

Gao et al. reveal that DCs shape a distinct pathogen-specific CD4 T cell transcriptome and discover an unexpected role for T cell–intrinsic caspase-1 in promoting Th17 differentiation.
Abstract
Dendritic cells (DCs) are critical for the differentiation of pathogen-specific CD4 T cells. However, to what extent innate cues from DCs dictate transcriptional changes in T cells remains elusive. Here, we used DCs stimulated with specific pathogens to prime CD4 T cells in vitro and found that these T cells express unique transcriptional profiles dictated by the nature of the priming pathogen. More specifically, the transcriptome of in vitro C. rodentium–primed Th17 cells resembled that of Th17 cells primed following infection in vivo but was remarkably distinct from cytokine-polarized Th17 cells. We identified caspase-1 as a unique gene up-regulated only in pathogen-primed Th17 cells and discovered a critical role for T cell–intrinsic caspase-1, independent of inflammasome, in optimal priming of Th17 responses. T cells lacking caspase-1 failed to induce colitis or confer protection against C. rodentium infection due to suboptimal Th17 cell differentiation in vivo. This study underlines the importance of DC-mediated priming in identifying novel regulators of T cell differentiation.
Graphical Abstract
Introduction
CD4 T helper (Th) cells play a central role in adaptive immunity by producing specific effector cytokines (Zhu et al., 2010). The differentiation of Th in vivo in response to a pathogen (henceforth referred to as pathogen-specific Th cells) results in a heterogeneous effector population (Sallusto, 2016). The frequency of naive precursors for specific epitopes is extremely low, ranging from 0.8 to 10 epitope-specific cells per million naive CD4 T cells (Jenkins and Moon, 2012). Due to the absence of tools available to identify all responding effector T cells, it has been challenging to detect, track, and analyze newly differentiated pathogen-specific Th cells following in vivo infections (O’Shea and Paul, 2010). Functionally, high degrees of heterogeneity and plasticity are the hallmark of pathogen-specific Th cells (Sallusto, 2016). Phenotypically plastic T cell subtypes (e.g. Th1/Th17 dual-lineage cells) emerge following infection and can perform either pathogenic or regulatory roles (McGeachy et al., 2007; Harbour et al., 2015), but identifying and tracing these cells following pathogen-specific activation and understanding their development remain to be explored.
Although dendritic cells (DCs) are major drivers of Th cell activation and differentiation in vivo, lineage-specific polarization by using defined cytokine cocktails has been a major approach to study CD4 T cell biology. However, a broad range of DC-derived cues that might affect differentiating T cells is not present in such an approach (Jain and Pasare, 2017). For example, DCs have been reported to impact the fate of Th cell differentiation by altering TCR signaling strength or by licensing the expression of transcription cofactors and regulatory microRNAs (Zhang et al., 2016; Tubo et al., 2013). Importantly, the influence of DCs on T cell fate goes beyond the initial activation phase. The generation of T cell heterogeneity and the formation of T cell memory rely on DC-derived cues (Sallusto, 2016; Iborra et al., 2016; Shen et al., 2010; MacLeod et al., 2010), but whether DCs alone are sufficient to elicit these changes in T cells remains unclear.
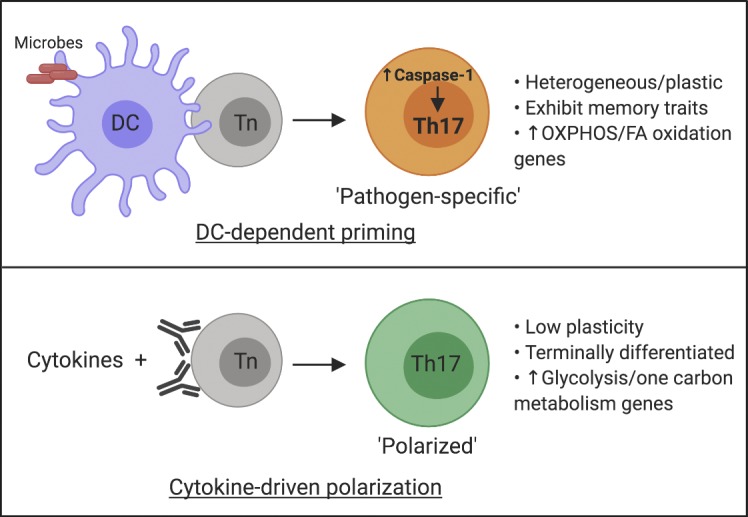
To understand if DCs exposed to different pathogens regulate the transcriptional profile of CD4 T cells during differentiation, we used an in vitro approach to prime naive CD4 T cells. This approach allows for an unbiased assessment of pathogen-specific clonal expansion and differentiation of naive CD4 T cells. We have found that in vitro priming was able to generate pathogen-specific Th cells and that the effector lineage commitment was dictated by the nature of the priming pathogen. Comparison of the transcriptional profile of cytokine-polarized and Citrobacter rodentium (Cr)–primedTh17 cells led to the identification of a unique gene cluster associated with DC-mediated priming. Among this cluster, we identified caspase-1 to be one of the unique genes up-regulated in pathogen-primed Th17 (ppTh17) cells and discovered that it functions in a T cell–intrinsic fashion to promote Th17 differentiation. This study establishes that DCs provide critical cues for transcriptional programming of CD4 T cells, which are absent during cytokine-driven polarization, and furthermore provides a framework for identifying novel regulators of CD4 T cell differentiation.
Results and discussion
In vitro priming system generates pathogen-specific Th cell responses
We aimed to study the transcriptional regulation of pathogen-specific Th cell differentiation. Recent reports have highlighted a system of in vitro priming and de novo differentiation of pathogen-specific human CD4 T cells by autologous monocytes, although the specificity and functionality of the primed T cells were not fully established (Zielinski et al., 2012; Becattini et al., 2015). Building on these previous ideas, we sought to validate a murine in vitro priming system to mimic in vivo priming of naive CD4 T cells following microbial infections. As DCs play a central role in T cell differentiation (Iwasaki and Medzhitov, 2015), we designed the in vitro system using splenic CD11c+ DCs (Flt3L dependent, ex vivo) to prime naive CD4 T cells isolated from the spleen and peripheral lymph nodes of WT mice (Fig. S1 A). Whole-cell lysates of bacteria were used to activate DCs. This bacterial lysate containing diverse pathogen-associated molecular patterns and proteins acted as pattern recognition receptor stimuli and the source of antigen without the addition of any exogenous peptide or recombinant cytokine. Subsequently, DCs were cocultured with naive CD4 T cells to initiate their differentiation (Fig. 1 A). We argue that this simple, reductionist system that contains core elements required for T cell priming should allow the differentiation of pathogen-specific Th cells. Indeed, a proportion of cocultured naive CD4 T cells proliferated (CFSE−CD90+), up-regulated activation markers, and produced effector cytokines, while unexpanded T cells (CFSE+CD90+) remained in a naive state (Fig. 1 B). A large proportion of CFSEhi cells suggested a very low frequency of naive T cells undergoing clonal expansion, in contrast to polyclonal priming by CD3 ligation (Fig. S1 B). Accordingly, in vitro T cell priming required peptide MHC class II (MHCII):TCR interaction, costimulation, and the innate cytokine milieu, indicating a stringent requirement for the three signals for T cell activation (Fig. S1, C and D).
Figure S1.

An in vitro priming approach to generate functional pathogen-specific Th cells. (A) Top: Purity of isolated naive CD4 T cells (CD4+MHCII−CD62L+CD44−) from two independent experiments. Bottom: Purity of CD11c+ DCs before or after isolation. (B) Overlay histograms of CFSE dilution curves from naive CD4 T cells primed with Cr-stimulated DCs (pathogen primed) or LPS stimulated DC + TCR ligation (LPS + anti-CD3). (C) CFSE−CD90+ percentage in Cr-primed CD4 T cells where 10 µg/ml blocking antibodies to mouse IA/IE (MHCII) or CD80/CD86 were added to the coculture. n = 2–3. (D) IFNγ+ percentage of Cr-primed CFSE−CD90+ T cells in the presence or absence of anti–IL-12p40 antibodies during differentiation. n = 3. (E) Proliferation of T cells in the priming system with the indicated pathogens, shown by CFSE dilution. Un, unstimulated. (F) Proliferation of pathogen- or commensal-specific T cells primed by DCs stimulated with heat-killed commensal bacteria Bf, La (MOI = 3), or 10 µg/ml Lm/Cr lysate. n = 2–3. (G and H) IFNγ (G) and IL-17A (H) quantities in the culture supernatant of CD4 T cells previously primed with Lm (left) or Cr (right) that were restimulated for 48 h with unstimulated (Un) or Lm/Cr-fed, irradiated B cells. Lm and Cr concentrations used for restimulation were titrated at 3, 10, and 30 µg/ml. Culture supernatants from anti-CD3 (30 ng/ml)-stimulated T cells were also assessed for IFNγ production as a positive control. n = 2 technical replicates. Data are representative of three independent experiments. (I) Histogram showing CD25+ percentage or mean fluorescence intensity (MFI) of CD69, ICOS, and CD44 (percentage and MFI shown on upper left corner) on the CD90+ T cells from the same experiments in G and H denoting up-regulation of indicated activation markers in response to Lm/Cr rechallenge. Lysate concentration = 10 µg/ml. (J) Quantified fold expansion of transferred (in vitro–primed with Lm or Cr) T cells (CD45.2+) in the peripheral lymph nodes (pLNs) of mice from the same experiments shown in Fig. 1 G. n = 7 mice per group. (K) Quantified MFI of surface ICOS on recipient endogenous CD45.1+ T cells from the same experiments shown in Fig. 1 G. n = 7 mice per group. Data are pooled from or representative of two to four independent experiments. Statistics represent mean ± SEM, and P values were determined by repeated-measures two-way ANOVA with Tukey correction (C), one-way ANOVA with Tukey correction (F), or paired Student’s t test (D and J). *, P < 0.05; **, P < 0.01.
Figure 1.

