

Abstract
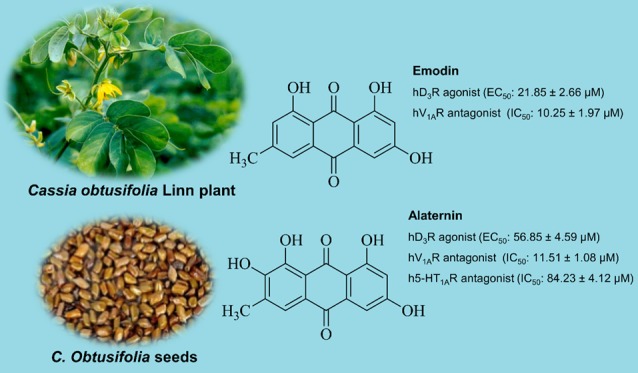
The present study examines the effect of human monoamine oxidase active anthraquinones emodin, alaternin (=7-hydroxyemodin), aloe-emodin, and questin from Cassia obtusifolia Linn seeds in modulating human dopamine (hD1R, hD3R, and hD4R), serotonin (h5-HT1AR), and vasopressin (hV1AR) receptors that were predicted as prime targets from proteocheminformatics modeling via in vitro cell-based functional assays, and explores the possible mechanisms of action via in silico modeling. Emodin and alaternin showed a concentration-dependent agonist effect on hD3R with EC50 values of 21.85 ± 2.66 and 56.85 ± 4.59 μM, respectively. On hV1AR, emodin and alaternin showed an antagonist effect with IC50 values of 10.25 ± 1.97 and 11.51 ± 1.08 μM, respectively. Interestingly, questin and aloe-emodin did not have any observable effect on hV1AR. Only alaternin was effective in antagonizing h5-HT1AR (IC50: 84.23 ± 4.12 μM). In silico studies revealed that a hydroxyl group at C1, C3, and C8 and a methyl group at C6 of anthraquinone structure are essential for hD3R agonist and hV1AR antagonist effects, as well as for the H-bond interaction of 1-OH group with Ser192 at a proximity of 2.0 Å. Thus, based on in silico and in vitro results, hV1AR, hD3R, and h5-HT1AR appear to be prime targets of the tested anthraquinones.
Introduction
Cassia obtusifolia Linn seeds have a long history of use in traditional Chinese medicine, where anthraquinones and naphthopyrones derivatives were reported as predominant constituents, particularly the glycosides (cassiaside, rubrofusarin gentiobioside, and cassiaside B).1 Seed extracts and their constituents have been reported for activities such as anti-Alzheimer’s disease,2−5 anti-Parkinson’s disease,6 antidiabetic and diabetic complications,7,8 hepatoprotection,9,10 anti-inflammation,2 neuroprotective activity,11,12 antibacterial,13 and antioxidant.14,15 In a previous study,16 100 μM emodin inhibited 4 nM (−)-epinephrine, 2 μM nicotinic acid, and 8 μM histamine-induced dynamic mass redistribution signals in human epidermoid carcinoma A431 cell, showing hydroxyl carboxylic acid receptor-2 (HCA-2), histamine receptor (H1R), and β2-adrenoceptor (β2-AR) as targets. Similarly, by upregulating glucocorticoid receptor (GR) and brain-derived neurotrophic factor (BDNF) levels in the hippocampus, emodin improved the depression-like behavior in chronic unpredictable mild stress-induced behavioral deficit (depression-like behavior) mice.17 Emodin at 30 μM concentration showed an antipsychotic effect in Schizophrenia model (epidermal growth factor challenged primary neuronal cultures) by attenuating the receptor activation of ErbB1 and ErbB2.18
Another anthraquinone, aloe-emodin, attenuated scopolamine-induced cognitive deficits by inhibiting the acetylcholinesterase activity (IC50 = 18.37 μg/mL) and modulating H2O2-induced oxidative stress in PC12 cells.19 Likewise, in subcutaneous human glioblastoma U87MG-implanted nude CG1 mice, i.p. administration of aloe-emodin at 50 mg/(kg day) for 15 days showed antiproliferative effect by decreasing Ki67 positive cells and proapoptotic effect by increasing P53 and caspase 6 in mouse brain.20 In the same study, aloe-emodin at 20 and 40 μM concentration induced cell cycle arrest in U87MG cells by increasing the expression levels of p53, p21, and the reduction of cyclin CDK2 in vitro.
More recently, we have reported the human monoamine oxidase (hMAO) inhibitory potential of Cassia seed-derived secondary metabolites21 and a possible role of rubrofusarin against comorbid diabetes and depression via protein tyrosine phosphatase 1B and hMAO inhibition.22 In that study,21 emodin, alaternin (7-hydroxyemodin), aloe-emodin, and questin inhibited hMAO enzyme activity with low micromolar IC50 values ranging from 0.17 to 23 μM.
Drugs have specific targets in the body through which they modulate the disease state. Modern drug discovery and development incorporates in silico prediction approaches to predict the potential target proteins to understand the mechanism of action of drugs in addition to in vitro and in vivo studies.23 G protein-coupled receptors (GPCRs) represent one of the most important drug targets with potential therapeutic benefits in the central nervous system (CNS) and endocrine systems. The concept of precise medication relies on GPCRs targeting, and to date, 34% of FDA-approved drugs are GPCR targets.24 So, the main objectives of this study were to: (a) predict the main targets for Cassia-derived secondary metabolites in CNS via proteocheminformatics modeling (PCM), (b) validate the PCM prediction by evaluating the modulatory effect on predicted receptors via cell-based functional GPCRs assays, and (c) look at the specific binding interactions of test ligands (Figure 1) and target receptors via molecular docking simulation.
Figure 1.

Structure of anthraquinones from Cassia obtusifolia seeds.
Results and Discussion
In Silico Target Prediction
From PCM, the highest-ranked 20-potential protein targets were predicted for the four anthraquinones. Table 1 enlists the target proteins with the normalization rate. As shown in the table, V1A receptor is on the top of the list followed by substance P and 5HT1A receptor, with the dopamine receptor also being placed within the 10 highest ranks. Based on this prediction, we then proceeded to validate the predictions in GPCRs cell-based functional assays with the dopamine (D1, D3, and D4), 5HT1A, and V1A receptor (Table 2).
Table 1. Twenty Most Highly Predicted Protein Targets Predicted from PCM Modeling for Cassia-Derived Anthraquinones in Neurodegenerative Diseasesa.
| organismal system | abbreviation | protein name | NR |
|---|---|---|---|
| STS | V1AR | vasopressin V1A receptor | 2.1742 |
| STS | SPR | substance P receptor | 1.9936 |
| NS | 5-HT-1A | 5-hydroxytryptamine receptor 1A | 1.9824 |
| STS | NKR | neuromedin-K receptor | 1.9311 |
| STS | OT-R | oxytocin receptor | 1.9099 |
| STS | MAPK14 | mitogen-activated protein kinase 14 | 1.8689 |
| STS, NS | 5-HT-6 | 5-hydroxytryptamine receptor 6 | 1.8408 |
| NS | DRD3 | D3 dopamine receptor | 1.8323 |
| STS, NS | 5-HT-2B | 5-hydroxytryptamine receptor 2B | 1.7906 |
| STS, NS | 5-HT-4 | 5-hydroxytryptamine receptor 4 | 1.7508 |
| NS | 5-HT-1B | 5-hydroxytryptamine receptor 1B | 1.7354 |
| STS | PI3K-α | phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic α-subunit | 1.7316 |
| STS, NS | ACM5 | muscarinic acetylcholine receptor M5 | 1.7278 |
| STS | NTR1 | neurotensin receptor type 1 | 1.7278 |
| STS, NS | DRD1 | D1 dopamine receptor | 1.7278 |
| STS | CysLTR1 | cysteinyl leukotriene receptor 1 | 1.7128 |
| STS, NS | 5-HT-2A | 5-hydroxytryptamine receptor 2A | 1.7128 |
| NS | 5-HT-3A | 5-hydroxytryptamine receptor 3A | 1.7128 |
| STS | HSP90 | heat shock protein HSP 90-α | 1.7054 |
| NS | ACM4 | muscarinic acetylcholine receptor M4 | 1.6980 |
NR: Normalization rate; NS: Nervous system; STS: Signaling transduction system.
