

Abstract
Background
The Lennox‐Gastaut syndrome (LGS) is an age‐specific disorder, characterised by epileptic seizures, a characteristic electroencephalogram (EEG), psychomotor delay and behavioural disorder. It occurs more frequently in males and onset is usually before the age of eight years, with a peak between three and five years of age. Late cases occurring in adolescence and early adulthood have rarely been reported. Language is frequently affected, with both slowness in ideation and expression in addition to difficulties of motor dysfunction. Severe behavioural disorders (e.g. hyperactivity, aggressiveness and autistic tendencies) and personality disorders are nearly always present. There is also a tendency for psychosis to develop with time. The long‐term prognosis is poor; although the epilepsy often improves, complete seizure freedom is rare and conversely the mental and psychiatric disorders tend to worsen with time.
Objectives
To compare the effects of pharmaceutical therapies used to treat LGS in terms of control of seizures and adverse effects. Many people who suffer from this syndrome will already be receiving other antiepileptic medications at the time of their entry into a trial. However, for the purpose of this review we will only consider the effect of the single therapeutic agent being trialled (often as add‐on therapy).
Search methods
We searched the Cochrane Epilepsy Group's Specialized Register (18 October 2012), the Cochrane Central Register of Controlled Trials (CENTRAL, The Cochrane Library Issue 10 of 12, 2012) and MEDLINE (1946 to October week 2, 2012). We also searched EMBASE (1980 to March 2003). We imposed no language restrictions. We searched the International Standard Randomised Controlled Trial Number (ISRCTN) register (18 October 2012) for ongoing trials and in addition, we contacted pharmaceutical companies and colleagues in the field to seek any unpublished or ongoing studies.
Selection criteria
All randomised controlled trials (RCTs) of the administration of drug therapy to patients with LGS.
Data collection and analysis
Two review authors independently extracted data. Analysis included assessing study quality, as well as statistical analysis of the effects on overall seizure rates and effects on specific seizure types (e.g. drop attacks), adverse effects and mortality.
Main results
We found nine RCTs, but were unable to perform any meta‐analysis, because each trial looked at different populations, different therapies and considered different outcomes.
Authors' conclusions
The optimum treatment for LGS remains uncertain and no study to date has shown any one drug to be highly efficacious; rufinamide, lamotrigine, topiramate and felbamate may be helpful as add‐on therapy, clobazam may be helpful for drop seizures. Until further research has been undertaken, clinicians will need to continue to consider each patient individually, taking into account the potential benefit of each therapy weighed against the risk of adverse effects.
Plain language summary
Treatment of Lennox‐Gastaut syndrome
The optimum treatment for Lennox‐Gastaut syndrome has yet to be established. Lennox‐Gastaut syndrome is a seizure (epilepsy) disorder that is commonly associated with behavioural and mental health problems. Many different treatments are currently used in the treatment of this disorder and many more have been tried in the past, often with little success. The review of trials found that there was no evidence to suggest that any one drug was more effective than another in the treatment of this disorder in terms of controlling the different seizure types. More research is needed to compare the therapies currently available.
Background
The Lennox‐Gastaut syndrome (LGS) is an age‐specific epileptic encephalopathy, characterised by epileptic seizures, slow spike‐waves in the waking electroencephalogram (EEG) and fast rhythmic bursts during sleep, psychomotor delay and personality disorders (Beaumanoir 2005). The incidence is not known but it has been estimated to account for 1% to 10% of all childhood epilepsies. It occurs more frequently in males, and onset is usually before the age of eight years, with a peak between three and five years of age. Late cases occurring in adolescence and early adulthood have rarely been reported. About one‐fifth of cases are preceded by West's syndrome (a triad of infantile spasms, hypsarrhythmia and psychomotor delay).
The LGS is an age‐dependent electroclinical diagnosis for which there may be multiple aetiologies, whether genetic, structural, metabolic or unknown (Berg 2011). Approximately one‐quarter have no prior history of epilepsy, neurological abnormality or developmental delay prior to the onset. Others have identical electrical and clinical features associated with such aetiologies as tuberous sclerosis and cortical dysplasia.
More recently a consensus approach reviewing the issues surrounding the diagnosis and treatment options for LGS has been published. This review also highlighted the limitations and difficulties of trials that have studied this syndrome to date, has proposed key considerations for future trials and the need for a comprehensive approach for the treatment of this syndrome (Arzimanoglou 2009).
The following seizure types and EEG findings are associated with LGS (Beaumanoir 2005).