Validation of an in vitro–priming approach to generate functional pathogen-specific Th cells. (A) Schematic overview of the priming system and workflow. (B) Top row: Representative CFSE dilution graph and cytokine (IFNγ and IL-17A) staining from CFSE+ and CFSE− populations. Bottom row: Histogram of CD25, CD44, and ICOS from CFSE+ and CFSE− populations. Cells were cocultured for 10–12 d before analysis. (C) Representative intracellular cytokine staining of IFNγ- and IL-17A–producing Th cells following priming by CD11c+ DCs stimulated with Lm, Cr, or Sa lysates. (D) Secreted protein levels of IFNγ, IL-17A, and IL-13 in the supernatants of cocultures as in C, measured by ELISA. n = 5. Un, unstimulated. (E) Scatter plot of mRNA expression values (log10[reads per kilobase of transcript per million mapped reads/RPKM]) obtained from RNA-seq of Lm- or Cr-specific CFSE− Th cells. Data were averaged from two independent samples. Blue data points indicate differentially expressed genes (fold change ≥2). (F) Percentages of IFNγ- and IL-17A–producing cells in pathogen- or commensal-specific Th cells. Naive WT CD4 T cells were primed by DCs stimulated with heat-killed commensal bacteria Bf, La (multiplicity of infection [MOI] = 3) or 10 µg/ml Lm/Cr lysate. Intracellular cytokine was examined at day 10. n = 2–4. (G) Experimental design for testing in vivo specificity (top) and the mismatch scheme (bottom). (H) Expansion of transferred (in vitro primed with Lm or Cr) CD4 T cells in the spleen at day 5 after infection of Lm (shown as fold change comparing CD45.2+ percentage of infected mouse to paired PBS control). n = 7 mice per group. (I) Mean fluorescence intensity (MFI) of surface ICOS on donor CD45.2+ T cells from the same experiment as G and H. n = 7 mice per group. Data are representative or combined from two to five independent experiments. All plots are pregated on live cells. Error bars represent mean ± SEM, and P values were determined by paired Student’s t test (D, H, and I) or two-way ANOVA with Tukey correction (F). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We tested whether this approach can differentiate functional pathogen-specific Th cells by using well-studied mouse pathogens: Listeria monocytogenes (Lm), Cr, and Staphylococcus aureus (Sa). All three induced significant T cell expansion (Fig. S1 E) but generated distinct cytokine profiles. Cr-primed T cells contained significantly more IL-17A+ cells, while Lm generated predominantly IFNγ+ T cells (Fig. 1, C and D). Sa-primed T cells contained both IFNγ+ and IL-17A+ subsets (Fig. 1, C and D). These results are consistent with previous in vivo observations (Lin et al., 2009; Hsieh et al., 1993; Collins et al., 2014). We further examined the transcriptome of Lm- and Cr-primed CFSE− T cells (Fig. 1 E). Cr-primed T cells exhibited a Th17-associated gene signature, while Lm-primed T cells contained a high level of Th1-associated transcripts. We also identified genes uniquely expressed in Lm- or Cr-primed T cells, indicating a pathogen-dictated T cell transcriptome (Fig. 1 E and Table S3). Furthermore, DCs stimulated with gut pathobiont Bacteroides fragilis (Bf) were able to induce robust proliferation of T cells and prime high levels of Th1/Th17 differentiation. In contrast, nonpathogenic commensal Lactobacillus acidophilus (La) resulted in significantly lower T cell responses (Figs. 1 F and S1 F). These data establish that the in vitro priming system faithfully recapitulates the in vivo abilities of pathogens and commensal bacteria to induce differential T cell responses.
Importantly, pathogen-primed Th (ppTh) cells were able to secrete cytokines (IFNγ/IL-17A) or up-regulate T cell activation markers only when restimulated in vitro with the priming pathogen, indicating their specificity (Fig. S1, G–I). We further tested the specificity of these T cells in vivo by transferring Lm- or Cr-primed CD45.2+ T cells into congenic recipients and challenged them with Lm 48 h after cell transfer (Fig. 1 G). Uninfected mice that received T cells served as baseline controls. Lm-primed, but not Cr-primed, donor T cells proliferated and up-regulated ICOS (inducible T cell costimulator) in response to Lm rechallenge (Fig. 1, H and I; and Fig. S1, J and K). In summary, these data demonstrate that the in vitro priming system generates a pathogen-specific Th cell response and, therefore, presents a reliable system to further interrogate the nature of these responses. Future work can be done to improve this in vitro priming system by including tissue-resident or migratory DC populations, such as lamina propria (LP) CD103+ DCs or skin Langerhans cells, or through the addition of tissue-specific microenvironmental cues, when investigating CD4 T cell responses to tissue invading or compartmentalized pathogens.
DC-mediated priming dictates the distinct transcriptional profile of Th17 cells
Th17 cells are sensitive to microenvironmental cues and display a high degree of plasticity and heterogeneity (Hu et al., 2011; Ghoreschi et al., 2011; Gagliani et al., 2015). This intrinsic fine-tuning of Th17 cells prompted us to dissect their transcriptional regulation during pathogen-specific differentiation. We generated 17A-fm mice (Il17aCRER26tdT) to fate map IL-17A–producing cells (Hirota et al., 2011). Cr-specific Th cells contained a higher percentage of tdTomato (tdT)+ population than Lm-specific Th cells, reflecting the same Th17 bias found previously (Fig. 1, C and D; and Fig. S2 A). Expression of Th17-associated cytokines and transcription factors (TFs) was highly enriched in the CFSE−tdT+ fraction, indicating consistent tracing of Th17 population (Fig. S2 B).
Figure S2.

Comparative transcriptional analysis reveals caspase-1 as a T cell–intrinsic regulator of Th17 differentiation. (A) tdT+ percentage of unstimulated (Un), Lm-primed, Cr-primed, or Th17-polarized 17A-fm CD4 T cells. Un, n = 4; Lm and Cr, n = 5; Th17, n = 2. (B) Heatmap of Th17-associated cytokine and TF expression in CFSE+, CFSE−tdT−, and CFSE−tdT+ populations from Cr-primed T cells. (C) Representative flow plots showing IFNγ-YFP+ percentage of CFSE−tdT+ population, from 17-γ double reporter T cells under ppTh17 (Cr-primed) or cdTh17 conditions at early (day 5) and late (day 10) stages of differentiation. (D) Representative flow plots showing IFNγ-YFP+ percentage of CD4+CD90+tdT+ population, from mLN of 17-γ double reporter mice at 10 dpi of Cr. 17-fm mice that do not carry the YFP allele were used as negative signal control. (E) Confirmation of representative differentially expressed genes shown in Fig. 2 by qRT-PCR, n = 2. (F) Expression levels of Casp1 and Casp11 in murine T cell populations, extracted from the Immgen database. n = 2–3; heatmap is shown as mean expression value. SPN, Spleen; sLN, skin-draining lymph node; PP, Peyer’s patches. (G) WT or Casp1Δ10 naive T cells were primed for 10 d with WT DCs stimulated with heat-killed Bf (MOI = 3). Flow plot shows the percentage of IL-17A– and IFNγ-producing cells in live CFSE−CD90+ population. n = 2–3. (H) Overlay of CFSE dilution histograms (left) 10–12 d after Cr-priming and quantified CD4 T cell proliferation (CFSE−%; right). n = 5. Δ10, Casp1Δ10. (I) tdT+ percentage in CFSE−CD90+ CD4 T cell population primed with Cr-stimulated DCs in vitro for the indicated time. (J) Western blot analysis of procaspase-1 (p45) and cleaved caspase-1 (p20) of Cr-primed CD4 T cells. Cells are sorted into CFSEhigh (hi), CFSEintermediate (int), and CFSEnegative (neg) populations and compared with WT macrophages (MΦ) undergoing inflammasome activation (4 h LPS + 30 min ATP [L+A]). (K) WT naive CD4 T cells were primed with Cr-stimulated WT DCs. At day 7, indicated inhibitors were added to the culture. Casp1 expression in FACS-sorted CFSE−CD90+ T cells was determined at day 10. Concentration of the inhibitors used were p38i (SB203580), 5 µM; JNKi (CAS 129–56-6), 2 µM; MEKi (U0126), 2 µM; JAKi (Ruxolitinib), 500 nM; IRAK1/4i (CAS 509093–47-4), 5 µM; IKKi (CAS 873225–46-8), 500 nM. n = 2–3. (L) IL-17A+ percentage of CFSE−CD90+ WT or Casp1Δ10 CD4 T cells primed with Cr-stimulated WT DCs in vitro. DMSO or Z-YVAD-fmk (20 µM) was present in the culture medium throughout the priming period. n = 3. (M) IL-17A+ percentage of CFSE−CD90+ WT or Casp1Δ10 CD4 T cells primed with Cr-stimulated WT DCs in vitro. Cultures were supplemented with 10 ng/ml IL-1α or IL-1β. n = 4. (N) Bicistronic hCD2 expression on the surface of Th17 cells transduced with indicated vector. Data are pooled from or representative of at least two independent experiments. Statistics represent mean ± SEM, and P values were determined by paired Student’s t test (A and H) or paired two-way ANOVA (L and M). ns, not significant; *, P < 0.05.
Th17 cells generated by TCR-activating antibodies and cytokine cocktails (cytokine differentiated, cdTh17), have been widely used for transcriptome profiling (Yosef et al., 2013; Ciofani et al., 2012). To understand transcriptional programming of pathogen-specific Th17 cells, we isolated tdT+ populations from cdTh17 cells or Cr-primed Th17 cells (ppTh17 cells) using 17A-fm mice and subjected these cells to RNA sequencing (RNA-seq) analysis (Fig. 2 A). We also interrogated the relationship between these in vitro–generated Th17 cells and Th17 cells primed in vivo following Cr infection (ex vivo tdT+; Fig. 2 A). Such a comparison would serve as an important validation of the in vitro priming system.
Figure 2.

Comparative transcriptional analysis reveals major divergence in programming between ppTh17 and cdTh17 cells. (A) Experimental design for transcriptional profiling of CFSE+ (naive), ppTh17 (Cr-specific, day 12), cdTh17 (day 5), or ex vivo tdT+ cells (10 dpi/peak of infection with 5 × 108 CFU of Cr). (B) Euclidean distance between global gene expression in naive CFSE+, ppTh17, cdTh17, or ex vivo tdT+ cells. CFSE+ and ex vivo tdT+, n = 2; ppTh17 and cdTh17, n = 3. (C) Number of genes shared or uniquely expressed by indicated three Th17 populations (compared with CFSE+ naive T cells) and their functional annotation enrichment analyzed by DAVID. FDR, false discovery rate. (D) Heatmap and hierarchical analysis of key T cell TFs, cytokines, and cytokine receptor expression from transcriptional profiling described in A. (E) Heatmap and hierarchical analysis of gene expression for gene cluster involved in in vivo T cell motility, migration, chemokine and chemokine receptor signaling, T cell positioning, and antigen sampling. (F) Heatmap and hierarchical analysis of gene expression for genes involved in metabolic processes. (G) Heatmaps of genes representing cell cycle/DNA-replication pathways and amino acid metabolism pathways. (H) GSEA analysis of ppTh17 and cdTh17 cells compared with Molecular Signature dataset of effector versus memory T cells. (I) Flow cytometry analysis of CD127 (IL-7Rα), CD215 (IL-15Rα), and c-Myc in ppTh17 and cdTh17 cells. Data are representative or combined from two to three independent experiments. In heatmaps, each row/column represents one independent sample. Hierarchical clustering was determined by Euclidean distance and pairwise average-linkage (B and D–G). Heatmap represents global expression z-score.
