

Abstract
D190N, a missense mutation in rhodopsin, causes photoreceptor degeneration in patients with autosomal dominant retinitis pigmentosa (adRP). Two competing hypotheses have been developed to explain why D190N rod photoreceptors degenerate: (a) defective rhodopsin trafficking prevents proteins from correctly exiting the endoplasmic reticulum, leading to their accumulation, with deleterious effects or (b) elevated mutant rhodopsin expression and unabated signaling causes excitotoxicity. A knock-in D190N mouse model was engineered to delineate the mechanism of pathogenesis. Wild type (wt) and mutant rhodopsin appeared correctly localized in rod outer segments of D190N heterozygotes. Moreover, the rhodopsin glycosylation state in the mutants appeared similar to that in wt mice. Thus, it seems plausible that the injurious effect of the heterozygous mutation is not related to mistrafficking of the protein, but rather from constitutive rhodopsin activity and a greater propensity for chromophore isomerization even in the absence of light.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03090-9) contains supplementary material, which is available to authorized users.
Keywords: D190N, GPCR, Rhodopsin, Mouse model, Retinitis pigmentosa, Retina, Excitotoxicity
Introduction
Patients with retinitis pigmentosa (RP), a heterogeneous group of inherited retinal degenerations, suffer from night blindness and a significantly constricted visual field. RP, with an incidence of 1:3500 [1], is characterized by progressive photoreceptor cell death, beginning in the periphery and marching towards the macula, and complete loss of central vision can occur in as little as 30 years [2, 3]. RP symptoms worsen with time and severely impact the ability of individuals to live independent lives.
RP can be caused by mutations in many different genes, although those in rhodopsin are the most prevalent, causing approximately 10% of all cases worldwide [4, 5] and at least 25% of all cases of autosomal dominant RP (adRP) [6]. Rhodopsin, which is contained in the membranous discs of the rod outer segments (ROS) [7], initiates phototransduction in rod photoreceptors [8, 9]. Rhodopsin is a G-protein-coupled receptor (GPCR) formed by the apoprotein opsin and the chromophore 11-cis-retinal [10], which isomerizes to all-trans-retinal after a photon impinges on the retina. This initiates a series of conformational changes that produce the active form of the molecule, metarhodopsin II (Meta II) [11]. Subsequently, Meta II activates transducin, and the visual cascade begins, enabling vision to transpire [12].
The substitution of asparagine in place of aspartate at position 190 (D190N) in the rhodopsin gene causes autosomal dominant RP (adRP) in humans [13–15]. There are two competing hypotheses describing how D190N may trigger retinal degeneration. The first proposes that defective rhodopsin trafficking initiates the unfolded protein response (UPR), inducing apoptosis [16]. The second theory claims that increased thermal instability may cause constitutive toxic rhodopsin-D190N signaling [17, 18]. Recent in vitro experiments support the thermal instability theory by comparing D190N mutant cells with S186W, another rhodopsin mutation [19].
We created a rhodopsin-D190N mouse model that faithfully mimics human disease based on electrophysiological, histological, and autofluorescent findings [20]. In the present article, we used this model to directly address the debate regarding whether defective rhodopsin trafficking or constitutive activity causes degeneration. Addressing this inquiry will yield insights in the search for mechanism-directed therapies to treat the disease in patients.
Materials and methods
Animals
Heterozygous (D190N/+) mice, homozygous (D190N/D190N) mice, and wild-type (wt) mice with 129XB6 backgrounds (a cross between 129/Sv and C57Bl/6) were used in accordance with the statement for the use of animals in ophthalmic and vision research of the Association for Research in Vision and Ophthalmology (ARVO), as well as the policy for the use of animals in research of the Society for Neuroscience. Animals were maintained on a 12-h light–dark cycle, with ad libitum access to food and water. All efforts were made to minimize the number of animals used and their suffering.
Structural modeling of mouse Rhodopsin
The structure of mouse rhodopsin was modeled off the previously solved bovine rhodopsin structure (PDB: 1U19) using MODELLER 9.14. The resultant model superimposed well with the template and had an RSMD of 0.131 angstroms over 323 Cα atoms. Conservation analysis was performed by submitting the homology model to the ConSurf server. PyMOL generated all structural figures.