1. Tonic axial seizures
These are the hallmark seizure type, and may be axial, appendicular or global, symmetrical, or unilateral. They consist of flexion of the neck and body, extension of the arms and legs, and contraction of the facial muscles. There may be associated apnoea, eye rolling and facial flushing. Consciousness is usually impaired. They are diurnal and nocturnal. They are usually brief, lasting seconds. The EEG shows discharges of fast bilateral bursts, particularly seen during sleep, predominately anteriorly or on the vertex.
2. Atypical absence seizures
These occur in the majority of cases and are frequently subtle. Loss of consciousness may be incomplete, allowing the individual to continue ongoing activities. However they are often accompanied by loss of muscle tone, myoclonic jerks and drooling (Aicardi 1996). EEG shows irregular diffuse slow spike‐wave activity at 2 to 2.5 Hz.
3. Atonic seizures
These are less frequent than the tonic axial seizures. They are manifested by a rapid loss of tone that may involve a sudden head drop or fall to the ground and are associated with polyspikes and slow waves, or diffuse spike waves on the EEG (Beaumanoir 2005).
4. Myoclonic seizures
These are considered rare and in children with prominent myoclonic jerks alternative diagnoses such as myoclonic astatic epilepsy or Dravet's syndrome should be considered. Rarely they may precede atonic attacks as myoclonic‐atonic attacks. They can be associated with slow waves, polyspike waves, diffuse rapid spike waves or brief discharges predominantly in the anterior regions.
5. Tonic‐clonic and partial‐onset seizures
These are less commonly seen in LGS than other epilepsies, but may nonetheless be present in a minority of cases.
6. Status epilepticus and non‐convulsive status
Occurs in approximately two‐thirds of patients and usually consists of continuous absence seizures punctuated by recurring tonic seizures; may be difficult to recognise.
EEG
This is abnormal in the vast majority of cases showing 2‐ to 2.5‐Hz slow spike‐wave discharges over both hemispheres with multifocal spikes and spike waves predominating in the frontal and temporal areas. In addition, the presence of fast (10 Hz) rhythms associated with tonic attacks or sometimes with minimal or no clinical manifestations, mainly during non‐REM sleep, is considered a necessary criteria by some authors. However, in some people the characteristic EEG abnormalities may be variable and even transient.
Prognosis
Psychomotor delay and neuropsychiatric symptoms occur in 90% of people with LGS. Some children have delayed development prior to the onset of their seizures as part of a predisposing condition, for example West's syndrome. Nevertheless even in these individuals further regression of development is often seen after the onset of LGS. Language is frequently affected, with both slowness in ideation and expression in addition to difficulties of motor dysfunction. Severe behavioural disorders are nearly always present. There is also a tendency for psychosis to develop with time. The long‐term prognosis is poor; although the epilepsy often improves, complete seizure freedom is rare and conversely the mental and psychiatric disorders tend to worsen with time (Beaumanoir 2005).
Treatment
Treatment is generally unsatisfactory and few randomised controlled trials (RCTs) have been undertaken. Treatment of the multiple seizure types often results in polypharmacy and poor seizure control with frequent episodes of status. Occasionally surgery (corpus callosotomy or insertion of a vagal nerve stimulator for intractable drop attacks (Patwardhan 2000) or resection of an underlying localised lesion) may be beneficial (Beaumanoir 2005). A ketogenic diet may also be considered in these patients. Psychotherapy and psychiatric interventions may be helpful in treating the psychiatric disorders.
Objectives
The aim is to compare the effects of single pharmaceutical therapies (either as first‐ or second‐line adjunctive therapy) used to treat LGS in terms of control of seizures and adverse effects. We did not consider the treatment of neuropsychiatric symptoms in this review. In addition, we did not consider surgery or alternative treatment (e.g. ketogenic diet, acupuncture or homeopathy) in this review.
The following hypotheses were tested.
Therapy A* is more effective in controlling the absence seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Therapy A* is more effective in controlling the tonic seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Therapy A* is more effective in controlling the atonic seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Where studies do not differentiate between atonic and tonic seizures but combine them under the category of "drop attacks" then the hypothesis that therapy A* is more effective in controlling the drop attacks (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B* will be tested.
Therapy A* is more effective in controlling the myoclonic seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Therapy A* is more effective in controlling the tonic‐clonic seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Therapy A* is more effective in controlling partial‐onset seizures (in terms of seizure cessation and reduction in total number of seizures) than placebo, no treatment or therapy B*.
Therapy A*/B* = adrenocorticotrophic hormone (ACTH), or carbamazepine, or clobazam, or clonazepam, or diazepam, or ethosuximide, or felbamate, or gabapentin, or lamotrigine, or levetiracetam, or nitrazepam, or oxcarbazepine, or phenobarbitone, or phenytoin, or prednisone, or pyridoxine (vitamin B6), or rufinamide, or tiagabine, or topiramate, or valproate, or vigabatrin, or zonisamide, or any other single pharmaceutical therapeutic agent studied in the literature.
