O. With the continuous injection and transfer of e–CB between Cu2O and TiO2-NAs (t = 3–4.5 ns), the intensity of transient PL peaks located at 385 and 431 nm is gradually stronger at the expense of emission intensity weaker centered at 558 nm. At the end stage of spectra recording (t = 6 ns), the transient PL peak emission at 385 nm slowly disappeared, concerning well with e–CB concentration in the CB of TiO2-NAs drastically decreased through Au NPs mediated CT, which demonstrated that the Z-scheme recombination for the interfacial CT channel should not be neglected. Briefly, the transient PL peak of NBE emission for TiO2 located at 385 nm disappeared in NTRT-PL spectra of Figure 4a,b, which mainly ascribed to the less concentration of e–CB in TiO2 induced by the Z-scheme CT route instead of the traditional type-II heterostructure CT manner.
NTRT-PL spectroscopy has proven to be a powerful exploration of the interfacial CT dynamics in nanoheterojunction and became a valuable resource for heterogeneous photoelectrochemistry, and it was performed for binary Cu2O/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs nanoheterojunctions with different Cu2O depositing times (20, 40, and 80 s) under irradiation of monochromatic wavelength at 400 nm (3.1 eV), which is close to the plasmon resonance wavelength of 541 nm (ca. 2.3 eV), as above proven in Figure 4b. For the specimens of Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs 80s, as seen in Figure 6a,d, there are three transient PL peaks, centering at 430, 535, and 675 nm, which originated from the shallow-trapping recombinative PL of single and doubly ionized OV defects58 in TiO2-NAs and Cu vacancies (Cuv) acceptor level in Cu2O,59 respectively. Simultaneously, as illustrated in Figure 6b,c, besides transient PL peaks at 430 and 675 nm stated above, the fresh emerging PL peaks are located at 570 nm (ca. 2.2 eV) and 560 nm (ca. 2.2 eV) for the samples of Cu2O/Au/TiO2-NAs with the Cu2O depositing times of 20 and 40 s. The emitted PL energy was identical to the band gap of Cu2O, both of which could be ascribed to the e–CB–h+VB pairs direct radiative recombination in Cu2O.
Figure 6.

NTRT-PL spectra for (a) binary Cu2O/TiO2-NAs nanoheterojunctions and ternary nanosized Cu2O/Au/TiO2-NAs nanoheterojunctions deposited Cu2O with different times of (b) 20 s, (c) 40 s, and (d) 80 s under irradiation of monochromatic wavelength at 400 nm, respectively.
In order to further evaluated the essence of Au NPs mediated interfacial CT transition between the interface of Cu2O and TiO2-NAs, we now present two applicable CT scenarios, including conventional type-II CT and interband transitions induced interfacial CT associated with Au NPs; the detailed schematic illustration is shown in Figure 7a–d. Figure 7a exhibits the band-gap structure of the Cu2O/TiO2-NAs integrated type-II heterojunction aerated in an atmosphere without light irradiation, O2 molecules can be much efficiently adsorbed on the surface of Cu2O/TiO2-NAs instantaneously. Owing to the difference in the work functions and Eg between semiconductor Cu2O and TiO2-NAs as stated above, it is plausible to believe that a type-II heterojunction barrier is formed in the interfacial region of the Cu2O and TiO2-NAs, resulting from the alignment of their different EF. Consequently, the ΔEc and ΔEv were found to be 0.81 and 1.8 eV, respectively, originating from the surface energy band bent of Cu2O/TiO2-NAs nanoheterojunction. For the Cu2O/TiO2-NAs nanocomplex irradiated by 400 nm light, as far as we known, most photogenerated electrons are promoted from the VB across the band gap to the CB, ascribing to the incident photon energy (i.e., 3.1 eV) being more than the Eg of Cu2O and TiO2-NAs, respectively. Similarly, there are two main reduction–oxidation reactions with the CT process between the nanointerface of Cu2O/TiO2-NAs, i.e., the photoexcited e–CB and h+VB simultaneously diffuse to the CB of Cu2O (−1.07 eV vs NHE) and the VB of TiO2-NAs (2.94 eV NHE) to combine with the O2 and OH–, respectively, leading to the generation of ·O2– and ·OH, which have a stronger oxidation–reduction ability compared with individual semiconductor. The e–CB in the CB of TiO2-NAs (−0.26 eV vs NHE) and the h+VB in the VB of Cu2O (1.13 eV vs NHE) cannot react with O2 and OH– to produce ·O2– and ·OH, owing to the potentials for the CB of TiO2-NAs and the VB of Cu2O that do not satisfy the reaction conditions of O2/·O2– (−0.33 eV vs NHE) and OH–/·OH (2.72 eV vs] NHE), respectively. These processes are schematically shown in Figure 7b. Transiently, after the 400 nm light irradiation, the photoinduced e––h+ pairs are no longer excited in Cu2O/TiO2-NAs nanocomposites. During the formation of the type-II heterojunction barrier, it is plausible to believe that the electrons and holes transferring are facilitated from the CB of Cu2O to TiO2-NAs and from the VB of TiO2-NAs to Cu2O with a driving force of the built-in electric field, respectively, i.e., ΔEc and ΔEv, until a new equilibrium is reached, resulting in both the e– and h+ that would move to lower-energy positions.
Figure 7.

Schematic illustration of (a) band gap structure for aerated Cu2O and TiO2-NAs after contact, (b) traditional type-II CT for binary Cu2O/TiO2-NAs nanoheterojunctions under 400 nm irradiation, (c) band gap structure for ternary Cu2O/Au/TiO2-NAs nanocomplex without no illumination, and (d) plasmon-induced interfacial CT (PICT) for ternary Cu2O/Au/TiO2-NAs nanohybrids irradiated by 400 nm.
We now present an applicable conventional type-II CT mechanism for the Cu2O/TiO2-NAs nanoheterojunction under 400 nm light irradiation based on the experimentally observed transient PL emission data, as shown in Figure 6a. In the beginning of 400 nm light excitation (t = 0 ns), since the concentration of e–CB excited by 3.1 eV was lower compared with photostimulated by 4.7 eV, it is reasonable to believe that e–CB preferentially transitions to the surface defect energy levels in Cu2O compared with direct recombination between e–CB with h+VB for Cu2O/TiO2-NAs under 266 nm irradiation. Predictably, the intensity of the transient PL peak located at 675 nm is quite weak, which associated with the e–CB recombination between Cuv electron-trapping states with h+VB in Cu2O. With spectral recording time after 1.5 ns (t = 1.5 ns), the intensity of the transient PL spectrum centered at 675 nm is boosted, resulting from the increase in concentration of e–CB for Cu2O. Concomitantly, the fresh appearance for transient PL peaks located at 430 and 535 nm is associated with the e–CB radiative recombination with OV defects in TiO2-NAs, which ascribed to the e–CB transfer from CB of Cu2O to TiO2 driven by ΔEc. The amount of e–CB increased with the increasing spectral recording time (t = 3 ns), resulting to the intensified intensity of all transient PL spectral. The processive consumption of e–CB with the evolution of the recording time (t = 4.5 ns), a larger percentage of e–CB radiative recombination with the Cuv, single and doubly ionized OV defects, radiating degraded 675, 430, and 535 nm transient PL emission, which caused by the transiently decreased electron concentration. Eventually, the intensity of all transient PL spectra are further depressed in the last period of spectral recording time (t = 6 ns), deriving from the intrinsic relation with the e–CB sustained depletion.