Histology
Control and mutant mice were sacrificed and hematoxylin–eosin (H–E) retinal sections were obtained. Eyes were subsequently enucleated and fixed in 0.5% Karnovsky’s fixative (2% paraformaldehyde, 1.25% glutaraldehyde, and 0.2 mol/L phosphate-buffered saline). Eyes were then embedded in paraffin and sectioned (4 mm), and H&E staining was performed. Pictures were taken under a Leica DM 5000B microscope (Leica Microsystems, Inc., Buffalo Grove, IL) at 40× magnification.
Immunohistochemistry
Wild-type and mutant animals were sacrificed. Globes were enucleated, fixed, and sectioned as previously described [20]. Sections were pre-incubated for 1 h at room temperature (RT) in blocking solution: 10% goat normal serum (GNS) and 0.1% Triton in PBS (PBST). Immunohistochemistry was performed overnight at 4 °C using the monoclonal anti-Rhodopsin antibody ID4 (1:200; Santa Cruz Biotech, Santa Cruz, CA) and the glycogen phosphorylase (glyphos, 1:500; kindly provided by Prof. Pfeiffer-Guglielmi). The tissue was rinsed with PBST and incubated (1 h, RT) with Alexa 555 or 488 goat anti-mouse secondary antibody (1:1000, Invitrogen, Carlsbad, CA), which were diluted in PBST. Sections were rinsed in PBS and mounted in Vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA). Sections were imaged under a Leica DM 5000B fluorescence microscope (Leica Microsystems).
Transmission electron microscopy
Retinae from wt and D190N mutant animals were fixed with 2.5% glutaraldehyde in 0.1 M Sorenson’s buffer (pH 7.2). Tissue was then post-fixed with 1% OsO4 in Sorenson’s buffer for 1 h. After dehydration, the tissue was embedded in Lx-112 (Ladd Research Industries, Inc., Williston, VT). Sections were prepared on the MT-7000 ultramicrotome (Boeckeler Instruments, Inc., Tucson, AZ) and cut to a thickness of 60 nm. Tissue was then stained with uranyl acetate and lead citrate and examined under a JEOL JEM-1200 EXII electron microscope (JEOL USA, Inc., Peabody, MA). Pictures were taken on an ORCA-HR digital camera (Hamamatsu, Bridgewater, NJ) and recorded with an AMT Image Capture Engine (Advanced Microscopy Techniques Corp., Danvers, MA).
Electroretinography
One-month-old control, D190N/+, and D190N/D190N animals were dark-adapted overnight. Pupils were dilated using 0.5% tropicamide and 2.5% phenylephrine hydrochloride, and animals were anesthetized by intraperitoneal injection of a ketamine/xylacine solution (Ketaset III, Fort Dodge, IA, USA; Lloyd Laboratories, Shenandoah, IA, USA). After anesthesia was applied, ERGs were recorded using a Espion ERG Diagnosys equipment (Diagnosys LLL, Littleton, MA, USA). A total of 40–60 responses were averaged for each trial. All further details on the ERG method have been described previously [20]. We measured scotopic and photopic ERG responses to assess rod-only, photoreceptor-specific, and cone-specific function. Maximal responses were taken from the Espion readout in microvolts.
Immunoblotting analyses
Neuroretinae from wt and D190N animals were collected and homogenized. Protein concentrations were measured by BCA protein assay. For glycosylation analyses, 5 µg of protein were treated with Endoglycosidase H (Endo-H, New England Labs, Ipswich, MA) or Peptide: N-Glycosidase F (PNGase F, New England Labs), according to the manufacturer’s protocol. Consequently, proteins were separated by SDS-PAGE in 12% acrylamide gels and transferred to PVDF membranes. Membranes were then blocked in dry skim milk and PBS Tween 0.1%. Later, membranes were incubated with mouse anti-Rhodopsin antibody (ID4, 1:5000, Santa Cruz Biotech) overnight at 4 °C. On the following day, membranes were incubated in goat anti-mouse secondary antibody (1:10,000, Santa Cruz Biotech) for 1 h at RT. Antibody complexes were visualized by chemiluminescence detection (Immobilon Western, Millipore Corporation, Billerica MA) using Kodak Biomax film (Kodak, Rochester, NY).
