

Supplemental Digital Content is available in the text.
Key Words: acalabrutinib, B-cell malignancies, CAR T-cell therapy, ibrutinib, liso-cel
Abstract
Chimeric antigen receptor (CAR) T-cell therapy is a promising treatment for patients with CD19+ B-cell malignancies. Combination strategies that improve CAR T-cell potency, limit tumor environment–mediated immune dysfunction, and directly reduce tumor burden may increase the potential for durable clinical benefit of CAR T-cell therapy. Lisocabtagene maraleucel (liso-cel) is a product therapy candidate being tested in patients with relapsed/refractory non-Hodgkin lymphoma or chronic lymphocytic leukemia. This study assessed the in vitro and in vivo functionality of CAR T cells transduced to express the anti-CD19 CAR of liso-cel in combination with ibrutinib or acalabrutinib. In prolonged stimulation assays, the presence of ibrutinib or acalabrutinib improved the CAR T-cell effector function. RNA-Seq analysis and surface marker profiling of these CAR T cells treated with ibrutinib but not acalabrutinib revealed gene expression changes consistent with skewing toward a memory-like, type 1 T-helper, Bruton tyrosine kinase phenotype. Ibrutinib or acalabrutinib improved CD19+ tumor clearance and prolonged survival of tumor-bearing mice when used in combination with CAR T cells. A combination of the defined cell product therapy candidate, liso-cel, with ibrutinib or acalabrutinib is an attractive approach that may potentiate the promising clinical responses already achieved in CD19+ B-cell malignancies with each of these single agents.
Molecular engineering of chimeric antigen receptors (CARs) generally unifies the antigen specificity of antibody single-chain variable fragment domains together with select components of the intracellular T-cell receptor signaling complexes. Transduction of CARs into T cells can redirect cytolytic activity to antigen-expressing targets, which may result in clearance of both tumor and normal cells that display the surface-expressed antigen of interest.1–3 To date, clinical responses with anti-CD19 CAR T cells in non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL) have been encouraging in patients with relapsed/refractory disease;4,5 however, opportunities exist to extend the duration of CAR T-cell–mediated responses.6–10 Lisocabtagene maraleucel (liso-cel) is an investigational, defined composition, 4-1BB-containing, anti-CD19 CAR T-cell product administered at a target dose of CD4+/CD8+ CAR T cells and is currently being investigated in phase 1/2 trials for the treatment of NHL and CLL (ClinicalTrials.gov NCT02631044, NCT03331198).4 In this study, we used cells expressing the anti-CD19 CAR of liso-cel, which includes CD3ζ signaling and 4-1BB costimulatory endodomains (Supplemental Digital Content 1a, http://links.lww.com/JIT/A549), to test whether a combinatorial approach utilizing ibrutinib or acalabrutinib would be suitable with this anti-CD19 CAR T-cell therapy.
During the pathogenesis of NHL and CLL, lymph node tumor microenvironments with immunosuppressive features emerge through malignant B-cell and stromal-cell interactions, thus limiting antitumor immune surveillance.11–13 In CLL, disease influence extends to the periphery, resulting in dysfunctional circulating T cells that are prognostic of disease progression.14–16 The immunosuppression associated with these diseases highlights potential adverse functional effects on adoptive T-cell therapeutics as well as an opportunity to overcome these barriers through combinatorial strategies.
Notably, Fraietta et al11 demonstrated that patients with CLL treated with the targeted inhibitor ibrutinib exhibited restored T-cell function relative to untreated patients. Ibrutinib is an irreversible inhibitor of Bruton tyrosine kinase (BTK) that has been approved for the treatment of several B-cell malignancies and for the graft-vs-host disease. Ibrutinib has shown beneficial activity in patients with the activated B cell–like subtype of diffuse large B-cell lymphoma (DLBCL), with best responses evidenced in patients with chronic active B-cell receptor signaling.17 Acalabrutinib is a highly selective, covalent BTK inhibitor in clinical development for the treatment of B-cell malignancies.18–20 It has recently received accelerated approval from the US Food and Drug Administration for the treatment of patients with mantle cell lymphoma who have received at least 1 prior therapy. Acalabrutinib has fewer off-target activities than ibrutinib; however, both agents influence the activity of kinases that could affect CAR T-cell performance.19,21 In a small cohort of patients with CLL treated with either acalabrutinib or ibrutinib, markers of exhaustion and tumor-mediated immunosuppression were reduced, and ibrutinib treatment was associated with increased effector-memory CD4 and CD8 T cells in some patients.22 Treatment with ibrutinib has also been shown to reduce immunosuppressive cell populations in patients, which is another factor that may influence T-cell functionality.23–28
One Tec-family kinase inhibited by ibrutinib is interleukin (IL) 2–inducible T-cell kinase (ITK), which is highly expressed in both CD4+ and CD8+ T cells and affects downstream T-cell receptor signaling.29 Treatment with ibrutinib was reported to skew the development of T cells toward a type 1 T-helper (Th1) immune phenotype through ITK inhibition during ex vivo treatments as well as murine models of antigen-mediated or pathogen-mediated T-cell skewing.30 A recent study from the same group showed that, in patients with CLL treated with ibrutinib, no change was observed in the percentages of Th1 or type 2 T-helper (Th2) cells, although an increase in IL-17-producing CD4 cells was noted.22 Acalabrutinib does not inhibit ITK and is less potent against other T cell–related kinases, including Src-family kinases as well as Tec-family kinases and Janus kinase 2, when tested head to head against ibrutinib.18,19 However, after acalabrutinib treatment in patients with CLL, reductions in interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and IL-4–producing CD4 cells were observed22 as well as decreases in plasma TNF-α, thymus and activation-regulated chemokine, and IL-10.20 Therefore, these 2 agents may have different in vivo effects, perhaps due to differential off-target kinase inhibition at clinically relevant concentrations.
No studies to date have been conducted to test the hypothesis that Th1 skewing and ITK inhibition are required for ibrutinib enhancement of T-cell function. As multiple potential mechanisms could influence the development of T cells (directly through cell-autonomous changes and indirectly through BTK inhibition in myeloid cells that establish the tumor environment), questions remain on the importance of off-target kinase activity in the mechanisms of ibrutinib enhancement of CAR T-cell function. The present study has been conducted to differentiate direct effects on the intrinsic biology of CAR T cells from pleiotropic effects on the tumor environment that act primarily in vivo. In addition, by comparing acalabrutinib, a highly selective agent, with ibrutinib, the relative contribution of ibrutinib off-target kinase activities, in particular, ITK inhibition, can be evaluated in CAR T-cell functional development.
Combinatorial therapies may uniquely influence the intrinsic functionality of the CAR T cell (positively or negatively) in addition to directly targeting tumor growth or potentiating an endogenous immune response. BTK inhibitors are becoming standard-of-care therapeutics in many B-cell malignancies. We evaluated to what extent these molecules influenced the CAR T-cell functionality of a CD19-targeted 4-1BB–based CAR composition with a defined CD4:CD8 ratio. Our results demonstrate that both ibrutinib and acalabrutinib positively influence CAR T cells in vitro and in vivo antitumor functionalities following long-term treatment. These results indicate the potential suitability of combination therapy using either a BTK inhibitor agent with CAR T-cell therapies.
MATERIALS AND METHODS
CAR T Cells
T cells obtained from peripheral blood apheresis samples from consenting adults (healthy human donors or patients with DLBCL) were engineered according to a process similar to that developed for liso-cel production (Supplemental Digital Content 1b, http://links.lww.com/JIT/A549).
CD4+ and CD8+ T-cell populations were isolated from the samples via immunoaffinity-based enrichment and separately activated, transduced with a lentiviral vector encoding the CD19-specific CAR, and expanded. Cells were then cryopreserved, stored at temperatures <−130°C, and thawed before use. To account for any variability in transduction efficiency, numbers of transduced cells were normalized across all experiments. Cells were used at a target CAR+CD4+:CAR+CD8+ ratio of 1:1 per the clinical formulation for liso-cel.
The research described in this publication did not involve a clinical investigation in human participants nor did it involve the use or collection of any identifiable personal information; therefore, institutional review board (IRB) review and approval was not required for the performance of the research. Human materials used in this research were received by the researchers in a fully de-identified manner from commercial repositories (Hemacare, Van Nuys, CA) or under unrelated IRB–approved clinical studies from adults who consented to the testing of their donated samples for future research purposes.
In Vitro Cytolytic CAR T-Cell Assay
A fluorescent CD19 expression tumor cell line was established using K562 cells, which are resistant to BTK inhibitor–mediated growth inhibition. K562 cells were transduced using a lentivirus vector containing the coding sequence for human CD19. CD19+ K562 cells were sorted twice for purity by flow cytometry. K562.CD19 cells were then transduced with a separate lentivirus vector containing NucLight Red (Essen BioScience, Ann Arbor, MI) (K562.CD19.NLR).