In the case of the Cu2O/Au/TiO2-NAs ternary integrated nanoheterojunctions with Cu2O depositing times of 20 and 40 s under no light irradiation. As we know, the EF for sole Cu2O, TiO2-NAs, and Au NPs are −4.7, −4.1, and −5.1 eV60 (vs Evac), respectively. On account of the mutual conversion relations between the Evac and ENHE, i.e., Evac = −ENHE – 4.44 (eV), the obtained EF for Cu2O, TiO2-NAs, and Au NPs are 0.26, 0.36, and 0.66 eV (vs. ENHE), respectively. Nevertheless, when the semiconductors and Au NPs are in contact with each other, the electrons are distributed between Cu2O, TiO2-NAs, and Au NPs until the ternary nanocomplex systems attain equilibration. Since the electron accumulation increases the EF of Au NPs (0.45 eV vs ENHE) to more negative potentials, the resultant EF of the Au NPs shifts closer to the CB of the semiconductors, leading to the formation of Schottky barriers at each S-M-S region boundary, in which the Schottky barrier heights (ϕB) between the interface of Cu2O/Au and Au/TiO2-NAs are 0.72 and 0.09 eV, respectively. Thus, it is reasonable to believe that the electron interfacial CT process is facilitated, thereby decreasing the charge recombination. It is also worth mentioning that the d-band electrons are about 2.3 eV below the EF for Au NPs,61 as the schematic illustration of the position for the EF and band gap structure vs ENHE presents in Figure 7c.
When the ternary Cu2O/Au/TiO2-NAs integrated nanoheterojunctions were irradiated by 400 nm femtosecond laser with a photon energy higher than the band gap energies of Cu2O and TiO2-NAs, electrons in the VB of Cu2O and TiO2-NAs can be excited to their CB, and the same amount of holes is simultaneously generated in the VB of Cu2O and TiO2-NAs, respectively. The band configuration of Au can be understood in terms of 6sp bands and 5d bands, which resembles the CB and VB of Au NPs, respectively.62 he interband transitions can be triggered between the filled d-band and the empty states of the sp-band under the excitation of appropriate light energy, which make Au NPs have the properties like semiconductor catalysts accompanied with the CT process.63,64 Consequently, the hot electrons in the 5d bands are excited and leap into the 6sp bands assisted by interband excitations under 400 nm light illumination, where the position of 6sp energy band lies 0.8 eV above the EF for Au NPs (6sp band position of Au = −0.35 eV vs ENHE). Transiently after the 400 nm light irradiation, the photogenerated electrons on the CB of Cu2O can be rapidly transferred to the 6sp band of Au NPs under the effect of inner electric field, coincident with the adsorbed O2 reacting with the e–CB to yield ·O2– sited at the CB of Cu2O and 6sp band, where their energy level structures are located at a more negative potential than the reduction potential of O2/·O2– (−0.33 eV vs ENHE). Analogously, for the contact interface between Au and TiO2-NAs, the hot electrons with energies higher than ϕB can overcome the Schottky barrier and transfer to the CB of TiO2-NAs, resulting in a charge separation. The electrons in the VB were excited to the CB of TiO2-NAs, with simultaneous generation of holes in the VB of TiO2, which can be trapped by adsorbed OH– to generate the ·OH species, owing to the VB energy level of TiO2-NAs that has a more positive potential than the oxidation potential of OH–/·OH (2.72 eV vs ENHE). By contrast, the h+VB in Cu2O cannot be captured by the OH– to produce the ·OH species because of the lack of redox potential. The photoinduced e––h+ pairs excited and interfacial CT processes for Cu2O/Au/TiO2-NAs are schematically shown in Figure 7d.
As we know, the PEC efficiency of the heterogeneous nanocomplex is determined by the overall population of the photogenerated carriers and the time scale of the interfacial CT process. Furube et al.65 announced that the time scale of hot-electron injection between Au and TiO2 occurs in less than 240 fs. It should be emphasized that hot-electron transfer competes with electron relaxation through rapid electron–electron scattering in the 6sp band on time scales of hundreds of femtoseconds,66 confirming fast injection, which is beyond our time resolution (ca. 0.1 ns). However, Serpone and co-workers28 demonstrated that the time scale for electrons trapped recombination associated with defect states and holes in VB is in the nanosecond order, which determines the photocatalytic activity of the heterojunction nanocomposites. Consequently, it is plausible to believe that NTRT-PL spectra of Cu2O/Au/TiO2-NAs with different Cu2O deposited times could be able to reveal the interfacial CT process related to the e–CB radiative recombination PL procedure.
The distinction for NTRT-PL spectra between Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs with different Cu2O depositing times of 20 and 40 s can be distinctly observed, including (i) the decrease of transient PL located at 430 nm at t = 6 ns; (ii) the transient PL sited at 535 and 675 nm that exist in the former but disappear in the latter. Undoubtedly, the embedding of Au NPs played a vital role for interband transitions induced interfacial CT process. Compared to the ternary Cu2O/Au/TiO2-NAs nanocomposites with binary Cu2O/TiO2-NAs nanocomplex excited by 400 nm irradiation, since the ϕB (0.72 eV)67 between the interface of Cu2O/Au is less than the ΔEc (0.81 eV) intermediate in Cu2O/TiO2-NAs, the driving force of built-in electric field of the former is smaller than that of the latter, resulting in the concentration of e–CB in Cu2O of the former that is more than that of the latter, which is responsible for the direct radiative recombination (570 nm) instead of Cuv defects trapped recombination (675 nm). Immediately, plenty of hot electrons in the 6sp band of Au are injected into the CB of TiO2-NAs driven by the Schottky barrier (ca. 0.1 eV) between the interfaces of Au/TiO2-NAs, resulting from the interband transitions of from the 5d to 6sp band in Au NPs. Consequently, the concentration of e–CB in TiO2 of the former is more than that of the latter, which boosted the PL intensity of the Vo defects trapping radiative recombination (430 nm). Simultaneously, hot-electron transfer competes with the generation of ·O2– in the 6sp band of Au, leading to ab amount of ·O2– of ternary Cu2O/Au/TiO2-NAs that is more than that of binary Cu2O/TiO2-NAs nanoheterojunctions, which showed that the PEC performance of the former is higher than that of the latter. Therefore, the implantation of Au NPs plays an essential influence on the population of charge carriers and time scales of CT in the ternary Cu2O/Au/TiO2-NAs nanoheterojunction system. In addition, it is explicitly revealed that the transient PL peak positions for Cu2O/Au/TiO2-NAs with a Cu2O depositing time of 80 s are identical with that of Cu2O/TiO2-NAs, yet quite different from that of Cu2O/Au/TiO2-NAs with Cu2O depositing times of 20 and 40 s, which ascribed to the diverse CT processes. With the increase of deposition time for Cu2O NPs to 80 s, the covering films of Cu2O NPs are too substantial to be penetrated by incident excitation light of 400 nm. Based on the above analysis, the interfacial CT for Cu2O/Au/TiO2-NAs with a Cu2O deposition time of 80 s should be predominantly attributed to the traditional type-II heterojunctions, while the hot electron excited by interband transitions of Au can be ignored.