Results
Proposed mechanism of the D190N mutation
Rhodopsin is a seven-transmembrane domain G-protein-coupled receptor (GPCR) located in rod photoreceptor cells (Fig. 1a, b). The seven-transmembrane helices form a binding pocket, where rhodopsin’s chromophore 11-cis-retinal is covalently bound to the seventh helix (TM7) through a protonated Schiff-base linkage (at residue K296). We modeled the structure of mouse rhodopsin using the crystal structure of bovine rhodopsin (PDB: 1U19) as a template (Fig. 1c). As described previously [17] an extracellular loop, termed the E-2 loop, gates the retinal-binding pocket of rhodopsin, forming a ‘retinal plug’ (Fig. 1d). This E-2 loop contains a critical ion pair between residues R177 and D190 that helps to stabilize the dark state of rhodopsin. Mutations in the E-2 loop (D190N) cause RP and have been previously shown to produce a thermally unstable rhodopsin protein (Fig. 1e). Our homology model suggests that the D190N mutation affects the mouse rhodopsin structure through the same mechanism. Next, we performed conservation analysis of the rhodopsin sequence and structure using ConSurf. This analysis revealed that the D190/R177 ion pair is 100% conserved in 150 homologous opsin sequences (ranging from rhodopsin to green-sensitive opsin 3 and short-wave sensitive opsin 2) and highlights it evolutionary importance in rhodopsin structure and function (Fig. 1f). Mutations in this region are not hypothesized to affect rhodopsin dimerization.
Fig. 1.

Mechanism of rhodopsin D190N mutation. a Membrane topological structure of rhodopsin. The E-2 loop is located on the extracellular side of the protein between helices TM4 and TM5. b Multiple sequence alignment highlighting the location and conservation of the D190N mutation. This residue is conserved multiple species. c Homology model of mouse rhodopsin based off the bovine rhodopsin crystal structure (PDB: 1U19). d Ion pair between D190 and R177 that stabilizes the E-2 loop. e D190N mutation disrupts the ion pair formed by D190/R177. f Conservation analysis in ConSurf reveals that R177/D190 ion pair is 100% conserved in 150 opsin sequences
Structural changes in retinas of D190N/+ and D190N/D190N mice
Histological observations in control, heterozygous, and homozygous D190N mice were conducted to compare retinal morphology. As was confirmed in the past studies, the heterozygous mice were morphologically similar to wt, with some decrease in overall layer thicknesses following retina development and remaining stable over time (Fig. S1) [20]. However, photoreceptor degeneration in D190N/D190N mice was overwhelming in comparison (Fig. 2a). At postnatal day (P) 12, the outer nuclear layer (ONL) of D190N/D190N animals presented fewer rows of photoreceptors compared to the control and the heterozygous mutant. At P21, the ONL was reduced to only two to three rows, and after 1 month, only a single row of dysmorphic photoreceptors was observed (Fig. 2a, third and fourth panels). Observations were conducted up to 7 months after birth (Fig. 2a, right panel), at which point only a few cells remained in the ONL of the homozygous animals.
Fig. 2.