Table 2. Efficacy Values (% Stimulation and % Inhibition) of Anthraquinones at Dopamine (D1, D3, and D4) and Serotonin (5HT1A) and Vasopressin (V1A) Receptors.
| emodin | alaternin | aloe-emodin | questin | reference drugs | |
|---|---|---|---|---|---|
| receptors | % stimulationa (% inhibitiona) | % stimulationa (% inhibitiona) | % stimulationa (% inhibitiona) | % stimulationa (% inhibitiona) | EC50b(IC50c) |
| D1 (h) | –3.55 ± 0.64 (−10.20 ± 6.36) | 0.01 ± 2.41 (−30.43 ± 8.95) | 0.15 ± 4.45 (−47.85 ± 9.69) | –2.10 ± 0.85 (10.95 ± 6.01) | 17 (3.6) |
| D3 (h) | 75.60 ± 12.87 (−10.8 ± 6.59) | 43.97 ± 4.76 (−9.8 ± 4.82) | –57.95 ± 32.88 (20.60 ± 6.79) | 34.85 ± 0.78 (1.25 ± 7.42) | 4.0 (13) |
| D4 (h) | 2.35 ± 2.76 (−0.35 ± 4.03) | 14.10 ± 0.31 (−2.4 ± 6.18) | -53.15 ± 7.00 (25.70 ± 15.70) | –38.15 ± 15.91 (4.50 ± 10.32) | 5.8 (320) |
| 5-HT1A (h) | –3.56 ± 0.21 (18.4 ± 3.89) | –5.46 ± 0.15 (34.50 ± 4.86) | 3.45 ± 0.21 (1.55 ± 3.61) | 2.45 ± 0.49 (0.70 ± 4.10) | 1.6 (4.4) |
| V1A (h) | –4.80 ± 0.69 (69.35 ± 1.34) | –11.0 ± 0.66 (59.20 ± 0.10) | 7.45 ± 1.20 (18.90 ± 9.05) | –3.40 ± 0.71 (16.30 ± 2.40) | 0.29 (4.5) |
% stimulation and % inhibition of control agonist response at 50 μM of anthraquinones.
EC50 (nM) values of standard agonists (D1, D3, and D4: dopamine, 5-HT1A:serotonin, V1A: AVP).
IC50 (nM) values of standard antagonists (D1: SCH-23390, D3: (+)-butaclamol, D4: clozapine, 5-HT1A: (S)-WAY-100635, V1A: [d(CH2)51, Tyr(Me)2]-AVP).
Emodin, Alaternin, and Questin as Human Dopamine D3 (hD3R) Agonists
The effect of test compounds on dopamine receptor was evaluated fluorimetrically by measuring the level of cAMP. Agonist effect was expressed as % of the control response to 300 nM dopamine and antagonist effect was expressed as % inhibition of control response to 10 nM dopamine. As tabulated in Table 2, 50 μM concentration of emodin, alaternin, and questin exhibited 75.6, 43.9, and 34.85% of control agonist response. The concentration-dependent agonist response is depicted in Figure 2A.
Figure 2.

Concentration-dependent percentage of control agonist effect (A) and percentage inhibition of control agonist effect (B, C) of emodin, alaternin, aloe-emodin, and questin on hD3R, hV1AR, and h5-HT1AR, respectively. ND: Not determined.
From the dose–response curve, half-maximal effective concentration (EC50) values of emodin and alaternin for hD3R were 21.85 ± 2.66 and 56.85 ± 4.59 μM, respectively. Dopamine had an EC50 value of 4 nM. The agonist effect of emodin at 12.5 μM was similar to that of 50 μM alaternin (i.e., approx. 45%). The potency of emodin was 2.5 times greater than that of alaternin as can be seen from the EC50 values of respective compounds.
Among central nervous system disorders, Parkinson’s disease (PD) is the second most common age-related neurodegenerative disorder with 1% prevalence rate in the population above 60 years of age and is characterized by rigidity, tremor, and bradykinesia. The administration of dopamine in its prodrug form, levodopa (l-dopa), in combination with peripheral DOPA decarboxylase inhibitor is the current therapeutic approach to treat PD. However, owing to the side effects of l-dopa and the development of dyskinesia (l-dopa-induced dyskinesia; LID) upon prolonged use, an alternative treatment approach is warranted. Various studies have discovered the involvement of dopamine D3 receptors in the etiology of PD and LID. In PD, dopamine D3 receptor expression decreases, while it increases in the brain region of LID patients.25−28
To interpret the result of the functional assay, emodin and alaternin were docked into the D3R co-crystal structure (PDB ID: 3PBL) in complex with eticlopride. The docking result (binding pose and interacting residues) was confirmed by redocking with the reference agonist dopamine and rotigotine, and antagonist eticlopride. As shown in Figure 3 and tabulated in Table 3, emodin was predicted to bind to the active site of dopamine D3R by forming three H-bonds with Ser196, Val111, and Thr115 at a distance of 2.0, 2.1, and 2.7 Å, respectively (shown by the blue lines in Figure 3B). In addition, the methyl group at C-6 formed a π–alkyl interaction with Phe345 (5.03 Å). Aromatic ring C formed a π–anion interaction with Asp110, while ring A formed π-sulfur and π–π T-shaped interactions with Cys114 and Phe346, respectively (Figure 3C).
Figure 3.

Molecular docking of human dopamine D3 receptor (hD3R) binding with emodin and alaternin along with positive controls (A). Emodin (B, C) and alaternin (D, E) docked into the active cavity of hD3R. Emodin, alaternin, dopamine, and eticlopride are presented as orange, green, red, and black sticks, respectively. Hydrogen-bond interactions are presented as blue lines (B, D).
Table 3. Binding Sites and Docking Scores of Emodin and Alaternin along with Reference Compounds in Human Dopamine D3 Receptor (hD3R).
| compounds | binding energy (kcal/mol) | no. of H-bonds | H-bond interaction residues | nonpolar interacting residues |
|---|---|---|---|---|
| dopaminea (agonist) | –5.84 | 5 | Asp110 (salt bridge), Val111, Thr115, Ser196 | Val111, Cys114, Phe346 |
| rotigotinea (agonist) | –9.23 | 2 | Asp110 (salt bridge), Ser192 | Val111, Phe345, His349, Val107, Cys181, Val111, Cys114 |
| eticlopridea (antagonist) | –8.50b | 3 | Asp110 (salt bridge), Asp110, His349 | Val111, Cys114, Val189, Phe346, His349, Val111, Ile183, Phe345 |
| emodin | –6.67 | 3 | Ser192, Thr115, Val111 | Tyr373, Thr369, Tyr365, His349, Phe345, Phe346, Ile183, Trp342, Asp110, Cys114, Ser196 |
| alaternin | –6.79 | 4 | Tyr365, Ser196 Thr115, Val111 | Tyr373, Thr369, Tyr365, His349, Phe345, Phe346, Ile183, Trp342, Asp110, Cys114, Ser192 |
Dopamine, rotigotine, and eticlopride were used as reference ligands.
