

Abstract
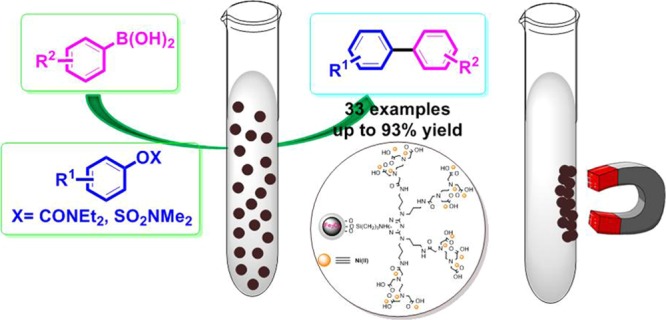
A highly efficient and air-, thermal-, and moisture-stable nickel-based catalyst with excellent magnetic properties supported on silica-coated magnetic Fe3O4 nanoparticles was successfully synthesized. It was well characterized by Fourier transform infrared spectroscopy, powder X-ray diffraction, transmission electron microscopy, field emission scanning electron microscopy, thermogravimetric analysis, dynamic light scattering (DLS), X-ray photoelectron spectroscopy, vibration sample magnetometry, energy-dispersive X-ray analysis, inductively coupled plasma analysis, and nitrogen adsorption–desorption isotherm analysis. The Suzuki–Miyaura coupling reaction between aryl carbamates and/or sulfamates with arylboronic acids was selected to demonstrate the catalytic activity and efficiency of the as-prepared magnetic nanocatalyst. Using the mentioned heterogeneous nanocatalyst in such reactions generated corresponding products in good to excellent yields in which the catalyst could easily be recovered from the reaction mixture with an external magnetic field to reuse directly for the next several cycles without significant loss of its activity.
Introduction
“Nanotechnology is the sixth truly revolutionary technology introduced in the modern world”, D. Allan Bromley (1926–2005).1 Nanotechnology has potentially changed our life and insights about it. Having many susceptibilities compared with other approaches, using nanotechnology is one of the main characteristics of modern scientific notions for the synthesis of novel structures.1,2 Nanoparticles (NPs) are interesting primary particles for scientists because of increased activity, modified structure, and surface area to volume ratio.3 Moreover, NPs have many advanced applications in electronic and optical industries, textile industries, drug delivery, medicine, and cosmetics.2−4
Magnetic NPs (MNPs) are the important groups of functional NPs that have been broadly pursued owing to their interesting magnetic properties and attractive potential.5,6 The study of such nontoxic and biocompatible particles opens new ways to figure out their great potential in technological or industrial applications including medical therapeutics and diagnostics, magnetic data storage, environmental remediation, hyperthermia-based therapy of cancer, magnetic resonance imaging (MRI), and catalysis.7 Although these particles especially Fe3O4 NPs have many advantages; unfortunately, they tends to aggregate quickly because of anisotropic dipolar attractions and their applications face a big challenge.7 Therefore, using stabilizers as the shell is vital to control their size increases.8,9
Among various shells, silica is one of the most trustable coating layers for Fe3O4 NPs adding to its chemical and thermal stability, high persistence in wide ranges of pH values, biocompatibility, and modified surface.8−11 Moreover, it enables organic compounds to be covalently attached to a library of intended NPs for interesting applications as a heterogeneous catalyst.12 As a result, magnetic core–shell structures offer a wide range of applications in optics, catalysis, biomedicine, materials, environmental science, and energy as the leading edge of modern research and hot topics.13,14 Besides, having excellent properties such as versatility, biocompatibility, controllability, stability, and being inexpensive, they have attracted outstanding interest.13−15 Furthermore, they can be easily recovered and reused from the reaction mixture several times by external magnetic fields resulting in a low cost and green situation for the reaction progress.8−16
The carbon–carbon bond-formations are the foremost challenges in the synthesis of a wide ranges of natural and artificial products for pharmaceutical and agrochemical applications.17,18 In recent decades, the transition-metal cross-coupling reactions have played a major role in the development of designing efficient reactions.19,20 In this regard, a number of highly reactive metal catalysts have been developed and introduced for cross-coupling reactions.21 Moreover, the correlation of these developments with the principles of green chemistry concepts is very necessary and important.22,23 Concerns over contamination of residual toxic transition metals such as palladium in products and the high price of this catalyst led scientists to proliferate the use of economical and safer metal catalysts for such cross-coupling reactions.24,25 Since the late twentieth century when the nickel-catalyzed cross-coupling reaction of aryl halides and arylboronic acids was reported by Miyaura and co-workers for the first time, this reaction has attracted a lot of attention in academic and industrial research and has become one of the most powerful synthetic tools for the C–C bond formations.26 Therefore, many efforts have recently been made to improve the efficiency and performance of this protocol.27−42 One of the most important achievements of these investigations is using phenol derivatives (aryl mesylates, tosylates, triflates, phosphates, sulfamates, carbonates, or carbamates) as the proper and effective alternatives to aryl halides in the C–C bond formation reactions.29−35 This achievement is so important because: (a) the aryl halides are generally not environmentally friendly and using such precursors produces halides as byproducts leading to environmental pollution, (b) the preparation of aryl halides often involves tedious steps, wasteful production processes, and harsh reaction conditions, and (c) the phenol derivatives are safer substrates, more accessible, and usually can be prepared on an industrial scale.29−40 However, this reaction still suffers from several limitations and drawbacks such as the necessity to employ additional quantities of ligands such as phosphine and carbenes, the high sensitivity of these ligands toward air and moisture, having expensive and multistep procedures for the synthesis of ligands, and high catalyst loading.41,42
Conventionally, because of the high sensitivity of the Ni(0) complexes against air and moisture that is the main catalytic limitation in such reactions,43,44 these catalysts generally are being generated from Ni(II) complexes in the presence of reducing agents via in situ reactions.45,46 It should be noted that the use of reducing toxic agents is another disadvantage of these methods.47 Fortunately, some effective methods without the necessity to apply any toxic reducing agent have been reported in recent years.48−52
Therefore, based on the aforementioned considerations and also the continuation of our efforts for preparing effective magnetic catalytic systems,53−56 herein, we have reported the synthesis, characterizations, and employment of nickel(II) NPs immobilized on EDTA-modified Fe3O4@SiO2 nanospheres (Fe3O4@SiO2–EDTA–Ni(II)) as a reusable and efficient catalyst for the Suzuki–Miyaura coupling of various aryl carbamates and/or sulfamates with arylboronic acids under mild conditions (Scheme 1).
Scheme 1. Fe3O4@SiO2–EDTA–Ni(II) as an Efficient and Recyclable Catalyst for the Suzuki Miyaura Cross-Coupling Reaction.

Results and Discussion
The nickel(II) particles were immobilized on EDTA-modified Fe3O4@SiO2 nanospheres (Fe3O4@SiO2–EDTA–Ni(II)) via the multistep procedure according to the procedure described in Scheme 2.
Scheme 2. Preparation of the Fe3O4@SiO2-EDTA-Ni(II) Nanocatalyst.

Subsequently, the prepared catalyst was well-characterized by the following instrumental techniques: Fourier transform infrared (FT-IR), powder X-ray diffraction (XRD), transmission electron microscopy (TEM), field emission scanning electron microscopy (FE-SEM), dynamic light scattering (DLS), energy dispersive X-ray (EDX), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), vibrating sample magnetometer (VSM), Brunauer–Emmett–Teller (BET), inductively coupled plasma (ICP), and elemental analysis
The successful functionalization of the MNP surface was confirmed by examination of the FT-IR spectra. Figure 1 shows the FT-IR spectra of the Fe3O4@SiO2, Fe3O4@SiO2–NH2, Fe3O4@SiO2–TCT, Fe3O4@SiO2–TCT–NH2, Fe3O4@SiO2–EDTA, and Fe3O4@SiO2–EDTA–Ni(II) MNPs. These materials showed broad bands around 3400 and 580 cm–1, which are the characteristic of O–H and Fe–O stretching bands, respectively.56,57 In the case of Fe3O4@SiO2 NPs, the sharp band at 1090 cm–1 is corresponded to Si–O–Si asymmetric stretching vibrations indicating the existence of silica in Fe3O4@SiO2 NPs (Figure 1a).58 The characteristic absorption bands at 2810–2986, 1489, 1123, and 576 cm–1 attributed to C-H (stretching vibration), CH2 (bending), Si–O–Si (stretching vibration), and Fe–O (stretching vibration) prove the existence of 3-aminopropyl(triethoxy)silane functional groups on the surface of the Fe3O4@SiO2 NPs. Furthermore, the weak peaks at about 3300–3400 cm–1 can be ascribed to NH2 stretching vibrations (Figure 1b). In the spectrum of Fe3O4@SiO2–TCT NPs, the characteristic adsorption at 1711, 1564, and 1511 cm–1 are attributed to C=N stretching vibrations (Figure 1c). The peak at 1091 cm–1 was related to the C–Cl groups of cyanuric chloride, which is overlapped by the stretching vibrations of Si–O=Si groups in silica shells (Figure 1c). The FT-IR spectra of Fe3O4@SiO2–TCT–NH2 NPs were characterized by the following absorption bands: stretching vibrations of C–N arising at 1257 cm–1, CH2 (bending) at 1451 cm–1, and the C–H (symmetric and asymmetric stretching vibrations) at 2871–3057 cm–1 (Figure 1d). As can be seen, the typical absorption peak at 3397 cm–1 indicates the overlapped stretching vibrations of N–H and O–H bonds (Figure 1d). According to Figure 1e, the successful Fe3O4@SiO2–TCT–NH2 surface modification with EDTA moieties was also verified. In the FT-IR spectra of Fe3O4@SiO2–EDTA (Figure 1e), basic characteristic vibrations were observed for C–H bands (asymmetric and symmetric stretching) at 2885–3070 cm–1, Si–O–Si asymmetric stretching and symmetric stretching at 1095 and 801 cm–1, respectively, and Fe–O (stretching vibration) at 578 cm–1. Furthermore, the characteristic bands of carbonyl groups were observed at 1736 cm–1 (C=O carboxylic acid stretching vibration) and 1629 cm–1 (C=O amide stretching vibration). Eventually, in terms of Fe3O4@SiO2–EDTA=Ni(II) (Figure 1f), a redshift of the band at 1736 cm–1 is observed (1736 cm–1 → 1724 cm–1), which is probably the characteristic of the carbonyl group after interaction with the nickel ions. Thus, the above results indicate that the functional groups were successfully grafted onto the surface of the magnetic Fe3O4@SiO2 NPs.