Sample-to-sample Euclidean distance indicates that ppTh17 cells were closely related to ex vivo tdT+ cells but highly divergent from cdTh17s (Fig. 2 B). We next focused on differentially regulated genes between the aforementioned three types of Th17 cells. The majority (871 genes) of the 1,133 ppTh17 signature genes (compared with cdTh17 cells) were also shared by ex vivo tdT+ cells (Fig. 2 C and Table S4). Less than 50% (935 of 1,938 genes) of cdTh17 up-regulated genes overlapped with either ppTh17 or ex vivo tdT+ cells, confirming that it deviated significantly from the other two Th17 populations (Fig. 2 C). Pathway analysis of these gene sets indicated that ppTh17/ex vivo tdT+ cells expressed higher levels of genes related to diverse T cell effector functions, while cdTh17s up-regulated cell cycle–related genes (Fig. 2 C and Table S5).
ppTh17 and ex vivo tdT+ cells exhibited lineage plasticity by expressing Th1- and Th2-associated TFs and cytokines (Tbx21, Gata3, Ifng, Il4, and Il10), while cdTh17 cells were characterized by polarized expression of Th17-lineage transcripts (Fig. 2 D). Indeed, ppTh17 and ex vivo tdT+ cells generated using 17-γ double reporter mice (Il17aCRER26tdTIfngYFP) contained a significant population of tdT+YFP+ cells, indicating Th1 trans-differentiation from committed Th17 lineage (Fig. S2, C and D). Overall, these data indicate a close functional relationship between ppTh17 and in vivo–primed Th17 cells and suggest that the cdTh17 does not resemble the physiologically generated Th17 population.
Additionally, ppTh17/ex vivo tdT+ cells expressed various chemokine receptor genes, potentially associated with their migration capacity (Fig. 2 E, Table S5, and Table S6). ppTh17/ex vivo tdT+ also highly expressed Il7r and Il15ra, suggesting that T cell memory formation was supported in these contexts (Fig. 2 E; Purton et al., 2007; Li et al., 2003). These cells also exhibited metabolic changes by switching to 5′ AMP-activated protein kinase (AMPK) pathway, fatty acid oxidation, and oxidative phosphorylation programs, whereas cdTh17 cells up-regulated c-Myc and amino acid metabolism genes that support proliferation (Fig. 2, F and G; Waickman and Powell, 2012; Wang et al., 2011). These transcriptional features indicate a lower activation amplitude in physiologically derived Th17 cells that typically maintain memory T cell functions, while cdTh17 cells resemble terminally differentiated effector T cells (Fig. 2, F and G; Patsoukis et al., 2016). Gene set enrichment analysis (GSEA) confirmed an enriched memory T cell signature in ppTh17s and an enriched effector signature in cdTh17s (Fig. 2 H and Table S7). The expression of specific ppTh17 and cdTh17 genes was validated by flow cytometry and quantitative RT-PCR (qRT-PCR; Figs. 2 I and S2 E). Cumulatively, these data suggest that DCs influence transcriptional programming of T cells that extends beyond the provision of activation signals and innate cytokines. CD4 T cells differentiated by pathogen-stimulated DCs, such as ppTh17s and ex vivo Th17s, showed transcriptional profiles that display lineage plasticity and features of memory differentiation, while Th17 differentiation driven by defined cytokines led to gene programs indicative of terminal differentiation and devoid of plasticity.
T cell–intrinsic caspase-1, independent of its enzymatic function, is critical for an optimal Th17 response
It remains unclear how DCs precisely regulate T cell differentiation beyond the provision of the classic three signals of T cell activation. To identify novel regulatory factors, we focused on genes uniquely expressed in ppTh17s but not in cdTh17s. Casp1 (caspase-1) emerged as a candidate, with 5–10-fold induction of expression in ppTh17/ex vivo tdT+ cells compared with cdTh17 or naive T cells (Fig. 3, A and B). Caspase-1 and caspase-11 are inflammatory caspases that have overlapping effector functions downstream of inflammasome activation, primarily in myeloid cells (Shi et al., 2014; Mariathasan et al., 2004). However, the role of these caspases in T cells is not well defined. Casp1 and Casp11 transcripts were up-regulated in memory T cells, but Casp11 levels were not significantly different between ppTh17s and cdTh17s, indicating that Casp11 may be up-regulated upon T cell activation, while Casp1 is specifically induced by interacting DCs (Figs. 3 A and S2 F). Therefore, we used Casp1Δ10 mice, which specifically lack caspase-1 but not caspase-11 (Rauch et al., 2017). We found defective Th17 differentiation in Casp1Δ10 T cells following in vitro priming with Cr- and Bf-stimulated WT DCs (Fig. 3, C and D; and Fig. S2 G). Interestingly, Th1/Th2 lineage commitments and proliferation were unaffected (Figs. 3 E and S2 H). These results prompted us to further examine the role for T cell–intrinsic caspase-1 in Th17 responses.
Figure 3.

Caspase-1 promotes the differentiation of Th17 lineage independent of its enzymatic activity or inflammasome activation. (A) Differentially expressed transcripts between ppTh17 and cdTh17 cells. Each dot represents the average of three independent experiments. Blue dots indicate differentially regulated genes (fold change >1.5, false discovery rate <0.05). Black dots indicate differentially regulated transcripts described in Fig. 2. Red dots indicate Casp1 and Casp11 transcripts. (B) mRNA expression of Casp1 relative to 18s rRNA in sorted naive (CFSE+), ppTh17, cdTh17, or ex vivo tdT+ cells from mLNs of Cr-infected 17A-fm mice (10 dpi), quantified by independent qRT-PCR experiments. n = 2. (C) Naive CD4 T cells from WT or Casp1Δ10 (Δ10) mice were primed in vitro by Cr lysate–stimulated WT splenic CD11c+ DCs, and IL-17A– and IFNγ-producing cells were measured by intracellular cytokine staining and flow cytometry analysis of CD90+CFSE− live CD4 T cells (left); IL-17A+ percentages were quantified (right). n = 7. (D) IL-17A in the supernatant from experiments in C, measured by ELISA. n = 3. Δ10, Casp1Δ10. (E) IFNγ+ and IL-13+ percentage of CD90+CFSE− live cells, quantified from experiments in C. n = 7. Δ10, Casp1Δ10. (F) Relative expression (normalized to 18s rRNA and relative to day 3) of Casp1 mRNA at indicated time points after Cr-priming. n = 4. (G) Western blot analysis of procaspase-1 (p45) and cleaved caspase-1 (p20) from naive CD4 T cells, sorted CD90+CFSE− Cr-primed Th cells, or WT bone marrow–derived macrophages (MΦ) that were unstimulated or under conventional inflammasome activation (4 h LPS + 30 min ATP [L+A]). (H) Naive WT, Casp1Δ10 (Δ10), Pycard−/−, Il1b−/− CD4 T cells were primed with Cr-stimulated WT DCs. IL-17A+ percentage of CFSE−CD90+ live cells was measured by intracellular cytokine staining and quantified. n = 5. (I) Casp1Δ10 CD4 T cells were differentiated to Th17 lineage and retrovirally reconstituted with MSCV-IRES-hCD2 alone (Vector), full-length Casp1 (FL), Casp1 deficient of CARD (Casp1ΔCARD), or enzymatically (Enz) inactive form of Casp1 (EnzDead, C284A) and quantified for IL-17A+ percentage (gated on live, hCD2+ population). n = 4. (J) Log2(fold change) of major Th cell cytokine and TF expression comparing WT and Casp1Δ10 Cr-specific Th cells. WT or Casp1Δ10 naive CD4 T cells were primed with Cr-stimulated WT DCs for 10 d. CFSE−CD90+ population was FACS-sorted and subjected to mRNA-seq and analysis. n = 2. (K) Heatmap and hierarchical clustering of differentially expressed (>1.5 fold) genes comparing WT and Casp1Δ10 (Δ10) Cr-specific Th cells. Genes of interest were labeled by the heatmap. Each column represents one independent replicate. (L) Activation z-score of pathways enriched in WT or Casp1Δ10 Cr-specific Th cells. Pathway analysis was performed using Ingenuity Pathway Analysis. Data are representative of or combined from two to seven independent experiments. Statistics represent mean ± SEM, and P values were determined by paired Student’s t test (C–E and H) or one-way ANOVA with Tukey correction (I). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The increase of Casp1 mRNA over time correlated with the expansion of the tdT+ signal, with the highest caspase-1 protein level being in the CFSE− population (Figs. 3 F and S2, I and J). The up-regulation of Casp1 was independent of NF-kB, MAPK, or JAK-STAT signaling (Fig. S2 K), suggesting a complex regulation mechanism that needs further investigation. Previous studies have reported inflammasome-mediated caspase-1 activation in virus-infected CD4 T cells (Doitsh et al., 2014; Galloway et al., 2015). Surprisingly, in Cr-primed T cells, we did not find the enzymatically active form of caspase-1 (p20; Figs. 3 G and S2 J). Consistent with this idea, inhibiting caspase-1 enzymatic activity by z-YVAD-fmk failed to normalize the differences between WT and Casp1Δ10 T cells (Fig. S2 L). We found normal Th17 differentiation following Cr-induced priming of Pycard (ASC)−/− and Il1b−/− CD4 T cells (Fig. 3 H). Supplementing IL-1α or IL-1β to Casp1Δ10 T cell cultures did not rescue the defect in Th17 differentiation (Fig. S2 M). Together, these data support the idea that the role of caspase-1 in Th17 differentiation is independent of inflammasome activation or the resulting release of IL-1 family cytokines from T cells.