Methods
Criteria for considering studies for this review
Types of studies
We included all RCTs of the administration of drug therapy to patients with LGS in this review, including trials that compare a therapy with none or placebo and trials that compare one drug with another.
Definition of RCT: trials in which participants are prospectively allocated to treatment groups by a random (e.g. random number generation, coin flips) or quasi‐random (e.g. by date of birth) process.
If the study was not an RCT, we did not include it in the review; we documented the existence of such studies.
We considered studies looking at drug therapy as second‐line therapy as well as those studies looking at drug therapies as first‐line therapy with intention to treat.
Types of participants
Any individual treated for LGS regardless of prior therapy or surgery.
Types of interventions
Any trial that compares at least one therapy against placebo treatment.
Any trial that compares at least one therapy against no therapy.
Any trial that compares at least one therapy against another therapy.
Therapies included: ACTH, or carbamazepine, or clobazam, or clonazepam, or diazepam, or ethosuximide, or felbamate, or gabapentin, or lamotrigine, or levetiracetam, or nitrazepam, or oxcarbazepine, or phenobarbitone, or phenytoin, or prednisone, or pyridoxine (vitamin B6), or rufinamide, or tiagabine, or topiramate, or valproate, or vigabatrin, or zonisamide, or any other single therapeutic agent studied in the literature.
Types of outcome measures
Cessation of seizures
Cessation of absence seizures: this is defined as total cessation of absence seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of tonic seizures: this is defined as total cessation of tonic seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of atonic seizures: this is defined as total cessation of atonic seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of drop attacks: this is defined as total cessation of drop attacks within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of myoclonic seizures: this is defined as total cessation of myoclonic seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of tonic‐clonic seizures: this is defined as total cessation of tonic‐clonic seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of partial‐onset seizures: this is defined as total cessation of partial‐onset seizures within the trial period. It is measured as a dichotomous variable (i.e. ceased/continuing).
Cessation of all seizures: this is defined as total cessation of all seizure types within the trial period.
Quantitative reduction of seizures
Quantitative reduction of absence seizures: this is measured as the number of absence seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of tonic seizures: this is measured as the number of tonic seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of atonic seizures: this is measured as the number of atonic seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of drop attacks: this is measured as the number of drop attacks occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of myoclonic seizures: this is measured as the number of myoclonic seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of tonic‐clonic seizures: this is measured as the number of tonic‐clonic seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of partial‐onset seizures: this is measured as the number of partial‐onset seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period and is measured as a continuous variable.
Quantitative reduction of all seizure types: this is measured as the number of all seizures occurring before treatment was commenced compared with the total number of seizures occurring at the end of the trial period.
Adverse effects
We measured only adverse effects that were considered severe enough to warrant discontinuation of the test treatment. We measured these as a dichotomous variable; that is therapy stopped versus therapy not stopped. We also qualitatively summarised them.
Deaths
We measured all deaths as a dichotomous variable (i.e. alive/deceased).
Search methods for identification of studies
Electronic searches
We searched the following databases. There were no language restrictions.
The Cochrane Epilepsy Group Specialized Register (18 October 2012).
The Cochrane Central Register of Controlled Trials (CENTRAL Issue 10 of 12, The Cochrane Library 2012), using 'Lennox Gastaut' as a free‐text search term.
MEDLINE (Ovid, 1946 to October week 2, 2012), using the search strategy outlined in Appendix 1.
We searched EMBASE in a similar manner from 1980 to March 2003, but we no longer have direct access to that database. However, a project to identify reports of trials in EMBASE is being carried out by the UK Cochrane Centre. This search is updated annually and these records are published in CENTRAL. These records are therefore available to us via our searches of CENTRAL.
We searched the International Standard Randomised Controlled Trial Number Register (ISRCTN) (www.controlled‐trials.com/isrctn/ searched on 18 October 2012) using 'Lennox Gastaut' as a free‐text search term for any ongoing or unpublished trials.
Searching other resources
We contacted pharmaceutical companies, authors of review articles and colleagues to try and identify unpublished data.
Data collection and analysis
Two review authors (Eleanor Hancock and Helen Cross) independently read relevant publications. We resolved discrepancies by discussion. There was no blinding of authorship or results. We considered all RCTs, published and unpublished. We also considered all non‐English studies and obtained a translation, where required.
1. Exclusion criteria
a. We excluded any trial that is not an RCT from our analysis, but documented it. b. We also excluded any trial in which there was doubt about the clinical diagnosis of LGS from our analysis, but documented it.