Figure 1.
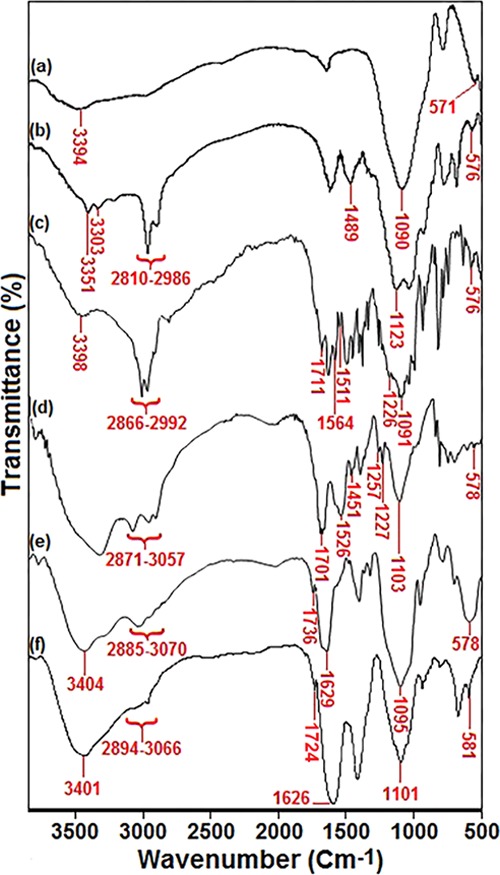
FT-IR spectra of (a) Fe3O4@SiO2, (b) Fe3O4@SiO2–NH2, (c) Fe3O4@SiO2–TCT, (d) Fe3O4@SiO2–TCT–NH2, (e) Fe3O4@SiO2–EDTA, and (f) Fe3O4@SiO2–EDTA–Ni(II) NPs.
The crystalline structure of MNPs was identified with the XRD technique. As illustrated in Figure 2a, the XRD pattern exhibited reflection peaks at around 2θ = 30.1, 35.4, 43.1, 53.4, 57.0, and 62.6° that can be indexed to (220), (311), (400), (422), (511), and (440) crystallographic planes of cubic Fe3O4 (JCPDS 88-0866).30 However, the peak positions of the composite sample remain unchanged, revealing that the crystal phase of magnetic cores is well-maintained after the coating process. However, the crystallinity of the samples decreases after the coating process and the catalyst went toward an amorphous structure (Figure 2b,c). These results provide further evidence for the successful functionalization of the nanomagnetic surfaces. Furthermore, the XRD pattern of Fe3O4@SiO2 shows an obvious diffusion peak between 10 and 20° that appears because of the presence of amorphous silica (Figure 2b). For Fe3O4@SiO2–EDTA–Ni(II) MNPs, the broad peak was transferred to lower angles due to the synergetic effect of amorphous silica and the dendrimer polymer (Figure 2c).
Figure 2.
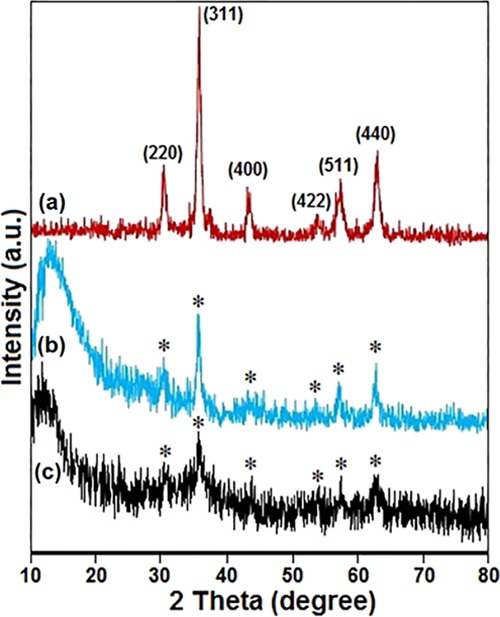
XRD diffraction pattern of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2–EDTA–Ni(II) NPs.
The morphologies, particle size, and structural features of the magnetic nanomaterials and the synthesized catalyst can be observed directly from the TEM and FE-SEM images (Figure 3a–c). The obtained magnetite particles possess a uniformly spherical shape and a mean diameter of ∼20 nm (Figure 3a,d). Coating silica over the Fe3O4 NPs was achieved via the well-known Stöber method. The TEM image of Fe3O4@SiO2 clearly shows the well-defined core–shell structure with a shell thickness of ∼10 nm (Figure 3b). Furthermore, the TEM image presented in Figure 3c indicates the structure of Fe3O4@SiO2–EDTA–Ni(II) NPs. After being coated with the organic layer, the particle size of Fe3O4@SiO2–EDTA–Ni(II) NPs was found to be 20 nm in diameter.
Figure 3.

TEM images of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2–EDTA–Ni(II); FE-SEM images of (d) Fe3O4, (e) Fe3O4@SiO2, and (f) Fe3O4@SiO2–EDTA–Ni(II); and size distributions of (g) Fe3O4, (h) Fe3O4@SiO2, and (i) Fe3O4@SiO2–EDTA–Ni(II) NPs.
The FE-SEM photographs demonstrate that the Fe3O4@SiO2 and Fe3O4@SiO2–EDTA–Ni(II) MNPs are in an almost regular spherical shape (Figure 3e,f). Moreover, these results are consistent with the particle-size distribution histogram of MNPs, which were in a narrow distribution in the range of 8–16, 16–24, and 23–37 nm and average size distribution of 12, 20, and 31 nm for Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–EDTA–Ni(II) NPs, respectively (Figure 3g–i).
The existence of nickel in the Fe3O4@SiO2–EDTA–Ni(II) catalyst was also confirmed by the EDX detector coupled to the SEM in which the presence of Fe, Si, C, N, and O can be confirmed clearly (Figure 4). The higher intensity of the Si peak compared with the Fe peaks indicates that the magnetite NPs were trapped by silica. According to the above analysis, it can be inferred that the Fe3O4@SiO2–EDTA–Ni(II) has been successfully synthesized.
Figure 4.
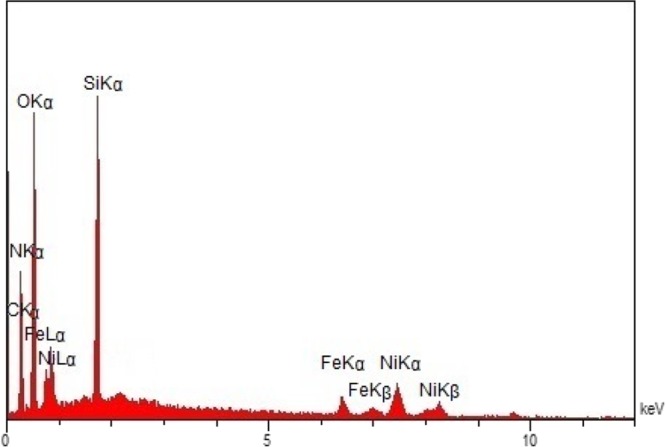
EDX spectrum of the Fe3O4@SiO2–EDTA–Ni(II) NPs.