Besides the enzymatic function, caspase-1 facilitates binding and scaffolding to various effector proteins by the caspase activation and recruitment domain (CARD; Park et al., 2007). We ectopically reconstituted Casp1Δ10 CD4 T cells with full-length, CARD-deficient (ΔCARD) or enzymatically inactive (EnzDead) caspase-1 and investigated their ability to induce Th17 lineage commitment (Figs. 3 I and S2 N). Expression of full-length and EnzDead caspase-1 increased IL-17A+ T cells compared with vector alone, but ΔCARD failed to promote Th17 differentiation (Fig. 3 I). This suggests that the role of caspase-1 in controlling Th17 differentiation is dependent on its CARD domain for scaffolding. Other CARD-containing proteins have important roles beyond cell death pathways and have been implicated in linking T cell receptor activation to signaling cascade (Shimada et al., 2018; Park et al., 2007). A variety of proteins, such as BCL10 and AutoImmune REgulator (AIRE), have been reported to contain core CARD domains, posing a wide possibility for the interaction partners for caspase-1 (Bertin et al., 2000; Ferguson et al., 2008). Previous reports have indicated that active caspase-1 processes IL-1α/β in T cells and mediates cell death during viral infections (Doitsh et al., 2014). The effector function of human Th1 cells has been reported to be enhanced by complement-driven, inflammasome-dependent caspase-1 activation (Arbore et al., 2016). However, consistent with our data, recent work has shown that ASC-caspase-8 inflammasome, rather than caspase-1, is activated during pathogenic Th17 generation (Martin et al., 2016). Interestingly, NLRP3 has been reported to promote Th2 differentiation independently of the inflammasome (Bruchard et al., 2015; Braga et al., 2019), hinting at standalone functions for proteins that have been implicated in inflammasome activation. Overall, these data provide compelling evidence that caspase-1 promotes Th17 differentiation in an inflammasome-independent manner.
Casp1Δ10 T cells exhibit increased fatty acid metabolism and decreased Th17 gene signature
To gain insight into how caspase-1 is regulating Th17 differentiation, we profiled in vitro–differentiated Cr-specific WT or Casp1Δ10 CD4 T cells by mRNA-seq. Consistent with the previous observation, we found reduced expression of Th17 cytokines and TFs in Casp1Δ10 T cells (Fig. 3 J). Ifng expression was also lower in Casp1Δ10 T cells, possibly due to a reduced IFNγ+IL-17A+ population, while Th1 TF Tbx21 was not significantly affected (Fig. 3 J). Th2 and T reg genes were increased, suggesting a shift in Th cell balance in the absence of caspase-1 (Fig. 3 J). We analyzed differentially expressed transcripts (Table S8), and found that genes involved in fatty acid uptake (Cd36) and utilization (Slc27a2, Slc27a5, Acat3, Cpt1b, and Acaa1b) were up-regulated in Casp1Δ10 T cells (Fig. 3 K). Indeed, PPARα and AMPK targets were enriched in Casp1Δ10 CD4 T cells (Fig. 3 L), suggesting a metabolic change to exogenous fatty acid metabolism that potentially inhibits Th17 differentiation (Berod et al., 2014). Additionally, iNOS response, reported to negatively regulate Th17 response (Yang et al., 2013), was also elevated in Casp1Δ10 T cells (Fig. 3 L). In summary, we find that Casp1Δ10 T cells exhibit an altered metabolic state that may negatively regulate Th17 differentiation. The detailed mechanism will require further investigation.
T cell–intrinsic caspase-1 is required for Th17-mediated host protection and autoinflammatory disease
Although the absence of T cell–intrinsic caspase-1 impaired the generation of ppTh17 cells, cdTh17 cells were unaffected, thus highlighting the differential regulation present in cdTh17 versus physiologically generated Th17 cells (Fig. S3, A and B). This prompted us to examine the in vivo function of T cell–intrinsic caspase-1 in host protection against Cr infection. To this end, we transferred CFSE-labeled WT or Casp1Δ10 naive CD4 T cells to Rag1−/− mice and subsequently challenged them with Cr (Fig. 4 A). Transferred WT or Casp1Δ10 T cells proliferated at a similar level (Fig. S3 C). However, Casp1Δ10 T cells showed defective commitment to Th17/Th22 cells as well as fewer IFNγ+IL-17A+ cells in the mesenteric lymph nodes (mLNs), but normal Th1 (IFNγ+IL-17A−) differentiation (Fig. 4, B and C). Consequently, transfer of WT CD4 T cells, but not Casp1Δ10 Τ cells, protected mice from Cr colonization compared with nontransferred mice (Fig. 4 D). Transfer of WT, but not Casp1Δ10 T cells, was also able to prevent stool inconsistency and increased stool fluidity (Fig. 4 E), a reported consequence of Cr-induced diarrhea (Viswanathan et al., 2009). Cytokine responses from the CD90+CD4− population (presumably innate lymphoid cells [ILCs]) were not significantly different between WT or Casp1Δ10 T cell transferred mice (Fig. S3 D), suggesting that T cell–intrinsic caspase-1 was specifically mediating protection against Cr infection.
Figure S3.

T cell–intrinsic caspase-1 is required for host protection and Th17-mediated autoinflammatory disease. (A) Naive CD4 T cells from WT or Casp1Δ10 (Δ10) mice were isolated and polarized to cdTh17 cells. Representative flow cytometry plot (left) and quantifications of IL-17A+ percentage (right) are shown. n = 4. (B) Representative histogram overlay of CFSE dilution from experiments in A. (C) CFSE− percentage of transferred CD4 T cells, examined at 10 dpi. n = 8–9 mice per group. Δ10, Casp1Δ10. (D) Percentage of cytokine-positive cells in CD90+CD4− population (ILCs) from the same experiments in Fig. 4. n = 5 mice per group. (E) Colonic crypt length measured from histology images in Fig. 5 D. n = 3–5 mice per group. NT, nontransferred (i.p. PBS). Δ10, Casp1Δ10. (F and G) WT and Casp1Δ10 CD45RBhi CD4 cells were transferred to Rag1−/− mice. 3 wk after transfer, LPL cells were isolated and stained with FAM-FLICA to detect activation of caspase-1. FLICA+PI+ indicates inflammasome-activated cells. WT and Casp1Δ10 bone marrow–derived DCs (BMDCs) that were treated with inflammasome-activating ligands (4 h LPS [100 ng/ml] and 30-min 5 mM ATP) were used as positive and negative controls for FLICA staining. Representative flow plot is shown in F and quantified in G; n = 2–4/group. (H and I) IL-17A+IFNγ− percentage (H) and IFNγ+IL-17A− percentage (I) of CD4+CD90+CD44+ T cells in the mLN or colonic LP (Colon-LPL) of Rag1−/− mice 4 wk after transfer of WT or Casp1Δ10 naive CD4 T cells. n = 8–13 mice per group. (J) Percentages and numbers of CD4+CD90+ T cells from Colon-LPL of Rag1−/− mice 4 wk after transfer of WT or Casp1Δ10 naive CD4 T cells. n = 6–7 mice per group. Data are representative of or pooled from two to four independent experiments. Statistics represent mean ± SEM, and P values were determined by paired Student’s t test (A), one-way ANOVA with Tukey correction (E), or unpaired Student’s t test (G–I). ns, not significant; *, P < 0.05; ***, P < 0.001.
Figure 4.

T cell–intrinsic caspase-1 is required for host protection against Cr. (A) Schematic of the experiment. (B) Representative intracellular cytokine staining of IFNγ, IL-22, and IL-17A in CD4+CD90+CD44+ T cells in the mLNs at 10 dpi of Cr. (C) Quantification of IL-17A+IL-22−, IL-17A+IL-22+, IL-17A−IL-22+ percentages (left) and IFNγ+IL-17A+%, IFNγ+IL-17A− percentages (right). n = 7–8 mice per group. (D) Cr burden (quantified as CFU) in the cecum at 10 dpi (left). Also shown is the Cr burden when normalized to cecum content weight (right). NT, nontransferred (i.v. PBS). n = 4–9 mice per group. (E) Representative image of stool appearance (left) and quantified stool water content (right, as percentage of water in each pellet). n = 4–7 mice per group. Data are representative of or combined from two independent experiments. Each data point represents one biological replicate. Statistics represent mean ± SEM, and P values were determined by unpaired Student’s t test (C) or one-way ANOVA (D and E). ns, not significant; *, P < 0.05; **, P < 0.01.
To examine the broad role of caspase-1 in Th17-mediated immune responses, we transferred WT, Casp1Δ10, or Il1b−/− CD45RBhi T cells to Rag1−/− mice to test their ability to induce colitis (Ostanin et al., 2009). In this model, transferred naive CD4 T cells differentiate in response to gut microbiota (Matsuda et al., 2000). Recipients of Casp1Δ10 T cells developed a mild disease, while WT or Il1b−/− T cell recipient mice developed measurable colitis, including significant weight loss (Fig. 5, A and B) and colon shortening (Fig. 5 C). Casp1Δ10 recipients also exhibited ameliorated colonic pathology, with less transmural infiltration of leukocytes and fewer submucosal morphological changes compared with WT (Figs. 5 D and S3 E). We isolated ex vivo CD90+CD4+ T cells from colonic LP 3 wk after transfer and did not find cleaved caspase-1 (detected by FAM-FLICA staining) in these cells (Fig. S3, F and G). Previous reports have suggested that caspase-1, in some cases, can contribute to the secretion of IL-1α as well as IL-1β that is independent of its cleavage and catalytic activity (Conos et al., 2016; Gross et al., 2012). We have excluded the role of IL-1α and IL-1β in vitro(Fig. S2 M) and T cell–derived IL-1β in vivo(Fig. 5 A). However, it is possible that IL-1α produced by T cells in vivocould contribute to colitis, and this remains to be investigated. Furthermore, the reported profile of caspase-1 substrates includes a broad range of cellular proteins and enzymes (Shao et al., 2007). Therefore, uncleaved caspase-1 could also mediate the release of unconventional substrates from T cells that act in cis and lead to Th17 differentiation.
Figure 5.