Root-mean-square deviation (RMSD) value: 0.48 Å.
Similarly, as shown by the blue lines in Figure 3D, four H-bond interactions with Tyr365, Val111, Ser196, and Thr115 were predicted at a distance of 2.1, 2.1, 2.4 and 2.8 Å, respectively, for alaternin. The methyl group involved in π-alkyl interactions with Phe345 (5.11 Å) and Tyr373 (4.34 Å). In addition, similar to emodin, aromatic ring C formed a π-anion interaction with Asp110, while ring A formed π-sulfur and π–π T-shaped interactions with Cys114 and Phe346, respectively (Figure 3E). From these docking results (Table 3), it was found that hydrophobic Phe345 and Phe346 residues of D3R are important for binding of ligands containing aromatic rings like emodin, alaternin, dopamine, and rotigotine. Conserved Asp and Ser residues also acted as bridges between emodin/alaternin and D3R via electrostatic and H-bond interaction, respectively.
Besides playing a central role in emotion and behavior, dopamine is responsible for the suppression of proinflammatory cytokines in macrophages, endothelial cells, neutrophils, mast cells, or glial cells, thereby regulating immune/inflammatory response.29−33 Likewise, in a recent study, dopamine suppressed inflammatory response and attenuated tissue injury in mice with acute pancreatitis34 and attenuated lipopolysaccharide/d-galactosamine-induced fulminant liver injury in mice by suppressing the production of TNF-α phosphorylation of c-jun-N-terminal kinase (JNK); cleavage of caspase-3; upregulation of hepatic caspase-3, caspase-8, and caspase-9 activities; and reducing the count of terminal deoxynucleotidyl transferase-mediated nucleotide nick-end labeling (TUNEL)-positive hepatocytes.35 Therefore, the hD3R agonist effect of emodin and alaternin might have a role for their reported anti-inflammatory effect.36,37
Emodin and Alaternin as Human Vasopressin 1A Receptor (hV1AR) Antagonists
In human recombinant Chinese hamster ovary (CHO) cells expressing the hV1A receptor, reference agonist arginine vasopressin (AVP) caused a concentration-dependent increase in intracellular Ca2+ concentration with an EC50 value of 0.29 nM (Figure 2B and Table 2). The test compounds emodin, alaternin, aloe-emodin, and questin did not show any effect on control agonist response. They did not show any agonist response up to 50 μM, and % of control agonist response was negative at 50 μM concentration (Table 2). However, for antagonist effect, emodin and alaternin showed a concentration-dependent inhibition of control response to 10 nM AVP (Figure 2B). Even at 12.5 μM concentration, emodin and alaternin inhibited the 10 nM AVP-induced intracellular Ca2+ concentration by >50%. The IC50 values for emodin and alaternin were 10.25 ± 1.97 and 11.51 ± 1.08 μM, respectively. The reference antagonist [d(CH2)51, Tyr(Me)2]-AVP inhibited AVP-induced Ca2+ response with an IC50 value of 4.5 nM. The antagonist effect of aloe-emodin and questin was very weak with an approximately 17% inhibition of control agonist response at 50 μM concentration.
Similarly, previous studies on natural emodin had reported platelet aggregation inhibition38 and vasorelaxant property.39 V1AR in vascular smooth muscles is responsible for vasoconstriction, myocardial contractility, platelet aggregation, and uterine contraction.40 Vasopressin receptor is another target for CNS drugs, and vasopressin antagonists represent a novel approach for the treatment of stress, mood, and behavioral disorders.41
Intraperitoneal injection of emodin at a dose of 25 g/(kg day) in cerebral ischemia/reperfusion (I/R) model rats improved neurological deficit scores and reduced blood–brain barrier (BBB) permeability and infarction area, suggesting the inhibition of the expressions of connexin 43 and aquaporin 4 (AQP4) as a probable mechanism.42 AQP4 is membrane water channel protein that plays an important role in the cerebral edema and brain water balance. A selective V1AR antagonist SR49059 prevented brain edema by suppressing injury-induced upregulation of GFAP, V1AR, and AQP4 after traumatic brain injury.43
Vasopressin has numerous peripheral roles. An increased VP level along with impaired renal water excretion and abnormal renal hemodynamics in a mouse model of CCl4-induced liver cirrhosis has previously been reported.44 Similarly, a recent study on ischemia-reperfusion injury mouse model45 identified upregulated V1R expression in hepatocytes and highlighted the importance of the hepatocyte V1R/Wnt/β-catenin/FoxO3a/Akt pathway in hepatoprotection. From PCM modeling, V1AR was predicted as a top-target for Cassia-anthraquinones. And further validation of PCM prediction via cell-based functional assays in transfected cells expressing human cloned V1AR (CHO-V1AR), emodin, and alaternin was characterized as V1AR antagonists (IC50 = 10.25 ± 1.97 and 11.51 ± 1.08 μM, respectively). Therefore, the reported vasorelaxant and antiedema property of emodin might be attributed to its V1AR antagonist effect.
To further interrogate the structural basis of the antagonist mechanism of emodin and alaternin on the vasopressin receptor, compounds were docked into the homology model of V1A receptor and the result (binding pose and interacting residues) was confirmed by redocking with the reference agonist arginine vasopressin (AVP) and antagonist SR49059. As shown in Figure 4 and tabulated in Table 4, AVP bound to the active site of V1AR with a binding score −9.14 kcal/mol by forming five H-bond interactions with Asp202, Glu54, Asp112, and Ile330. Similarly, antagonist SR49059 displayed H-bond interactions with Gln131, Gln108, and Lys128 with a binding score of −8.98 kcal/mol. Emodin involved in four H-bond interactions with Met135, Gln131, Ala101, and Gly337 with a bond length of 2.0–2.6 Å (shown by the blue lines in Figure 4B).
Figure 4.

Molecular docking of human vasopressin V1A receptor (hV1AR) binding with emodin and alaternin along with positive controls (A). Emodin (B, C) and alaternin (D, E) docked into the active cavity of hV1AR. Emodin, alaternin, and SR49059 are presented as orange, green, and black sticks, respectively. Agonist, AVP is presented as a red ribbon. Hydrogen-bond interactions are presented as blue lines (B, D).
Table 4. Binding Sites and Docking Scores of Emodin and Alaternin along with Reference Compounds in the Modeled Human Vasopressin V1A Receptor (hV1AR).
| compounds | binding energy (kcal/mol) | no. of H-bonds | H-bond interaction residues | nonpolar interacting residues |
|---|---|---|---|---|
| AVPa (agonist) | –9.14 | 5 | Asp202, Glu54, Asp112, Ile330 | Trp204, Ile330, Ala101, Ala334, Val132, Met135 |
| SR49059a (antagonist) | –8.98 | 5 | Gln131, Gln108, Lys128 | Phe307, Trp204, Val132, Met135, Met220, Ala334, Ala205, Gln131, Thr333 |
| emodin | –6.36 | 4 | Met135, Gln131, Ala101, Gly337 | Val100, Ala101, Ala334, Phe307, Trp304, Met135 |
| alaternin | –6.40 | 5 | Lys128, Gln131, Ser213, Val217, Gln311 | Lys128, Met135, Met220, Phe189, Ala205 |
Arginine vasopressin (AVP) and (2S)1-{(2R,3S)-5-chloro-3-(2-chloro-phenyl)-1-(3,4-dimethoxybenzene-sulfonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl}pyrrolidine-2-carboxamide (SR49059) were used as reference ligands.