Photoreceptor degeneration in retinas of control and D190N animals. a Hematoxylin–eosin-stained paraffin sections of wild type (wt, at P30), heterozygous (D190N/+, at P30), and homozygous (D190N/D190N) mice at P12, 21, 30, and 210. At P12, D190N/D190N mice have 5–6 rows of photoreceptors. By P21, only 2 rows worth of cells remained in the ONL. At P210, only a single row of photoreceptors was observed. b Anti-rhodopsin-stained cryosections (red) showed that in D190N/+ mice, rhodopsin appears to localize correctly in the OS of the retina. In the D190N homozygotes, rhodopsin appears mislocalized within the photoreceptor nuclei, and its expression continues to decline with time. c Anti-Glyphos antibody (green) was used to stain for cones. At P12, cones resembled that of wt mice, although the IS/OS were not completely formed. The INL and the outer plexiform layer are partially stained. At P21, staining was decreased, although some remained in the ONL. At P210, only a few cells stained positively for glyphos in the ONL. Outer segments, OS; inner segments, IS; outer nuclear layer, ONL; inner nuclear layer, INL; ganglion cell layer, GCL. DAPI (blue) was used to counterstain nuclei. Scale bar: 50 μm. n = 3 for each genotype and each time point
Rhodopsin localization was performed by immunostaining (Fig. 2b). Wt and heterozygous D190N animals expressed rhodopsin in the outer segments (OS) of the photoreceptor layer at all observed ages (Fig. 2b, first and second panels), as previously described [20]. In contrast, homozygous D190N mice presented mutant rhodopsin in the IS/OS, neither of which appeared to have developed completely in the model (P12, Fig. 2b, third panel). Rhodopsin was also mislocalized all along the ONL. At P21, when the ONL was almost completely deteriorated, there remained a trace of rhodopsin staining, particularly in the area adjacent to the RPE, where the OS used to reside (Fig. 2b, fourth panel). From P30 to P210, rhodopsin staining was no longer visible (Fig. 2b, last two panels). Cone photoreceptors were also studied using an antibody against glycogen phosphorylase (Glyphos). Anti-Glyphos staining of M- and S-cones presented photoreceptors with well-organized segments at P12 (Fig. 2c). At P21, the Glyphos staining had decreased, but was still observable in the ONL, surrounding surviving cones, until P210, at which point minimal cones could be detected, with only a single row of cells in the ONL. Together, these findings demonstrate that D190N/+ experience slow degeneration, while D190N/D190N mice undergo rapid disease progression.
The morphology of the retina was also studied by transmission electron microscopy (TEM, Fig. 3). Although the OS seemed correctly organized in heterozygous D190N/+ animals compared to wt controls, it appeared shortened on TEM (Fig. 3a, b). Homozygous retinas appeared disorganized, and the OS layer already lacked the outer fragment by P12, the youngest age observed (Fig. 3c). Membranous discs in the OS of heterozygous animals grew correctly, although they presented a slightly disordered morphology (Fig. 3e) compared to control OS (Fig. 3d). Homozygous D190N/D190N retinas presented some photoreceptors entering apoptosis (Fig. 3f). Regular elongated mitochondria were found in wt animals (Fig. 3g). Impaired swollen mitochondria were prominently seen in D190N/+ retinas (Fig. 3h) and in D190N/D190N mice (Fig. 3i), where disrupted cilia epithelium were also detected.
Fig. 3.

D190N/+ mice show relatively normal outer segment discs compared to D190N/D190N mice, which degenerate sooner. OS of wt, D190N/+, and D190N/D190N were observed with electron microscopy. Compared to wt (a), shortened OS were observed in D190N/+ mice at P21 (b) and in D190N/D190N mice at P12, for which the OS and IS were indistinguishable (c). D190N/+ OS were not only shortened, but also presented an irregular morphology (e) compared to wt (d). Photoreceptors undergoing apoptosis were observed in D190N/D190N at P12 (f). Regular elongated mitochondria in wt animals (arrows in g) were observed, while swollen mitochondria were seen in D190N/+ (arrows in h) at P21. Accumulation of dysmorphic mitochondria was observed also in the D190/D190N background (arrows in i), and disrupted cilia epithelium was observed (arrowheads in i). RPE retinal pigment epithelium, ONL outer nuclear layer, OS outer segments, IS inner segments, N nucleus
Retinal function
Electroretinograms (ERGs) were recorded in dark-adapted control, heterozygous, and homozygous mice. Three different experiments were performed: scotopic dim light rod-specific recordings, scotopic maximum recordings, and photopic maximum cone-specific recordings. When the rod-specific response was studied, D190N/+ animals did not show a significant difference compared to control mice, whereas D190N/D190N animals already showed a significant difference by P30 (Fig. 4a). The maximum response traces of heterozygous and wt mice seemed similar, but in the homozygous animal, a- and b-waves were extremely reduced (Fig. 4b), likely because the ONL was almost completely collapsed by this point. When the cone-specific response was studied, wt and heterozygous traces were very similar, while the homozygous trace was again exceptionally decreased (Fig. 4c).
Fig. 4.