The TGA of the magnetic nanocatalyst was performed over the temperature range of 25–700 °C. As shown in Figure 5A, the first weight loss for all samples, which occurred below 150 °C, was attributed to the loss of adsorbed water molecules on the surface of nanostructured materials, and the second step occurred between about 150 °C and nearly 600 °C that is attributed to the decomposition of coating organic layers in the nanocomposite. As shown in Figure 5A(d), a significant weight loss of nearly 42.7% in the range of 150–600 °C was observed for the sample catalyst due to the elimination of organic material over Fe3O4@SiO2 NPs.
Figure 5.

(A) TGA spectrum of (a) Fe3O4@SiO2–NH2, (b) Fe3O4@SiO2–TCT, (c) Fe3O4@SiO2–TCT–NH2, and (d) Fe3O4@SiO2–EDTA–Ni(II) NPs; (B) magnetic hysteresis loops of (a) Fe3O4 and (b) Fe3O4@SiO2–EDTA–Ni(II) NPs; (C) good dispersity and easy separation of the catalyst by an external magnetic field.
To study the magnetic properties of MNPs, the hysteresis loops of magnetite and functionalized magnetite NPs at room temperature were investigated by using a VSM. As shown in Figure 5B, no hysteresis was observed in the hysteresis loops of three materials, and the remanence and coercivity were nearly zero, exhibiting typical superparamagnetic behavior. The magnetizations of Fe3O4 and Fe3O4@SiO2–EDTA–Ni(II) MNPs were 64.8 and 28.7 emu/g, respectively (Figure 5B). The silica shell and other organic compounds cause a reduction in the magnetic strength of the composite owing to the weight contribution from the nonmagnetic portion. Nevertheless, Fe3O4@SiO2–EDTA–Ni(II) possesses excellent magnetic responsibility and suitable magnetization values, which can quickly respond to the external magnetic field and quickly disperse again when the external magnetic field is removed (Figure 5C). These results exhibit the good magnetic properties of the nanocomposite, which is an advantage in our catalytic system for separation.
N2 adsorption–desorption isotherms were obtained to investigate the porous structure and surface area of the NPs. The measured specific surface areas were 480, 430.3, and 392.6 m2/g for Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–EDTA–Ni(II), respectively (Table 1). Also, the particle sizes of magnetite calculated using the Scherrer equation were 11.33, 12.64, and 14.97 nm for Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–EDTA–Ni(II), respectively (Table 1).
Table 1. Selected Properties of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–EDTA–Ni(II) NPs.
| sample | Fe3O4 crystal structure | specific surface area (m2/g)a | magnetite particle size (nm)b |
|---|---|---|---|
| Fe3O4 | cubic spinel | 480 | 11.33 |
| Fe3O4@SiO2 | cubic spinel | 430.3 | 12.64 |
| Fe3O4@SiO2–EDTA–Ni(II) | cubic spinel | 371.6 | 14.97 |
Calculated by the BJH method.
Calculated by the Scherrer equation based on XRD patterns.
Elemental analyses for Fe3O4@SiO2–NH2, Fe3O4@SiO2–TCT, Fe3O4@SiO2–TCT–NH2, and Fe3O4@SiO2–EDTA–Ni(II) were carried out, and the data are tabulated in Table 2 which are in good agreement with the result obtained from TGA. These obtained results displayed that the contents of C, H, and N for Fe3O4@SiO2–EDTA–Ni(II) are 21.61, 2.87, and 9.232%, respectively.
Table 2. TGA and Elemental Analysis for Fe3O4@SiO2–NH2, Fe3O4@SiO2–TCT, Fe3O4@SiO2–TCT–NH2, and Fe3O4@SiO2–EDTA–Ni(II) NPs.
| sample | C (%) | H (%) | N (%) | total (%)a | |
|---|---|---|---|---|---|
| Fe3O4@SiO2–NH2 | TGA (wt %) | 6.746 | 1.501 | 2.627 | 10.874 |
| EA (wt %) | 6.614 | 1.475 | 2.532 | 10.621 | |
| Fe3O4@SiO2–TCT | TGA (wt %) | 7.377 | 0.717 | 5.736 | 3.830 |
| EA (wt %) | 7.468 | 0.750 | 5.627 | 13.845 | |
| Fe3O4@SiO2–TCT–NH2 | TGA (wt %) | 16.172 | 2.918 | 10.564 | 29.654 |
| EA (wt %) | 15.946 | 2.871 | 10.377 | 29.194 | |
| Fe3O4@SiO2–EDTA–Ni(II) | TGA (wt %) | 22.821 | 2.853 | 9.112 | 34.786 |
| EA (wt %) | 21.611 | 2.871 | 9.232 | 33.714 |
Total (%) = C (%) + H (%) + N (%).
Additionally, Ni loading of the catalyst was confirmed by the ICP analyzer. For this purpose, the catalyst (1 g) was stirred in aq. HCl (37%), then the magnetic nanocomposite was separated by an external magnetic field and the remaining solution was analyzed by ICP to determine the content of nickel. The amount of Ni on the support was determined as (0.55) mmol per gram of the catalyst.
After the preparation and detailed characterization of Fe3O4@SiO2–EDTA–Ni(II), the efficiency of the synthesized nanocatalyst on cross-coupling reactions to form C–C bonds was evaluated. To find the effective reaction conditions, the model reaction between phenyl carbamate (A1) with phenylboronic acid (B1) using the proposed catalytic system was investigated (Table 3). Various factors such as solvents, bases, catalyst loadings, temperature, and reaction time were tested, and the results have been depicted in Table 3. The solvent evaluation for the model reaction showed that using ethylene glycol (EG) as the solvent presents the highest yield (Table 3, entry 4). Moreover, the desired product, C1 was not observed when no base was added (Table 3, entry 15). This experiment showed that the presence of a base is crucial to have an efficient reaction. Therefore, we decided to investigate the series of organic and inorganic bases that are commonly used in the coupling reactions (Table 3, entries 4, 7–14). Based on these experiments, the best efficiency and performance were observed in the presence of EG monosodium salt (NaOC2H4OH) as the base (Table 3, entry 4). Also, it seems that the reaction efficiency depends on the amount of NaOC2H4OH and the highest yields were observed when its amount was 2 mmol (Table 3, entry 4). It should be noted that higher amount of the base did not present better yields of the desired product (Table 3, entries 18, 19). To improve the yield of the cross-coupling reaction, we also checked the catalyst loading (Table S1), temperature (Table S3), and reaction time (Table S3) on the model reaction and the best results have been obtained using 0.02 g of Fe3O4@SiO2–EDTA–Ni(II) catalyst (1 mol % Ni(II)) at 100 °C after 6 h (Table 3, entry 4).
Table 3. Optimization Studies for Cross-Coupling of Phenyl Carbamate with Phenylboronic Acida.

| entry | base (equiv) | solvent | temp (°C) | yield (%)b |
|---|---|---|---|---|
| 1 | NaOC2H4OH(2.0) | H2O | reflux | 43 |
| 2 | NaOC2H4OH(2.0) | ethanol | reflux | 56 |
| 3 | NaOC2H4OH(2.0) | n-PrOH | reflux | 52 |
| 4 | NaOC2H4OH(2.0) | EG | 100 | 91 |
| 5 | NaOC2H4OH(2.0) | glycerol | 100 | 73 |
| 6 | NaOC2H4OH(2.0) | dioxane | reflux | 37 |
| 7 | K3PO4 (2.0) | EG | 100 | 83 |
| 8 | NaOH (2.0) | EG | 100 | 72 |
| 9 | K2CO3 (2.0) | EG | 100 | 80 |
| 10 | Cs2CO3 (2.0) | EG | 100 | 79 |
| 11 | DBU (2.0) | EG | 100 | 69 |
| 12 | DABCO (2.0) | EG | 100 | 66 |
| 13 | NaOtBu (2.0) | EG | 100 | 89 |
| 14 | NaOC2H5 (2.0) | EG | 100 | 84 |
| 15 | EG | 100 | 0 | |
| 16 | NaOC2H4OH(1.0) | EG | 100 | 70 |
| 17 | NaOC2H4OH(1.5) | EG | 100 | 79 |
| 18 | NaOC2H4OH(2.5) | EG | 100 | 90 |
| 19 | NaOC2H4OH(3.0) | EG | 100 | 91 |
Reaction conditions: phenyl carbamate (1 mmol), phenylboronic acid (1 mmol), base, Fe3O4@SiO2–EDTA–Ni(II) catalyst(0.018 g, 1 mol %), solvent, 6 h
Isolated yield.