For each cell-sample preparation from individual donors, the cryopreserved CD4+ and CD8+ CAR-expressing T cells were thawed and plated in triplicate on Corning poly-D-lysine–coated plates (Fisher Scientific, Hampton, NH). K562.CD19.NLR tumor cells were coincubated at 37°C and 5% CO2, with the CAR-expressing effector cells at an effector:target ratio of 2.5:1. Titrations of ibrutinib (Selleck Chemicals, Houston, TX) and acalabrutinib (Acerta Pharma, Redwood City, CA) were evaluated encompassing a range of concentrations above and below the total serum maximum concentration (227 nM and 1.8 µM, respectively), as previously reported.19,31 CAR T-cell cytolytic activity was measured by assessing the loss of red fluorescent target cells using an IncuCyte Live Cell Analysis System (Essen BioScience). Normalized target cell numbers were generated by dividing target cell counts to cell counts at the start of each culture. To capture the cytolytic activity of CAR T cells against target cells over time, the area under the curve (AUC) was calculated. To generate the killing index, the 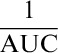 values from treatment conditions were normalized using GraphPad Prism 7 v7.02 (GraphPad Software, La Jolla, CA). Baselines of 0 and 100 were established using
values from treatment conditions were normalized using GraphPad Prism 7 v7.02 (GraphPad Software, La Jolla, CA). Baselines of 0 and 100 were established using 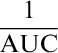 values from target cells alone and CAR+ T cells cocultured with target cells in vehicle control, respectively. Values above or below 100 indicate increased or decreased cytolytic activity over the duration of the assay, respectively.
values from target cells alone and CAR+ T cells cocultured with target cells in vehicle control, respectively. Values above or below 100 indicate increased or decreased cytolytic activity over the duration of the assay, respectively.
Agonistic liso-cel Antibody Stimulation Assays (Short-Term and Long-Term)
CAR T cells were stimulated with 30 µg/mL plate-coated anti–liso-cel agonistic antibody (Juno Therapeutics Inc.) in the presence of ibrutinib (0.5–500 nM) or acalabrutinib (50–5000 nM). After 2 days, culture supernatants were taken for cytokine analysis. For the long-term stimulation assay, CAR T cells were treated with 30 µg/mL plate-coated agonistic antibody in the presence of ibrutinib or acalabrutinib for 6 days. After 6 days, CAR T cells were washed twice in media and replated in new media without BTK inhibitor on new agonistic-antibody–coated plates (30 µg/mL). The culture supernatant was sampled at 24 hours for cytokine analysis. Day 6 treated CAR T cells were also utilized in a cytolytic assay with K562.CD19.NLR tumor cells at an effector:target ratio of 2.5:1.
In Vitro Immunophenotyping
CAR T cells were analyzed by flow cytometry (BD LSRFortessa; BD Biosciences, San Jose, CA). Cells were stained for surface markers CD69, CD107a, programmed cell death-1 (PD-1), CD25, CD38, CD39, CD95, CD62L, CCR7, and CD45RO (BD Biosciences). T-distributed stochastic neighbor embedding (t-SNE) high-dimensional analysis was performed using FlowJo v10.1 software (FlowJo, Ashland, OR).
Cytokine Measurements
CAR T cells were stimulated without inhibitor or in the presence of ibrutinib or acalabrutinib as previously shown. Cytokines were measured from culture supernatants at indicated timepoints using Meso Scale Discovery cytokine kits (Meso Scale Diagnostics, Rockville, MD).
Serial Stimulations
A serial stimulation assay was used to evaluate the ability of CAR T cells cultured under various conditions to expand ex vivo following multiple rounds of antigen stimulation, which can correlate with in vivo function, fitness, and/or persistence capacity.1 Concentrations of ibrutinib (5, 50, and 500 nM) or acalabrutinib (15, 158, and 1581 nM) were added to CAR-expressing cells plated in triplicate at 1×105 cells/well on 96-well poly-D-lysine–coated plates. Irradiated targets (K562.CD19.NLR) were added at an effector:target ratio of 2.5:1. Live cell numbers were counted every 3–4 days using acridine orange and propidium iodide dye on the Celigo imaging platform (Nexcelom Bioscience, Lawrence, MA). To reset culture conditions, 1×105 CAR T cells from each counted well were replated in fresh media with BTK inhibitor and newly thawed target cells.
Library Preparation and RNA-Seq
RNA was isolated from CAR-expressing T cells (donors 1–3) treated for 18 days in a serial stimulation assay in the presence of ibrutinib (50 or 500 nM), acalabrutinib (158 or 1581 nM), or control (0 nM). RNA isolation was performed using the RNeasy Micro Kit (Qiagen, Valencia, CA). Strand-specific, bar-coded cDNA libraries were prepared using the NEBNext Ultra RNA Library Prep Kit with the NEBNext Poly(A) mRNA Magnetic Isolation Module for mRNA enrichment (New England Biolabs, Ipswich, MA). Samples were sequenced on a NextSeq. 500 (Illumina, San Diego, CA) to yield 75-base, single-end reads. Demultiplexing and FASTQ generation were performed using Illumina’s bcl2fastq (v2.17.1.14) software.
Transcriptomic Analyses
RNA-Seq reads were mapped to the human genome (GRCh38) and aligned to the GENCODE Release 24 gene model using the OSA aligner and Array Studio software (OmicSoft, Cary, NC).32,33 Gene-level differential expression analyses were performed in R (version 3.4) using the DESeq. 2 package (version 1.16.1)34 by comparing ibrutinib-treated samples with reference controls taking donor, treatment, and batch into account and by comparing acalabrutinib-treated samples with reference controls taking donor and treatment into account. Before differential expression analysis, the gene set was filtered for protein-coding genes, excluding genes with zero counts across all samples. Differentially expressed genes were identified by imposing a log2 fold change (FC) cutoff of±0.5 and a Benjamini-Hochberg adjusted false discovery rate (FDR) cutoff of 0.05. A gene ontology (pathway) analysis was performed in Ingenuity Pathway Analysis (Qiagen) using an absolute log2 FC cutoff of 0.5 and an FDR cutoff of 0.05.
Nalm-6 Xenograft Tumor Model
A Nalm-6 disseminated BTK inhibitor–resistant CD19+ tumor model was developed for evaluating CAR T-cell function in vivo (Supplemental Digital Content 2, http://links.lww.com/JIT/A549). NOD.Cg-PrkdcscidIL-2rgtm1Wjl/SzJ mice (NSG) (Jackson Laboratory, Bar Harbor, ME) were injected intravenously with 5×105 Nalm-6-FFLuc tumor cells on day 0. For the initial ibrutinib studies, starting at day 4 posttumor implant, mice were dosed orally daily with 25 mg/kg ibrutinib or 0.5% methyl cellulose vehicle. For studies with ibrutinib and acalabrutinib, mice received an oral loading dose (25 mg/kg) at day 4 posttumor implant followed by continuous administration of the drug in drinking water starting on day 5 (0.15 mg/mL of ibrutinib or acalabrutinib in 2% hydroxypropyl-β-cyclodextrin in sterile water pH 7). On the basis of average daily water consumption in mice, this is estimated to deliver a dose of 25 mg/kg/d.35 On day 5, a suboptimal dose of 5×105 CAR T cells was injected intravenously into mice. A suboptimal dose was selected to enable the interpretation of a negative or positive BTK inhibitor combination effect. Tumor burden was monitored by bioluminescence imaging (IVIS Spectrum; PerkinElmer, Waltham, MA) following intraperitoneal injection with luciferin (15 µg/g) (PerkinElmer, Norwalk, CT). Tissues were harvested at day 7, 12, and 19 post-CAR T-cell transfer. Levels of the CAR T-cell surface markers CD45RA, CD62L, CD154, CXCR3, CXCR4, and PD-1 were assessed by flow cytometry using appropriate antibodies. The t-SNE analysis was performed using FlowJo software.
All animal studies were strictly conducted in accordance with Omeros’ Animal Care and Use Committee (protocol 15-06). Mice were housed communally, 4–5 per microisolator cage, at Omeros Corporation (Seattle, WA) for the study duration unless precluded by study activities. Primary enclosures are as specified in the US Department of Agriculture Animal Welfare Act (9 CFR, parts 1, 2, and 3) and as described in the Guide for the Care and Use of Laboratory Animals. Food (PicoLab Rodent diet 20–5053) and water were freely available to mice, except during study activities. Humane endpoints were used during the animal survival study. Because untreated leukemia causes hind-limb paralysis, weight loss, and death, mice were monitored for clinical signs and weight approximately twice weekly. Mice were euthanized by carbon dioxide inhalation upon hind-limb weakness or paralysis, when moribund or with hunched back, when dermatitis developed, or when body weight was 20% less than baseline. The primary endpoint was overall survival, defined as the time from the first dose of CAR T cells to death. The secondary endpoint was tumor-growth inhibition monitored by bioluminescence imaging. The exploratory endpoint was in-life quantitation of CAR T cells in mouse blood, spleen, and bone marrow.