In addition, other nonpolar interactions such as π–π T-shaped (Phe307 and Trp304) and π-alkyl (Ala334, 5.25 Å) interactions were observed for aromatic rings A and C (Figure 4C). In the case of alaternin, five H-bond interactions with Lys128, Gln131, Ser213, Val217, and Gln311 at a distance of 1.8–3.0 Å were predicted with a binding score of −6.40 kcal/mol.
Aromatic rings were involved in π-alkyl interactions with Lys128 (5.43 Å) and Ala205 (4.73 Å), and π-sulfur interaction with Met220. Although the number of H-bond interactions for alaternin was greater than that of emodin, both showed similar binding energies. The binding sites for antagonist SR49059 and test compounds overlapped—i.e., interaction with polar residue Lys128 and Gln131 in TM III, nonconserved residue Ala334 in TM VII, and aromatic residue Phe307 at the bottom of the binding pocket in TM VI. The interaction with Ala334 and Phe307 plays an important role in binding V1AR selective ligands.46
Alaternin as Human Serotonin-1A Receptor (h5-HT1AR) Antagonists
The antagonist effect of alaternin on h5-HT1A receptor was evaluated fluorimetrically by measuring the free cytosolic Ca2+-ion concentration in response to 30 nM serotonin. Figure 2C illustrates a concentration-dependent inhibitory effect of alaternin on the control agonist (30 nM serotonin) response along with the 50% inhibitory concentration. As shown in Table 2, alaternin showed 34.5% inhibition of 30 nM serotonin effect at 50 μM and gave an IC50 value of 84.23 ± 4.12 μM (Figure 2C). The reference antagonist (S)-WAY-100635 had an IC50 value of 4.4 nM. Emodin, aloe-emodin, and questin remained ineffective in antagonizing the h5-HT1A receptor activity.
In a recent study, emodin improved cycloheximide-induced amnesia in rats, and the authors suggested the blocking of serotonin release or activating the presynaptic 5-HT1A receptor and muscarinic receptor as a possible mechanism.47 However, for the particular effect measured here, we did not observe an effect on 5HT1A receptor. In a previous study,48Cassia seed extract at 20 μg/mL showed a significant antiallergic effect in IgE-mediated mast cells and anaphylactic models by inhibiting the production of IL4 (p < 0.05), TNF (p < 0.01), PGE2 secretion (p < 0.01), and histamine release (p < 0.01). Serotonin is one of the putative inflammatory mediators that is able to induce dose-dependent nociceptive behaviors when injected into the paw, and also appears to interact synergistically with other inflammatory mediators to generate pain.49 In a study on subcutaneous formalin-injected paw model of pain,50 coadministration of 5-HT1A receptor antagonist WAY 100 135 (450 μg/paw) inhibited the phase 2 (long lasting) intense flinching behavior significantly (P < 0.001). Similarly, the analgesic effect of electroacupuncture on inflammatory pain in the rat model of collagen-induced arthritis was blocked by a 5-HT1A receptor antagonist spiroxatrine (1 mg/kg i.p.).51
In a previous report, alaternin attenuated neuronal cell death in transient cerebral hypoperfused mice via anti-inflammatory responses by preventing nitrotyrosine and lipid peroxidation as well as inhibiting nitric oxide synthase expression.37 There has been a well-reported correlation between 5-HT1A receptor mRNA expression and neuroinflammation.52,53 The expression of the 5-HT1A receptor mRNA was enhanced in spinal GABA and enkephalin neurons after inflammation.54 We observed a moderate antagonist effect of alaternin in 5-HT1AR (IC50: 84.23 ± 4.12 μM). This corroborates the reported anti-inflammatory effect of alaternin.
In the functional assay, only alaternin showed a moderate antagonist effect on h5-HT1AR for which we hence performed docking. As tabulated in Table 5, the 3-OH group of emodin and alaternin displayed a H-bond interaction with Cys187 (Figure 5B,D). Similarly, the same π–π T-shaped interaction with Phe112, π–π stacked interaction with Phe361, and π-anion interaction with Asp116 were observed for both the test compounds (Figure 5C,E). However, additional nonpolar interaction between the 7-OH group and Trp358 was observed for alaternin (Figure 5B). This additional interaction could explain the potency of alaternin compared to that of emodin. However, further experimental analysis is needed to confirm the role of Trp358 in h5-HT1A antagonism.
Table 5. Binding Sites and Docking Scores of Emodin and Alaternin along with Reference Compounds in the Modeled of Human 5-hydroxytryptamine 1A Receptor (h5HT1AR).
| compounds | binding energy (kcal/mol) | no. of H-bonds | H-bond interaction residues | nonpolar interacting residues |
|---|---|---|---|---|
| serotonina (agonist) | –6.77 | 4 | Asp116 (salt bridge), Thr200, Thr121 | Ala203, Val117, Phe362, Phe361, Cys120 |
| WAY 100635a (antagonist) | –11.22 | 3 | Asp116 (salt bridge), Asn386 | Ala93, Tyr96, Thr200, Phe112, Cys120, Ala203, Pbe362, Phe361 |
| emodin | –7.01 | 1 | Cys187 | Asp116, Ile189, Phe112, Phe361 |
| alaternin | –6.77 | 1 | Cys187 | Asp116, Ile189, Phe112, Phe361, Trp358 |
Serotonin and N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]-ethyl}-N-(2-pyridinyl) cyclohexanecarboxamide (WAY 100635) were used as reference ligands.
Figure 5.

Molecular docking of human 5-hydroxytryptamine1A receptor (h5HT1AR) binding with emodin and alaternin along with positive controls (A). Emodin (B, D) and alaternin (C, E) docked into the active cavity of h5HT1AR. Emodin, alaternin, serotonin, and WAY 00635 are presented as orange, green, red, and black sticks, respectively. Hydrogen-bond interactions are presented as blue lines (B, D).
Drug-likeness and ADME Prediction
Drug-likeness was predicted for emodin, alaternin, aloe-emodin, and questin. The results in Table 6 suggested that these compounds have good druglike properties, as they adhered to the MDDR-like rule55 and Lipinski’s rule.56 All anthraquinones were predicted midstructures in MDDR-like rule and suitable for drug development from Lipinski’s rule. In ADME prediction, all compounds were predicted with strong plasma protein binding (91–100%), good human intestinal absorption (HIA) (75–94%), and good lipophilicity (log Po/w value range, 1.89–2.69), indicating the suitability for CNS delivery. Likewise, the blood–brain barrier (BBB) penetration values ([brain]/[blood]) were >0.45% indicating moderate absorption by the CNS.
Table 6. Drug-likeness and ADME Characteristics as Predicted by PreADMET.
| drug-likeness |
ADME
characteristics |
|||||
|---|---|---|---|---|---|---|
| compounds | MDDR-like rule | Lipinski’s rule | log Po/wa | PPBb | HIAc | in vivo BBB penetration ([brain]/[blood])d |
| emodin | midstructure | suitable | 2.56 | 100 | 90.43 | 0.668 |
| alaternin | midstructure | suitable | 2.57 | 98.17 | 75.71 | 0.459 |
| aloe-emodin | midstructure | suitable | 1.89 | 91.11 | 90.64 | 0.492 |
| questin | midstructure | suitable | 2.69 | 96.06 | 94.04 | 0.730 |
The log of the coefficient of solvent partitioning between 1-octanol and water.
Plasma protein binding (PPB) (<90% represents weak binding, and >90% represents strong binding).
Human intestinal absorption (HIA) (0–20%: poor, 20–70%: moderate, and 70–100%: good).
Absorption by the CNS <0.1: low, 0.1–2.0: moderate, and >2.0: high.