After getting the optimized conditions, we tested different phenol-based electrophiles in Ni-catalyzed Suzuki–Miyaura cross-coupling reactions to determine the high proficiency and generality of this catalytic system (Table 4). Although the usage of aryl methyl ether did not lead to the production of the desired product under the optimized reaction conditions (Table 4, entry 1), moderate yields were observed when the phenol derivatives such as phenyl acetate, phenyl pivalate, phenyl mesylate, phenyl tosylate, and phenyl triflate were used as the electrophile (Table 4, entries 2–6). Accordingly, phenyl carbonate furnished the desired product in good yields (Table 4, entry 7). Finally, it was determined that the corresponding carbamate and sulfamate substrates introduced the coupling product in the highest yield (Table 4, entries 8 and 9). Thus, we elected the aryl carbamates and sulfamates as the suitable electrophiles for continuing the research.
Table 4. Survey of Pseudo Halide Substratesa.

| entry | X | yield (%)b |
|---|---|---|
| 1 | Me | 0 |
| 2 | Ac | 43 |
| 3 | Piv | 47 |
| 4 | Ms | 54 |
| 5 | Ts | 62 |
| 6 | Tf | 57 |
| 7 | CO2tBu | 75 |
| 8 | SO2NEt2 | 89 |
| 9 | CONEt2 | 91 |
Reaction conditions: phenol-based electrophiles(1 mmol), phenylboronic acid (1 mmol), NaOC2H4OH (2.0 mmol), Fe3O4@SiO2–EDTA–Ni(II) catalyst (0.018 g, 1 mol %), EG (3 mL), 100 °C, 6 h.
Isolated yield.
The optimal reaction conditions were identified and then, we decided to investigate the scope, generality, and efficiency of this method for C–C bond formation. At first, we started from the treatment of a series of aryl carbamates and sulfamates in the reaction with phenylboronic acid (Table 5). Satisfyingly, both electron-donating and withdrawing groups on the phenyl ring of aryl carbamates and sulfamates efficiently produced the desired products in good to excellent yields (Table 5). It should be noted that a lower yield was observed in the case of ortho-substituted aryl carbamates and sulfamates in comparison to meta- and para-substituted ones, which might be due to steric factors (Table 5, entries C4 and C9). It is also noteworthy that the naphthyl carbamates and sulfamates display high reactivity leading to the corresponding cross-coupled biaryl products in high yields (Table 5, entries C5 and C6). Although pyridinyl and pyrimidinyl derivatives generally deactivate the transition-metal catalysts, especially in the nickel-based catalysts, our system was also suitable for these kinds of substrates and the desired coupling products were produced in significant yields (Table 5, entries C16–C18).
Table 5. Substrate Scope of Aryl Carbamates and Sulfamatesa.


Reaction conditions: aryl carbamates or sulfamates(1 mmol), phenylboronic acid (1 mmol), NaOC2H4OH (2.0 mmol), Fe3O4@SiO2–EDTA–Ni(II) catalyst (0.018 g, 1 mol %), EG (3 mL), 100 °C, 6 h.
Isolated yield.
Inspired from interesting results achieved, we also decided to examine the effects of different substituents on the phenyl ring of aryl boronic acids under the optimal reaction conditions (Table 6). Arylboronic acids bearing various functional groups were compatible with this reaction system and the corresponding products were obtained in good to excellent yields (Table 6, entries C19–C33). However, the precursors with an electron-withdrawing group are somehow less effective than other arylboronic acids under these reaction conditions (Table 6, entries C26–C29). It is noteworthy that in the case of 2-methylphenylboronic acid, lower yield was obtained and it may be due to steric hindrance (Table 6, entry C22). Interestingly, we found that the heterocyclic boronic acids such as thiophen-2-ylboronic acid also worked efficiently under this catalytic system (Table 6, entries C30–C33).
Table 6. Substrate Scope of Arylboronic Acidsa.


Reaction conditions: aryl carbamates or sulfamates(1 mmol), aryl boronic acids (1 mmol), NaOC2H4OH (2.0 mmol), Fe3O4@SiO2–EDTA–Ni(II) catalyst (0.018 g, 1 mol %), EG (3 mL), 100 °C, 6 h.
Isolated yield.
According to the reported research in the literature, we proposed a plausible catalytic mechanism for the Suzuki–Miyaura coupling of aryl carbamates or aryl sulfamates with arylboronic acids (Scheme 3).59−62 Initially, the process begins with the reduction of Ni(II) with EG as the reducing agent to provide the active Ni(0) species. Subsequently, the catalytic cycle starts with the oxidative addition of Ni(0) to aryl carbamates or aryl sulfamates to generate the intermediate, (1) in situ. The transmetalation step occurs by the conversion of intermediate (1) to a nucleophilic intermediate, (2) in the presence of the base EG monosodium salt (NaOC2H4OH). This complex subsequently reacts with an organoboron compound which facilitates the aryl group transfer to reach the diaryl intermediate, (3). Finally, the formation of a C–C bond through the reductive elimination step generates the Ni (0) catalyst.
Scheme 3. Proposed Mechanism of Suzuki–Miyaura Cross-Coupling of Aryl Carbamates and Aryl Sulfamates.

To determine the oxidation state of nickel, high-resolution XPS spectra of Ni 2p core levels were obtained from the catalyst before and after the reaction (Figure 6). According to the fitted data, the deconvoluted peaks at the binding energies 855.9 and 861.8 eV are attributed to Ni 2p3/2 and peaks at 872.2 and 879.8 eV are attributed to Ni 2p1/2 for the fresh catalyst that can be indexed to Ni2+ (Figure 6a).63 Besides, the XPS patterns of the recovered catalyst show the peaks of both Ni2+ and Ni(0) (Figure 6b). The peaks at around 852.4 and 872.2 eV were assigned to the Ni 2p3/2 and Ni 2p1/2 levels in the Ni(0) which are in good agreement with the literature report.64 The above phenomena supported that the reaction proceeded via the traditional Ni2+/Ni(0) cycle mechanism.
Figure 6.
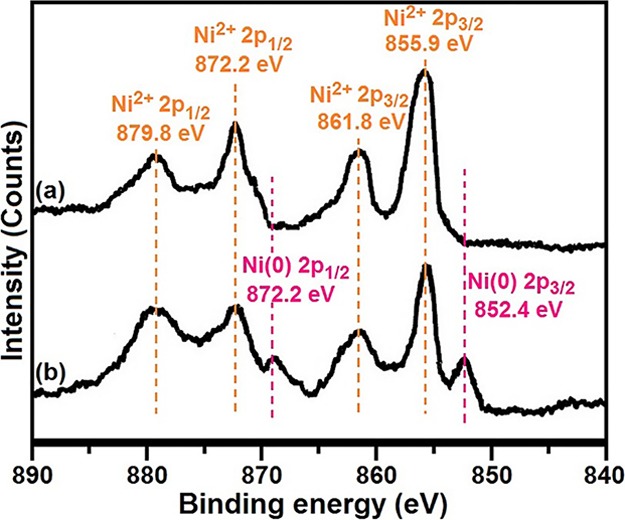
XPS spectra of the (a) fresh and (b) reused catalyst.
The recyclability and high stability of the economically and eco-friendly catalytic systems are very important in the industry and designing green and effective synthetic pathways. Therefore, we investigated these important factors in our catalyst. First, the reusability of the catalyst in the model reaction was studied and tested (Figure 7a). After completion of the model reaction, the catalyst was easily separated by using an external magnetic field, washed twice with ethanol, and dried in an oven. Then, the recovered catalyst was used for the next runs with the same substrates. As shown in Figure 7a, the recovered Fe3O4@SiO2–EDTA–Ni(II) exhibited almost constant catalytic activity for at least six runs in the model reaction and significant reduction in catalytic efficiency was not observed. Additionally, the TEM micrographs for the MNPs after the sixth cycle are displayed in Figure 7b. As revealed in this figure, almost all Fe3O4@SiO2–EDTA–Ni(II) particles have the morphology and size the same as the fresh catalyst, indicating that the aggregation of NPs is venial. Eventually, the hydrodynamic diameter of the catalyst was investigated by the DLS technique (Figure 7c) in which this size distribution is centered at a value of 33 nm.
Figure 7.
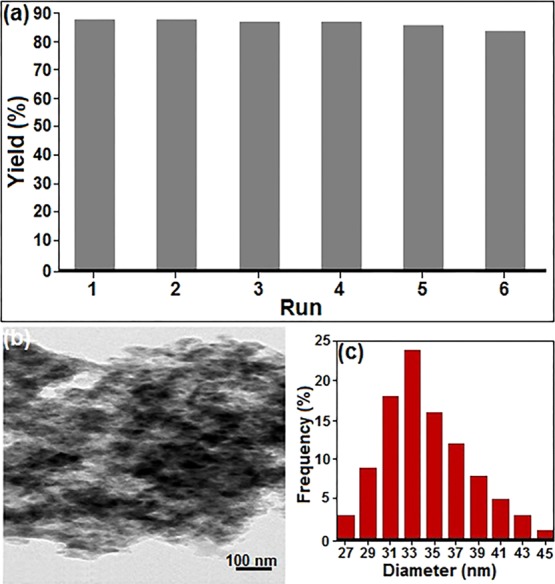
(a) Reusability of the catalyst for the Suzuki–Miyaura cross-coupling; (b) TEM and (c) DLS images of Fe3O4@SiO2–EDTA–Ni(II) after the sixth recycling experiment.