Statistical Analyses
Statistical analysis and graph generation for the animal studies were performed using GraphPad Prism 7 v7.02 (GraphPad Software, La Jolla, CA). Data are presented as means±standard error of the mean (SEM). When appropriate, a Mann-Whitney U test and a 1-way or 2-way analysis of variance were used to compare experimental groups. The log-rank (Mantel-Cox) test was used to compare Kaplan-Meier curves. A difference was considered significant if P-value <0.05.
RESULTS
Off-BTK Effects of Ibrutinib and Acalabrutinib in Short-Term Stimulation With Agonistic Antibody Influence Anti-CD19 CAR T-Cell Cytokine Secretion
After confirming minimal expression of BTK in CAR T cells (Supplemental Digital Content 3, http://links.lww.com/JIT/A549), in vitro studies were initiated to understand whether the off-BTK activities of ibrutinib and acalabrutinib19,36 influenced intrinsic anti-CD19 CAR T-cell functionality. Because BTK inhibitors are likely to affect malignant B-cell biology, initial studies were conducted independent of CD19-expressing target cells. An agonistic, anti–liso-cel antibody was used for CAR T-cell stimulation in the presence of either ibrutinib or acalabrutinib. Following 48 hours of short-term stimulation, CAR T-cell effector cytokine production was measured. Ibrutinib treatment dose-dependently decreased CAR T-cell cytokine production with the highest concentration tested (500 nM), decreasing IFN-γ production by a mean of 36% (92 ng/mL) (P<0.0001) across 3 different anti-CD19 CAR T-cell donors. Diminished production of IL-2 and TNF-α was also observed in these experimental conditions (Fig. 1A, Supplemental Digital Content 4a, http://links.lww.com/JIT/A549). Acalabrutinib had an inconsistent effect on reducing cytokine production across CAR T-cell donors and concentrations (Fig. 1B, Supplemental Digital Content 4b, http://links.lww.com/JIT/A549).
FIGURE 1.
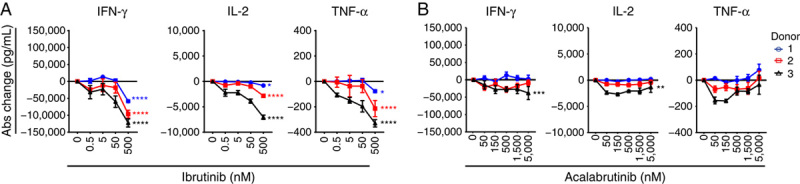
Off-BTK effects of ibrutinib and acalabrutinib in short-term stimulation with agonistic antibody contextually influence anti-CD19 CAR T cytokine secretion. Absolute change in cytokine concentrations 2 days after stimulation with plate-bound 30 µg/mL agonistic antibody of CAR T cells treated with ibrutinib (A) or acalabrutinib (B) compared with untreated controls. Data from 2 independent experiments and 3 different CAR T-cell donors (mean±SEM). Statistically significant differences from each donor are indicated for ibrutinib 500 nM and acalabrutinib 5000 nM as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. Abs indicates absolute; BTK, Bruton tyrosine kinase; CAR, chimeric antigen receptor; IFN-γ, interferon-gamma; IL-2, interleukin 2; TNF-α, tumor necrosis factor-alpha.
Short-Term CAR T-Cell Function Against CD19+ Tumor Cells Is Not Negatively Affected by Ibrutinib or Acalabrutinib
After observing the intrinsic effects of ibrutinib or acalabrutinib on CAR T cells using agonistic antibody stimulation, we wanted to understand whether BTK inhibitor influenced CAR T-cell function in an activation model using CD19-expressing tumor cells. K562 cells were selected because their growth was previously shown to be insensitive to the effects of ibrutinib,26 a result that was reproduced here together with acalabrutinib (Fig. 2A). CAR T cells were stimulated with tumor cells in the presence of ibrutinib or acalabrutinib for 2 days, and secreted cytokine concentrations were measured. Although the magnitude of responses differed between donors, ibrutinib did not inhibit cytokine production at concentrations <500 nM (Fig. 2B, Supplemental Digital Content 4c, http://links.lww.com/JIT/A549). However, at ibrutinib 500 nM, an inhibitory effect was observed on IL-2 secretion in all 3 donors, with a mean decrease of 19.6% (1200 pg/mL; P<0.05; Fig. 2B). In contrast, acalabrutinib modestly increased cytokine production (Fig. 2C, Supplemental Digital Content 4d, http://links.lww.com/JIT/A549) in some donors at the highest concentration tested (5000 nM).
FIGURE 2.
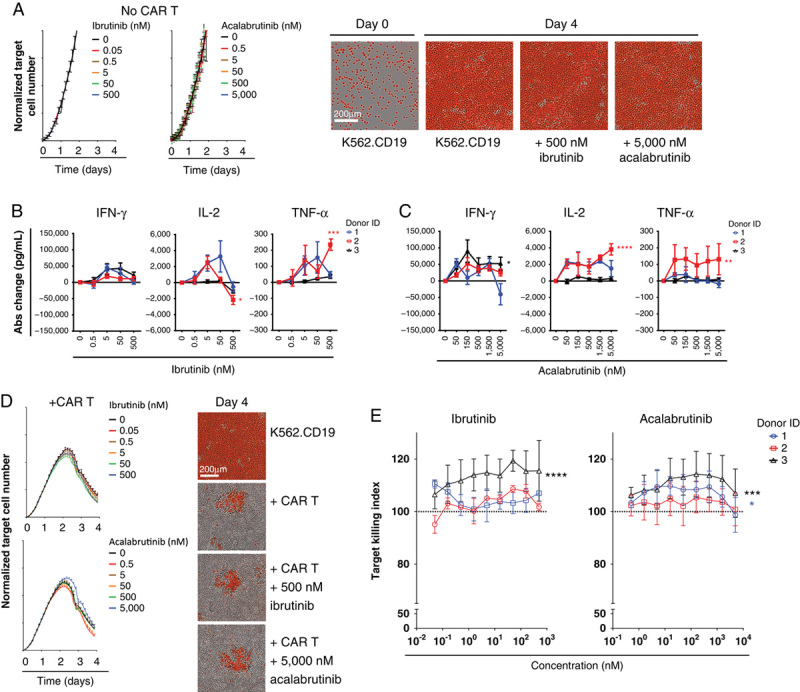
Cytotoxic function and cytokine production of chimeric antigen receptor (CAR) T cells were not affected by Bruton tyrosine kinase inhibitor when stimulated with CD19+ tumor cells. A, Target cell numbers normalized to time 0, from triplicate wells without CAR T cells with ibrutinib or acalabrutinib (mean±SEM). Representative images show red target cells at day 0 and day 4 of treatment. Absolute (Abs) change in cytokine concentration compared with untreated controls of interferon-gamma (IFN-γ), interleukin 2 (IL-2), and tumor necrosis factor-alpha (TNF-α) 2 days after stimulation with target cells at an effector:target ratio of 2.5:1 and ibrutinib (B) or acalabrutinib (C). Data from 2 independent experiments and 3 CAR T-cell donors (mean±SEM). Statistical significance from each donor indicated for acalabrutinib 5000 nM and ibrutinib 500 nM. D, Target cell numbers (effector:target ratio, 2.5:1) normalized to time 0, from triplicate wells cocultured with CAR T cells with ibrutinib or acalabrutinib (mean±SEM). Representative images show red target cells on day 4 of the cytotoxic assay. E, Dose effect of ibrutinib or acalabrutinib on cytolytic activity (effector:target ratio, 2.5:1) of CAR T cells, normalized to untreated control (100). Data from 3 independent experiments and 3 CAR T-cell donors (mean±SEM). Statistically significant differences are indicated as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
To further understand the BTK inhibitor effect on cytolytic activity, an in vitro cytolytic assay was performed against target K562.CD19.NLR tumor cells with ibrutinib or acalabrutinib added at the start of cocultures. After an initial period of target cell growth, CAR T cells from all donors reduced fluorescent tumor cell numbers over a period of 4 days, demonstrating effective killing in the assay (Fig. 2D). High concentrations of ibrutinib (500 nM) or acalabrutinib (5000 nM) treatment did not significantly affect the in vitro cytolytic activity of CAR T cells. Modest increased target cell killing was observed in donor 3 when treated with ibrutinib (P<0.0001) and acalabrutinib (P<0.001; Fig. 2E).