To check the feasibility of as-proposed CT mechanisms, the photocatalytic performances of pristine TiO2-NAs, bare Cu2O NPs films, binary Cu2O/TiO2-NAs type-II nanoheterojunctions, and ternary Cu2O/Au/TiO2-NAs nanohybrids with different Cu2O depositing amounts were conducted under the UV–vis light irradiation, respectively. In the experiment, the catalytic performance of the different samples was carried out by using MO as a simulated pollutant owing to its universal use and good long-term stability. For the Cu2O/TiO2-NAs type-II dual heterojunction photocatalyst, the expected active charge carriers excited, and photocatalytic processes for the MO photodegradation under UV–vis light irradiated, which would most likely resemble with ternary Cu2O/Au/TiO2-NAs nanocomposites, as depicted pictorially in Scheme 1. The excited e–CB transfer to the CB of TiO2-NAs; meanwhile, h+VB reside in the VB of Cu2O, respectively. Subsequently, the h+VB was trapped by the surface hydroxyl groups (or H2O) at the catalyst surface to yield ·OH. Dissolved O2 react with the e–CB to yield·O2– and react with water and generate the hydroperoxyl radicals (·HO2), producing oxydol (H2O2) and·OH, which are strong oxidizing agents to decompose the MO organic dye. These chemical reactions for the degradation of MO can be proposed as follows:
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
Scheme 1. Schematic Diagram for the Photodegradation of MO over the Cu2O/TiO2-NAs Type-II Heterojunction System under UV–Vis Light Irradiation.

The UV–vis photocatalytic activity investigations for binary and ternary heterojunction nanohybrids were performed under irradiation by a standard solar spectrum simulation light source. All degradation experiments were done using a concentration of 10 mg/L MO. In the first step, a blank reaction was performed, i.e., the MO dye in solution was exposed to the UV–vis radiation without a catalyst, excluding the photobleaching effect of MO and ascertain that it is only the photocatalytic activity of the nanocomposite that is responsible for the MO photodegradation. Making sure that the adsorption–desorption of the dye on the sample reaches equilibrium, the photocatalytic material mixed with the MO solution was stirred in the dark condition for an hour, and the maximum adsorption of MO on the nanocomposites was established by recording the UV–vis absorption spectra before carrying out the photocatalysis reaction.
Figure 8 exhibits the photodegradation rate (η) experimental results for MO aqueous solution, the pristine TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs with different Cu2O deposition times of 20, 40, and 80 s under a UV–vis lamp irradiation for 4 h, respectively. The UV–vis absorption spectra recorded showed that even at the end of 4 h, minimal degradation (≤5%) of MO was observed. The η of MO was calculated by the equation:68 η = (C0 – C)/C0 × 100%, where C0 and C represent the initial and the concentration of MO after degradation, respectively. The pure TiO2-NAs exhibit the poor degradation ability of MO (ca. 27%), which is desired to carry out an overall investigation in order to explore the effect of the structural modulation and decoration on the photoactivity. The Cu2O/TiO2-NAs show stronger photodegradation activity of MO (ca. 71%) than the pure TiO2-NAs, which ascribed to the higher reductive/oxidative potential of type-II nanoheterostructure in comparison with pristine TiO2-NAs. With the increase in Cu2O deposition times from 20 to 40 s, the photodegradation activities of the Cu2O/Au/TiO2-NAs are enhanced from 78 to 90%. Yet, with the Cu2O depositing time to 80 s, the photodegradation ability for Cu2O/Au/TiO2-NAs declined to 83%. Thus, it can be concluded that the photodegradation performance of the ternary Cu2O/Au/TiO2-NAs nanohybrids greatly depends on the Cu2O NPs deposition amount.
Figure 8.

Photodegradation rate η of MO, the pristine TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs with different Cu2O times (20, 40, and 80s) irradiated with a UV–vis lamp, respectively.
In order to quantitatively examine the reaction dynamics, we presumed that the photocatalytic behavior for MO aqueous solution followed the pseudo-first-order kinetic model plotted by the equation of ln(C0/Ct) = kt,69 in which k, C0, and Ct stand for the reaction rate constant, initial concentration, and concentration at time t of the MO concentration, respectivelys, as displayed in Figure 9. This plot also includes the blank reaction, i.e., without any materials with photocatalytic activity, and the rate constant can be determined by plotting ln(C0/Ct) as a function of time. All the traces show the maximum value of the pseudo-first-order rate constant for ternary Cu2O/Au/TiO2-NAs with a Cu2O deposition time of 40 s, implying that it is the optimum composition for the best photodegradation performance amongst the various heterojunction nanocompositions studied toward the MO degradation under UV–vis light irradiation.
Figure 9.

Plot of ln(C0/Ct) versus irradiation time of MO degradation for MO, the pristine TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs with different Cu2O times (20, 40, and 80 s) irradiated with a UV–vis lamp.
It is considered that the recycling usability and stability are also vital factors other than photocatalytic efficiency as it can improve its financial feasibility and reduce impact to the environment. Thus, cyclic photodegradation experiments were performed to investigate the reusability of the binary Cu2O/TiO2-NAs and ternary Cu2O/Au/TiO2-NAs heterojunction nanocomposites irradiated by UV–vis light under the same conditions six times successively, as exhibited in Figure 10. For each time, the specimens were washed with deionized water, dried in an oven overnight, and then reused. The results revealed that the photodegradation activities of Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs nanocomposites for MO removal had a slight decline as can be expected, perhaps resulting from inevitable weight loss in the process of collecting and washing. Even after six consecutive cycles, the photodegradation efficiency deterioration is less than 8%, fully demonstrating that as-prepared ternary Cu2O/Au/TiO2-NAs nanocomposites were highly stable S-M-S photocatalytic nanohybrids.
Figure 10.

Cyclic photodegradation efficiency of the MO, the pristine TiO2-NAs, Cu2O/TiO2-NAs, and Cu2O/Au/TiO2-NAs with different Cu2O deposition times (20, 40, and 80 s) heterojunction nanocomplex irradiated by UV–vis light under the same conditions six times, respectively.
As from above experimental analysis, it can be distinctly observed that the sequence of photocatalytic activities for as-prepared binary and ternary heterojunction nanocomposites was connected well with the trend of PEC performance measurements, including the amperometric I–t curve, Voc–t curve, and EIS test. For Cu2O/Au/TiO2-NAs nanohybrids, the embedded Au NPs served as a solid-state electron mediator and hot electrons provider to enhance the interfacial CT process. The enhancement of the amperometric I–t curve and Voc–t curve for ternary Cu2O/Au/TiO2-NAs compared with binary Cu2O/TiO2-NAs nanocomposites significantly retarded the recombination of photoinduced e––h+ pairs and boosted the of active charge carriers, which could prolong charge lifetime and enhance the photocatalytic performance. What is more, EIS Nyquist plots indicated that the sample of Cu2O/Au/TiO2-NAs with a Cu2O depositing time of 40 s exhibited a smaller arc radius than those of Cu2O/Au/TiO2-NAs with Cu2O depositing times of 20 and 80 s nanocomposites, and its smaller arc radius showed a smaller CT resistance and faster interfacial transfer rate of photoinduced charge carriers. Based on the number of Cu2O/Au/TiO2-NAs heterojunctions formed by the surface coverage of Cu2O on Au/TiO2-NAs, initially increasing the amount (deposition time is 20 s) of Cu2O leads to an increase in the formation of ternary nanoheterojunctions, and thus the photocatalytic activity is seen to increase. With the further increase in Cu2O NPs depositing time to 40 s, an appropriate amount of Cu2O decorating could effectively inhibit the recombination of photoinduced carriers, thereby displaying a superior CT capability and exhibiting an outstanding photocatalytic performance. For the depositing time of 80 s, an excess amount of Cu2O NPs results in aggregate with each other and shadow the surface of Au/TiO2-NAs from the incident light photons, bringing about weakened light exposure of Cu2O/Au/TiO2-NAs ternary nanoheterostructure for photoinduced energetic carrier production. Additionally, the experiment results as mentioned above are also in agreement with the analysis of NTRT-PL spectra.