Additionally, the catalyst after the last run was investigated by ICP analysis to determine the amount of nickel leaching. Accordingly, the amount of loaded nickel on the recovered catalyst was measured to be 0.54 mmol/g. Propitiously, the ICP analysis after the seventh run showed less than 1% nickel leaching. Moreover, to determine the stability of the catalytic systems in the model reaction and the responsibility of nickel moiety for carrying out the model reaction, a hot filtration test was performed. When the reaction time of the model reaction reached the half time of reaction quenching, the catalyst NPs were taken out from the reaction mixture by an external magnetic field, the residue was allowed to stir under the reaction conditions. The monitoring of the reaction mixture by TLC did not show any considerable progress. These results showed that only a few species of nickel may exist in the solution phase and the main responsible species that catalyzes the model reaction is the Fe3O4@SiO2–EDTA–Ni(II) NPs. All of these data confirmed the high stability and reusability of the catalyst under these reaction conditions.
According to the wonderful results obtained, the application of the presented efficient strategy was also checked out for the preparation of biphenyl (C1) in different scales (1, 50, and 200 mmol). The obtained results are presented in Table 7 in which the capability of this method was proved to find efficient large-scale laboratory syntheses with the yield of trials at small scales.
Table 7. Comparison of Cross-Coupling of Phenyl Carbamate or Sulfamate with Phenylboronic Acid at Different Scales.


Isolated yield.
In the end, we decided to compare the capability of the proposed nanocatalyst in the synthesis of aryl carbamates and sulfamates with those reported in the literature (Table 8). As can be seen, almost all reports have applied toxic solvents with more reaction times and temperatures. In addition, they showed less reactivity against such reactions and also, fewer yields have been observed (Table 8, entries 1–4, 7–9). Although there are lower reaction times reported (Table 8, entries 5, 6), the observed yields are somehow less than the present work. Moreover, the reaction conditions for these are harsher. From the point of view of the recovery and reusability of the catalyst, this catalyst can be recovered and reused for consequent reactions but other reports applied the catalyst with no recovery ability.
Table 8. Comparison with Reported Results for Suzuki–Miyaura Cross-Coupling Reactions between Phenylboronic Acid and 4-Methoxyphenyl Carbamates or Sulfamates.

| entry | X | catalyst | reaction conditions | time | yield (%) | reusability | refs |
|---|---|---|---|---|---|---|---|
| 1 | CONEt2 | triazine-based Ni(II) PNP pincer complexes (2 mol %) | toluene/K3PO4/120–135 °C | 16 h | 84 | (65) | |
| 2 | CONEt2 | NiCl2(PCy3)2 (10 mol %) | toluene/K3PO4/110 °C | 24 h | 41 | (66) | |
| 3 | SO2NMe2 | NiCl2(PCy3)2 (10 mol %) | toluene/K3PO4/110 °C | 24 h | 80 | (66) | |
| 4 | SO2NMe2 | dppf Ni(II) (o-tol) (Cl) (2.5 mol %) | toluene/K3PO4/80 °C | 24 h | 72 | (52) | |
| 5 | CONEt2 | Ni(PCy3)2Cl2 (5 mol %) | toluene/K3PO4/MW, 180 °C | 10 min | 67 | (67) | |
| 6 | SO2NMe2 | Ni(PCy3)2Cl2 (5 mol %) | toluene/K3PO4/MW, 180 °C | 10 min | 70 | (67) | |
| 7 | SO2NMe2 | NiCl2(dppp) (2 mol %) | dioxane/K3PO4/110 °C | 24 h | 35 | (68) | |
| 8 | SO2NMe2 | NiCl2(dppf) (5 mol %) | toluene/K3PO4/110 °C | 24 h | 68 | (69) | |
| 9 | CONMe2 | NiCl2(dppf) (5 mol %) | toluene/K3PO4/110 °C | 24 h | 19 | (69) | |
| 10 | CONEt2 | Fe3O4@SiO2–EDTA–Ni(II)(1 mol %) | EG/NaOC2H4OH/100 °C | 6 | 86 | Yes | this work |
| 11 | SO2NMe2 | Fe3O4@SiO2–EDTA–Ni(II)(1 mol %) | EG/NaOC2H4OH/100 °C | 6 | 87 | Yes | this work |
Conclusions
In summary, we have proposed a facile and effective method to prepare a magnetic nanocatalyst (Fe3O4@SiO2–EDTA–Ni(II)) with characterization by various techniques. The catalyst demonstrated great performance in the Suzuki–Miyaura coupling reaction of aryl carbamates and/or sulfamates with arylboronic acids under mild reaction conditions. The methodology complements the more established methods without using external ligands or reducing agents. Also, a wide range of substrate scope was applied, even the presence of electron-rich substrate. Additionally, the catalyst can be easily recovered in a very short time using an external magnet and directly reused for six cycles with no significant loss in catalyst activity. Considering the improved economic and environmental benefits, this methodology was presented here to show its attractive features in using sulfamate and carbamate substrates as highly regarded precursors, especially in the pharmaceutical industry.
Experimental Section
Chemicals and Instrumentation
Chemical materials with high purity were purchased from Fluka and Merck. The reaction monitoring was accomplished by thin-layer chromatography (TLC) on gel F254 plates. Melting points were obtained with a micro melting point apparatus (electrothermal, BUCHI 510). NMR spectra were recorded in CDCl3 on a Bruker Avance DPX 250 spectrometer using TMS as the internal standard. FT-IR spectra were collected on a Shimadzu FT-IR 8300 spectrometer using KBr pellets. XRD patterns were recorded in a Bruker AXS D8-advance powder X-ray diffractometer using Cu Kα radiation. The TEM images were recorded using a Philips EM208 transmission electron microscope operated at 80 kV accelerating voltage. FE-SEM characterization was performed on a Hitachi S-4160 operated at a 20 kV accelerating voltage. DLSs were performed using a HORIBA-LB550. The surface composition was investigated using XPS on XR3E2 (VG Microtech) spectrometer using a Mg and Al twin anode X-ray gun with a multichannel detector and a hemispherical analyzer with a resolution of 1.0 eV. The BET surface area of the material was measured by the nitrogen adsorption isotherm method (BET; Micromeritics ASAP 2000). The magnetic properties of Fe3O4 MNPs and Fe3O4@SiO2–EDTA–Ni magnetic nanocomposites were analyzed using the MDK instrument model 7400 VSM. The TGA was performed on a NETZSCH STA 409 PC/PG instrument. The nickel amount on the carriers was measured by ICP–atomic emission spectrometry (ICP–AES). The elemental analyses (C, H, N) were obtained using a Flash EA-1112 CHNSO analyzer. Therefore, all products were identified by comparison of their spectral data and physical properties such as 1H and 13C NMR, CHNS, and melting points with those of the authentic sample and all yields refer to isolated products.
Synthesis of Silica-Coated MNPs (Fe3O4@SiO2NPs)
Magnetic (Fe3O4) NPs were synthesized according to our previous reports.54−56 In brief, FeCl3·6H2O (1.3 g, 4.8 mmol), FeCl2·4H2O (0.9 g, 4.5 mmol), and poly(vinyl alcohol) (PVA 15,000, 1 g) as the surfactant were dissolved in 30 mL of deionized water with vigorous mechanical stirring at 80 °C for 30 min. Then, hexamethylenetetramine (1.0 mol) was slowly added to the solution and the solution pH was maintained at 10. The mixture was held at 80 °C in a water bath for 2 h with constant stirring and then, the black magnetite was collected by applying an external magnetic field and was rinsed with ethanol several times and dried under vacuum at 80 °C for 10 h. In the next step, for the synthesis of Fe3O4@SiO2, 0.5 g of the synthesized Fe3O4 NPs were dispersed in a mixture containing ethanol (50 mL), deionized water (5 mL), and tetraethoxysilane (0.20 mL) in a glass reactor by using ultrasound irradiation.55−58 Then, NaOH (10 wt %, 5 mL) was added to the mixture and stirred at room temperature for 30 min. The resulting Fe3O4@SiO2 NPs were collected by an external magnet, washed with distilled water and ethanol, and dried in a vacuum oven at 80 °C for 10 h.
Synthesis of Fe3O4@SiO2–NH2NPs58
To a suspension of SiO2-coated magnetite particles (1 g) in ethanol (10 mL), 3-aminopropyl(triethoxy)silane (1 mmol, 0.25 mL) was added. The mixture was refluxed for 12 h, and then, it was cooled to room temperature to obtain a brown precipitate. The obtained solid was separated with an external magnet, washed with water and ethanol (1:1) to remove unreacted species, and dried under vacuum at 80 °C.