Overall, drug-likeness and ADME prediction results demonstrated that the test anthraquinones are suitable for CNS delivery. In general, relatively higher lipophilicity provides better CNS penetration; however, too high values may enhance nonspecific plasma protein binding.57 The lipophilicity values log P/log D ranging from 1.7 to 2.8 demonstrate the highest CNS penetration,58,59 and the values for the test anthraquinones in the present study fall within this range. The HIA rate was predicted to be good for all of the compounds, indicating the suitability for oral administration. However, in a previous report,60 oral administration of emodin at doses of 20 and 40 mg/kg rapidly underwent phase II metabolism to form its glucuronide, and the parent form of emodin was almost undetectable in vivo. Therefore, oral administration would not be the best method of application of emodin because of fast elimination and low bioavailability in vivo. All of these predicted results will be helpful for the optimization of druglike properties.
High-throughput screening of chemical compounds had predicted quinone derivatives as reactive and pan assay interference compounds (PAINS) that could show a false biological effect.61,62 However, this effect cannot be generalized for all of the quinone derivatives and structure–activity relationships would be evidence.61 The basic chemical structure of the test compounds is anthracene-9,10-dione with different substituents at ortho-, meta-, and para-positions of two side rings (Figure 1). The effect of the test compounds on test receptors varied with substitution (Table 2). When an anthracene-9,10-dione moiety had a polyhydroxy group at positions C1, C3, and C8 with methylation at position C6 (emodin), it showed potent agonist effect at dopamine hD3R and antagonist effect at hV1AR. However, an additional hydroxyl group at position C7 of emodin (as in alaternin) retarded the hD3R agonist effect without altering the antagonist effect at hV1AR. Besides, a moderate antagonist effect at h5-HT1AR was observed for alaternin, which was not observed for emodin. Similarly, a dihydroxy group at position C1 and C8, and a hydroxymethyl group at C6 position in an anthracene-9,10-dione moiety (aloe-emodin) showed mild antagonist effect at hD3R and hD4R. The hD3R agonist effect and hV1AR antagonist effect showed by emodin and alaternin was completely abolished in aloe-emodin. Interestingly, when an anthracene-9,10-dione moiety had a dihydroxy group at position C3 and C8, methyl group at position C6 and methoxy group at the C1 position, questin selectively modulated the hD3R agonist effect. From this structure–activity relation, the following insights can be drawn—(1) hydroxyl group at C1, C3, and C8 and a methyl group at C6 are essential for hD3R agonist and hV1AR antagonist effects; (2) an additional hydroxyl group at C7 of emodin is functional for the h5-HT1AR antagonist effect.
If all quinone derivatives are PAINS indeed, then all of the test anthraquinones of the present study should show functional effect in all of the tested receptors. However, only emodin and alaternin (7-hydroxyemodin) showed a selective agonist effect on dopamine D3R and antagonist effect on vasopressin V1AR, meaning these two compounds selectively bind to the particular receptor for functional effect. This clearly shows that the functional effect of emodin and alaternin is attributed to the substituents in the anthraquinone structure rather than quinone itself. Still, these effects need to be proved in vivo. Still, these effects need to be proved in vivo.
Conclusions
In conclusion, the present study demonstrates the effect of emodin, alaternin (7-hydroxyemodin), aloe-emodin, and questin from C. obtusifolia seeds on various GPCRs (hD1R, hD3R, hD4R, h5-HT1AR, and hV1AR) modulation via cell-based functional assays and corroborate with the PCM prediction. Results characterize emodin and alaternin as dopamine D3R agonists and vasopressin V1A antagonists. Questin showed a moderate hD3R agonist effect, and aloe-emodin showed mild hD3R antagonist effect. Only alaternin was effective in antagonizing h5-HT1AR, and the remaining compounds remained ineffective. Thus, we conclude that anthraquinones, especially emodin and alaternin appear to be an attractive therapeutic route for neuroprotection that has a beneficial effect on the aminergic pathways involved in neurodegeneration.
Materials and Methods
Chemicals and Reagents
A murine interleukin-3-dependent pro-B (Ba/F3) and a transfected Chinese hamster ovary (CHO) cell lines were obtained from Eurofins Scientific (Le Bois I’Eveque, France). Dulbecco’s modified Eagle’s medium (DMEM) buffer, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, and Hank’s balanced salt solution (HBSS) buffer were purchased from Invitrogen (Carlsbad, CA). The reference agonists (dopamine, serotonin, and arginine vasopressin) and antagonists (SCH-23390, (+)-butaclamol, clozapine, (S)-WAY-100635, and [d(CH2)5,1 Tyr(Me)2]-AVP) were obtained from Sigma-Aldrich (St. Louis, MO). All of the drugs, chemicals, and reagents were of the highest grade available.
Isolation of Compounds
Details on plant material, extraction, fractionation, isolation, and identification have been described in our recent work.21 The purity of these compounds was considered to be >98%, as evidenced by spectroscopic data (NMR and MS).
In Silico Prediction of Targets
To predict potential protein targets for the four anthraquinones, a proteocheminformatics modeling (PCM) in silico target prediction method was employed. The model was trained on the chemical and biological similarities of 55 079 compounds active and inactive against 99 human proteins (11 537 active pairs vs 43 542 inactive pairs). Machine learning algorithm, in this case, Parzen Rosenblatt Windows (PRW)63,64 was utilized to evaluate the patterns that differentiate between active and inactive complexes. Based on the patterns established, the activity of novel compounds against the 99 protein targets can be predicted. Chemical structures were represented as ECFP_4 fingerprint,65 and chemical similarities were calculated using Aitchison–Aitken kernel.66 Protein vectors were represented by their full sequence where a protein sequence is denoted as a string of characters, and each character represents an amino acid that is part of the protein. Prior to calculating the similarities between two protein sequences, the sequences are subjected to alignment using MUSCLE,67 performed using the bio3d package.68 The model was internally and externally validated by sensitivity values of 0.6837 and 0.4492, respectively. For full information on the model, readers are directed to a previous report.23
GPCR Functional Assay for Human Dopamine Receptor
The effect of test compounds at human dopamine (D1, D3, and D4) receptors expressed in CHO cells was evaluated by measuring their effect on cAMP modulation via homogeneous time-resolved fluorescence (HTRF), as described previously.69 Agonist activity was determined by measuring the effect on cAMP modulation, and antagonist activity by measuring the effect on agonist-induced cAMP modulation using the HTRF detection method. The cellular agonist effect was calculated as the percentage of the control response to dopamine for each receptor (D1, D3, and D4) targets, and the cellular antagonist effect was calculated as the percentage inhibition of the dopamine response for each target. To validate the result, reference antagonist SCH-23390, (+)-butaclamol, and clozapine were used for D1, D3, and D4 receptors, respectively.
GPCR Functional Assay for Human Serotonin 5-HT1A and Vasopressin V1A Receptor
The agonist activity of test compounds at the human 5-HT1A receptor expressed in Ba/F3 cells and V1A receptor expressed in transfected CHO cells was determined by measuring their effect on cytosolic Ca2+-ion mobilization using a fluorimetric detection method described in our previous reports.70,71 For antagonist activity, the effect on agonist-induced cytosolic Ca2+-ion mobilization was measured.
Cellular agonist effect at 5-HT1A receptor was calculated as the percentage of the control response to serotonin (2.5 μM), and the antagonist effect was calculated as the percentage inhibition of the control response to 30 nM serotonin. To validate the result, reference antagonist (S)-WAY-100635 was employed. Similarly, for the cellular agonist at V1A receptor, the percentage of the control response to 1 μM AVP was determined, and for antagonist effect, percentage inhibition of control response to 10 nM AVP was recorded. The standard reference antagonist [d(CH2)51 Tyr(Me)2]-AVP was used to validate the result.