Long-Term CAR T-Cell Exposure to Ibrutinib or Acalabrutinib With Agonistic Antibody Stimulation Influences T-Cell Activation and Enhances Cytokine Secretion
Next, long-term treatment effects of both ibrutinib and acalabrutinib on anti-CD19 CAR T-cell function were assessed following 6 days of chronic anti–liso-cel antibody stimulation. This chronic stimulation of CAR T cells reduces their ability to produce cytokines on restimulation compared with the freshly thawed cells in the previous short-term stimulation assay (Supplemental Digital Content 4e, http://links.lww.com/JIT/A549). Following the 6-day stimulation period in the presence or absence of BTK inhibitor, CAR T cells were washed, counted, replated, and restimulated with agonistic antibody in the absence of further BTK inhibitor treatment. After 24 hours of restimulation, CAR T-cell cytokine production was measured. On restimulation, ibrutinib-treated and acalabrutinib-treated CAR T cells secreted more IFN-γ, IL-2, and TNF-α relative to control untreated cells. Improved cytokine production for both BTK inhibitors occurred generally in a dose-dependent manner. Across 3 different CAR T-cell donors, IFN-γ production increased by a mean of 86% (10,000 pg/mL; P<0.0001) and 142% (15,000 pg/mL; P<0.0001) for ibrutinib (500 nM) and acalabrutinib (1500 nM), respectively (Figs. 3A, B, Supplemental Digital Content 4f and g, http://links.lww.com/JIT/A549). To gain further insight into how BTK inhibitors improved anti-CD19 CAR T-cell cytokine production following chronic stimulation, phenotypic markers associated with T-cell activation and differentiation state were assessed by flow cytometry. Multivariate t-SNE analysis from 3 different donors revealed that ibrutinib or acalabrutinib treatment increased the percentage of CD8+ CAR T cells that were positive for the following activation markers: CD69, CD38, PD-1, and CD107a relative to control conditions (Supplemental Digital Content 5, http://links.lww.com/JIT/A549).
FIGURE 3.
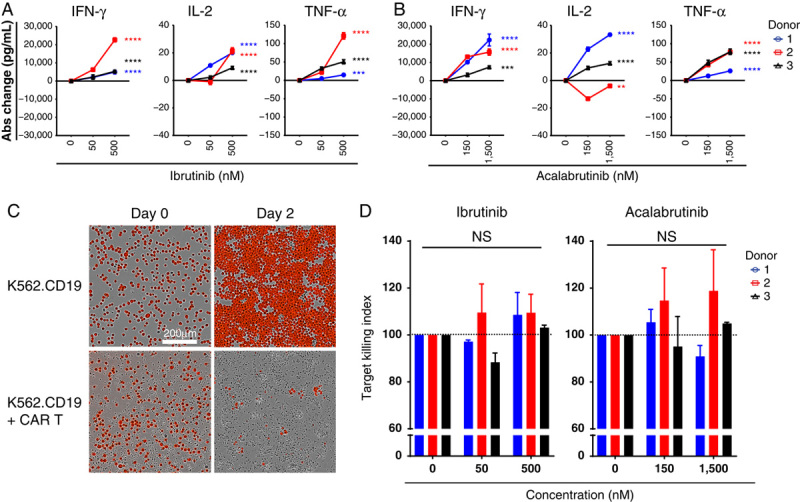
Effects of ibrutinib and acalabrutinib in long-term stimulation with agonistic antibody on CAR T-cell cytokine secretion and cytotoxic function. Before restimulation, CAR T cells were stimulated with plate-bound 30 µg/mL agonistic antibody for 6 days with ibrutinib (A) or acalabrutinib (B). Cells were then restimulated for 24 hours in fresh plate-bound 30 µg/mL agonistic antibody in the absence of BTK inhibitor. Abs change in cytokines compared with untreated controls was measured. Representative data from 2 independent experiments and 3 different CAR T-cell donors (mean±SEM). Statistical significance for each donor indicated for acalabrutinib 1500 nM and ibrutinib 500 nM. C, Day 6 treated CAR T cells were also cocultured with K562.CD19.NLR (NucLight Red K562.CD19) target cells at an effector:target ratio of 2.5:1 in the absence of additional BTK inhibitor. Representative images showing red target cells at day 0 and day 2 of the cytotoxic assay. D, Cytolytic activity of CAR T cells in the presence of ibrutinib or acalabrutinib, normalized to untreated control (100%). Data from 2 independent experiments and 3 different CAR T-cell donors (mean±SEM). Statistically significant differences are indicated as **P<0.01, ***P<0.001, and ****P<0.0001. Abs indicates absolute; BTK, Bruton tyrosine kinase; CAR, chimeric antigen receptor; IFN-γ, interferon-gamma; IL-2, interleukin 2; NS, not significant; TNF-α, tumor necrosis factor-alpha.
To further examine how prolonged ibrutinib and acalabrutinib treatment influenced anti-CD19 CAR T-cell function following 6 days of stimulation, an in vitro cytolytic assay was performed against target K562.CD19.NLR cells. Following chronic stimulation with the anti–liso-cel antibody in the presence of BTK inhibitor, CAR T cells were then cocultured with K562.CD19.NLR tumor cells in the absence of additional BTK inhibitor. Cytolytic activity was monitored over a period of 2 days (Fig. 3C). Normalized target cell counts using AUC calculations showed that the ability of CAR T cells to lyse K562.CD19.NLR cells was not altered by pretreatment with ibrutinib or acalabrutinib (Fig. 3D).
Ibrutinib or Acalabrutinib Minimally Affected Surface Activation and Memory Marker Expression During Early CAR T-Cell Activation
Given subtle changes in phenotype observed with ibrutinib and acalabrutinib during long-term agonistic stimulation, we investigated the potential effects of ibrutinib and acalabrutinib on CAR T-cell phenotype following activation by CD19+ tumor cells. A panel of activation and differentiation markers were evaluated on CAR+, CD4+, and CD8+ cells (from 3 donors) daily over a 4-day coculture period with K562.CD19 target cells. Following stimulation, neither ibrutinib nor acalabrutinib were observed to have significant effects on CD25, CD38, CD39, or CD95 expression (Supplemental Digital Content 6, http://links.lww.com/JIT/A549), nor were there effects on central or effector-memory subsets as assessed by the expression of CCR7 and CD45RA (Supplemental Digital Content 7, http://links.lww.com/JIT/A549). However, ibrutinib and acalabrutinib subtly decreased the percentage of CAR T cells expressing CD69, CD107a, and PD-1 from some donors (Supplemental Digital Content 8, http://links.lww.com/JIT/A549).
CAR T-Cell Counts Increased During Serial Stimulation in the Presence of Ibrutinib or Acalabrutinib
The increase in cytokine production in the presence of BTK inhibitor following treatment with the agonistic antibody for 6 days (Figs. 3A, B) warranted examination of whether BTK inhibitor could influence anti-CD19 CAR T-cell performance in a long-term serial stimulation assay. This assay differs from the 6-day chronic stimulation assay by the use of irradiated CD19-expressing tumor cells. Tumor cells are lysed by CAR T cells within 48 hours of the assay (data not shown), and this removal of antigen provides a period of rest for the CAR T cells that differs from the chronic stimulation provided by the agonistic antibody. Anti-CD19 CAR T cells generated from 3 donors were restimulated every 3–4 days with irradiated K562.CD19 target cells in the presence of either ibrutinib or acalabrutinib. Expansion kinetics were similar in all treatment groups after 3 rounds of stimulation (day 11), as observed by FC and the number of population doublings (Figs. 4A, B). However, after 5 rounds of stimulation (day 18), CAR T cell counts from donor 2 (treated with either ibrutinib or acalabrutinib at the highest concentrations) had increased significantly (P<0.0.05) relative to control cells (Figs. 4C, D). Furthermore, treatment with BTK inhibitor did not affect CAR T-cell viability or the ratio of CD4:CD8 (data not shown). When assessing cell counts across control conditions, cells derived from donors 2 and 3 had reduced expansion relative to donor 1 cells in this assay. Notably, these donors with lower proliferation benefited from ibrutinib and acalabrutinib treatment in this assay by experiencing an increase in proliferation.
FIGURE 4.