Synthesis of Fe3O4@SiO2–TCT NPs
In a typical procedure, 1 g of the as-prepared Fe3O4@SiO2–NH2 NPs was first dispersed in 10 mL of THF containing 1 mmol (0.185 g) cyanuric chloride (TCT) and 1 mmol (0.17 mL) of diisopropylethylamine (DIPEA). Then, the obtained crude was stirred at room temperature for 16 h. Next, the modified NPs by cyanuric chloride were collected by magnetic field and washed 3 times with distilled water and ethanol solution. Finally, the resultant was dried at 60 °C for 4 h.
Synthesis of Fe3O4@SiO2–TCT–NH2 NPs
In a typical preparation procedure, bis(3-aminopropyl)amine (2 mmol, 0.25 mL) and DIPEA (2 mmol, 0.35 mL) were added to DMF (5 mL) containing cyanuric chloride-immobilized Fe3O4@SiO2–NH2NPs (1.0 g). After stirring at 80 °C for 12 h, the precipitate (Fe3O4@SiO2–TCT–NH2) was collected from the solution using a magnet, washed with water and ethanol several times, and dried at 70 °C for 4 h.
Synthesis of Fe3O4@SiO2–EDTA–Ni(II) NPs
First, EDTA (2 mmol) was dispersed in DMSO (15 mL). Then, the solution of thionyl chloride (SOCl2) (2 mmol) in DMSO (5 mL) was added dropwise into the solution under vigorous stirring. Afterward, Fe3O4@SiO2–TCT–NH2 (1.5 g) was quickly added and the mixture was stirred or 2 h at room temperature. The mixture was then separated by an external magnet, washed with deionized water, sodium carbonate solution (0.1 mol L–1), and acetone and dried at 60 °C to give Fe3O4@SiO2–EDTA NPs. To synthesis the Fe3O4@SiO2–EDTA–Ni NPs, 1.5 g of the as-prepared Fe3O4@SiO2–EDTA nanocomposite and 1 g of nickel(II) acetate tetrahydrate (4 mmol) were dispersed in 10 mL of THF by ultrasonication and stirred for 4 h under reflux conditions. Finally, the products (Fe3O4@SiO2–EDTA–Ni NPs) were collected by a magnet, washed with water and ethanol several times, and dried at 60 °C overnight.
EG Monosodium Salt
According to the approach reported previously,70 300 g of EG (4.83 mol) and 176 g of NaOH (4.39 mol) were first mixed in xylenes (3 L). Then, the mixture was stirred under reflux conditions with a Dean–Stark trap until H2O separation was finished. Next, the obtained mixture was cooled to room temperature and the precipitate was filtered, washed with xylenes (3 × 100 mL) and t-BuOMe (3 × 125 mL), dried in vacuo, and finally, stored under argon.
General Procedure for Suzuki–Miyaura Reactions Using Fe3O4@SiO2–EDTA–Ni(II) NPs
Aryl carbamate and/or aryl sulfamate (1 mmol), arylboronic acid (1 mmol), sodium monoethylene glycolate (0.17 g, 2 equiv), Fe3O4@SiO2–EDTA–Ni(II) NPs (0.018 g, 1 mol % Ni(II)), and EG (3 mL) were added to a 10 mL round-bottom flask. The flask was heated in an oil bath at 100 °C for an appropriate time with stirring under nitrogen atmosphere. The progress of the reaction was monitored by TLC analysis. After the completion of the reaction, the crude was cooled to room temperature and then the catalyst was separated with an external magnet, washed with ethanol, dried under vacuum, and used directly for the subsequent reaction runs. The organic layer was washed with water to get product precipitates and subsequently, the precipitates were separated by filtration and washed with distilled water 3 times. For more purification in some cases, the impure product (checked by TLC) was extra purified by column chromatography (petroleum ether/ethyl acetate = 10:1 (v/v)) to afford the pure product.
Acknowledgments
The authors gratefully acknowledge the financial support of this work by the research council of Shiraz University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04450.
Reaction optimization, general experimental methods, chemicals, IR, 1H NMR, 13C NMR spectra, and elemental analysis for all compounds, C1–C33 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kumar S.; Yadav I.; Aswal V. K.; Kohlbrecher J. Structure and Interaction of Nanoparticle-Protein Complexes. Langmuir 2018, 34, 5679–5695. 10.1021/acs.langmuir.8b00110. [DOI] [PubMed] [Google Scholar]
- Macnaghten P.; Kearnes M. B.; Wynne B. Nanotechnology, governance, and public deliberation: what role for the social sciences?. Sci. Commun. 2005, 27, 268–291. 10.1177/1075547005281531. [DOI] [Google Scholar]
- Kango S.; Kalia S.; Celli A.; Njuguna J.; Habibi Y.; Kumar R. Surface modification of inorganic nanoparticles for development of organic-inorganic nanocomposites-A review. Prog. Polym. Sci. 2013, 38, 1232–1261. 10.1016/j.progpolymsci.2013.02.003. [DOI] [Google Scholar]
- Dewan A.; Sarmah M.; Thakur A. J.; Bharali P.; Bora U. Greener Biogenic Approach for the Synthesis of Palladium Nanoparticles Using Papaya Peel: An Eco-Friendly Catalyst for C-C Coupling Reaction. ACS Omega 2018, 3, 5327–5335. 10.1021/acsomega.8b00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi B.; Tavakolian M.; Mansouri F.; Vali H. Nanopalladium on Magnetic Ionic Nanoparticle Network (MINN) as an Efficient and Recyclable Catalyst with High Ionic Density and Dispersibility. ACS Sustainable Chem. Eng. 2018, 7, 3811–3823. 10.1021/acssuschemeng.8b04566. [DOI] [Google Scholar]
- Wu L.; Mendoza-Garcia A.; Li Q.; Sun S. Organic phase syntheses of magnetic nanoparticles and their applications. Chem. Rev. 2016, 116, 10473–10512. 10.1021/acs.chemrev.5b00687. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Arco L.; Rodriguez I. A.; Carriel V.; Bonhome-Espinosa A. B.; Campos F.; Kuzhir P.; Duran J. D. G.; Lopez-Lopez M. T. Biocompatible magnetic core-shell nanocomposites for engineered magnetic tissues. Nanoscale 2016, 8, 8138–8150. 10.1039/c6nr00224b. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zumin Q.; Huang J. Preparation and analysis of Fe3O4 magnetic nanoparticles used as targeted-drug carriers. Chin. J. Chem. Eng. 2008, 16, 451–455. 10.1016/s1004-9541(08)60104-4. [DOI] [Google Scholar]
- Sardarian A. R.; Zangiabadi M.; Inaloo I. D. Fe3O4@SiO2/Schiff base/Pd complex as an efficient heterogeneous and recyclable nanocatalyst for chemoselective N-arylation of O-alkyl primary carbamates. RSC Adv. 2016, 6, 92057–92064. 10.1039/c6ra17268g. [DOI] [Google Scholar]
- Deng Y.; Cai Y.; Sun Z.; Liu J.; Liu C.; Wei J.; Li W.; Liu C.; Wang Y.; Zhao D. Multifunctional mesoporous composite microspheres with well-designed nanostructure: a highly integrated catalyst system. J. Am. Chem. Soc. 2010, 132, 8466–8473. 10.1021/ja1025744. [DOI] [PubMed] [Google Scholar]
- Gawande M. B.; Monga Y.; Zboril R.; Sharma R. K. Silica-decorated magnetic nanocomposites for catalytic applications. Coord. Chem. Rev. 2015, 288, 118–143. 10.1016/j.ccr.2015.01.001. [DOI] [Google Scholar]
- Liu R.; Guo Y.; Odusote G.; Qu F.; Priestley R. D. Core–shell Fe3O4 polydopamine nanoparticles serve multipurpose as drug carrier, catalyst support and carbon adsorbent. ACS Appl. Mater. Interfaces 2013, 5, 9167–9171. 10.1021/am402585y. [DOI] [PubMed] [Google Scholar]
- Li J.-F.; Zhang Y.-J.; Ding S.-Y.; Panneerselvam R.; Tian Z.-Q. Core-Shell Nanoparticle-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117, 5002–5069. 10.1021/acs.chemrev.6b00596. [DOI] [PubMed] [Google Scholar]