Homology Modeling and Molecular Docking
The primary sequence of the human 5HT1AR and human V1AR was obtained from UniProt (ID: P08908 and P37288, respectively). 5-HT1B receptor (PDB 5V54) and μ-opioid receptor (4DKL) structures were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) and used as a template for homology modeling of 5HT1A and V1A receptors, respectively. Modeling was conducted through SWISS-MODEL and refined through ModRefiner server (RMSD = 1.264 Å for 5HT1AR and 0.645 Å for V1AR).72 Likewise, X-ray crystallography of a human dopamine D3 receptor (hD3R)–eticlopride complex (PDB ID: 3PBL) at a resolution 2.89 Å was obtained from the PDB.73 The three-dimensional (3D) structures of emodin, alaternin, aloe-emodin, and questin were obtained from the PubChem Compound database (NCBI), with compound CIDs of 3220, 12548, 10207, and 160717, respectively. The docking of the target proteins and active compounds was successfully simulated using the AutoDock 4.2 program.74 Automated docking simulations were performed using AutoDockTools (ADT) to assess appropriate binding orientations. For the docking calculations, Gasteiger charges were added by default, rotatable bonds were set by ADT, and all torsions were allowed to rotate. Grid maps were generated by AutoGrid. The docking protocol for rigid and flexible ligand docking consisted of 20 independent genetic algorithms; the other parameters used were the ADT defaults. The results were visualized and analyzed using Discovery Studio (v17.2, Accelrys, San Diego, CA) and PyMOL (v1.7.4, Schrödinger, LLC, Cambridge, MA).
Drug-likeness and ADME Prediction
Drug-likeness predictions were carried out with PreADMET (v2.0, Yonsei University, Seoul, Korea). This web-based server can be used to predict absorption, distribution, metabolism, and excretion (ADME) data and build a drug-likeness library in silico.
Statistics
All results are expressed as mean ± standard deviation (SD) of triplicate samples. Statistical significance was analyzed using one-way analysis of variance (ANOVA) (Systat Inc., Evaston, IL) and was noted at p < 0.05.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2019R1I1A3A01049380).
The authors declare no competing financial interest.
References
- Su H.; Zhuju W.; Liying T. Simultaneous determination of 4 major components in semen cassiae obtusifoline by HPLC. China J. Chin. Mater. Med. 2011, 36, 1327–1329. 10.4268/cjcmm20111017. [DOI] [PubMed] [Google Scholar]
- Yi J. H.; Park H. J.; Lee S.; Jung J. W.; Kim B. C.; Lee Y. C.; Ryu J. H.; Kim D. H. Cassia obtusifolia seed ameliorates amyloid β-induced synaptic dysfunction through anti-inflammatory and Akt/GSK-3β pathways. J. Ethnopharmacol. 2016, 178, 50–57. 10.1016/j.jep.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Jung H. A.; Ali M. Y.; Jung H. J.; Jeong H. O.; Chung H. Y.; Choi J. S. Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J. Ethnopharmacol. 2016, 191, 152–160. 10.1016/j.jep.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Yoon B. H.; Kim Y.-W.; Lee S.; Shin B. Y.; Jung J. W.; Kim H. J.; Lee Y. S.; Choi J. S.; Kim S. Y.; et al. The seed extract of Cassia obtusifolia ameliorates learning and memory impairments induced by scopolamine or transient cerebral hypoperfusion in mice. J. Pharmacol. Sci. 2007, 105, 82–93. 10.1254/jphs.FP0061565. [DOI] [PubMed] [Google Scholar]
- Shrestha S.; Seong S. H.; Paudel P.; Jung H. A.; Choi J. S. Structure related inhibition of enzyme systems in cholinesterases and BACE1 in vitro by naturally occurring naphthopyrone and its glycosides isolated from Cassia obtusifolia. Molecules 2018, 23, 69. 10.3390/molecules23010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S. K.; Narasingappa R. B.; Joshi C. G.; Girish T. K.; Vincent B. Neuroprotective effects of Cassia tora against paraquat-induced neurodegeneration: relevance for Parkinson’s disease. Nat. Prod. Res. 2018, 32, 1476–1480. 10.1080/14786419.2017.1353504. [DOI] [PubMed] [Google Scholar]
- Jung H. A.; Ali M. Y.; Choi J. S. Promising inhibitory effects of anthraquinones, naphthopyrone, and naphthalene glycosides from Cassia obtusifolia on α-glucosidase and human protein tyrosine phosphatases 1B. Molecules 2017, 22, 28. 10.3390/molecules22010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S.; Paudel P.; Seong S. H.; Min B. S.; Seo E. K.; Jung H. A.; Choi J. S. Two new naphthalenic lactone glycosides from Cassia obtusifolia L. seeds. Arch. Pharm. Res. 2018, 41, 737–742. 10.1007/s12272-018-1044-0. [DOI] [PubMed] [Google Scholar]
- Ali M. Y.; Jannat S.; Jung H. A.; Min B. S.; Paudel P.; Choi J. S. Hepatoprotective effect of Cassia obtusifolia seed extract and constituents against oxidative damage induced by tert-butyl hydroperoxide in human hepatic HepG2 cells. J. Food Biochem. 2018, 42, e12439. 10.1111/jfbc.12439. [DOI] [Google Scholar]
- Paudel P.; Jung H. A.; Choi J. S. Anthraquinone and naphthopyrone glycosides from Cassia obtusifolia seeds mediate hepatoprotection via Nrf2-mediated HO-1 activation and MAPK modulation. Arch. Pharm. Res. 2018, 41, 677–689. 10.1007/s12272-018-1040-4. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Kim S.; Jung W. Y.; Park S. J.; Park D. H.; Kim J. M.; Cheong J. H.; Ryu J. H. The neuroprotective effects of the seeds of Cassia obtusifolia on transient cerebral global ischemia in mice. Food Chem. Toxicol. 2009, 47, 1473–1479. 10.1016/j.fct.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Drever B. D.; Anderson W. G.; Riedel G.; Kim D. H.; Ryu J. H.; Choi D.-Y.; Platt B. The seed extract of Cassia obtusifolia offers neuroprotection to mouse hippocampal cultures. J. Pharmacol. Sci. 2008, 107, 380–392. 10.1254/jphs.08034FP. [DOI] [PubMed] [Google Scholar]
- Hatano T.; Uebayashi H.; Ito H.; Shiota S.; Tsuchiya T.; Yoshida T. Phenolic constituents of Cassia seeds and antibacterial effect of some naphthalenes and anthraquinones on methicillin-resistant Staphylococcus aureus. Chem. Pharm. Bull. 1999, 47, 1121–1127. 10.1248/cpb.47.1121. [DOI] [PubMed] [Google Scholar]
- Choi J. S.; Lee H. J.; Kang S. S. Alatemin, cassiaside and rubrofusarin gentiobioside, radical scavenging principles from the seeds of Cassia tora on 1, 1-diphenyl-2-picrylhydrazyl (DPPH) radical. Arch. Pharm. Res. 1994, 17, 462–466. 10.1007/BF02979126. [DOI] [PubMed] [Google Scholar]
- Yen G.-C.; Chen H.-W.; Duh P.-D. Extraction and identification of an antioxidative component from Jue Ming Zi (Cassia tora L.). J. Agric. Food Chem. 1998, 46, 820–824. 10.1021/jf970690z. [DOI] [Google Scholar]
- Zhang P.; Wang J.; Zhao Y.; Zhang X.; Qu L.; Wang C.; Feng J.; Wang A.; Zhou W.; Liu Y.; et al. Discovery of novel antagonists on β2-adrenoceptor from natural products using a label-free cell phenotypic assay. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 1411–1420. 10.1007/s00210-018-1555-8. [DOI] [PubMed] [Google Scholar]