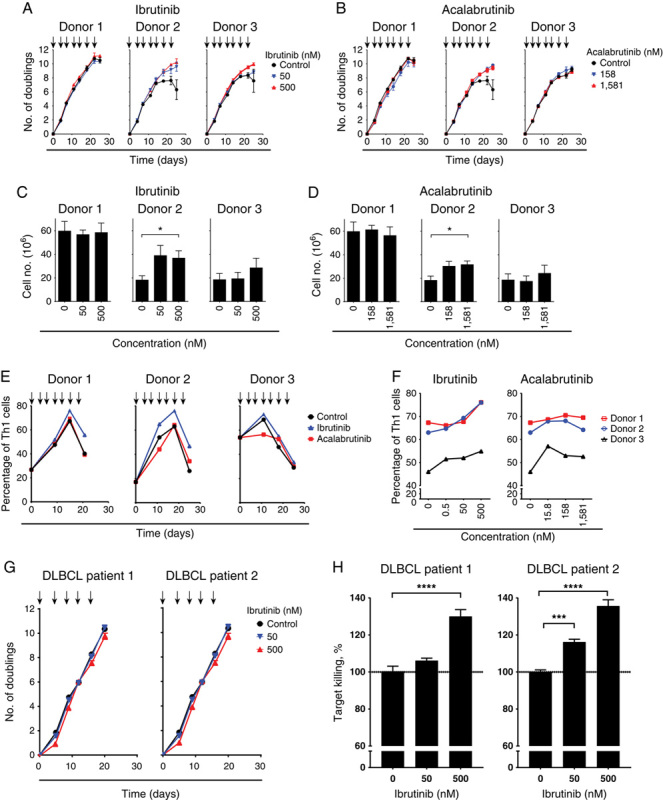
Bruton tyrosine kinase inhibitor enhances chimeric antigen receptor (CAR) T-cell expansion and the proportion of type 1 T-helper (Th1) cells in samples from some donors after serial stimulation. Representative plots showing a number of population doublings of CAR T cells from 3 donors stimulated in triplicate wells with irradiated target cells (NucLight Red K562.CD19) at an effector:target ratio of 2.5:1 together with ibrutinib (A) or acalabrutinib (B) during a 25-day serial stimulation. Arrows indicate the timepoint of each restimulation at which CAR T cells were counted and new target cells along with ibrutinib or acalabrutinib were added. Number of CAR T cells from 3 donors treated with ibrutinib (C) or acalabrutinib (D) for 18 days and after 5 rounds of restimulation, *P<0.05. Data from 2 independent experiments (mean±SEM). E, Percentage of Th1 (CXCR3+CRTH2−) cells within CD4+ CAR T cells over time during serial stimulations for 3 donors treated with ibrutinib (500 nM) or acalabrutinib (1581 nM). F, Percentage of Th1 cells within CD4+ CAR T cells after 18 days of serial stimulation and treatment with ibrutinib or acalabrutinib. Representative data from 2 independent experiments. G, Number of population doublings during the 21-day culture period for CAR T cells from patients with diffuse large B-cell lymphoma (DLBCL). Arrows indicate the timepoint of each restimulation at which CAR T cells were counted and new target cells along with ibrutinib were added. H, Cytotoxicity killing assay of K562.CD19.NLR target cells after 16 days of restimulation with CAR T cells from patients with DLBCL. Percentage of killing normalized to untreated control (100%). Data are shown as mean±SEM from replicate wells. Statistically significant differences are indicated as *P<0.05, ***P<0.001, and ****P<0.0001.
Long-Term Ibrutinib Treatment During Serial Stimulation of CAR T Cells Resulted in Greater Th1 Differentiation
The increased cell counts observed for 1 donor in the serial stimulation assay warranted additional phenotypic exploration. Ibrutinib has been observed to limit Th2 CD4 T-cell activation and proliferation in vitro and in mouse models through the inhibition of ITK.28 Acalabrutinib had no activity against ITK at concentrations up to 200 times the half-maximal inhibitory concentration for ibrutinib in a functional kinase assay,18,19 suggesting that ibrutinib-mediated Th1 skewing could be influenced by off-target ITK activity. Percentages of CAR T CD4 Th1 (CXCR3+CRTH2−) and Th2 (CXCR3−CRTH2+) populations36 were evaluated following serial stimulation with or without ibrutinib or acalabrutinib. During the 18-day serial stimulation period, the percentage of CD4+ CAR T Th1 cells increased for all 3 donors under control conditions (Fig. 4E). Treatment with ibrutinib 500 nM during the serial stimulation further enhanced the percentage of Th1 cells at day 18 (P<0.01) (Fig. 4F), whereas no significant effects were observed with acalabrutinib treatment, although a trend was observed for donor 3 (Figs. 4E, F).
No significant effects of either inhibitor on additional CAR T-cell activation or memory markers were observed in CAR T cells isolated from the serial stimulation assay (Supplemental Digital Content 9, http://links.lww.com/JIT/A549). Of note, subtle changes were observed for CD62L, for which expression at day 18 was increased following ibrutinib treatment in 2 of the 3 donors.
Ibrutinib Treatment of CAR T-Cell Clinical Samples Derived From 2 Patients With DLBCL-enhanced Cytolytic Function In Vitro Following Serial Stimulation
To further understand the effects of BTK inhibitor during long-term serial stimulation, we conducted an ex vivo evaluation of CAR T-cell clinical samples generated from 2 patients with DLBCL who were enrolled in the TRANSCEND clinical trial evaluating liso-cel (the anti-CD19 CAR T-cell product candidate with a 4-1BB endodomain).4 These evaluations were conducted to assess the effect of BTK inhibitors on patient-derived CAR T cells, as these patients could potentially have lower immune competence compared with healthy donors. CAR T cells generated from 2 DLBCL patient samples were assessed using the serial stimulation assay with ibrutinib; however, ibrutinib did not inhibit the proliferation of CAR T cells derived from either patient (Fig. 4G). Furthermore, an increase in cytolytic activity after 16 days of serial stimulation was observed with the addition of ibrutinib 500 nM in cells from both patients (P<0.001) and with ibrutinib 50 nM in patient 2 (P<0.01; Fig. 4H).
Molecular Signature of Ibrutinib-treated Cells Suggests the Emergence of a Th1 Memory-like T-Cell Phenotype
Identification of subtle functional effects on a defined composition of anti-CD19 CAR T cells in the presence of ibrutinib warranted evaluation of whether long-term treatment influenced gene expression. RNA-Seq analysis was performed on CAR T cells from the 3 donors treated with or without ibrutinib for 18 days in the serial stimulation assay. Ibrutinib 500 nM significantly (FDR<0.05, abslog2FC>0.5) altered the expression of 41 protein-coding genes (Figs. 5A, C). In a separate experiment under similar culture conditions, only 3 genes were significantly altered (FDR<0.05, abslog2FC>0.5) with acalabrutinib treatment (1581 nM) (Fig. 5B). Moreover, some of the genes with altered expression in response to ibrutinib 500 nM, including granzyme A, showed changes at the lower ibrutinib (50 nM) concentration (Fig. 5D), whereas the lower concentration of acalabrutinib (158 nM) did not show any significant changes in gene expression (data not shown).
FIGURE 5.
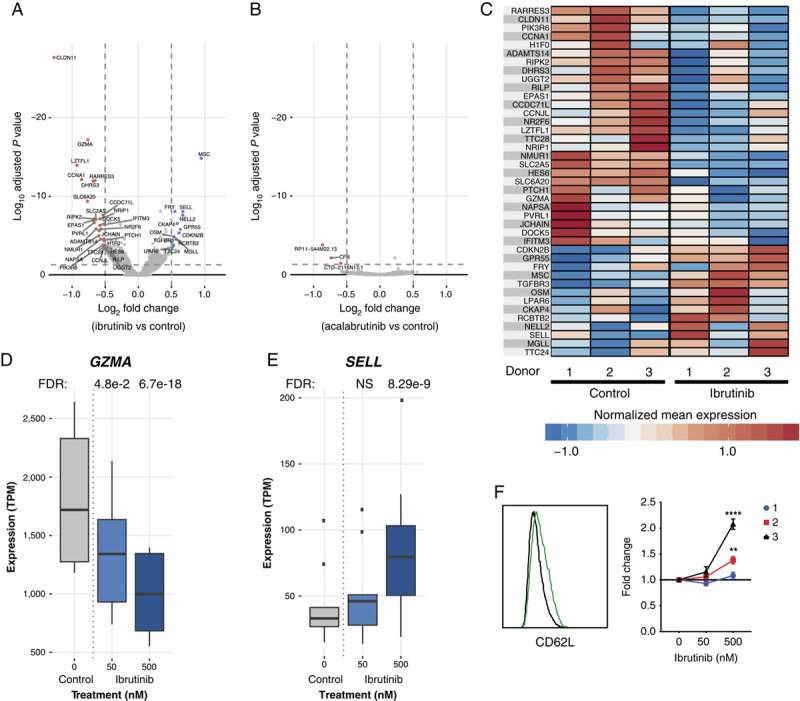
Molecular signature of ibrutinib-treated cells suggests the emergence of type 1 T-helper memory-like T-cell phenotype. A, Volcano plot depicting differentially expressed genes from day 18 serially stimulated CAR T cells with ibrutinib 500 nM compared with control. Significantly differentially upregulated and downregulated genes (FDR<0.05, abslog2FC>0.5) are labeled and colored in red and blue, respectively. B, Volcano plot depicting differentially expressed genes from day 18 serially stimulated CAR T cells with acalabrutinib 1581 nM compared with control. Significantly differentially upregulated and downregulated genes (FDR<0.05, abslog2FC>0.5) are labeled and colored in red and blue, respectively. C, Gene expression profiles of differentially expressed genes across 3 donors. Heat map depicts normalized expression (mean transcripts per million per donor+condition, z-score normalized per gene) of the differentially expressed genes between the ibrutinib 500 nM and control treatments. D and E, GZMA and SELL (CD62L) exhibit an ibrutinib dose-dependent decrease and increase in expression, respectively. Gene expression (transcripts per million) profiles are summarized across donors and experiments per condition. F, Histogram of CD62L expression in donor 3 after 18 days of serial stimulation; FC in the percentage of CD62L+ CAR T cells normalized to control measured by flow cytometry. Data from 2 independent experiments (mean±SEM). Statistically significant differences are indicated as **P<0.01 and ****P<0.0001. CAR indicates chimeric antigen receptor; FC, fold change; FDR; false discovery rate; NS, not significant.