T cell–intrinsic caspase-1 is required for Th17-mediated colitis. (A) Weight change of Rag1−/− mice that received WT, Casp1Δ10, or Il1b−/− naive CD4 T cells (CD45RBhi) at indicated time points (n = 6–10 mice for each group). NT, nontransferred (i.p. PBS). (B) Percentage of initial body weight 4 wk after transfer. n = 12–19 mice per group. (C) Representative image of colons (left) and measured colon length (right) of Rag1−/− mice at 4–5 wk after transfer. n = 4–8 mice per group. (D) Representative H&E staining of colon sections from NT, WT, or Casp1Δ10 (Δ10) naive T cell–transferred Rag1−/− mice (left) and histology score (right). Images are displayed at 20× magnification. Ruler bar represents 100 µm. n = 12–14 mice per group. (E) Representative flow plots showing the percentages of IL-17A+IFNγ+ of CD4+CD90+CD44+ T cells in the mLNs or colonic LP (Colon-LPL) of Rag1−/− mice 4 wk after transfer of WT or Casp1Δ10 (Δ10) naive CD4 T cells. n = 8–13 mice per group. (F) The number of CD4+CD90+IL-17A+ T cells in the spleens of the Rag1−/− mice that received WT or Casp1Δ10 (Δ10) naive CD4 T cells. n = 6 per group. (G) Serum IL-17A levels at the indicated time points from the mice in Fig. 5 F. n = 5 mice per group. Data are representative of or combined from two to four independent experiments. Each data point represents one biological replicate. Statistics represent mean ± SEM and P values were determined by two-way repeated ANOVA with Bonferroni correction (A), one-way ANOVA (B and C), Mann-Whitney U test (D), unpaired Student’s t test (E and F) or multiple t tests with Holm-Sidak correction (G). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
In the mLN and colon LP, we found a reduced frequency of pathogenic IL-17A+IFNγ+ cells in Casp1Δ10 compared with WT T cell recipients, consistent with the established role of these cells in the induction of T cell–mediated colitis and the development of pathology (Fig. 5 E; Harbour et al., 2015). Of note, there was no difference in the proportion of IL-17A+IFNγ− nonpathogenic Th17 or IL-17A−IFNγ+ Th1 cells between WT and Casp1Δ10 recipients (Fig. S3, H and I). The total percentage and numbers of CD4+CD90+ cells were comparable between the two genotypes (Fig. S3 J), confirming that caspase-1 deficiency did not affect T cell proliferation or cell death. Additionally, we observe significantly less splenic expansion of total IL-17A+ Casp1Δ10 cells (Fig. 5 F), consistent with the reduced circulating IL-17A levels throughout the course of the disease (Fig. 5 G). Collectively, these data extend the function of caspase-1 to the differentiation of autoinflammatory Th17 cells. Earlier studies have reported ameliorated autoimmune disease in caspase-1–deficient animals and attributed it to abrogated IL-1β and IL-18 production during inflammasome activation (Furlan et al., 1999; Siegmund et al., 2001). Our results suggest that impaired Th17 commitment, resulting from T cell–intrinsic deficiency of caspase-1, could also contribute to these phenotypes. Since the majority of these studies used whole-body knockout mice, it would be necessary to dissect the specific roles of caspase-1 in innate and adaptive immune compartments in regulating Th17 differentiation and associated disease outcomes.
Taken together, our work highlights a novel workflow for studying pathogen-specific Th cell differentiation. By combining the in vitro priming system with genetic fate mapping and mRNA-seq, we were able to profile the transcriptome of Cr-specific Th17 cells. Conceptually, the dataset from this study provides experimental evidence for the importance of DCs in dictating global transcriptional programming of pathogen-specific Th cells. Integration of a systems biology approach into this in vitro priming system will empower high-throughput analysis of antimicrobial T cell responses to discover novel players, such as caspase-1, in CD4 T cell differentiation.
Materials and methods
Animals
C57BL/6J (WT), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+), and B6.129S7-Rag1tm1Mom/J (Rag1−/−) mice were obtained from Jackson Laboratory and maintained in the University of Texas (UT) Southwestern mouse breeding core facility. STOCK Il17atm1.1(icre)Stck/J (Il17aCRE) and B6.129S4-Ifngtm3.1Lky/J GREAT (IfngYFP) mice were obtained from Jackson Laboratory and bred in-house to B6.Cg-Gt(ROSA)26Sortm14(CAG-tdT)Hze/J (R26tdT) mice (a gift from Morrison Laboratory, UT Southwestern). Casp1Δ10 mice were a kind gift from Drs. Russell Vance and Isabella Rauch (University of California, Berkeley, CA) and were maintained on Jackson Laboratory C57/BL6 background. Pycard−/− mice were provided by Dr. Vishva Dixit (Genentech, South San Francisco, CA). Il1b−/− mice were provided by Dr. Fayyaz S. Sutterwala (Cedars Sinai Medical Center, Los Angeles, CA). Unless specified, mice were bred and maintained at the specific pathogen–free facility of UT Southwestern Medical Center and provided with sterilized food and water ad libitum. Mice used for infection experiments were kept at a conventional animal facility and provided with nonautoclaved food and water ad libitum. Age- and sex-matched mice 6–12 wk of age were used for all experiments. Both female and male mice were used in experiments. All mouse experiments were performed as per protocols approved by Institutional Animal Care and Use Committee at UT Southwestern Medical Center.
Bacterial strains and infection
Lm (LM 10403 serotype 1, a gift from Dr. James Forman, UT Southwestern Medical Center, Dallas, TX), Cr (strain ICC168, Nalidixic acid-resistant) and Sa (ATCC-25923) were cultured in agar plate of brain-heart infusion, Luria-Bertani, with 30 µg/ml nalidixic acid, and tryptic soy broth, respectively. A single colony was chosen and secondarily expanded in the respective liquid broth with appropriate antibiotics. Bf and La were cultured as described previously (Fan et al., 2015). For Lm infection, bacteria were grown to log phase (OD600 = 0.6–1) on the day of infection, extensively washed, and resuspended in PBS. Mice were injected i.p. with 2 × 104 CFU of Lm. Tissues were harvested 5 d postinfection (dpi). For Cr infection, mice were intragastrically administered 1% sodium bicarbonate and, 20–30 min later, infected with 2–5 × 108 CFU of Cr. mLNs were harvested at 10 dpi.
Extraction of bacterial lysates and protein quantification
Bacterial cultures were grown to late log phase (OD600 = 0.8–1.2), washed extensively, and resuspended in PBS. Bacteria suspensions were subjected to three cycles of snap-freeze-thaw in liquid nitrogen and lysed by repeated sonication. Lysates were centrifuged at 4,000 rpm for 20 min to remove insoluble components, and the supernatant was sterilized through a 0.22-µm membrane filter. Protein concentration was measured for cleared lysate by Bradford assay.
Expansion of mouse splenic DCs by B16-FLT3L implant
B16-FLT3L mouse melanoma cell lines were maintained in 10% complete RPMI (RPMI [Hyclone], 10% FBS [Sigma-Aldrich]), l-glutamine, penicillin-streptomycin, sodium pyruvate, and β-mercaptoethanol [Sigma-Aldrich]). For the generation of spleen-derived DCs, 5 × 106 B16-FLT3L cells were injected subcutaneously into the flank of mice. After 10–16 d, spleens were harvested and used for CD11c+ DC isolation. B16-FLT3L injection did not induce CD11c+ DC maturation, as reported previously (Dudziak et al., 2007).
Flow cytometry and FACS
Antibodies used for flow cytometry are listed in Table S2. For surface marker staining, cells were blocked with Fc block (anti-mouse CD16/CD32, BioLegend) for 10 min and then incubated with antibodies for 30 min, with extensive washes with FACS buffer (PBS, 2% FCS, and 2 mM EDTA). For intracellular cytokine analysis, cells were stimulated with 50 ng/ml PMA (Sigma-Aldrich) and 1 µM ionomycin (Sigma-Aldrich) in the presence of 5 µg/ml brefeldin A (BioLegend) for 4–6 h. Cell permeabilization and intracellular staining were performed according to manufacturer’s protocol (eBioscience Foxp3/TF staining buffer set). Samples were analyzed using an LSR Fortessa flow cytometer (BD) or Novocyte 3001 (ACEA Biosciences). Cells were gated on singlets, and dead cells were excluded using Zombie Yellow live/dead staining (BioLegend). Data were analyzed using FlowJo software (BD). For FACS sorting, cells were stained in sterile FACS buffer. Dead cells were excluded by DAPI or propidium iodide staining. Cells were sorted by FACS Aria (BD) into FACS buffer or directly into TriZol LS Reagent (Invitrogen) for RNA extraction.
Isolation of mouse lymphocyte populations
Spleen and lymph nodes were harvested from 6–12-wk-old mice. Single-cell suspension was obtained by dissociation using sterile frosted slides and passing through a 70-µm cell strainer. Red blood cell lysis was performed as needed. Naive CD4 T cells were isolated according to MojoSort kit protocol (BioLegend). The purity of naive CD4 T cells was constantly monitored and maintained at >95% CD4+MHCII−CD62L+CD44−. Splenic DCs were isolated from the spleen of a mouse injected with B16-FLT3L melanoma. Splenocytes were blocked with Fc block (anti-CD32/CD16) and stained with CD11c-biotin (BioLegend), then with anti-Biotin beads (Miltenyi), and isolated using AutoMacs magnetic selection (Miltenyi). The purity of isolated splenic DCs was maintained at >98% CD11c+. For all experiments, DC donor mice and naive CD4 T cell donor mice were age and sex matched. B cells were isolated from sex- and age-matched naive mouse spleen by isolating the CD19+ population using CD19-biotin (BD) and AutoMacs (Miltenyi). LP lymphocytes (LPLs) were isolated as previously described (Hu et al., 2011).
Pathogen-specific Th cell priming
X-VIVO15 serum-free media (Lonza) was used to avoid T cell activity to bovine serum proteins. CD11c+ DCs were pulsed with pathogen lysate (10 µg/ml, dose titrated to induce the maximum response and minimum cell death across the panel) at 106/ml for 5 h and then extensively washed. Naive T cells were labeled with CFSE (5 µM, BioLegend). DCs and T cells were cocultured at a 1:5 ratio for 5–12 d, depending on the experiments.
Recall of pathogen-specific T cell response by B cell–mediated restimulation
Pathogen-specific T cells generated using the approach described previously were rested with the provision of a low dose of IL-2 (10 units/ml, BioLegend) for an additional 2 d until active cytokine production waned. B cells were either pulsed with pathogen lysate or blasted with CpG (Keck Oligonucleotide Synthesis Facility, Yale University, New Haven, CT) for 18–24 h in X-VIVO15 serum-free medium, extensively washed, irradiated at a dose of 12 Gy using an x-ray irradiator (X-RAD320, Precision X-Ray), and cocultured with T cells at a 2:1 ratio. T cell responses were assessed 48 h later.