- Li M.; Fu Q.; Li Y.; Li S.; Xue J.; Ma S. Emodin opposes chronic unpredictable mild stress induced depressive-like behavior in mice by upregulating the levels of hippocampal glucocorticoid receptor and brain-derived neurotrophic factor. Fitoterapia 2014, 98, 1–10. 10.1016/j.fitote.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Mizuno M.; Kawamura H.; Takei N.; Nawa H. The anthraquinone derivative emodin ameliorates neurobehavioral deficits of a rodent model for schizophrenia. J. Neural Transm. 2008, 115, 521–530. 10.1007/s00702-007-0867-5. [DOI] [PubMed] [Google Scholar]
- Tao L.; Xie J.; Wang Y.; Wang S.; Wu S.; Wang Q.; Ding H. Protective effects of aloe-emodin on scopolamine-induced memory impairment in mice and H2O2-induced cytotoxicity in PC12 cells. Bioorg. Med. Chem. Lett. 2014, 24, 5385–5389. 10.1016/j.bmcl.2014.10.049. [DOI] [PubMed] [Google Scholar]
- Arcella A.; Oliva M. A.; Staffieri S.; Sanchez M.; Madonna M.; Riozzi B.; Esposito V.; Giangaspero F.; Frati L. Effects of aloe emodin on U87MG glioblastoma cell growth: In vitro and in vivo study. Environ. Toxicol. 2018, 33, 1160–1167. 10.1002/tox.22622. [DOI] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Shrestha S.; Jung H. A.; Choi J. S. In vitro and in silico human monoamine oxidase inhibitory potential of anthraquinones, naphthopyrones, and naphthalenic lactones from Cassia obtusifolia Linn seeds. ACS Omega 2019, 4, 16139–16152. 10.1021/acsomega.9b02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Jung H. A.; Choi J. S. Rubrofusarin as a dual protein tyrosine phosphate 1B and human monoamine oxidase-A inhibitor: An in vitro and in silico study. ACS Omega 2019, 4, 11621–11630. 10.1021/acsomega.9b01433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauzi F. M.; John C. M.; Karunanidhi A.; Mussa H. Y.; Ramasamy R.; Adam A.; Bender A. Understanding the mode-of-action of Cassia auriculata via in silico and in vivo studies towards validating it as a long term therapy for type II diabetes. J. Ethnopharmacol. 2017, 197, 61–72. 10.1016/j.jep.2016.07.058. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Chavali S.; Masuho I.; Jahn L. J.; Martemyanov K. A.; Gloriam D. E.; Babu M. M. Pharmacogenomics of GPCR drug targets. Cell 2018, 172, 41–54.e19. 10.1016/j.cell.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bézard E.; Ferry S.; Mach U.; Stark H.; Leriche L.; Boraud T.; Gross C.; Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 2003, 9, 762–767. 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Guigoni C.; Aubert I.; Li Q.; Gurevich V.; Benovic J.; Ferry S.; Mach U.; Stark H.; Leriche L.; Håkansson K.; et al. Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat. Disord. 2005, 11, S25–S29. 10.1016/j.parkreldis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Aviles-Olmos I.; Kefalopoulou Z.; Foltynie T. Understanding and prevention of “Therapy-” induced dyskinesias. Parkinson’s Dis. 2012, 2012, 640815. 10.1155/2012/640815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi S.; Lévesque D.; Blanchet P. J. Upregulation of dopamine D3, not D2, receptors correlates with tardive dyskinesia in a primate model. Mov. Disord. 2014, 29, 1125–1133. 10.1002/mds.25909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchinetti F.; Del Giudice E.; Furegato S.; Passarotto M.; Arcidiacono D.; Leon A. Dopamine inhibits responses of astroglia-enriched cultures to lipopolysaccharide via a β-adrenoreceptor-mediated mechanism. J. Neuroimmunol. 2004, 150, 29–36. 10.1016/j.jneuroim.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Elenkov I. J.; Haskó G.; Kovács K. J.; Vizi E. S. Modulation of lipopolysaccharide-induced tumor necrosis factor-α production by selective α-and β-adrenergic drugs in mice. J. Neuroimmunol. 1995, 61, 123–131. 10.1016/0165-5728(95)00080-L. [DOI] [PubMed] [Google Scholar]
- Basu S.; Dasgupta P. S. Dopamine, a neurotransmitter, influences the immune system. J. Neuroimmunol. 2000, 102, 113–124. 10.1016/S0165-5728(99)00176-9. [DOI] [PubMed] [Google Scholar]
- Xue L.; Geng Y.; Li M.; Jin Y.-F.; Ren H.-X.; Li X.; Wu F.; Wang B.; Cheng W.-Y.; Chen T.; Chen Y.-J. The effects of D3R on TLR4 signaling involved in the regulation of METH-mediated mast cells activation. Int. Immunopharmacol. 2016, 36, 187–198. 10.1016/j.intimp.2016.04.030. [DOI] [PubMed] [Google Scholar]
- Sookhai S.; Wang J. H.; Winter D.; Power C.; Kirwan W.; Redmond H. P. Dopamine attenuates the chemoattractant effect of interleukin-8: a novel role in the systemic inflammatory response syndrome. Shock 2000, 14, 295–299. 10.1097/00024382-200014030-00009. [DOI] [PubMed] [Google Scholar]
- Han X.; Li B.; Ye X.; Mulatibieke T.; Wu J.; Dai J.; Wu D.; Ni J.; Zhang R.; Xue J.; et al. Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-κB signalling pathway. Br. J. Pharmacol. 2017, 174, 4751–4770. 10.1111/bph.14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Tang L.; Yang Y.; Lin L.; Dai J.; Ge P.; Ai Q.; Jiang R.; Zhang L. Dopamine alleviated acute liver injury induced by lipopolysaccharide/d-galactosamine in mice. Int. Immunopharmacol. 2018, 61, 249–255. 10.1016/j.intimp.2018.06.004. [DOI] [PubMed] [Google Scholar]
- Meng G.; Liu Y.; Lou C.; Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-κB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br. J. Pharmacol. 2010, 161, 1628–1644. 10.1111/j.1476-5381.2010.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin B. Y.; Kim D. H.; Hyun S. K.; Jung H. A.; Kim J. M.; Park S. J.; Kim S. Y.; Cheong J. H.; Choi J. S.; Ryu J. H. Alaternin attenuates delayed neuronal cell death induced by transient cerebral hypoperfusion in mice. Food Chem. Toxicol. 2010, 48, 1528–1536. 10.1016/j.fct.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Yun-Choi H. S.; Kim J. H.; Takido M. Potential inhibitors of platelet aggregation from plant sources, V. Anthraquinones from seeds of Cassia obtusifolia and related compounds. J. Nat. Prod. 1990, 53, 630–633. 10.1021/np50069a014. [DOI] [PubMed] [Google Scholar]
- Huei-Chen H.; Chai-Rong L.; Pei-Dawn Lee C.; Ching-Chow C.; Shu-Hsun C. Vasorelaxant effect of emodin, an anthraquinone from a Chinese herb. Eur. J. Pharmacol. 1991, 205, 289–294. 10.1016/0014-2999(91)90912-A. [DOI] [PubMed] [Google Scholar]