Decreased visual function in homozygous D190N animals. a Scotopic dim light rod-specific ERG traces for a B6 control mouse (blue), a D190N/+ (orange) and a D190N/D190N mouse (red) at 3–4 weeks of age. Quantification of dim light scotopic a-waves did not show any difference among the groups, whereas the b-wave of the homozygous mice was significantly decreased when compared to the wt and the heterozygous mice (b). Scotopic maximum ERG traces were recorded. Heterozygous and wt a- and b-waves were similar, but the homozygous trace was significantly flatter. c Photopic cone-specific traces were recorded. Again, wt and heterozygous signals were similar, while the homozygous trace was significantly diminished. (Error bars show SEM for each time point. Significance was calculated using a ratio paired t test analysis. n ≥ 3 mice. *p < 0.05; **p < 0.01; ***p < 0.001)
Rhodopsin glycosylation state
To investigate glycosylation status of D190N rhodopsin, retinal homogenates of wt and D190N/+ animals were treated with Endo-H and PNGase F deglycosylation enzymes. The D190N/+ mice presented less intense bands than the wt controls (Fig. 5), indicating that less protein was present, most likely due to a reduced number of cells. Glycosylated rhodopsin was found at ~ 35 kD in both mutant and wt homogenates. After Endo-H treatment, ~ 35 kD protein was also found in both homogenates. Rhodopsin from both wt and mutant mice shifted to a lower molecular weight (~ 32 kD) after PNGase F treatment. Rhodopsin from mutant mice appeared to behave similar to wt rhodopsin after deglycosylation, which is consistent with correct folding of the protein.
Fig. 5.
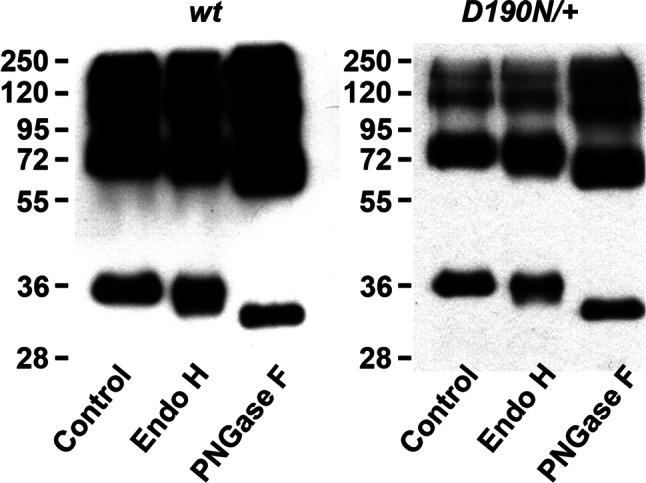
Glycosylation state of rhodopsin. Retinas from wt and D190N/+ animals at P30 were collected and homogenized. After treating with Endo-H or PNGase, proteins were separated by electrophoresis. Blots from both wt and D190N/+ were found at the same level
Discussion
As we discussed in our previous publication [20], the D190N heterozygous mouse is an accurate model for studying human RP caused by a dominant mutation in that gene. In fact, heterozygotes experienced slow degeneration, much alike to the human phenotype. Heterozygous D190N/+ displayed shortened ROS by P21, but by P210, the ROS remained, even though the retina had lost several photoreceptor cells. Shortened ROS were also reported in the P23H mouse, another mutant rhodopsin model that generates retinal degeneration [21]. In classic models, such as the rd1 mouse, where the mutation is held in the gene that encodes for PED6B, the OS are still present, even though photoreceptor cell death occurs very fast [22]. The homozygous mutant presented rhodopsin in the ONL at P12 and displayed absent OS and severe photoreceptor loss. In D190N/D190N at P30, rhodopsin is not visible in the retina, meaning that probably, all the rod photoreceptor cells are gone.
Proper rhodopsin localization in ROS in D190N/+ was inconsistent with the defective trafficking hypothesis suggested in the past [16]. Furthermore, glycosylation status appeared to be unaffected in D190N/+ animals. Endo-H glycosidase recognized immature high-mannose sugars in mis-traffic proteins retained in the ER and/or Golgi [21]. Endo-H digestion generated similar (~ 35 kDa) products from both D190N/+ and control retinal extracts. Endo-H insensitivity was consistent with the fact that mutant rhodopsin did not accumulate in the ER and localized correctly in the OS in heterozygous animals. Whereas correct trafficking of rhodopsin was observed in the heterozygous animal, the homozygous presented an obvious mislocalization and was observed throughout the ONL. In young animals (P12), the protein was seen in the segments too, most probably IS, since the OS were absent, as was perceived under the electron microscope. The functional studies of the animal, made at 3–4 weeks of age, showed us that rod-specific traces were flat due to the absence of rod photoreceptors. Nevertheless, there is some signal, probably due to the activity of the remaining cones.