T cell polarization (cytokine-differentiated T cells)
Tissue culture-treated plates were coated with 5 µg/ml of anti-mouse CD3 (BioLegend) and anti-mouse CD28 (Tonbo) for 2–4 h. 1 × 106/ml naive T cells were polarized for 5 d under Th17 polarization (cdTh17) conditions with 10 µg/ml anti-IFNγ (BioLegend), 10 µg/ml anti–IL-4 (BioLegend), 20 ng/ml IL-6 (Peprotech), 5 ng/ml TGFβ1 (Peprotech), 10 ng/ml IL-1β (Peprotech), and 20 ng/ml IL-23 (BioLegend) or Th1 polarization conditions with 10 µg/ml anti–IL-4, 50 units/ml IL-2, and 10 ng/ml IL-12 (Peprotech). For some experiments, polarized cells were removed from all polarizing cytokines and plate-bound anti-CD3/CD28, washed, and cultured with 10 units/ml IL-2 for an additional 5 d.
Retroviral transduction of Th17 cells
Retrovirus was prepared from 106 Platinum-E cells transfected with 2.5 µg vector and 0.63 µg pCL-Eco using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific). Viral supernatant was harvested from Platinum-E cultures after 48 and 72 h of transfection. 50 units/ml of IL-2 (BioLegend) and 10 µg/ml of protamine sulfate (Sigma-Aldrich) were added to the viral supernatant before transduction. Naive CD4 T cells were prepared and differentiated as described earlier. 24 and 48 h after activation, 106 T cells were transduced with 1 ml of viral supernatant under spin-infection of 2,500 rpm for 90 min at 32°C. T cells were returned to the original activation medium after spin-infection. 5 d after T cell activation, cells were harvested and stained for hCD2 as a transduction efficiency marker, as well as intracellular staining.
In vitro neutralization and recombinant cytokine supplement
In vivo–grade anti-mouse IA/IE, anti-mouse CD86, anti-mouse CD80, or anti-mouse IL-12/23p40 antibodies (BioLegend) are listed in Table S2. Purified DCs were pulsed and extensively washed as indicated previously. DCs were preincubated with Fc block for 10 min and subsequently with blocking antibodies (10 µg/ml) for 30 min. Then DCs were cocultured, without further washing steps, with labeled naive T cells as described above.
Adoptive transfer of in vitro–primed, pathogen-specific T cells
ppTh cells were extensively washed with plain RPMI 1640 without serum or additives and injected intravenously into the recipient mice (2 × 106 CD25+ICOS+ cells/mouse). Recipient mice were cohoused with their respective controls and experimental groups for ≥2 wk before the experiment and during the experimental period. Typically, one pair of mice represents one transferred-uninfected and one transferred-infected (cohoused and transferred at the same time) subject. Fold expansion was calculated as normalized to the cohoused uninfected control.
Naive T cell transfer and Cr infection
Naive T cells (CD4+MHCII−CD62L+CD44−) from indicated genotypes were injected into recipient (age- and sex-matched) Rag1−/− mice via tail vein (2 × 106 cells/mouse) 24 h before infection. Mice were then gavaged with 2 × 108 CFU/mouse Cr as described in the previous section. At 10 dpi, mLNs were harvested for measuring CD4 T cell responses. Ceca were dissected, weighed, and exudated of content. Cecum contents were homogenized in sterile PBS and plated on Luria-Bertani plates with nalidixic acid in serial dilution. Bacterial burden was calculated by averaging colony counts from countable dilutions. Mouse fecal pellet from the distal colon was collected and weighed as initial stool weight. The feces were allowed to completely dry for 24 h at 37°C and weighed as dry stool weight. Stool water content was calculated as [(initial stool weight − dry stool weight)/initial stool weight] × 100%.
T cell transfer model of colitis
5 × 105 CD4+CD62LhiCD44loCD45RBhiCD25− T cells were FACS sorted from spleens and lymph nodes of mice of each genotype, washed, and injected i.p. into Rag1−/− mice. Weights were measured weekly when weight >100% initial weight and biweekly when weight loss progressed (weight <100% initial weight). Serum was collected every week via submandibular bleeding. Mice were sacrificed 4–5 wk after transfer or when weight loss exceeded 20% of initial weight, and colon length was measured at the time of sacrifice. Colons were removed from the cecum to anus, photographed, fixed with formalin, and submitted to the UT Southwestern Molecular Pathology Core for paraffin embedding, sectioning, and H&E staining. Digital images were obtained using a Zeiss Axiovert 100 Inverted Microscope with Jenoptik Gryphax Camera. Histology scores were blind-scored by Dr. Bret Evers at UT Southwestern. The scoring criteria are modified from Ostanin et al. (2009): (a) the degree of inflammatory infiltrate in the LP, range 1–3; (b) goblet cell loss as a marker of mucin depletion, range 0–2; (c) reactive epithelial hyperplasia/atypia with nuclear changes, range 0–3; (d) the number of intraepithelial lymphocytes in the epithelial crypts, range 0–3; (e) abnormal crypt architecture (distortion, branching, atrophy, crypt loss), range 0–3; (f) number of crypt abscesses, range 0–2; (g) mucosal erosion to frank ulcerations, range 0–2; and (h) submucosal spread to transmural involvement, range 0–2; for a maximum score of 20. LPL cells were isolated from colons of diseased mice, and intracellular staining was performed as described in Flow cytometry and FACS.
ELISA
Briefly, coating antibodies were diluted and coated in an ELISA plate overnight at 4°C, then blocked with PBS containing 10% FCS or 1% BSA (Sigma-Aldrich). Samples were loaded in duplicate, diluted in blocking buffer, and incubated overnight. Detection antibodies were used according to manufacturer’s instructions. Protein concentrations were quantified using 3,3′,5,5′-tetramethylbenzidine or o-phenylenediamine dihydrochloride colorimetric assay. Plates were washed extensively in between steps with PBS and 0.05% Tween-20.
Western blot analysis
Cells were extensively washed with PBS on ice and directly lysed in boiled SDS-containing 2× Laemmli buffer. Protein concentration was measured by detergent-resistant Bradford assay. 5–10 µg of each lysate was loaded to SDS-PAGE, and immunoblotting was performed using standard protocols. Antibodies used for Western blot are listed in Table S2.
Inflammasome activation assay and FAM-FLICA staining
For detection of caspase-1 processing following inflammasome activation, bone marrow–derived macrophages were incubated with LPS (100 ng/ml) for 4 h and pulsed with ATP (5 mM) during the last 30 min. Following stimulation, cells were immediately incubated on ice and washed with cold PBS, and boiled 2× Laemmli SDS sample buffer was directly added to each well to lyse cells. The lysate was harvested and boiled for 10 min before Western blot analysis.
For detection of caspase-1 processing by flow cytometry, bone marrow–derived DCs were incubated with 100 ng/ml LPS at 5 × 106/500 µl in complete medium in a polypropylene tube for 4 h at 37°C. Cells were spun down, 200 µl medium was removed, and cells were resuspended in the remaining 300 µl medium. 5 mM ATP and FAM-FLICA dye were directly added to the medium and incubated at 37°C for an additional 30 min. Cells were extensively washed in apoptosis buffer provided in the FAM-FLICA staining kit and resuspended in propidium iodide–containing FACS buffer for flow cytometry analysis. For staining of LPL T cells for FAM-FLICA, purified LPL cells were not pretreated with LPS or ATP. LPL cells were directly costained with FAM-FLICA and antibodies against CD90 and CD4 to discriminate LPL CD4 T cells.
Plasmids
MSCV-IRES-hCD2tm* vector was described previously (Furlan et al., 2014). Full-length caspase-1 or caspase-1 deficient of CARD was cloned into backbone using XhoI and NotI loci and primers listed in Table S1. Enzymatically inactive caspase-1 was cloned by mutating cysteine (TGC) 284 to alanine (GCT) of the full-length caspase-1.
RNA isolation and quantification and qRT-PCR
Cells were sorted into either Trizol LS reagent or complete medium and lysed with Trizol reagent. RNA was extracted using miRNeasy (Qiagen) and treated with DNase I (Qiagen), according to the manufacturer’s instructions. Quantities of extracted RNA were determined using NanoDrop2000 for cDNA synthesis or Agilent Bioanalyzer 2100 before RNA-Seq Library preparation. All RNA sent for library preparation qualified for RNA integrity number >8.
cDNA synthesis was performed using Moloney murine leukemia virus reverse transcription (Invitrogen) in the presence of RNase inhibitor (Promega). qRT-PCR primers used in this study are listed in Table S1. qRT-PCR was performed using SYBR Green MasterMix (Applied Biosystems) and QuantStudio 7 Flex Real-Time PCR System.
RNA-seq analysis
RNA-seq libraries were prepared with the Illumina TruSeq Stranded RNA Sample Preparation kit (Illumina) according to the manufacturer’s protocol. Libraries’ quality was validated on an Agilent Bioanalyzer 2100. This included using standard protocols for cDNA synthesis, fragmentation, addition of adaptors, size selection, amplification, and quality control (Illumina). The amplified libraries were size selected, and libraries were quantified by PicoGreen assay (Life Technologies). SE85 single-end sequencing was performed using NextSeq SE-75 High Output V2 flow cell with an average of 23 million reads/sample. Alignment of RNA-seq reads on mouse reference genome mm10 was performed on a CLC Genomics Workbench 7 (Qiagen). The expression of the genes in all samples was calculated as reads/fragments per kilobase of transcript per million mapped reads. Methods for data normalization and analysis are based on the use of internal standards that characterize some aspects of the system’s behavior, such as technical variability, as presented elsewhere (Dozmorov and Lefkovits, 2009; Dozmorov and Centola, 2003). The comparison of these methods with some other normalization and analysis procedures was described previously (Dozmorov et al., 2010). Created initially for the analysis of microarray data, they were slightly modified to the needs of RNA-seq data analysis. The two-step normalization procedure and the associative analysis functions were implemented in MatLab (Mathworks). These algorithms are also obtainable from an R package diffGeneAnalysis, available as a part of Bioconductor packages (http://www.bioconductor.org/packages/2.5/bioc/html/diffGeneAnalysis.html).