Conclusions
In conclusion, a unique ternary nanoheterostructured comprising Cu2O/Au/TiO2-NAs has been fabricated by strategic integration of Au NPs with well-defined binary Cu2O/TiO2-NAs type-II heterojunctions through a three-step electrodepositing method. The as-fabricated ternary Cu2O/Au/TiO2-NAs nanoheterostructures are expected to exhibit progressively enhanced PEC performance and photocatalytic activity toward degradation of MO compared with binary Cu2O/TiO2-NAs type-II nanoheterojunctions under UV–vis irradiation, ascribing to the synergistic effect of Cu2O/TiO2-NAs associated with Au NPs. Mechanistic studies demonstrated that the CT pattern of the Cu2O/Au/TiO2-NAs is remarkably different from that of the conventional type-II Cu2O/TiO2-NAs heterostructure, which was confirmed by NTRT-PL spectroscopy. The embedding Au NPs into the interface of Cu2O and TiO2 were verified to assist exciton transport at the interface and improve the hot electrons generation. As a result, it is the synergistic interaction of multiple charge-carrier transfer channels including the Z-scheme and interband transitions for enhancement interfacial CT dynamics process. Therefore, it is plausible that the ternary Cu2O/Au/TiO2-NAs nanohybrids not only provides new insights into the CT mechanism for aa high-activity photocatalyst but also explores new prospects for the development of semiconductor heterojunction devices.
Experimental Section
Synthesis of Pristine TiO2-NAs Substrate
Anodic oxidation method was used to prepare uniform and stable TiO2-NAs with vertical alignment. Prior to the anodization process, the polished titanium (Ti) foil (99.99% purity with a dimension of 30 × 15 × 0.2 mm) was ultrasonically cleaned and degreased with acetone, absolute ethyl alcohol, and deionized water for 30 min successively and then blow dried with nitrogen. The anodization was performed in a self-made two-electrode electrochemical cell with the Ti foil as the working electrode and graphite sheet as the counter electrode at room temperature (30 °C). The electrolyte consisted of NH4F (0.45 wt %), ethanediol (98 vol %), and 2 g of deionized water, which were mixed and stirred for 1 h. A constant voltage of 60 V was applied to the two electrodes for 2 h by using a direct current (DC) power supply. In order to ensure that the tube mouth is neat and tidy, as-fabricated samples were carried out by sonicating in ethanol for 30s.
Preparation of TiO2-NAs Films Decorated with Au NPs
Coating of ultra-thin Au film for 30 s (thickness of less than 10 nm) on air-dried TiO2-NAs films was performed using an ion-sputtering system. Well-controlled sample preparations are critical to form nice structures of well-separated Au nanoparticles on the surface of TiO2-NAs, which will result in suppression of aggregation of Au NPs, just as shown in the following morphological characterization.
Fabrication of Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs Nanoheterojunctions
The above-prepared Au/TiO2-NAs nanohybrids created on the Ti foils were electrochemically deposited with Cu2O NPs. Scheme 2 illustrates the synthetic procedures for sample preparation of Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs nanoheterojunctions. Electrochemical deposition of Cu2O-NPs films was carried out in a three-electrode cell equipped with a saturated calomel electrode (SCE) as the reference electrode, a Au/TiO2-NAs nanocomposite as the working electrode, and Pt plate as the counter electrode. The deposition solution of Cu2O-NPs films was composed of 0.05 M CuSO4 and stabilized with 0.05 M citric acid as a chelating agent, while the pH value was adjusted to 11 by addition of 4 M sodium hydroxide (NaOH) solution. The electrolyte solution was stirred constantly by a magnetic stirrer throughout electrodeposition and kept at a fixed temperature of 60 °C. A constant potential of −0.6 V was applied vs the Ag/AgCl reference electrode using a potentiostatic model of the CHI660E electrochemistry workstation (CH Instruments Co. Ltd., China). The constant potential was applied for different duration times of 20, 40, and 80 s, respectively, in order to deposit Cu2O-NPs films for different thicknesses. After the deposition process was finished, the as-formed specimens were rinsed with deionized water for several times to remove any solutions from the surface and afterward dried using nitrogen. As the reference for characterization of optical and PEC properties, the dual Cu2O/TiO2-NAs nanoheterojunctions were prepared by an electrochemical depositing route with a duration time of 20 s, and the other experimental parameters were set the same as described above. For obtaining the desired crystallographic phase of Cu2O/TiO2-NAs and Cu2O/Au/TiO2-NAs heterojunction nanohybrids, the as-fabricated samples were annealed in a furnace in dry air at 450 °C for 0.5 h with 10 °C/min of ramp up and cool down rate.
Scheme 2. Synthetic Procedures for Preparation of (a) Dual Type-II Cu2O/TiO2-NAs Nanoheterojunctions and (b) Ternary Cu2O/Au/TiO2-NAs Nanohybrids, respectively.

Characterization
The cross-sectional morphologies and microstructures of the Cu2O/Au/TiO2-NAs heterostructure nanocomposites were investigated using scanning electron microscopy (SEM, Hitachi S4200) operating at an accelerating voltage of 15.0 kV. By using the UV–vis spectrophotometer (UV-1800, Shimadzu), the UV–vis light absorption measurements were performed in air at room temperature. High-resolution transmission electron microscopy (HRTEM) observation was carried out on a transmission electron microscope (TEM, JEOL JEM-2100) working at 200 kV. The phase purity and crystal structure of the prepared samples were characterized by an X-ray diffractometer (XRD, Shimadzu XRD-600) using Cu kα radiation (λ = 15.418 nm) as the X-ray source and employing a scanning rate of 0.02° s–1 in the 2θ range of 20–80°. The oxidation states of Ti and Cu elements at the Cu2O/Au/TiO2-NAs composite nanoheterostructure surface were examined by X-ray photoelectron spectroscopy (XPS, ESCALAB 250, Thermo Fisher Scientific Ltd.). The X-ray source was the Al anode emitting the Kα (1486.6 eV) radiation of 150 W. The crystal structure and chemical bonding states of Cu2O/Au/TiO2-NAs nanocomposites are investigated by using a Micro-Raman spectroscopy system (Horiba JY-HR800) equipped with a confocal microscope with an Ar+ laser operating at 532 nm at an excitation power level of 6 mW at room temperature. Quantitative elemental analysis of as-prepared samples was performed using a Quant’X (Thermo Fisher Scientific Inc., USA) energy-dispersive X-ray fluorescence spectrometer (EDXRF) and UniQuant software.