The constitutive GPCR signaling in the D190N/+ mice appeared to render the retinae in a perpetual light-adapted state, as reflected by the desensitized scotopic photoreceptor a-waves from dark-reared heterozygotes (intensity series) seen previously [20]. The equivalent light hypothesis attributed retinal degeneration to nearly constant, constitutive photoreceptor signaling [23–25] resulting from thermal isomerization of rhodopsin in darkness. Thermal isomerization of rhodopsin mimicked the change caused by a photon, so it triggered the phototransduction cascade in vivo exactly as a photon would. Studying monkey retinas in 1984, Baylor and collaborators found that discrete, photon-like events occurred at a rate of approximately 0.0063 s−1, or the rough equivalent of one photon every 3 min [26]. They estimated that there were 1.2 × 108 molecules of rhodopsin in a rod cell and thereby calculated the half-life of rhodopsin in vivo to be approximately 420 years. Given the large number of D190N molecules presented in dark-adapted rod photoreceptors, there would be sufficient isomerization-like events to create the impression of constant light. Because the photo-activation would deplete intracellular Ca2+, this constitutive light signal depressed intracellular Ca2+ to cytotoxic levels. Together with the localization and glycosylation assays, the results of the ERGs support the “equivalent light hypothesis” of D190N-mediated photoreceptor death.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We greatly appreciate the assistance of the members of the Bernard & Shirlee Brown Glaucoma laboratory, especially to Chun-Wei Hsu for technical support. SHT is a Burroughs-Wellcome Program in Biomedical Sciences Fellow, and is also supported by the Charles E. Culpeper-Partnership for Cures 07-CS3, Crowley Research Fund, Schneeweiss Stem Cell Fund, New York State N09G-302, Foundation Fighting Blindness [TA-NMT-0116-0692-COLU] (Owings Mills, MD), TS080017 from US Department of Defense, NIH Grants [P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437], Research to Prevent Blindness (New York, NY), and Joel Hoffmann Scholarship. CSL is the Homer McK. Rees Scholar. JSP is a BEST2016 awardee (BEST/2016/030, Conselleria de Educación, Investigación, Cultura y Deporte; Generalitat Valenciana) and his research is supported by a Prometeo Grant (PROMETEO/2016/094; Conselleria de Educación, Investigación, Cultura y Deporte; Generalitat Valenciana) and by internal funds from Universidad Católica de Valencia San Vicente Mártir (2018-128-001). VBM is supported by NIH Grants K08EY020530, R01EY016822, The Doris Duke Charitable Foundation Grant #2013103, and Research to Prevent Blindness (New York, NY); GV is supported by NIH Grants [F30EYE027986 and T32GM007337].
Author contributions
JSP and XC ran most of the experiments: the histology, immunostainings, electron microscopy, ERGs, and glycosilation; WL helped with glycosylation experiments; YT, WS, SK, and CH ran part of the ERG recordings and the western blotting; IW assisted and advised with the experiments; GV, AGB, and VBM created the modeling for D190N rhodopsin and prepared Fig. 1; CSL created the animal model; SHT planned and supervised the experiments; JSP, SJ, and KSP wrote the main document; all authors reviewed the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Javier Sancho-Pelluz and Xuan Cui contributed equally to this paper.