Global hierarchical clustering and differential expression presented in Fig. 2 (B and C) were performed with DESeq2 and clustered by Euclidean distance. Heatmaps and hierarchical clustering were further generated using GenePattern with list of genes of interest. Briefly, data were log2 transformed. Z-score was used for row normalization and calculated as described previously (Cheadle et al., 2003). Row/column distances were measured by Pearson correlation and clustered using the Euclidean distance method. Heatmaps were visualized by HierarchicalClusteringImage with global z-score scale. Functional annotation of differentially regulated genes was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) or Ingenuity Pathway Analysis, as described in each figure. Activation z-score or fold enrichment >1.5 were considered significantly enriched pathways.
GSEA analysis
The GSEA method has been described previously (Subramanian et al., 2005). 11,868 genes that were considered significantly expressed were used to create a ranked list ordered by comparing differential expression between ppTh17 and cdTh17 cells. This list was then used to compare with established memory T cell gene signatures downloaded from the Molecular Signature Database in GSEA server. The molecular signature dataset used by this study was derived from a publication by Luckey et al. (2006) (GSE1000002_1582_200_DN and GSE1000002_1582_200_UP), where the author extracted the top 200 enriched genes in effector versus memory CD8 T cells. The table containing common genes between query list and molecular signature dataset can be found in Table S7.
Quantification and statistical analysis
Statistical analyses performed are indicated in the figure legends, analyzed by GraphPad Prism 7. P values <0.05 were considered statistically significant. Significantly differentially expressed genes in RNA-seq experiments were determined using encoded MatLab or Gene Pattern DESeq2 function described in RNA-seq analysis. Sample sizes were not predetermined by statistical methods.
Data and software availability
Illustrations were created with Biorender (https://biorender.com). Matlab code used for RNA-seq analysis is described in RNA-seq analysis and is available on request. The sequencing data reported in this paper have been deposited to the Gene Expression Omnibus with the accession nos. GSE141054, GSE141061, and GSE141062.
Online supplemental material
Fig. S1 demonstrates the de novo differentiation of pathogen-specific Th cells using the in vitro–priming system, their dependence on three-signal activation, and specificity to the priming pathogen. Fig. S2 shows the divergent transcriptional profile between ppTh17/ex vivo Th17 and cdTh17 cells and also specific up-regulation of caspase-1 in DC-primed T cells and its role in Th17 differentiation independent of catalytic activity. Fig. S3 shows that T cell–intrinsic caspase-1 is required for host protection and Th17-mediated autoinflammatory disease. Table S1 and Table S2 list the primers and reagents used in this study. Table S3 lists differentially expressed genes between Lm- and Cr-primed CD90+CFSE− Th cells shown in Fig. 1. Table S4 and Table S5 shows a list of genes differentially expressed between ppTh17/ex vivo tdT+ and cdTh17 cells and their functional annotations determined by DAVID. Table S6 lists normalized expression values for genes shown in the heatmaps in Fig. 2. Table S7 shows a GSEA-determined list of differentially expressed genes in ppTh17 and cdTh17 cells that are enriched in memory or effector T cell signatures. Table S8 shows differentially regulated genes and their expression in WT and Casp1Δ10 Cr-specific Th cells.
Supplementary Material
lists primers used in this study.
lists reagents.
is a list of genes differentially expressed between Lm-primed and Cr-primed CD90+CFSE− CD4 T cells, along with normalized expression values.
is a list of genes differentially expressed between ppTh17, cdTh17, and ex vivo tdT+ cells.
shows significantly enriched functional annotations in differentially expressed genes in ppTh17/ex vivo tdT+ or cdTh17 cells, determined by DAVID.
lists normalized expression values for genes shown in Fig. 2.
is a list of differentially expressed genes in ppTh17 and cdTh17 cells that are enriched in memory or effector T cell signatures, determined by GSEA.
is a list of differentially expressed genes in WT and Casp1Δ10 Cr-specific Th cells.
Acknowledgments
We thank all members of the Pasare laboratory, especially Ms. Margaret McDaniel, for helpful discussions and critical reading of the manuscript and Mrs. Linley Riediger Pierce for mouse colony management and genotyping. We thank the members of Genomics and Microarray Core at UT Southwestern Medical Center for their help with the RNA-seq experiments and analysis. Russell E. Vance, PhD, at University of California, Berkeley (Berkeley, CA), generously provided Casp1Δ10 mice. Fayyaz S. Sutterwala, MD, PhD, at Cedars-Sinai Medical Center (Los Angeles, CA), kindly provided Il1b−/− mice. Vishva Dixit, MD, kindly provided Pycard−/− mice.
This work was supported by grants from the National Institutes of Health (AI113125, GM120196, and AI123176) to C. Pasare and the Edwin L. Cox Distinguished Chair in Immunology and Genetics to E.K. Wakeland. K. Deason was supported by American Heart Association grant 17PRE33410075.
Author contributions: Y. Gao, E.K. Wakeland, and C. Pasare conceived and designed the study and interpreted all the data; Y. Gao performed all in vitro experiments and in vivo experiments with the help of K. Deason; Y. Gao and I. Dozmorov analyzed the RNA-seq data; A. Jain and R.A. Irizarry-Caro provided experimental support; L.A. Coughlin and A.Y. Koh provided the commensal microbe; I. Rauch provided the Casp1Δ10 mice; B.M. Evers performed blind histopathology scoring; Y. Gao and C. Pasare wrote the manuscript; E.K. Wakeland and C. Pasare acquired and provided funding sources for this study.
References
- Arbore G., West E.E., Spolski R., Robertson A.A.B., Klos A., Rheinheimer C., Dutow P., Woodruff T.M., Yu Z.X., O’Neill L.A., et al. 2016. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 352:aad1210 10.1126/science.aad1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini S., Latorre D., Mele F., Foglierini M., De Gregorio C., Cassotta A., Fernandez B., Kelderman S., Schumacher T.N., Corti D., et al. 2015. T cell immunity. Functional heterogeneity of human memory CD4+ T cell clones primed by pathogens or vaccines. Science. 347:400–406. 10.1126/science.1260668 [DOI] [PubMed] [Google Scholar]
- Berod L., Friedrich C., Nandan A., Freitag J., Hagemann S., Harmrolfs K., Sandouk A., Hesse C., Castro C.N., Bähre H., et al. 2014. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20:1327–1333. 10.1038/nm.3704 [DOI] [PubMed] [Google Scholar]
- Bertin J., Guo Y., Wang L., Srinivasula S.M., Jacobson M.D., Poyet J.-L., Merriam S., Du M.-Q., Dyer M.J.S., Robison K.E., et al. 2000. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-κ B. J. Biol. Chem. 275:41082–41086. 10.1074/jbc.C000726200 [DOI] [PubMed] [Google Scholar]
- Braga T.T., Brandao W.N., Azevedo H., Terra F.F., Melo A.C.L., Pereira F.V., Andrade-Oliveira V., Hiyane M.I., Peron J.P.S., and Camara N.O.S.. 2019. NLRP3 gain-of-function in CD4+ T lymphocytes ameliorates experimental autoimmune encephalomyelitis. Clin. Sci. (Lond.). 133:1901–1916. 10.1042/CS20190506 [DOI] [PubMed] [Google Scholar]
- Bruchard M., Rebé C., Derangère V., Togbé D., Ryffel B., Boidot R., Humblin E., Hamman A., Chalmin F., Berger H., et al. 2015. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol. 16:859–870. 10.1038/ni.3202 [DOI] [PubMed] [Google Scholar]
- Cheadle C., Vawter M.P., Freed W.J., and Becker K.G.. 2003. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 5:73–81. 10.1016/S1525-1578(10)60455-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M., Madar A., Galan C., Sellars M., Mace K., Pauli F., Agarwal A., Huang W., Parkhurst C.N., Muratet M., et al. 2012. A validated regulatory network for Th17 cell specification. Cell. 151:289–303. 10.1016/j.cell.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J.W., Keeney K.M., Crepin V.F., Rathinam V.A.K., Fitzgerald K.A., Finlay B.B., and Frankel G.. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat. Rev. Microbiol. 12:612–623. 10.1038/nrmicro3315 [DOI] [PubMed] [Google Scholar]
- Conos S.A., Lawlor K.E., Vaux D.L., Vince J.E., and Lindqvist L.M.. 2016. Cell death is not essential for caspase-1-mediated interleukin-1β activation and secretion. Cell Death Differ. 23:1827–1838. 10.1038/cdd.2016.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsh G., Galloway N.L.K., Geng X., Yang Z., Monroe K.M., Zepeda O., Hunt P.W., Hatano H., Sowinski S., Muñoz-Arias I., and Greene W.C.. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 505:509–514. 10.1038/nature12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov I., and Centola M.. 2003. An associative analysis of gene expression array data. Bioinformatics. 19:204–211. 10.1093/bioinformatics/19.2.204 [DOI] [PubMed] [Google Scholar]
- Dozmorov I., and Lefkovits I.. 2009. Internal standard-based analysis of microarray data. Part 1: analysis of differential gene expressions. Nucleic Acids Res. 37:6323–6339. 10.1093/nar/gkp706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov M.G., Guthridge J.M., Hurst R.E., and Dozmorov I.M.. 2010. A comprehensive and universal method for assessing the performance of differential gene expression analyses. PLoS One. 5:e12657 10.1371/journal.pone.0012657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudziak D., Kamphorst A.O., Heidkamp G.F., Buchholz V.R., Trumpfheller C., Yamazaki S., Cheong C., Liu K., Lee H.-W., Park C.G., et al. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science. 315:107–111. 10.1126/science.1136080 [DOI] [PubMed] [Google Scholar]
- Fan D., Coughlin L.A., Neubauer M.M., Kim J., Kim M.S., Zhan X., Simms-Waldrip T.R., Xie Y., Hooper L.V., and Koh A.Y.. 2015. Activation of HIF-1α and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 21:808–814. 10.1038/nm.3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B.J., Alexander C., Rossi S.W., Liiv I., Rebane A., Worth C.L., Wong J., Laan M., Peterson P., Jenkinson E.J., et al. 2008. AIRE’s CARD revealed, a new structure for central tolerance provokes transcriptional plasticity. J. Biol. Chem. 283:1723–1731. 10.1074/jbc.M707211200 [DOI] [PubMed] [Google Scholar]
- Furlan R., Martino G., Galbiati F., Poliani P.L., Smiroldo S., Bergami A., Desina G., Comi G., Flavell R., Su M.S., and Adorini L.. 1999. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J. Immunol. 163:2403–2409. [PubMed] [Google Scholar]