- Narayen G.; Mandal S. N. Vasopressin receptor antagonists and their role in clinical medicine. Indian J. Endocrinol. Metab. 2012, 16, 183–191. 10.4103/2230-8210.93734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon N. G.; Guillon C.; Fabio K.; Heindel N. D.; Lu S.-f.; Miller M.; Ferris C. F.; Brownstein M. J.; Garripa C.; Koppel G. A. Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Pat. CNS Drug Discovery 2008, 3, 77–93. 10.2174/157488908784534586. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xu Q.-Q.; Shan C.-S.; Shi Y.-H.; Wang Y.; Zheng G.-Q. Combined use of emodin and ginsenoside Rb1 exerts synergistic neuroprotection in cerebral ischemia/reperfusion rats. Front. Pharmacol. 2018, 9, 943. 10.3389/fphar.2018.00943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmarou C. R.; Liang X.; Abidi N. H.; Parveen S.; Taya K.; Henderson S. C.; Young H. F.; Filippidis A. S.; Baumgarten C. M. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89–102. 10.1016/j.brainres.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linas S. L.; Anderson R. J.; Guggenheim S. J.; Robertson G. L.; Berl T.; Dickmann D. C. Role of vasopressin in impaired water excretion in conscious rats with experimental cirrhosis. Kidney Int. 1981, 20, 173–180. 10.1038/ki.1981.119. [DOI] [PubMed] [Google Scholar]
- Liu X.; Luo G.; Jiang J.; Ma T.; Lin X.; Jiang L.; Cheng J.; Tao R. Signaling through hepatocyte vasopressin receptor 1 protects mouse liver from ischemia-reperfusion injury. Oncotarget 2016, 7, 69276. 10.18632/oncotarget.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotte N.; Balestre M. N.; Aumelas A.; Mahé E.; Phalipou S.; Morin D.; Hibert M.; Manning M.; Durroux T.; Barberis C.; Mouillac B. Conserved aromatic residues in the transmembrane region VI of the V1a vasopressin receptor differentiate agonist vs. antagonist ligand binding. Eur. J. Biochem. 2000, 267, 4253–4263. 10.1046/j.1432-1033.2000.01472.x. [DOI] [PubMed] [Google Scholar]
- Lu M.-C.; Hsieh M.-T.; Wu C.-R.; Cheng H.-Y.; Hsieh C.-C.; Lin Y.-T.; Peng W.-H. Ameliorating effect of emodin, a constitute of Polygonatum multiflorum, on cycloheximide-induced impairment of memory consolidation in rats. J. Ethnopharmacol. 2007, 112, 552–556. 10.1016/j.jep.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kim M.; Lim S. J.; Lee H.-J.; Nho C. W. Cassia tora seed extract and its active compound aurantio-obtusin inhibit allergic responses in IgE-mediated mast cells and anaphylactic models. J. Agric. Food Chem. 2015, 63, 9037–9046. 10.1021/acs.jafc.5b03836. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Abbott F. V. Behavioural effects of intraplantar injection of inflammatory mediators in the rat. Neuroscience 1994, 63, 827–836. 10.1016/0306-4522(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Parada C. A.; Tambeli C. H.; Cunha F. Q.; Ferreira S. H. The major role of peripheral release of histamine and 5-hydroxytryptamine in formalin-induced nociception. Neuroscience 2001, 102, 937–944. 10.1016/S0306-4522(00)00523-6. [DOI] [PubMed] [Google Scholar]
- Baek Y. H.; Choi D. Y.; Yang H. I.; Park D. S. Analgesic effect of electroacupuncture on inflammatory pain in the rat model of collagen-induced arthritis: Mediation by cholinergic and serotonergic receptors. Brain Res. 2005, 1057, 181–185. 10.1016/j.brainres.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Q.; Gao X.; Huang Y.-L.; Wu G.-C. Expression of 5-HT1A receptor mRNA in rat dorsal raphe nucleus and ventrolateral periaqueductal gray neurons after peripheral inflammation. NeuroReport 2000, 11, 3361–3365. 10.1097/00001756-200010200-00020. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Q.; Gao X.; Yang Z.-L.; Huang Y.-L.; Wu G.-C. Expression of 5-HT1A receptor mRNA in rat nucleus raphe magnus neurons after peripheral inflammation. Brain Res. 2000, 887, 465–468. 10.1016/S0006-8993(00)03037-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Q.; Gao X.; Ji G.-C.; Huang Y.-L.; Wu G.-C.; Zhao Z.-Q. Expression of 5-HT1A receptor mRNA in rat lumbar spinal dorsal horn neurons after peripheral inflammation. Pain 2002, 98, 287–295. 10.1016/S0304-3959(02)00026-X. [DOI] [PubMed] [Google Scholar]
- Oprea T. I. Property distribution of drug-related chemical databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. 10.1023/A:1008130001697. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Arnott J. A.; Kumar R.; Planey S. L. Lipophilicity indices for drug development. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 31–36. [Google Scholar]
- Gabathuler R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases.. Neurobiol. Dis. 2010, 37, 48–57. 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Gharagheizi F.; Abbasi R.; Tirandazi B. Prediction of Henry’s law constant of organic compounds in water from a new group-contribution-based model. Ind. Eng. Chem. Res. 2010, 49, 10149–10152. 10.1021/ie101532e. [DOI] [Google Scholar]
- Shia C.-S.; Hou Y.-C.; Tsai S.-Y.; Huieh P.-H.; Leu Y.-L.; Chao P.-D. L. Differences in pharmacokinetics and ex vivo antioxidant activity following intravenous and oral administrations of emodin to rats. J. Pharm. Sci. 2010, 99, 2185–2195. 10.1002/jps.21978. [DOI] [PubMed] [Google Scholar]
- Baell J. B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Lowe R.; Glen R. C.; Mitchell J. B. O. Predicting phospholipidosis using machine learning. Mol. Pharm. 2010, 7, 1708–1714. 10.1021/mp100103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsoukas A.; Lowe R.; KalantarMotamedi Y.; Mussa H. Y.; Klaffke W.; Mitchell J. B. O.; Glen R. C.; Bender A. In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass Naïve Bayes and Parzen-Rosenblatt window. J. Chem. Inf. Model. 2013, 53, 1957–1966. 10.1021/ci300435j. [DOI] [PubMed] [Google Scholar]
- Nigsch F.; Bender A.; Jenkins J. L.; Mitchell J. B. O. Ligand-target prediction using Winnow and naive Bayesian algorithms and the implications of overall performance statistics. J. Chem. Inf. Model. 2008, 48, 2313–2325. 10.1021/ci800079x. [DOI] [PubMed] [Google Scholar]
- Aitchison J.; Aitken C. G. Multivariate binary discrimination by the kernel method. Biometrika 1976, 63, 413–420. 10.1093/biomet/63.3.413. [DOI] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P.; ElSawy K. M.; McCammon J. A.; Caves L. S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Jung H. A.; Choi J. S. Characterizing fucoxanthin as a selective dopamine D3/D4 receptor agonist: Relevance to Parkinson’s disease. Chem.-Biol. Interact. 2019, 310, 108757. 10.1016/j.cbi.2019.108757. [DOI] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Wu S.; Park S.; Jung H. A.; Choi J. S. Eckol as a potential therapeutic against neurodegenerative diseases targeting dopamine D3/D4 receptors. Mar. Drugs 2019, 17, 108. 10.3390/md17020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong S. H.; Paudel P.; Choi J.-W.; Ahn D. H.; Nam T.-J.; Jung H. A.; Choi J. S. Probing multi-target action of phlorotannins as new monoamine oxidase inhibitors and dopaminergic receptor modulators with the potential for treatment of neuronal disorders. Mar. Drugs 2019, 17, 377. 10.3390/md17060377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y. T.; Liu W. T.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. . [DOI] [PubMed] [Google Scholar]