A Ti:sapphire femtosecond laser system (Spectra-Physics)
was used to excite the NTRT-PL. The excitation laser beams with wavelengths
centered at 266 and 400 nm are acquired accordingly by frequency doubling
of the 800 nm (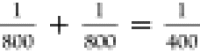 ) and sum frequency of 800 and
400 nm (
) and sum frequency of 800 and
400 nm (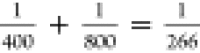 ) laser beams through a BBO (beta-BaB2O4, barium metaborate) crystal, with a pulse duration
of 130 fs, repetition rate of 1 kHz, and spot size of 1 mm. The PL
emission is collected by a spectrometer (Bruker Optics 250 IS/SM)
coupled with an intensified charge-coupled device (CCD) detector (IStar
740, Andor). Nanosecond time-resolved experiments were carried out
by adjusting the delay time of the CCD shutter. A laser pulse gave
an external trigger signal to open the 100 ms and 0.5 ns temporal
gates of the intensified CCD through a synchronization/delay generator
(SDG) and digital delay/pulse generator (DG 535), and then the transient
PL spectra at different decay times could then be recorded by the
CCD detector. Figure 11 gives a schematic diagram of the experimental setup. The time-resolved
PL data were measured in a customized single photon counting system,
in which a sub-nanosecond pulsed diode laser (λex = 375 nm, PicoQuant, PLD 375) was installed as the excitation source.
) laser beams through a BBO (beta-BaB2O4, barium metaborate) crystal, with a pulse duration
of 130 fs, repetition rate of 1 kHz, and spot size of 1 mm. The PL
emission is collected by a spectrometer (Bruker Optics 250 IS/SM)
coupled with an intensified charge-coupled device (CCD) detector (IStar
740, Andor). Nanosecond time-resolved experiments were carried out
by adjusting the delay time of the CCD shutter. A laser pulse gave
an external trigger signal to open the 100 ms and 0.5 ns temporal
gates of the intensified CCD through a synchronization/delay generator
(SDG) and digital delay/pulse generator (DG 535), and then the transient
PL spectra at different decay times could then be recorded by the
CCD detector. Figure 11 gives a schematic diagram of the experimental setup. The time-resolved
PL data were measured in a customized single photon counting system,
in which a sub-nanosecond pulsed diode laser (λex = 375 nm, PicoQuant, PLD 375) was installed as the excitation source.
Figure 11.

Experiment setup of nanosecond time-resolved transient PL (NTRT-PL) measurements.
In the PEC performance test, the electrochemical impedance spectra (EIS), photocurrent density versus testing time curve (amperometric I–t curve), open-circuit potential versus testing time curve (Voc–t curve) were examined using a CHI660E electrochemical workstation. The electrochemical test electrolyte is 0.5 M Na2SO4 aqueous solution. The standard three-electrode configuration, the as-fabricated samples, Pt plate, and SCE were used as the working electrode, counter electrode and reference electrode, respectively. A 150 W xenon lamp (SS150A, ZOLIX, China) with an AM 1.5G cutoff filter was employed as the light source in the PEC measurements. Moreover, the photocatalytic activities of the Cu2O/Au/TiO2-NAs heterostructure films were investigated using the photodegradation of MO under a standard solar simulator (AM 1.5) powered by a 300 W xenon lamp to provide UV–vis light irradiation. MO (5 mg) was dissolved in 500 mL of deionized water to yield a concentration of 10 mg/L. The supernatant was transferred to a quartz cuvette to measure its absorption spectrum. The catalyst-containing solution was exposed to UV–vis irradiation for different durationss at room temperature. The concentration of MO solution was checked every 0.5 h using a UV–is spectrophotometer by monitoring the intensity variation for the characteristic absorption peak at 465 nm.
Acknowledgments
We are thankful to the financial support of the National Natural Science Foundation of China (NSFC) (grants nos. 61474009 61575029) and Research project of Liaoning provincial educational bureau (project no. LZ2016002). We thank the Institute of Condensed Matter Physics from Harbin Institute of Technology for providing facilitate conditions on the nanosecond time-resolved transient photoluminescence (NTRT-PL) spectra experiment.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00299.
HRTEM image, XRD patterns, UV–vis light absorbance spectra, XPS characterizations, Nyquist plots, amperometric I–t curve, Voc–t curve, and EDXRF spectrum data of the samples (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fujishima A.; Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- Chiu Y.-H.; Chang T.-F. M.; Chen C.-Y.; Sone M.; Hsu Y.-J. Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts. Catalysts 2019, 9, 430. 10.3390/catal9050430. [DOI] [Google Scholar]
- Hsieh P.-Y.; Chiu Y.-H.; Lai T.-H.; Fang M.-J.; Wang Y.-T.; Hsu Y.-J. TiO2 Nanowire-Supported Sulfide Hybrid Photocatalysts for Durable Solar Hydrogen Production. ACS Appl. Mater. Interfaces 2019, 11, 3006–3015. 10.1021/acsami.8b17858. [DOI] [PubMed] [Google Scholar]
- Chiu Y.-H.; Lai T.-H.; Chen C.-Y.; Hsieh P.-Y.; Ozasa K.; Niinomi M.; Okada K.; Chang T.-F. M.; Matsushita N.; Sone M.; Hsu Y.-J. Fully Depleted Ti–Nb–Ta–Zr–O Nanotubes: Interfacial Charge Dynamics and Solar Hydrogen Production. ACS Appl. Mater. Interfaces 2018, 10, 22997–23008. 10.1021/acsami.8b00727. [DOI] [PubMed] [Google Scholar]
- Gao D.; Wang Q.; Chang Y.; Jia Y.; Wang L.; Wang C. Solvothermal ion exchange preparation of Bi11VO19 to sensitize TiO2 NTs with high photoelectrochemical performances. Sep. Purif. Technol. 2020, 233, 116011. 10.1016/j.seppur.2019.116011. [DOI] [Google Scholar]