Decreases in genes such as granzyme A (Fig. 5D) and increases in SELL/CD62L (Fig. 5E) suggest that ibrutinib dampens terminal effector–like genes while enhancing genes associated with memory development. In support of the RNA-Seq results, we observed significant increases in CD62L expression by flow cytometry after 18 days of serial stimulation with ibrutinib 500 nM in 2 donors (Fig. 5F). Furthermore, RNA-Seq revealed that genes associated with promoting Th1 differentiation were altered by ibrutinib: upregulation of MSC, known to suppress Th2 programming,37 and downregulation of NRIP1, LZTFL1, and RARRES3, which are associated with the ATRA/retinoic acid signaling pathway and inhibit Th1 development (Figs. 5A, C).38–40 Indeed, using an unbiased approach, at the pathway level, differentially expressed genes in the presence of 500 nM ibrutinib were significantly enriched in the Th1 (P=6.2e−4) and Th2 (P=1.6e−4) pathways, with z-scores indicating an upregulation of Th1-related pathways and a downregulation of Th2-related pathways (z=−1.633, z=0.816 for Th1 and Th2 canonical pathways, respectively).
Addition of Ibrutinib or Acalabrutinib in Combination With a Suboptimal Dose of CAR T Cells Resulted in Increased Tumor Clearance and Survival in a Disseminated CD19+ Tumor Model
The effects of ibrutinib or acalabrutinib on anti-CD19 CAR T cells in vivo were evaluated using the disseminated CD19+ Nalm-6 xenogeneic tumor model. For the initial ibrutinib studies, Nalm-6-FFLuc tumor-bearing NSG mice were treated once daily with ibrutinib (25 mg/kg orally). CAR T cells from 2 different donors were transferred intravenously into mice at a suboptimal dose that has been observed to delay tumor growth but not fully eliminate tumor burden. The combination of CAR T cells and ibrutinib significantly (P<0.001) delayed tumor growth and increased survival compared with CAR T cells and vehicle (Figs. 6A–C).
FIGURE 6.
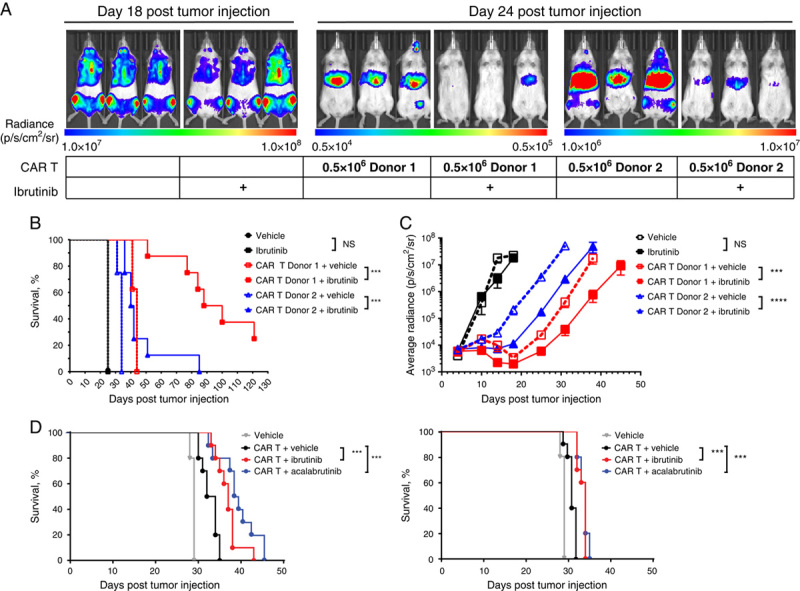
Ibrutinib and acalabrutinib enhanced CD19-directed CAR T-cell–mediated antitumor activity in the disseminated Nalm-6 tumor model. A, Nalm-6 tumor-bearing NOD.Cg-PrkdcscidIL-2rgtm1Wjl/SzJ (NSG) mice were treated daily with PO ibrutinib 25 mg/kg. A suboptimal dose of 0.5×106 CAR T cells/mouse was transferred intravenously into mice 5 days posttumor injection. N=10 mice per group. Representative bioluminescence images of mice at day 18 (no CAR T-cell treatment mice) and day 24 posttumor transfer. Scales indicate the levels of radiance measured (p/s/cm2/sr) for each group of mice. B, Kaplan-Meier curves showing the survival of tumor-bearing mice treated with PO ibrutinib 25 mg/kg and CAR T cells from 2 different donors. C, Tumor growth over time as indicated by measuring average radiance by bioluminescence from mice treated with PO ibrutinib 25 mg/kg and CAR T cells from 2 different donors. D, Kaplan-Meier curves showing the survival of tumor-bearing mice treated with ibrutinib or acalabrutinib in drinking water (equivalent to PO dose of 25 mg/kg/d) and CAR T cells from 2 different donors. N=8 mice per group were monitored for tumor burden. Statistically significant differences are indicated as ***P<0.001 and ****P<0.0001. CAR indicates chimeric antigen receptor; NS, not significant; p/s/cm2/sr, photons per second per centimeter squared per steradian; PO, oral.
In a series of separate experiments with CAR T cells from 2 different donors, Nalm-6 tumor-bearing NSG mice were treated with ibrutinib or acalabrutinib in drinking water (equivalent to 25 mg/kg/d). A bridging experiment confirmed that administration of ibrutinib via drinking water in combination with CAR T cells showed antitumor activity similar to that of oral gavage administration (data not shown). Ibrutinib or acalabrutinib, administered in combination with CAR T cells, significantly (P<0.001) increased survival (Fig. 6D) and decreased tumor growth (Supplemental Digital Content 10, http://links.lww.com/JIT/A549) compared with the CAR T cells administered with vehicle alone.
Ibrutinib and Acalabrutinib Each Altered the Phenotypic Distribution of CAR T Cells Within Tumors While Increasing Cell Numbers and Persistence in the Periphery
Pharmacokinetic analysis of the preclinical CAR T-cell product was performed on blood and bone marrow. Samples were analyzed for the presence of CAR+ T cells (Fig. 7A) and CD19+ tumor cells (Fig. 7B). Tumor cells were significantly decreased in the blood and bone marrow of mice that received ibrutinib or acalabrutinib in addition to CAR T cells (Fig. 7B). In addition, mice treated with either ibrutinib or acalabrutinib in drinking water exhibited a greater expansion of CAR T cells in the blood (Figs. 7A, C). Multivariate t-SNE fluorescence-activated cell sorting analysis identified 4 distinct population clusters (Fig. 7D). Twelve days after CAR T-cell transfer, we observed in the bone marrow of mice treated with ibrutinib or acalabrutinib an increase in population 2 (CD8+ CXCR3hi CD45RAlo CD62Lhi) and population 4 (CD4+ CXCR3int CD45RAhi CD62Lhi) (Figs. 7D–F). Furthermore, acalabrutinib-treated mice exhibited a greater enrichment of population 2 compared with that observed in ibrutinib-treated animals (representing 29.2% and 8.4% of CAR T cells in these mice, respectively), whereas a greater enhancement of population 4 was observed in ibrutinib-treated animals (15.2% compared with 4.4% of CAR T cells in acalabrutinib-treated mice; Fig. 7F).
FIGURE 7.