- Furlan S.N., Mandraju R., Brewer T., Roybal K., Troutman T.D., Hu W., Palm N.W., Unni A., and Pasare C.. 2014. Enhancement of anti-tumor CD8 immunity by IgG1-mediated targeting of Fc receptors. MAbs. 6:108–118. 10.4161/mabs.27052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliani N., Amezcua Vesely M.C., Iseppon A., Brockmann L., Xu H., Palm N.W., de Zoete M.R., Licona-Limón P., Paiva R.S., Ching T., et al. 2015. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 523:221–225. 10.1038/nature14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway N.L., Doitsh G., Monroe K.M., Yang Z., Muñoz-Arias I., Levy D.N., and Greene W.C.. 2015. Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells. Cell Reports. 12:1555–1563. 10.1016/j.celrep.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K., Laurence A., Yang X.-P., Hirahara K., and O’Shea J.J.. 2011. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 32:395–401. 10.1016/j.it.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O., Yazdi A.S., Thomas C.J., Masin M., Heinz L.X., Guarda G., Quadroni M., Drexler S.K., and Tschopp J.. 2012. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 36:388–400. 10.1016/j.immuni.2012.01.018 [DOI] [PubMed] [Google Scholar]
- Harbour S.N., Maynard C.L., Zindl C.L., Schoeb T.R., and Weaver C.T.. 2015. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA. 112:7061–7066. 10.1073/pnas.1415675112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K., Duarte J.H., Veldhoen M., Hornsby E., Li Y., Cua D.J., Ahlfors H., Wilhelm C., Tolaini M., Menzel U., et al. 2011. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12:255–263. 10.1038/ni.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C.S., Macatonia S.E., Tripp C.S., Wolf S.F., O’Garra A., and Murphy K.M.. 1993. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 260:547–549. 10.1126/science.8097338 [DOI] [PubMed] [Google Scholar]
- Hu W., Troutman T.D., Edukulla R., and Pasare C.. 2011. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity. 35:1010–1022. 10.1016/j.immuni.2011.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra S., Martínez-López M., Khouili S.C., Enamorado M., Cueto F.J., Conde-Garrosa R., Del Fresno C., and Sancho D.. 2016. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1+ Dendritic Cells. Immunity. 45:847–860. 10.1016/j.immuni.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., and Medzhitov R.. 2015. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16:343–353. 10.1038/ni.3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., and Pasare C.. 2017. Innate Control of Adaptive Immunity: Beyond the Three-Signal Paradigm. J. Immunol. 198:3791–3800. 10.4049/jimmunol.1602000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins M.K., and Moon J.J.. 2012. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 188:4135–4140. 10.4049/jimmunol.1102661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Huston G., and Swain S.L.. 2003. IL-7 promotes the transition of CD4 effectors to persistent memory cells. J. Exp. Med. 198:1807–1815. 10.1084/jem.20030725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.M., Hu W., Troutman T.D., Jennings M., Brewer T., Li X., Nanda S., Cohen P., Thomas J.A., and Pasare C.. 2014. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. U S A. 111:775–780. 10.1073/pnas.1320294111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L., Ibrahim A.S., Xu X., Farber J.M., Avanesian V., Baquir B., Fu Y., French S.W., Edwards J.E. Jr., and Spellberg B.. 2009. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 5:e1000703 10.1371/journal.ppat.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckey C.J., Bhattacharya D., Goldrath A.W., Weissman I.L., Benoist C., and Mathis D.. 2006. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 103:3304–3309. 10.1073/pnas.0511137103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod M.K.L., Kappler J.W., and Marrack P.. 2010. Memory CD4 T cells: generation, reactivation and re-assignment. Immunology. 130:10–15. 10.1111/j.1365-2567.2010.03260.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S., Newton K., Monack D.M., Vucic D., French D.M., Lee W.P., Roose-Girma M., Erickson S., and Dixit V.M.. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 430:213–218. 10.1038/nature02664 [DOI] [PubMed] [Google Scholar]
- Martin B.N., Wang C., Zhang C.J., Kang Z., Gulen M.F., Zepp J.A., Zhao J., Bian G., Do J.S., Min B., et al. 2016. T cell-intrinsic ASC critically promotes T(H)17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 17:583–592. 10.1038/ni.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda J.L., Gapin L., Sydora B.C., Byrne F., Binder S., Kronenberg M., and Aranda R.. 2000. Systemic activation and antigen-driven oligoclonal expansion of T cells in a mouse model of colitis. J. Immunol. 164:2797–2806. 10.4049/jimmunol.164.5.2797 [DOI] [PubMed] [Google Scholar]
- McGeachy M.J., Bak-Jensen K.S., Chen Y., Tato C.M., Blumenschein W., McClanahan T., and Cua D.J.. 2007. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 8:1390–1397. 10.1038/ni1539 [DOI] [PubMed] [Google Scholar]
- O’Shea J.J., and Paul W.E.. 2010. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 327:1098–1102. 10.1126/science.1178334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostanin D.V., Bao J., Koboziev I., Gray L., Robinson-Jackson S.A., Kosloski-Davidson M., Price V.H., and Grisham M.B.. 2009. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 296:G135–G146. 10.1152/ajpgi.90462.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.H., Lo Y.-C., Lin S.-C., Wang L., Yang J.K., and Wu H.. 2007. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25:561–586. 10.1146/annurev.immunol.25.022106.141656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N., Bardhan K., Weaver J., Herbel C., Seth P., Li L., and Boussiotis V.A.. 2016. The role of metabolic reprogramming in T cell fate and function. Curr. Trends Immunol. 17:1–12. [PMC free article] [PubMed] [Google Scholar]
- Purton J.F., Tan J.T., Rubinstein M.P., Kim D.M., Sprent J., and Surh C.D.. 2007. Antiviral CD4+ memory T cells are IL-15 dependent. J. Exp. Med. 204:951–961. 10.1084/jem.20061805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch I., Deets K.A., Ji D.X., von Moltke J., Tenthorey J.L., Lee A.Y., Philip N.H., Ayres J.S., Brodsky I.E., Gronert K., and Vance R.E.. 2017. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 46:649–659. 10.1016/j.immuni.2017.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F. 2016. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu. Rev. Immunol. 34:317–334. 10.1146/annurev-immunol-032414-112056 [DOI] [PubMed] [Google Scholar]
- Shao W., Yeretssian G., Doiron K., Hussain S.N., and Saleh M.. 2007. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 282:36321–36329. 10.1074/jbc.M708182200 [DOI] [PubMed] [Google Scholar]
- Shen C.-H., Talay O., Mahajan V.S., Leskov I.B., Eisen H.N., and Chen J.. 2010. Antigen-bearing dendritic cells regulate the diverse pattern of memory CD8 T-cell development in different tissues. Proc. Natl. Acad. Sci. USA. 107:22587–22592. 10.1073/pnas.1016350108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., and Shao F.. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 514:187–192. 10.1038/nature13683 [DOI] [PubMed] [Google Scholar]
- Shimada K., Porritt R.A., Markman J.L., O’Rourke J.G., Wakita D., Noval Rivas M., Ogawa C., Kozhaya L., Martins G.A., Unutmaz D., et al. 2018. T-Cell-Intrinsic Receptor Interacting Protein 2 Regulates Pathogenic T Helper 17 Cell Differentiation. Immunity. 49:873–885.e7. 10.1016/j.immuni.2018.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B., Lehr H.A., Fantuzzi G., and Dinarello C.A.. 2001. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA. 98:13249–13254. 10.1073/pnas.231473998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., and Mesirov J.P.. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo N.J., Pagán A.J., Taylor J.J., Nelson R.W., Linehan J.L., Ertelt J.M., Huseby E.S., Way S.S., and Jenkins M.K.. 2013. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. 153:785–796. 10.1016/j.cell.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan V.K., Hodges K., and Hecht G.. 2009. Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat. Rev. Microbiol. 7:110–119. 10.1038/nrmicro2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waickman A.T., and Powell J.D.. 2012. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 249:43–58. 10.1111/j.1600-065X.2012.01152.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Dillon C.P., Shi L.Z., Milasta S., Carter R., Finkelstein D., McCormick L.L., Fitzgerald P., Chi H., Munger J., and Green D.R.. 2011. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 35:871–882. 10.1016/j.immuni.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang R., Lu G., Shen Y., Peng L., Zhu C., Cui M., Wang W., Arnaboldi P., Tang M., et al. 2013. T cell–derived inducible nitric oxide synthase switches off Th17 cell differentiation. J. Exp. Med. 210:1447–1462. 10.1084/jem.20122494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N., Shalek A.K., Gaublomme J.T., Jin H., Lee Y., Awasthi A., Wu C., Karwacz K., Xiao S., Jorgolli M., et al. 2013. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 496:461–468. 10.1038/nature11981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Liu S.-Q., Li C., Lykken E., Jiang S., Wong E., Gong Z., Tao Z., Zhu B., Wan Y., and Li Q.-J.. 2016. MicroRNA-23a Curbs Necrosis during Early T Cell Activation by Enforcing Intracellular Reactive Oxygen Species Equilibrium. Immunity. 44:568–581. 10.1016/j.immuni.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Yamane H., and Paul W.E.. 2010. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28:445–489. 10.1146/annurev-immunol-030409-101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski C.E., Mele F., Aschenbrenner D., Jarrossay D., Ronchi F., Gattorno M., Monticelli S., Lanzavecchia A., and Sallusto F.. 2012. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 484:514–518. 10.1038/nature10957 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
lists primers used in this study.
lists reagents.
is a list of genes differentially expressed between Lm-primed and Cr-primed CD90+CFSE− CD4 T cells, along with normalized expression values.
is a list of genes differentially expressed between ppTh17, cdTh17, and ex vivo tdT+ cells.
shows significantly enriched functional annotations in differentially expressed genes in ppTh17/ex vivo tdT+ or cdTh17 cells, determined by DAVID.
lists normalized expression values for genes shown in Fig. 2.
is a list of differentially expressed genes in ppTh17 and cdTh17 cells that are enriched in memory or effector T cell signatures, determined by GSEA.
is a list of differentially expressed genes in WT and Casp1Δ10 Cr-specific Th cells.
Data Availability Statement
Illustrations were created with Biorender (https://biorender.com). Matlab code used for RNA-seq analysis is described in RNA-seq analysis and is available on request. The sequencing data reported in this paper have been deposited to the Gene Expression Omnibus with the accession nos. GSE141054, GSE141061, and GSE141062.