- Pu Y.-C.; Wang G.; Chang K.-D.; Ling Y.; Lin Y.-K.; Fitzmorris B. C.; Liu C.-M.; Lu X.; Tong Y.; Zhang J. Z.; Hsu Y.-J.; Li Y. Au Nanostructure-Decorated TiO2 Nanowires Exhibiting Photoactivity Across Entire UV-visible Region for Photoelectrochemical Water Splitting. Nano Lett. 2013, 13, 3817–3823. 10.1021/nl4018385. [DOI] [PubMed] [Google Scholar]
- Chiu Y.-H.; Lai T.-H.; Kuo M.-Y.; Hsieh P.-Y.; Hsu Y.-J. Photoelectrochemical cells for solar hydrogen production: Challenges and opportunities. APL Mater. 2019, 7, 080901 10.1063/1.5109785. [DOI] [Google Scholar]
- Zhuang H.; Liu X.; Li F.; Xu W.; Lin L.; Cai Z. Construction of CdSe@TiO2 core-shell nanorod arrays by electrochemical deposition for efficient visible light photoelectrochemical performance. Int. J. Energy Res. 2019, 43, 7197–7205. 10.1002/er.4744. [DOI] [Google Scholar]
- Qu X.; Liu M.; Li L.; Wang R.; Sun H.; Shi L.; Du F. BiOBr flakes decoration and structural modification for CdTe/TiO2 spheres: Towards water decontamination under simulated light irradiation. Mater. Sci. Semicond. Process. 2019, 93, 331–338. 10.1016/j.mssp.2019.01.006. [DOI] [Google Scholar]
- Fang M.-J.; Tsao C.-W.; Hsu Y.-J. Semiconductor nanoheterostructures for photoconversion applications. J. Phys. D: Appl. Phys. 2020, 53, 143001. 10.1088/1361-6463/ab5f25. [DOI] [Google Scholar]
- Chang Y.-S.; Choi M.; Baek M.; Hsieh P.-Y.; Yong K.; Hsu Y.-J. CdS/CdSe co-sensitized brookite H:TiO2 nanostructures: Charge carrier dynamics and photoelectrochemical hydrogen generation. Appl. Catal., B 2018, 225, 379–385. 10.1016/j.apcatb.2017.11.063. [DOI] [Google Scholar]
- Ma S.; Li K.; Xu H.; Zhu J.; Zhu H.; Wu H. Lattice-Mismatched PbTe/ZnTe Heterostructure with High-Speed Midinfrared Photoresponses. ACS Appl. Mater. Interfaces 2019, 11, 39342–39350. 10.1021/acsami.9b13154. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Fei H.; Wang D. MoS2/Cu2O nanohybrid as a highly efficient catalyst for the photoelectrocatalytic hydrogen generation. Mater. Lett. 2019, 256, 126622. 10.1016/j.matlet.2019.126622. [DOI] [Google Scholar]
- Pu Y.-C.; Chou H.-Y.; Kuo W.-S.; Wei K.-H.; Hsu Y.-J. Interfacial charge carrier dynamics of cuprous oxide-reduced graphene oxide (Cu2O-rGO) nanoheterostructures and their related visible-light-driven photocatalysis. Appl. Catal., B 2017, 204, 21–32. 10.1016/j.apcatb.2016.11.012. [DOI] [Google Scholar]
- Zhou S.; Chen M.; Lu Q.; Hu J.; Wang H.; Li K.; Li K.; Zhang J.; Zhu Z.; Liu Q. Design of hollow dodecahedral Cu2O nanocages for ethanol gas sensing. Mater. Lett. 2019, 247, 15–18. 10.1016/j.matlet.2019.03.085. [DOI] [Google Scholar]
- Li J.-M.; Cheng H.-Y.; Chiu Y.-H.; Hsu Y.-J. ZnO–Au–SnO2 Z-scheme photoanodes for remarkable photoelectrochemical water splitting. Nanoscale 2016, 8, 15720–15729. 10.1039/C6NR05605A. [DOI] [PubMed] [Google Scholar]
- Pu Y.-C.; Lin W.-H.; Hsu Y.-J. Modulation of charge carrier dynamics of NaxH2–xTi3O7-Au-Cu2O Z-scheme nanoheterostructures through size effect. Appl. Catal., B 2015, 163, 343–351. 10.1016/j.apcatb.2014.08.009. [DOI] [Google Scholar]
- Lu Y.; Chu Y.; Zheng W.; Huo M.; Huo H.; Qu J.; Yu H.; Zhao Y. Significant tetracycline hydrochloride degradation and electricity generation in a visible-light-driven dual photoelectrode photocatalytic fuel cell using BiVO4/TiO2 NT photoanode and Cu2O/TiO2 NT photocathode. Electrochim. Acta 2019, 320, 134617. 10.1016/j.electacta.2019.134617. [DOI] [Google Scholar]
- Meng A.; Zhang L.; Cheng B.; Yu J. Dual Cocatalysts in TiO2 Photocatalysis. Adv. Mater. 2019, 31, 1807660. 10.1002/adma.201807660. [DOI] [PubMed] [Google Scholar]
- Trang T. N. Q.; Tu L. T. N.; Man T. V.; Mathesh M.; Nam N. D.; Thu V. T. H. A high-efficiency photoelectrochemistry of Cu2O/TiO2 nanotubes based composite for hydrogen evolution under sunlight. Composites, Part B 2019, 174, 106969. 10.1016/j.compositesb.2019.106969. [DOI] [Google Scholar]
- Li J.-M.; Wang Y.-T.; Hsu Y.-J. A more accurate, reliable method to evaluate the photoelectrochemical performance of semiconductor electrode without under/over estimation. Electrochim. Acta 2018, 267, 141–149. 10.1016/j.electacta.2018.02.015. [DOI] [Google Scholar]
- Chiu Y.-H.; Chang K.-D.; Hsu Y.-J. Plasmon-mediated charge dynamics and photoactivity enhancement for Au-decorated ZnO nanocrystals. J. Mater. Chem. A 2018, 6, 4286–4296. 10.1039/C7TA08543E. [DOI] [Google Scholar]
- Lin W.-H.; Chiu Y.-H.; Shao P.-W.; Hsu Y.-J. Metal-Particle-Decorated ZnO Nanocrystals: Photocatalysis and Charge Dynamics. ACS Appl. Mater. Interfaces 2016, 8, 32754–32763. 10.1021/acsami.6b08132. [DOI] [PubMed] [Google Scholar]
- Seemala B.; Therrien A. J.; Lou M.; Li K.; Finzel J. P.; Qi J.; Nordlander P.; Christopher P. Plasmon-Mediated Catalytic O2 Dissociation on Ag Nanostructures: Hot Electrons or Near Fields?. ACS Energy Lett. 2019, 4, 1803–1809. 10.1021/acsenergylett.9b00990. [DOI] [Google Scholar]
- Aguirre M. E.; Zhou R.; Eugene A. J.; Guzman M. I.; Grela M. A. Cu2O/TiO2 heterostructures for CO2 reduction through a direct Z-scheme: Protecting Cu2O from photocorrosion. Appl. Catal., B 2017, 217, 485–493. 10.1016/j.apcatb.2017.05.058. [DOI] [Google Scholar]
- Li J.-M.; Tsao C.-W.; Fang M.-J.; Chen C.-C.; Liu C.-W.; Hsu Y.-J. TiO2-Au-Cu2O Photocathodes: Au-Mediated Z-Scheme Charge Transfer for Efficient Solar-Driven Photoelectrochemical Reduction. ACS Appl. Nano Mater. 2018, 1, 6843–6853. 10.1021/acsanm.8b01678. [DOI] [Google Scholar]
- Colombo D. P.; Bowman R. M. Does Interfacial Charge Transfer Compete with Charge Carrier Recombination? A Femtosecond Diffuse Reflectance Investigation of TiO2 Nanoparticles. J. Phys. Chem. 1996, 100, 18445–18449. 10.1021/jp9610628. [DOI] [Google Scholar]