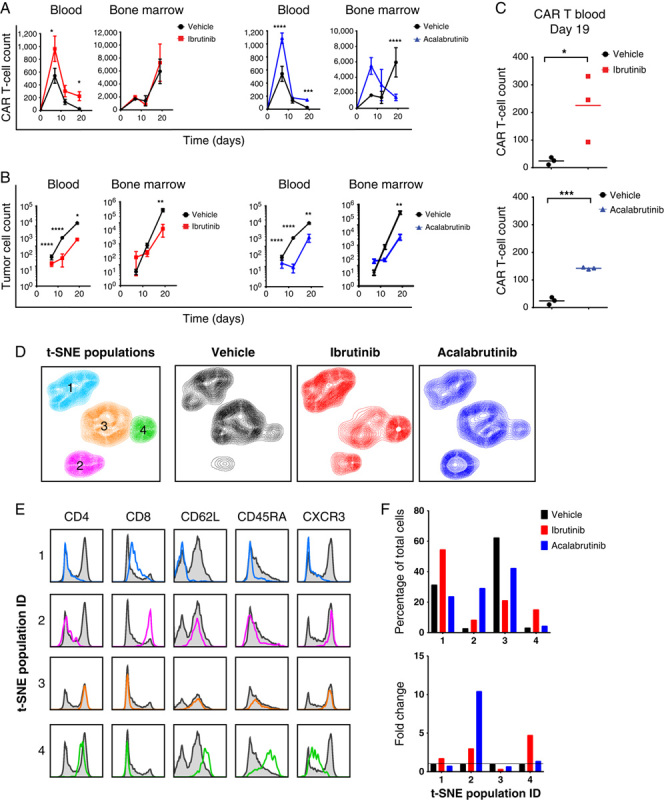
Ibrutinib or acalabrutinib addition altered the phenotype of chimeric antigen receptor (CAR) T cells within tumors while enhancing proliferation and persistence. A and B, Blood and bone marrow from mice that received a suboptimal dose of CAR T cells (donor 2) with vehicle control, ibrutinib, or acalabrutinib delivered via drinking water were analyzed for the presence of CAR T cells (A) and CD19+ tumor cells (B) at days 7, 12, 19, and 26 post-CAR T-cell transfer. N=3 mice per timepoint per group (mean±SEM). C, Number of cells in the blood of mice at day 19 post-CAR T-cell transfer. D, Four populations derived from T-distributed stochastic neighbor embedding (t-SNE) analysis of CAR T cells from the bone marrow of mice day 12 post-CAR T-cell transfer. Pooled analysis from 3 mice per group. E, Histograms showing the individual expression profiles of CD4, CD8, CD62L, CD45RA, and CXCR3 from the 4 gated t-SNE populations overlaid on the expression of the total population (gray histogram). F, Percentage and fold change of each t-SNE population from the control and ibrutinib-treated and acalabrutinib-treated groups. Statistically significant differences are indicated as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
DISCUSSION
On the basis of early evidence from the phase 1 TRANSCEND NHL 001 study (NCT02631044), a defined composition of anti-CD19 CAR T cells with a 4-1BB endodomain has demonstrated durable responses in heavily pretreated patients with aggressive NHL.4 Combination approaches may provide an opportunity to extend and broaden durable response rates from anti-CD19 CAR T-cell therapies such as liso-cel in patients with relapsed/refractory NHL or CLL by reducing tumor burden directly, reducing the immunosuppressive microenvironment and limiting or reversing the onset of T-cell dysfunction. Noncurative,41 durable clinical responses observed with BTK inhibitors in B-cell malignancies have resulted in part from inhibition of oncogenic B-cell receptor signaling; however, modulation of the microenvironment and improved patient T-cell state suggest that efficacious responses driven by these small molecules may be multifaceted.20,28 In normal T cells, BTK expression is silenced via epigenetic mechanisms; thus, the effects of BTK inhibitor on the function of T cells are thought to derive either from alterations in the function of accessory cells such as antigen-presenting cells or from potential off-target activities of the small-molecule agents.30 BTK expression was confirmed to be negligible in the cells tested here that utilized the anti-CD19 CAR of liso-cel (Supplemental Digital Content 3, http://links.lww.com/JIT/A549). The off-BTK activity of ibrutinib on ITK has been proposed as a mechanism supporting the enhancement of certain immunotherapeutic approaches.27,42 The additional off-BTK activity of BTK inhibitors may be consequential for directly influencing intrinsic CAR T-cell biology in a manner unique to the individual therapeutic cell product and may, therefore, be differentiated from endogenous T-cell responses.
Here we identified that direct, cell-autonomous effects on CAR T cells could be measured following treatment with BTK inhibitor. Such experiments are often confounded by the use of target cell lines within the assay that likely respond, in some form, to BTK inhibitor; however, use of an agonistic anti–liso-cel antibody for activation enabled investigation of the intrinsic biological response of the CAR T-cell product alone. Short-term stimulation under these experimental conditions led to a decrease in the secretion of IFN-γ, IL-2, and TNF-α, consistent with previous studies that evaluated the effects of ibrutinib on T-cell cytokine production.26 Alternatively, in the context of a single stimulation with CD19+ tumor cells, neither ibrutinib nor acalabrutinib had any notable negative effects on CAR T-cell effector cytokine production. Furthermore, due to the differences in response between short-term agonistic antibody stimulation and single stimulation with CD19+ tumor cells, our data suggest that the intrinsic effects of BTK inhibitor on CAR T cells may be influenced by activation stimulus. Alternatively, while the tumor cell lines utilized here are resistant to BTK inhibitor–mediated growth inhibition, it is possible that a BTK inhibitor effect on tumor cell biology may influence the pharmacological function of CAR T cells.
Importantly, we demonstrated that the presence of BTK inhibitor during long-term stimulation with the agonistic antibody resulted in superior functional performance of the anti-CD19 CAR T-cell product in the form of increased cytokine secretion and T-cell activation, and did not negatively affect the ability of CAR T cells to kill CD19+ tumor cells. The long-term beneficial effects of BTK inhibitor treatment were again observed for serial-stimulated CAR T cells cocultured with CD19+ target cells. CAR T donor lots with poorer expansion potential exhibited increased cell counts when treated with BTK inhibitor, suggesting that ibrutinib or acalabrutinib intrinsically improved CAR T-cell proliferative potential or survival capacity within the assay. Reports by Zhao et al43 suggest that CAR T-cell expansion and survival in vitro following repeated stimulation may predict CAR T-cell fitness and in vivo efficacy.
Notably, a number of gene expression alterations were identified for CAR T cells during long-term in vitro treatment with ibrutinib, but not acalabrutinib, suggesting that the effects on gene expression may not necessarily inform the functional benefit derived from the 2 different BTK inhibitors. In vivo efficacy and pharmacokinetic phenotyping of the anti-CD19 CAR T cells isolated from the bone marrow reinforced the observation that both BTK inhibitors may improve CAR T-cell functionality and facilitate a memory-like phenotype.
Although both ibrutinib and acalabrutinib improved CAR T-cell–mediated antitumor clearance and survival in murine xenograft studies, differences in the effect of BTK inhibitor on CAR T-cell pharmacokinetics were noted. The initial study, in which mice received a suboptimal dose of CAR T cells and once-daily oral dosing of ibrutinib, showed no increase in CAR T-cell counts in blood relative to controls. However, the timepoint of peak CAR T-cell expansion was not measured (Supplemental Digital Content 11, http://links.lww.com/JIT/A549). In subsequent studies with different donor-derived CAR T cells, CAR T-cell numbers were increased in the blood with either ibrutinib or acalabrutinib treatment delivered via drinking water. One donor CAR T-cell lot treated with BTK inhibitor was also more persistent and enriched for memory (CD62L+) CD8+ T cells relative to untreated controls. Because of multiple variables across experiments (CAR T-cell donor, BTK inhibitor dose schedule), it is unclear whether increased CAR T-cell counts in the periphery of mice accounted for the enhanced in vivo antitumor efficacy observed across all in vivo models treated with combined CAR T cells and BTK inhibitor.
Because acalabrutinib has limited ITK activity and did not affect CAR T-cell Th1 skewing based on surface markers or significantly altered gene expression, yet still provides benefit to the CAR T cells tested here, it is not clear mechanistically how BTK inhibitors improve functionality. Rather, we observed similar but not statistically significant trends in the directionality of genes that were commonly changed by both acalabrutinib and ibrutinib. The coordination of many pleiotropic factors is likely involved in influencing the response. Inhibition of additional Tec-family kinases could potentially tune the activation response and influence the strong stimulation input provided in these studies. In patients with CLL treated with either ibrutinib or acalabrutinib, decreased PD-1 on CD8+ T-cell subsets and decreased T-lymphocyte–associated protein 4 on CD4+ T-cell subsets were observed after treatment.22 Subtle decreases in activation could delay the onset of terminally differentiated CAR T cells, increasing the potential for a more persistent phenotype. This premise is consistent with our findings that treatment with BTK inhibitor leads to enhanced long-term CAR T-cell function and persistence. Furthermore, the increased number of CD62L cells observed in liso-cel in both the in vitro and in vivo ibrutinib-treated conditions aligns with the hypothesis that a higher percentage of memory-like adoptively transferred T cells may increase the durability of response.44
The microenvironment of lymphomatous nodes and bone marrow can also provide stromal support for the survival of malignant B cells.13 Recent reports demonstrate a high degree of clinical efficacy of CAR T-cell therapies in acute B-cell leukemia compared with CLL, DLBCL, and other B-cell malignancies with significant lymphadenopathy.45 More effective CAR T-cell encounters might occur during periods of BTK inhibitor–induced lymphocytosis, either due to enhanced activation of CAR T-cell responsiveness or increased sensitivity of the malignant B cells outside the protective microenvironment.