- Ghosh Chaudhuri R.; Paria S. Core/shell nanoparticles: classes, properties, synthesis mechanisms, characterization, and applications. Chem. Rev. 2011, 112, 2373–2433. 10.1021/cr100449n. [DOI] [PubMed] [Google Scholar]
- Polshettiwar V.; Luque R.; Fihri A.; Zhu H.; Bouhrara M.; Basset J.-M. Magnetically recoverable nanocatalysts. Chem. Rev. 2011, 111, 3036–3075. 10.1021/cr100230z. [DOI] [PubMed] [Google Scholar]
- Wang D.; Astruc D. Fast-growing field of magnetically recyclable nanocatalysts. Chem. Rev. 2014, 114, 6949–6985. 10.1021/cr500134h. [DOI] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Rygus J. P. G.; Crudden C. M. Enantiospecific and Iterative Suzuki-Miyaura Cross-Couplings. J. Am. Chem. Soc. 2017, 139, 18124–18137. 10.1021/jacs.7b08326. [DOI] [PubMed] [Google Scholar]
- Han F.-S. Transition-metal-catalyzed Suzuki-Miyaura cross-coupling reactions: a remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. 10.1039/c3cs35521g. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Nova A. Designing Pd and Ni catalysts for cross-coupling reactions by minimizing off-cycle species. ACS Catal. 2018, 8, 3499–3515. 10.1021/acscatal.8b00230. [DOI] [Google Scholar]
- Fuentes-Rivera J. J.; Zick M. E.; Düfert M. A.; Milner P. J. Overcoming Halide Inhibition of Suzuki-Miyaura Couplings with Biaryl Monophosphine-Based Catalysts. Org. Process Res. Dev. 2019, 23, 1631–1637. 10.1021/acs.oprd.9b00255. [DOI] [Google Scholar]
- Saptal V. B.; Saptal M. V.; Mane R. S.; Sasaki T.; Bhanage B. M. Amine-Functionalized Graphene Oxide-Stabilized Pd Nanoparticles (Pd@APGO): A Novel and Efficient Catalyst for the Suzuki and Carbonylative Suzuki-Miyaura Coupling Reactions. ACS Omega 2019, 4, 643–649. 10.1021/acsomega.8b03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Guo X.; Jian F.; Wei G. Highly efficient method for Suzuki reactions in aqueous media. ACS Omega 2018, 3, 4418–4422. 10.1021/acsomega.8b00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi M.; Nishiwaki N. Selective Synthesis of (Benzyl)biphenyls by Successive Suzuki-Miyaura Coupling of Phenylboronic Acids with 4-Bromobenzyl Acetate under Air Atmosphere. ACS Omega 2017, 2, 7767–7771. 10.1021/acsomega.7b01450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayan R.; Karak N. Photo-Assisted Synthesis of a Pd-Ag@CQD Nanohybrid and Its Catalytic Efficiency in Promoting the Suzuki-Miyaura Cross-Coupling Reaction under Ligand-Free and Ambient Conditions. ACS Omega 2017, 2, 8868–8876. 10.1021/acsomega.7b01504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S.; Oh-tani S.; Miyaura N. Synthesis of Biaryls via a Nickel(0)-Catalyzed Cross-Coupling Reaction of Chloroarenes with Arylboronic Acids. J. Org. Chem. 1997, 62, 8024–8030. 10.1021/jo9707848. [DOI] [PubMed] [Google Scholar]
- Dehghani M.; Tadjarodi A.; Chamani S. Synthesis and Characterization of Magnetic Zeolite Y-Palladium-Nickel Ferrite by Ultrasonic Irradiation and Investigating Its Catalytic Activity in Suzuki-Miyaura Cross-Coupling Reactions. ACS Omega 2019, 4, 10640–10648. 10.1021/acsomega.9b00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatupheeraphat A.; Liao H.-H.; Srimontree W.; Guo L.; Minenkov Y.; Poater A.; Cavallo L.; Rueping M. Ligand-controlled chemoselective C (acyl)–O bond vs C (aryl)–C bond activation of aromatic esters in nickel catalyzed C (sp2)–C(sp3) cross-couplings. J. Am. Chem. Soc. 2018, 140, 3724–3735. 10.1021/jacs.7b12865. [DOI] [PubMed] [Google Scholar]
- Schwarzer M. C.; Konno R.; Hojo T.; Ohtsuki A.; Nakamura K.; Yasutome A.; Takahashi H.; Shimasaki T.; Tobisu M.; Chatani N.; Mori S. Combined Theoretical and Experimental Studies of Nickel-Catalyzed Cross-Coupling of Methoxyarenes with Arylboronic Esters via C-O Bond Cleavage. J. Am. Chem. Soc. 2017, 139, 10347–10358. 10.1021/jacs.7b04279. [DOI] [PubMed] [Google Scholar]
- Chen H.; Huang Z.; Hu X.; Tang G.; Xu P.; Zhao Y.; Cheng C.-H. Nickel-catalyzed cross-coupling of aryl phosphates with arylboronic acids. J. Org. Chem. 2011, 76, 2338–2344. 10.1021/jo2000034. [DOI] [PubMed] [Google Scholar]
- Harris M. R.; Hanna L. E.; Greene M. A.; Moore C. E.; Jarvo E. R. Retention or inversion in stereospecific nickel-catalyzed cross-coupling of benzylic carbamates with arylboronic esters: control of absolute stereochemistry with an achiral catalyst. J. Am. Chem. Soc. 2013, 135, 3303–3306. 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardarian A. R.; Inaloo I. D. 4-Dodecylbenzenesulfonic acid (DBSA) promoted solvent-free diversity-oriented synthesis of primary carbamates, S-thiocarbamates and ureas. RSC Adv. 2015, 5, 76626–76641. 10.1039/c5ra14528g. [DOI] [Google Scholar]
- Inaloo I. D.; Majnooni S. Eco-Efficient Ultrasonic-Responsive Synthesis of Primary O-Alkyl and O-Aryl Thiocarbamates Using Brønsted Acid Ionic Liquid [H-NMP][HSO4] in Aqueous Media at Room Temperature. ChemistrySelect 2018, 3, 4095–4100. 10.1002/slct.201800107. [DOI] [Google Scholar]
- Modarresi-Alam A. R.; Inaloo I. D.; Kleinpeter E. Synthesis of primary thiocarbamates by silica sulfuric acid as effective reagent under solid-state and solution conditions. J. Mol. Struct. 2012, 1024, 156–162. 10.1016/j.molstruc.2012.05.033. [DOI] [Google Scholar]
- Antoft-Finch A.; Blackburn T.; Snieckus V. N,N-DiethylO-Carbamate: Directed Metalation Group and Orthogonal Suzuki–Miyaura Cross-Coupling Partner. J. Am. Chem. Soc. 2009, 131, 17750–17752. 10.1021/ja907700e. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wu S.-B.; Shi W.-J.; Shi Z.-J. C-O/C-H Coupling of Polyfluoroarenes with Aryl Carbamates by Cooperative Ni/Cu Catalysis. Org. Lett. 2016, 18, 2548–2551. 10.1021/acs.orglett.6b00819. [DOI] [PubMed] [Google Scholar]
- Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardarian A. R.; DindarlooInaloo I.; Zangiabadi M. Selective Synthesis of Secondary Arylcarbamates via Efficient and Cost Effective Copper-Catalyzed Mono Arylation of Primary Carbamates with Aryl Halides and Arylboronic Acids. Catal. Lett. 2018, 148, 642–652. 10.1007/s10562-017-2277-0. [DOI] [Google Scholar]
- Inaloo I. D.; Majnooni S. Ureas as safe carbonyl sources for the synthesis of carbamates with deep eutectic solvents (DESs) as efficient and recyclable solvent/catalyst systems. New J. Chem. 2018, 42, 13249–13255. 10.1039/c8nj02624f. [DOI] [Google Scholar]
- Sardarian A. R.; Inaloo I. D.; Modarresi-Alam A. R. Highly efficient synthesis of alkyl and aryl primary thiocarbamates and dithiocarbamates under metal- and solvent-free conditions. Mol. Diversity 2018, 22, 863–878. 10.1007/s11030-018-9831-6. [DOI] [PubMed] [Google Scholar]
- Ohtsuki A.; Yanagisawa K.; Furukawa T.; Tobisu M.; Chatani N. Nickel/N-Heterocyclic Carbene-Catalyzed Suzuki-Miyaura Type Cross-Coupling of Aryl Carbamates. J. Org. Chem. 2016, 81, 9409–9414. 10.1021/acs.joc.6b01627. [DOI] [PubMed] [Google Scholar]
- Muto K.; Hatakeyama T.; Yamaguchi J.; Itami K. C-H arylation and alkenylation of imidazoles by nickel catalysis: solvent-accelerated imidazole C-H activation. Chem. Sci. 2015, 6, 6792–6798. 10.1039/c5sc02942b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehle A. R.; Zhou Q.; Watson M. P. Nickel(0)-Catalyzed Heck Cross-Coupling via Activation of Aryl C-OPiv Bonds. Org. Lett. 2012, 14, 1202–1205. 10.1021/ol203322v. [DOI] [PubMed] [Google Scholar]