- Serpone N.; Lawless D.; Khairutdinov R.; Pelizzetti E. Subnanosecond Relaxation Dynamics in TiO2 Colloidal Sols (Particle Sizes Rp = 1.0-13.4 nm). Relevance to Heterogeneous Photocatalysis. J. Phys. Chem. 1995, 99, 16655–16661. 10.1021/j100045a027. [DOI] [Google Scholar]
- Wang C.; Zhao Y.; Xu H.; Li Y.; Wei Y.; Liu J.; Zhao Z. Efficient Z-scheme photocatalysts of ultrathin g-C3N4-wrapped Au/TiO2-nanocrystals for enhanced visible-light-driven conversion of CO2 with H2O. Appl. Catal., B 2020, 263, 118314. 10.1016/j.apcatb.2019.118314. [DOI] [Google Scholar]
- Zhen W.; Jiao W.; Wu Y.; Jing H.; Lu G. The role of a metallic copper interlayer during visible photocatalytic hydrogen generation over a Cu/Cu2O/Cu/TiO2 catalyst. Catal. Sci. Technol. 2017, 7, 5028–5037. 10.1039/C7CY01432E. [DOI] [Google Scholar]
- Ibrahim M. M.; Mezni A.; El-Sheshtawy H. S.; Abu Zaid A. A.; Alsawat M.; El-Shafi N.; Ahmed S. I.; Shaltout A. A.; Amin M. A.; Kumeria T.; Altalhi T. Direct Z-scheme of Cu2O/TiO2 enhanced self-cleaning, antibacterial activity, and UV protection of cotton fiber under sunlight. Appl. Surf. Sci. 2019, 479, 953–962. 10.1016/j.apsusc.2019.02.169. [DOI] [Google Scholar]
- Mezni A.; Ibrahim M. M.; El-Kemary M.; Shaltout A. A.; Mostafa N. Y.; Ryl J.; Kumeria T.; Altalhi T.; Amin M. A. Cathodically activated Au/TiO2 nanocomposite synthesized by a new facile solvothermal method: An efficient electrocatalyst with Pt-like activity for hydrogen generation. Electrochim. Acta 2018, 290, 404–418. 10.1016/j.electacta.2018.08.083. [DOI] [Google Scholar]
- Wang X.; Dong H.; Hu Z.; Qi Z.; Li L. Fabrication of a Cu2O/Au/TiO2 composite film for efficient photocatalytic hydrogen production from aqueous solution of methanol and glucose. Mater. Sci. Eng., B 2017, 219, 10–19. 10.1016/j.mseb.2017.02.011. [DOI] [Google Scholar]
- Rekeb L.; Hamadou L.; Kadri A.; Benbrahim N.; Chainet E. Highly broadband plasmonic Cu film modified Cu2O/TiO2 nanotube arrays for efficient photocatalytic performance. Int. J. Hydrogen Energy 2019, 44, 10541–10553. 10.1016/j.ijhydene.2019.02.188. [DOI] [Google Scholar]
- Huang H.; Hou X.; Xiao J.; Zhao L.; Huang Q.; Chen H.; Li Y. Effect of annealing atmosphere on the performance of TiO2 nanorod arrays in photoelectrochemical water splitting. Catal. Today 2019, 330, 189–194. 10.1016/j.cattod.2018.04.011. [DOI] [Google Scholar]
- Wongratanaphisan D.; Kaewyai K.; Choopun S.; Gardchareon A.; Ruankham P.; Phadungdhitidhada S. CuO-Cu2O nanocomposite layer for light-harvesting enhancement in ZnO dye-sensitized solar cells. Appl. Surf. Sci. 2019, 474, 85–90. 10.1016/j.apsusc.2018.05.037. [DOI] [Google Scholar]
- Fu X.; Li G. G.; Villarreal E.; Wang H. Hot carriers in action: multimodal photocatalysis on Au@SnO2 core–shell nanoparticles. Nanoscale 2019, 11, 7324–7334. 10.1039/C9NR02130B. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Jia K.; He X.; Wei S.; Wang P.; Liu X. Assembly of carboxylated zinc phthalocyanine with gold nanoparticle for colorimetric detection of calcium ion. J. Mater. Sci.: Mater. Electron. 2018, 29, 8380–8389. 10.1007/s10854-018-8849-y. [DOI] [Google Scholar]
- Shao Z.; Tian Z.; Pang J.; Feng G.; Guo B.; Zeng C.; Yang Y.; Liu S.; Wang Q. Optically modulated charge transfer in TiO2-Au nano-complexes. Mater. Res. Express 2014, 1, 045033 10.1088/2053-1591/1/4/045033. [DOI] [Google Scholar]
- Pham V. V.; Bui D. P.; Tran H. H.; Cao M. T.; Nguyen T. K.; Kim Y. S.; Le V. H. Photoreduction route for Cu2O/TiO2 nanotubes junction for enhanced photocatalytic activity. RSC Adv. 2018, 8, 12420–12427. 10.1039/C8RA01363B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q.; Zhang H.; Cui Y.; Deng X.; Guo R.; Cheng X.; Xie M.; Cheng Q. Fabrication of Cu2O/TiO2 nano-tube arrays photoelectrode and its enhanced photoelectrocatalytic performance for degradation of 2,4,6-trichlorophenol. J. Ind. Eng. Chem. 2018, 57, 181–187. 10.1016/j.jiec.2017.08.020. [DOI] [Google Scholar]
- Yang L.; Wang W.; Zhang H.; Wang S.; Zhang M.; He G.; Lv J.; Zhu K.; Sun Z. Electrodeposited Cu2O on the {101} facets of TiO2 nanosheet arrays and their enhanced photoelectrochemical performance. Sol. Energy Mater. Sol. Cells 2017, 165, 27–35. 10.1016/j.solmat.2017.02.026. [DOI] [Google Scholar]
- Sarkar S.; Akshaya R.; Ghosh S. Nitrogen doped graphene/CuCr2O4 nanocomposites for supercapacitors application: Effect of nitrogen doping on coulombic efficiency. Electrochim. Acta 2020, 332, 135368. 10.1016/j.electacta.2019.135368. [DOI] [Google Scholar]
- El-Shaer A.; Ismail W.; Abdelfatah M. Towards low cost fabrication of inorganic white light emitting diode based on electrodeposited Cu2O thin film/TiO2 nanorods heterojunction. Mater. Res. Bull. 2019, 116, 111–116. 10.1016/j.materresbull.2019.04.005. [DOI] [Google Scholar]
- Thompson T. L.; Yates J. T. Surface Science Studies of the Photoactivation of TiO2 New Photochemical Processes. Chem. Rev. 2006, 106, 4428–4453. 10.1021/cr050172k. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang F.; Yang M.; Wang Z.; Ren Y.; Cui J.; Zhao Y.; Du J.; Li K.; Wang W.; Kang D. J. Synthesis of porous MoS2/CdSe/TiO2 photoanodes for photoelectrochemical water splitting. Microporous Mesoporous Mater. 2019, 284, 403–409. 10.1016/j.micromeso.2019.04.055. [DOI] [Google Scholar]
- Bai X.; Ma L.; Dai Z.; Shi H. Electrochemical synthesis of p-Cu2O/n-TiO2 heterojunction electrode with enhanced photoelectrocatalytic activity. Mater. Sci. Semicond. Process. 2018, 74, 319–328. 10.1016/j.mssp.2017.10.049. [DOI] [Google Scholar]