We propose that BTK inhibitor–mediated effects on CAR T-cell functionality may improve performance by up to 3 distinct mechanisms. First, although not directly tested due to the BTK inhibitor–resistant tumors in mouse studies presented here, B-cell receptor–mediated signals in malignant B cells are inactivated by BTK inhibition, resulting in loss of growth/survival signals and reduced tissue homing.46 A reduction in tumor burden and disruption of the tumor microenvironment are likely to delay the onset of CAR T-cell exhaustion and dysfunction. Second, although also not directly tested here, inhibition of BTK reduces immunosuppressive cell populations, including myeloid and regulatory T and B cells in patients.23,24,28,47 Third, we demonstrated here that in the less active of the preclinical CAR T-cell product lots tested in this study, performance in serial stimulation and cytotoxicity assays during long-term cultures improved with BTK inhibitor treatment. This result suggests that acalabrutinib or ibrutinib has the potential to counteract intrinsic factors that may otherwise dampen CAR T-cell function over time upon encounter with antigen. Furthermore, the subtle phenotypic changes that were observed for CAR T cells with concurrent BTK inhibitor treatment in vitro manifested in the in vivo studies, strengthening the observation that BTK inhibitor can directly influence CAR T-cell biology.
Improvements in expansion and functionality observed in the long-term assays also suggest a potential treatment paradigm for patients undergoing anti-CD19 CAR T-cell therapy. Dosing with BTK inhibitor before CAR T-cell engraftment could serve to reduce tumor size and/or normalize immune functions and microenvironment conditions. Concurrent dosing with BTK inhibitor after administration of CD19-targeted CAR therapies, such as liso-cel, may mitigate any potential in vivo CAR T-cell dysfunction, delay potential exhaustion, or improve CAR T-cell expansion, particularly for CAR T-cell compositions engineered from cells with inferior intrinsic performance. It is important to note that ibrutinib monotherapy is not a curative approach for patients with CLL.41 As a treatment for relapsed/refractory CLL or for patients with DLBCL with incomplete responses to standard-of-care therapy, an autologous T-cell therapy with a defined ratio of CD4+ and CD8+ T cells engineered to express a second-generation, CD19-directed 4-1BB CAR could be combined with BTK inhibition to provide deeper responses, improve the duration of responses, and target BTK inhibitor–resistant clones before the emergence of progressive disease.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
ACKNOWLEDGMENTS
The authors thank the “T shop” for the manufacture of CAR T cells. They also thank the rest of the preclinical team for scientific discussion, and the protein science team, analytical development team, clinical team, and patients for providing valuable samples and material. The authors thank Tim Ingallinera for acalabrutinib formulation analysis and Bart van Lith and Diana Mittag (Acerta Pharma) for BTK occupancy assays to confirm the Nalm-6 dosing regimen for acalabrutinib. They also thank Allard Kaptein (Acerta Pharma), Hyam Levitsky, Amy Mills, and the rest of the Juno review team for reviewing the manuscript and providing insight.
Acknowledgments
Conflicts of Interests/Financial Disclosures
Supported by Juno Therapeutics, a Celgene company, and Acerta Pharma, a member of the AstraZeneca Group. J.S.Q., T.G.J., A.B., R.J.H., S.P.R., C.R.C., J.C.J., R.P., R.A.S., and M.O.P. are employed by, and have equity interest in, Juno Therapeutics, a Celgene company. C.M.K. is employed by, and has equity interest in, Acerta Pharma, a member of the AstraZeneca Group.
REFERENCES
- 1.Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor‐modified T cells. Immunol Rev. 2014;257:127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riviere I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther. 2017;25:1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36:494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abramson JS, Siddiqi T, Palomba ML, et al. High durable CR rates and preliminary safety profile for JCAR017 in R/R aggressive b-NHL (TRANSCEND NHL 001 Study): a defined composition CD19-directed CAR T-cell product with potential for outpatient administration. J Clin Oncol. 2018;36:120. [Google Scholar]
- 5.Siddiqi T, Dorritie KA, Soumerai JD, et al. TRANSCEND CLL 004: minimal residual disease (MRD) negative responses after lisocabtagene maraleucel (Liso-Cel; JCAR017), a CD19-directed CAR T cell product, in patients (pts) with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL). J Clin Oncol. 2019;37:7501. [Google Scholar]
- 6.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraietta JA, Beckwith KA, Patel PR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreira J, Rabe K, Cerhan JR, et al. Infectious complications among individuals with clinical monoclonal B-cell lymphocytosis (MBL): a cohort study of newly diagnosed cases compared to controls. Leukemia. 2013;27:136–141. [DOI] [PubMed] [Google Scholar]
- 13.Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. [DOI] [PubMed] [Google Scholar]
- 14.Bonyhadi M, Frohlich M, Rasmussen A, et al. In vitro engagement of CD3 and CD28 corrects T cell defects in chronic lymphocytic leukemia. J Immunol. 2005;174:2366–2375. [DOI] [PubMed] [Google Scholar]
- 15.D’Arena G, Laurenti L, Minervini MM, et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res. 2011;35:363–368. [DOI] [PubMed] [Google Scholar]
- 16.Palmer S, Hanson CA, Zent CS, et al. Prognostic importance of T and NK‐cells in a consecutive series of newly diagnosed patients with chronic lymphocytic leukaemia. Br J Haematol. 2008;141:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363:240–252. [DOI] [PubMed] [Google Scholar]
- 19.Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014;10:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long M, Beckwith K, Do P, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017;127:3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton tyrosine kinase–dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honda F, Kano H, Kanegane H, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13:369–378. [DOI] [PubMed] [Google Scholar]
- 25.Long M, Beckwith KA, Maddocks KJ, et al. Ibrutinib treatment reduces both T-regulatory cells and B-regulatory cell phenotype in malignant B cells in chronic lymphocytic leukemia patients. Blood. 2015;126:2940. [Google Scholar]
- 26.Ruella M, Kenderian SS, Shestova O, et al. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22:2684–2696. [DOI] [PubMed] [Google Scholar]
- 27.Sagiv-Barfi I, Kohrt HE, Czerwinski DK, et al. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci USA. 2015;112:E966–E972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stiff A, Trikha P, Wesolowski R, et al. Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016;76:2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg LJ, Finkelstein LD, Lucas JA, et al. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol. 2005;23:549–600. [DOI] [PubMed] [Google Scholar]
- 30.Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J, Ge H, Newman M, et al. OSA: a fast and accurate alignment tool for RNA-Seq. Bioinformatics. 2012;28:1933–1934. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Hu J, Newman M, et al. RNA-seq analysis pipeline based on oshell environment. IEEE/ACM Trans Comput Biol Bioinform. 2014;11:973–978. [DOI] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman SE, Montraveta A, Niemann CU, et al. The Bruton tyrosine kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu C, Chen Z, Dardalhon V, et al. The transcription factor musculin promotes the unidirectional development of peripheral T reg cells by suppressing the T H 2 transcriptional program. Nat Immunol. 2017;18:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heim KC, White KA, Deng D, et al. Selective repression of retinoic acid target genes by RIP140 during induced tumor cell differentiation of pluripotent human embryonal carcinoma cells. Mol Cancer. 2007;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Promchan K, Lin B-R, et al. LZTFL1 upregulated by all-trans retinoic acid during CD4+ T cell activation enhances IL-5 production. J Immunol. 2016;196:1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zirn B, Samans B, Spangenberg C, et al. All-trans retinoic acid treatment of Wilms tumor cells reverses expression of genes associated with high risk and relapse in vivo. Oncogene. 2005;24:5246–5251. [DOI] [PubMed] [Google Scholar]
- 41.Woyach JA, Furman RR, Liu T-M, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kokhaei P, Jadidi-Niaragh F, Sotoodeh Jahromi A, et al. Ibrutinib-A double-edge sword in cancer and autoimmune disorders. J Drug Target. 2016;24:373–385. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Z, Condomines M, van der Stegen SJ, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Naranjo A, Brown CE, et al. Phenotypic and functional attributes of lentivirus modified CD19-specific human CD8+ central memory T cells manufactured at Clinical Scale. J Immunother. 2012;35:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turtle CJ, Hanafi L-A, Berger C, et al. High rates of durable complete response in ALL, NHL, and CLL after immunotherapy with optimized lymphodepletion and defined composition CD19 CAR-T cells (JCAR014). J Clin Oncol. 2016;34:102. [Google Scholar]
- 46.Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31:119–132. [DOI] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.