- Sardzinski L. W.; Wertjes W. C.; Schnaith A. M.; Kalyani D. Nickel-Catalyzed Decarboxylative Cross-Coupling of Perfluorobenzoates with Aryl Halides and Sulfonates. Org. Lett. 2015, 17, 1256–1259. 10.1021/acs.orglett.5b00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Yuan X.; Li H.; Ren L.; Sun Z.; Hou Y.; Chu W. Nickel-catalysed Suzuki-Miyaura cross-coupling reactions of aryl halides with arylboronic acids in ionic liquids. Catal. Commun. 2015, 58, 154–157. 10.1016/j.catcom.2014.08.037. [DOI] [Google Scholar]
- Ke H.; Chen X.; Zou G. N-Heterocyclic Carbene-Assisted, Bis(phosphine)nickel-Catalyzed Cross-Couplings of Diarylborinic Acids with Aryl Chlorides, Tosylates, and Sulfamates. J. Org. Chem. 2014, 79, 7132–7140. 10.1021/jo501291y. [DOI] [PubMed] [Google Scholar]
- Tobisu M.; Xu T.; Shimasaki T.; Chatani N. Nickel-Catalyzed Suzuki-Miyaura Reaction of Aryl Fluorides. J. Am. Chem. Soc. 2011, 133, 19505–19511. 10.1021/ja207759e. [DOI] [PubMed] [Google Scholar]
- Magano J.; Monfette S. Development of an air-stable, broadly applicable nickel source for nickel-catalyzed cross-coupling. ACS Catal. 2015, 5, 3120–3123. 10.1021/acscatal.5b00498. [DOI] [Google Scholar]
- a Park N. H.; Teverovskiy G.; Buchwald S. L. Development of an air-stable nickel precatalyst for the amination of aryl chlorides, sulfamates, mesylates, and triflates. Org. Lett. 2014, 16, 220–223. 10.1021/ol403209k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields J. D.; Gray E. E.; Doyle A. G. A Modular, Air-Stable Nickel Precatalyst. Org. Lett. 2015, 17, 2166–2169. 10.1021/acs.orglett.5b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattmann L.; Saeb R.; Nöthling N.; Cornella J. An air-stable binary Ni(0)-olefin catalyst. Nat. Catal. 2020, 3, 6–13. 10.1038/s41929-019-0392-6. [DOI] [Google Scholar]
- Mohadjer Beromi M.; Nova A.; Balcells D.; Brasacchio A. M.; Brudvig G. W.; Guard L. M.; Hazari N.; Vinyard D. J. Mechanistic Study of an Improved Ni Precatalyst for Suzuki-Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc. 2017, 139, 922–936. 10.1021/jacs.6b11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardarian A. R.; Dindarloo Inaloo I.; Modarresi-Alam A. R.; Kleinpeter E.; Schilde U. Metal-Free Regioselective Monocyanation of Hydroxy-, Alkoxy-, and Benzyloxyarenes by Potassium Thiocyanate and Silica Sulfuric Acid as a Cyanating Agent. J. Org. Chem. 2019, 84, 1748–1756. 10.1021/acs.joc.8b02191. [DOI] [PubMed] [Google Scholar]
- Inaloo I. D.; Majnooni S. A Fe3O4@ SiO2/Schiff Base/Pd Complex as an Efficient Heterogeneous and Recyclable Nanocatalyst for One-Pot Domino Synthesis of Carbamates and Unsymmetrical Ureas. Eur. J. Org. Chem. 2019, 2019, 6359–6368. 10.1002/ejoc.201901140. [DOI] [Google Scholar]
- Sardarian A. R.; Dindarloo Inaloo I. D.; Zangiabadi M. An Fe3O4@SiO2/Schiff base/Cu (ii) complex as an efficient recyclable magnetic nanocatalyst for selective mono N-arylation of primary O-alkyl thiocarbamates and primary O-alkyl carbamates with aryl halides and arylboronic acids. New J. Chem. 2019, 43, 8557–8565. 10.1039/c9nj00028c. [DOI] [Google Scholar]
- Inaloo I. D.; Majnooni S.; Esmaeilpour M. Superparamagnetic Fe3O4 Nanoparticles in a Deep Eutectic Solvent: An Efficient and Recyclable Catalytic System for the Synthesis of Primary Carbamates and Monosubstituted Ureas. Eur. J. Org. Chem. 2018, 2018, 3481–3488. 10.1002/ejoc.201800581. [DOI] [Google Scholar]
- Esmaeilpour M.; Javidi J.; Zahmatkesh S. One-pot synthesis of 1-and 5-substituted 1H-tetrazoles using 1, 4-dihydroxyanthraquinone–copper (II) supported on superparamagnetic Fe3O4@SiO2 magnetic porous nanospheres as a recyclable catalyst. Appl. Organomet. Chem. 2016, 30, 897–904. 10.1002/aoc.3518. [DOI] [Google Scholar]
- Esmaeilpour M.; Zahmatkesh S.; Fahimi N.; Nosratabadi M. Palladium nanoparticles immobilized on EDTA-modified Fe3O4@SiO2 nanospheres as an efficient and magnetically separable catalyst for Suzuki and Sonogashira cross-coupling reactions. Appl. Organomet. Chem. 2018, 32, e4302 10.1002/aoc.4302. [DOI] [Google Scholar]
- Quasdorf K. W.; Antoft-Finch A.; Liu P.; Silberstein A. L.; Komaromi A.; Blackburn T.; Ramgren S. D.; Houk K. N.; Snieckus V.; Garg N. K. Suzuki–Miyaura Cross-Coupling of Aryl Carbamates and Sulfamates: Experimental and Computational Studies. J. Am. Chem. Soc. 2011, 133, 6352–6363. 10.1021/ja200398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Liu L. Recent advances in mechanistic studies on Ni catalyzed cross-coupling reactions. Chin. J. Catal. 2015, 36, 3–14. 10.1016/s1872-2067(14)60217-5. [DOI] [Google Scholar]
- Cheung M. S.; Sheong F. K.; Marder T. B.; Lin Z. Computational Insight into Nickel-Catalyzed Carbon-Carbon versus Carbon-Boron Coupling Reactions of Primary, Secondary, and Tertiary Alkyl Bromides. Chem.—Eur. J. 2015, 21, 7480–7488. 10.1002/chem.201500110. [DOI] [PubMed] [Google Scholar]
- Nowrouzi N.; Zarei M. NiCl2Ě6H2O: an efficient catalyst precursor for phosphine-free Heck and Sonogashira cross-coupling reactions. Tetrahedron 2015, 71, 7847–7852. 10.1016/j.tet.2015.08.023. [DOI] [Google Scholar]
- Adhikari S.; Madras G. Role of Ni in hetero-architectured NiO/Ni composites for enhanced catalytic performance. Phys. Chem. Chem. Phys. 2017, 19, 13895–13908. 10.1039/c7cp01332a. [DOI] [PubMed] [Google Scholar]
- Yung T.-Y.; Huang L.-Y.; Chan T.-Y.; Wang K.-S.; Liu T.-Y.; Chen P.-T.; Chao C.-Y.; Liu L.-K. Synthesis and characterizations of Ni-NiO nanoparticles on PDDA-modified graphene for oxygen reduction reaction. Nanoscale Res. Lett. 2014, 9, 444. 10.1186/1556-276x-9-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastalir M.; Stöger B.; Pittenauer E.; Allmaier G.; Kirchner K. Air-Stable Triazine-Based Ni(II) PNP Pincer Complexes As Catalysts for the Suzuki-Miyaura Cross-Coupling. Org. Lett. 2016, 18, 3186–3189. 10.1021/acs.orglett.6b01398. [DOI] [PubMed] [Google Scholar]
- Quasdorf K. W.; Riener M.; Petrova K. V.; Garg N. K. Suzuki–Miyaura Coupling of Aryl Carbamates, Carbonates, and Sulfamates. J. Am. Chem. Soc. 2009, 131, 17748–17749. 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghbanzadeh M.; Pilger C.; Kappe C. O. Rapid Nickel-Catalyzed Suzuki–Miyaura Cross-Couplings of Aryl Carbamates and Sulfamates Utilizing Microwave Heating. J. Org. Chem. 2011, 76, 1507–1510. 10.1021/jo1024464. [DOI] [PubMed] [Google Scholar]
- Chen G.-J.; Han F.-S. An efficient Suzuki–Miyaura coupling of aryl sulfamates and boronic acids catalyzed by NiCl2 (dppp). Eur. J. Org. Chem. 2012, 2012, 3575–3579. 10.1002/ejoc.201200444. [DOI] [Google Scholar]
- Li X.-J.; Zhang J.-L.; Geng Y.; Jin Z. Nickel-Catalyzed Suzuki-Miyaura Coupling of Heteroaryl Ethers with Arylboronic Acids. J. Org. Chem. 2013, 78, 5078–5084. 10.1021/jo4005537. [DOI] [PubMed] [Google Scholar]
- Grygorenko O. O.; Bondarenko A. V.; Tolmachev A. A.; Vashchenko B. V. Synthesis of Functionalized 1, 4-Dioxanes with an Additional (Hetero) Aliphatic Ring. Synthesis 2018, 50, 3696–3707. 10.1055/s-0037-1610195. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.