- Meng X.-B.; Sheng J.-L.; Tang H.-L.; Sun X.-J.; Dong H.; Zhang F.-M. Metal-organic framework as nanoreactors to co-incorporate carbon nanodots and CdS quantum dots into the pores for improved H2 evolution without noble-metal cocatalyst. Appl. Catal., B 2019, 244, 340–346. 10.1016/j.apcatb.2018.11.018. [DOI] [Google Scholar]
- Hosseini-Sarvari M.; Jafari F.; Mohajeri A.; Hassani N. Cu2O/TiO2 nanoparticles as visible light photocatalysts concerning C(sp2)–P bond formation. Catal. Sci. Technol. 2018, 8, 4044–4051. 10.1039/C8CY00822A. [DOI] [Google Scholar]
- Chen B.; Zhou L.; Tian Y.; Yu J.; Lei J.; Wang L.; Liu Y.; Zhang J. Z-scheme inverse opal CN/BiOBr photocatalysts for highly efficient degradation of antibiotics. Phys. Chem. Chem. Phys. 2019, 21, 12818–12825. 10.1039/C9CP01495K. [DOI] [PubMed] [Google Scholar]
- Wang P.; Chen J.; Bai Y.; Yang P.; Du Y.; Zhang H. Preparation of a novel Z-scheme AgI/Ag/Bi24O31Cl10 catalyst with enhanced photocatalytic performance via an Ag0 electron transfer intermediate. J. Mater. Sci.: Mater. Electron. 2019, 30, 10606–10618. 10.1007/s10854-019-01405-x. [DOI] [Google Scholar]
- Kaviyarasan K.; Vinoth V.; Sivasankar T.; Asiri A. M.; Wu J. J.; Anandan S. Photocatalytic and photoelectrocatalytic performance of sonochemically synthesized Cu2O@TiO2 heterojunction nanocomposites. Ultrason. Sonochem. 2019, 51, 223–229. 10.1016/j.ultsonch.2018.10.022. [DOI] [PubMed] [Google Scholar]
- Low J.; Yu J.; Jaroniec M.; Wageh S.; Al-Ghamdi A. A. Heterojunction Photocatalysts. Adv. Mater. 2017, 29, 1601694. 10.1002/adma.201601694. [DOI] [PubMed] [Google Scholar]
- Chen N.-N.; Jin G.; Wang L.-J.; Sun H.-N.; Zeng Q.-S.; Yang B.; Sun H.-Z. Highly Efficient Aqueous-Processed Hybrid Solar Cells: Control Depletion Region and Improve Carrier Extraction. Adv. Energy Mater. 2019, 9, 1803849. 10.1002/aenm.201803849. [DOI] [Google Scholar]
- Shao Z.-F.; Yang Y.-Q.; Liu S.-T.; Wang Q. Transient competition between photocatalysis and carrier recombination in TiO2 nanotube film loaded with Au nanoparticles. Chin. Phys. B 2014, 23, 096102–096109. 10.1088/1674-1056/23/9/096102. [DOI] [Google Scholar]
- Subramanian V.; Wolf E. E.; Kamat P. V. Catalysis with TiO2/Gold Nanocomposites. Effect of Metal Particle Size on the Fermi Level Equilibration. J. Am.Chem. Soc. 2004, 126, 4943–4950. 10.1021/ja0315199. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Lv H.; Yuan M.; Chen Z.; Chen Z.; Wang X. In Situ Hydrothermal Construction of Direct Solid-State Nano-Z-Scheme BiVO4/Pyridine-Doped g-C3N4 Photocatalyst with Efficient Visible-Light-Induced Photocatalytic Degradation of Phenol and Dyes. ACS Omega 2017, 2, 2728–2739. 10.1021/acsomega.7b00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury B.; Choudhury A. Luminescence characteristics of cobalt doped TiO2 nanoparticles. J. Lumin. 2012, 132, 178–184. 10.1016/j.jlumin.2011.08.020. [DOI] [Google Scholar]
- Hou X.; Sun H.; Liu L.; Jia X.; Liu H. Unexpected large room-temperature ferromagnetism in porous Cu2O thin films. J. Magn. Magn. Mater. 2015, 382, 20–25. 10.1016/j.jmmm.2015.01.041. [DOI] [Google Scholar]
- Chiu Y.-H.; Hsu Y.-J. Au@Cu7S4 yolk@shell nanocrystal-decorated TiO2 nanowires as an all-day-active photocatalyst for environmental purification. Nano Energy 2017, 31, 286–295. 10.1016/j.nanoen.2016.11.036. [DOI] [Google Scholar]
- Govorov A. O.; Zhang H.; Demir H. V.; Gun’ko Y. K. Photogeneration of hot plasmonic electrons with metal nanocrystals: Quantum description and potential applications. Nano Today 2014, 9, 85–101. 10.1016/j.nantod.2014.02.006. [DOI] [Google Scholar]
- Tian B.; Lei Q.; Tian B.; Zhang W.; Cui Y.; Tian Y. UV-driven overall water splitting using unsupported gold nanoparticles as photocatalysts. Chem. Commun. 2018, 54, 1845–1848. 10.1039/C7CC09770K. [DOI] [PubMed] [Google Scholar]
- Dunklin J. R.; Rose A. H.; Zhang H.; Miller E. M.; van de Lagemaat J. Plasmonic Hot Hole Transfer in Gold Nanoparticle-Decorated Transition Metal Dichalcogenide Nanosheets. ACS Photonics 2020, 7, 197–202. 10.1021/acsphotonics.9b01393. [DOI] [Google Scholar]
- Al-Zubeidi A.; Hoener B. S.; Collins S. S. E.; Wang W.; Kirchner S. R.; Hosseini Jebeli S. A.; Joplin A.; Chang W.-S.; Link S.; Landes C. F. Hot Holes Assist Plasmonic Nanoelectrode Dissolution. Nano Lett. 2019, 19, 1301–1306. 10.1021/acs.nanolett.8b04894. [DOI] [PubMed] [Google Scholar]
- Furube A.; Du L.; Hara K.; Katoh R.; Tachiya M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. 10.1021/ja076134v. [DOI] [PubMed] [Google Scholar]
- Wu K.; Rodríguez-Córdoba W. E.; Yang Y.; Lian T. Plasmon-Induced Hot Electron Transfer from the Au Tip to CdS Rod in CdS-Au Nanoheterostructures. Nano Lett. 2013, 13, 5255–5263. 10.1021/nl402730m. [DOI] [PubMed] [Google Scholar]
- Xue J.; Elbanna O.; Kim S.; Fujitsuka M.; Majima T. Defect state-induced efficient hot electron transfer in Au nanoparticles/reduced TiO2 mesocrystal photocatalysts. Chem. Commun. 2018, 54, 6052–6055. 10.1039/C8CC02853B. [DOI] [PubMed] [Google Scholar]
- El Mragui A.; Zegaoui O.; Daou I.; Esteves da Silva J. C. G. Preparation, characterization, and photocatalytic activity under UV and visible light of Co, Mn, and Ni mono-doped and (P,Mo) and (P,W) co-doped TiO2 nanoparticles: a comparative study. Environ. Sci. Pollut. Res. 2019, 10.1007/s11356-019-04754-6. [DOI] [PubMed] [Google Scholar]
- Charoenthai N.; Yomma N. Effect of Annealing Temperature and Solvent on the Physical Properties and Photocatalytic activity of Zinc Oxide Powder Prepared by Green Synthesis Method. Mater. Today: Proc. 2019, 17, 1386–1395. 10.1016/j.matpr.2019.06.159. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.