References
- 1.Boughman JA, Conneally PM, Nance WE. Population genetic studies of retinitis pigmentosa. Am J Hum Genet. 1980;32:223–235. [PMC free article] [PubMed] [Google Scholar]
- 2.Berson EL. Retinitis pigmentosa: the friedenwald lecture. Invest Opthal Vis Sci. 1993;34:1655–1676. [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Rivolta C, Sharon D, De Angelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- 5.Wilson JH, Wensel TG. The nature of dominant mutations of rhodopsin and implications for gene therapy. Mol Neurobiol. 2003;28:149–158. doi: 10.1385/MN:28:2:149. [DOI] [PubMed] [Google Scholar]
- 6.Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filipek S, Stenkamp RE, Teller DC, Palczewski K. G protein-coupled receptor rhodopsin: a prospectus. Annu Rev Physiol. 2003;65:851–879. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jager S, Palczewski K, Hofmann KP. Opsin/all-trans-retinal complex activates transducin by different mechanisms than photolyzed rhodopsin. Biochemistry. 1996;35:2901–2908. doi: 10.1021/bi9524068. [DOI] [PubMed] [Google Scholar]
- 9.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 11.Smith SO. Structure and activation of the visual pigment rhodopsin. Annu Rev Biophys. 2010;39:309–328. doi: 10.1146/annurev-biophys-101209-104901. [DOI] [PubMed] [Google Scholar]
- 12.Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Tsui I, Chou CL, Palmer N, Lin CS, Tsang SH. Phenotype-genotype correlations in autosomal dominant retinitis pigmentosa caused by RHO, D190N. Curr Eye Res. 2008;33:1014–1022. doi: 10.1080/02713680802484645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park SP, Lee W, Bae EJ, Greenstein V, Sin BH, Chang S, Tsang SH. Early structural anomalies observed by high-resolution imaging in two related cases of autosomal-dominant retinitis pigmentosa. Ophthalmic Surg Lasers Imaging Retina. 2014;45:469–473. doi: 10.3928/23258160-20140908-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai YT, Wu WH, Lee TT, Wu WP, Xu CL, Park KS, Cui X, Justus S, Lin CS, Jauregui R, Su PY, Tsang SH. Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology. 2018;125:1421–1430. doi: 10.1016/j.ophtha.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 17.Janz JM, Fay JF, Farrens DL. Stability of dark state rhodopsin is mediated by a conserved ion pair in intradiscal loop E-2. J Biol Chem. 2003;278:16982–16991. doi: 10.1074/jbc.M210567200. [DOI] [PubMed] [Google Scholar]
- 18.Yan EC, Kazmi MA, Ganim Z, Hou JM, Pan D, Chang BS, Sakmar TP, Mathies RA. Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc Natl Acad Sci USA. 2003;100:9262–9267. doi: 10.1073/pnas.1531970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu MY, Liu J, Mehrotra D, Liu Y, Guo Y, Baldera-Aguayo PA, Mooney VL, Nour AM, Yan EC. Thermal stability of rhodopsin and progression of retinitis pigmentosa: comparison of S186W and D190N rhodopsin mutants. J Biol Chem. 2013;288:17698–17712. doi: 10.1074/jbc.M112.397257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sancho-Pelluz J, Tosi J, Hsu CW, Lee F, Wolpert K, Tabacaru MR, Greenberg JP, Tsang SH, Lin CS. Mice with a D190N mutation in the gene encoding rhodopsin: a model for human autosomal-dominant retinitis pigmentosa. Mol Med. 2012;18:549–555. doi: 10.2119/molmed.2011.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286:10551–10567. doi: 10.1074/jbc.M110.209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zencak D, Schouwey K, Chen D, Ekström P, Tanger E, Bremner R, van Lohuizen M, Arsenijevic Y. Retinal degeneration depends on Bmi1 function and reactivation of cell cycle proteins. Proc Natl Acad Sci USA. 2013;110:593–601. doi: 10.1073/pnas.1108297110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fain GL, Lisman JE. Photoreceptor degeneration in vitamin A deprivation and retinitis pigmentosa: the equivalent light hypothesis. Exp Eye Res. 1993;57:335–340. doi: 10.1006/exer.1993.1132. [DOI] [PubMed] [Google Scholar]
- 24.Fain GL, Lisman JE. Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest Ophthalmol Vis Sci. 1999;40:2770–2772. [PubMed] [Google Scholar]
- 25.Lisman J, Fain G. Support for the equivalent light hypothesis for RP. Nat Med. 1995;1:1254–1255. doi: 10.1038/nm1295-1254. [DOI] [PubMed] [Google Scholar]
- 26.Baylor DA, Nunn BJ, Schnapf JL. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J Physiol. 1984;357:575–607. doi: 10.1113/jphysiol.1984.sp015518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.