2. Assessment of methodological quality
a. Selection bias: we assessed the studies as to whether allocation concealment was adequate, unclear or inadequate. b. Performance bias: we assessed the studies as to whether recipients and those measuring outcome were unaware of the assigned therapy. c. Attrition bias: we assessed the studies for loss to follow‐up. Using the above criteria, we then divided studies into (i) those with a low risk of bias; (ii) those with a moderate risk of bias and (iii) those with a high risk of bias. In studies where it was unclear whether the above criteria had been met, we endeavoured to obtain additional information by contacting the first author on up to three occasions. We documented this. If it remained unclear whether the criteria had been met, we recorded this as "unclear".
Two review authors (Eleanor Hancock and Helen Cross) independently extracted the data. We endeavoured to resolve any discrepancies by discussion; if this was not possible then we contacted an external referee.
I. Participants (i.e. those characteristics of the population that may affect outcome regardless of treatment)
a. Sex (male, female). b. Age at onset. c. Age at diagnosis. d. Age at start of treatment (mean, median and range).
II. Interventions
a. Type of pharmaceutical agent used (e.g. lamotrigine, felbamate, vigabatrin, valproate). b. Dose is measured in the internationally accepted units (e.g. milligrams, micrograms). c. Frequency (this was measured as the number of times the pharmaceutical agent is given in a 24‐hour period). d. Route of administration (i.e. oral, intravenously). e. Length of treatment.
III. Outcome measures
a. Cessation of seizure types (e.g. absence, tonic etc.). This was recorded as a yes or no variable and was subsequently analysed as dichotomous data. b. Reduction in seizures (e.g. number of absences, tonic seizures etc.). This was measured as the seizure rate (frequency) occurring prior to treatment compared with the seizure rate (frequency) occurring after the treatment. Where possible we have measured it for each individual participant. If data are not given individually but are given for each treatment group, then we have measured it for each treatment group. We have analysed it as continuous data. c. Adverse effects. This was recorded as the number of participants in whom the adverse effects were severe enough to warrant stopping the treatment and those in whom treatment continued; it was subsequently presented as dichotomous data. We also collected details of the types of adverse effects and qualitative information as reported by the investigators. d. Deaths. This was recorded as a yes or no variable and will subsequently be presented as dichotomous data.
Again when data are missing from the published report, we have attempted to contact the first author on up to three occasions. If the data remain unavailable, we have recorded it as "not reported".
3. Analysis plan
a. Study quality: this was done by a visual plot of met/unmet criteria for selection and performance bias.
b. Dichotomous data: for each item of data requiring dichotomous analysis, we recorded the following: number of participants who experienced the event (or outcome) in each group for each comparison and the total number in each group.
c. Continuous data: for each item of data requiring continuous data analysis, we recorded the following: number of participants in each group; the mean value for the outcome in each group and the standard deviation for each mean. Where these values are not stated in the study reviewed, we have attempted to obtain missing data (where possible) from the investigators. We have analysed any such data using mean differences.
d. When data for the same outcome are presented in some studies as dichotomous and in others as continuous data then we have endeavoured to obtain continuous data from the investigators. If it is not possible to obtain continuous data, for example because it was not recorded, then we have either analysed the data as dichotomous, with a "cut off" point agreed by the two review authors, or a mixture of dichotomous and continuous data using two separate tables.
e. We also looked for sources of heterogeneity between trials of methodological and clinical differences, for example age at trial entry. We have also looked for and reported, where applicable, subgroup analysis of differences in drug dosages and timing and length of treatment.
Results
Description of studies
The literature search of the Cochrane Epilepsy Group's Specialized Register (October 2012), MEDLINE (1946 to October 2012), EMBASE (1980 to March 2003) and ISRCTN (October 2012) found a total of 13 possible RCTs. We excluded one study (Vajda 1985) because only some of the participants had a diagnosis of LGS and the results were not given independently for those participants. Two further studies remain under review. Jensen 1994 appears to include the same cohort of participants as Anonymous 1993; we are contacting the authors in order to confirm or refute this possibility. Vassella 1978: this paper is written in German and the results remain unclear after translation; again we hope to contact the authors in order to clarify the results. Glauser 2009 included the same cohort of patients as Glauser 2008 but with different long‐term follow‐up results. This gave a total of nine studies to be evaluated for inclusion into this review, with a total of 979 participants looking at seven different pharmacological agents: cinromide, clobazam, felbamate, thyrotrophin‐releasing hormone (TRH), lamotrigine, rufinamide and topiramate. We intend to continue correspondence with authors, colleagues and drug companies and to update our literature searches of the above databases regularly, so that we may update this review as future studies are completed.
Anonymous 1989
Anonymous 1989 was a randomised, double‐blind, placebo‐controlled multicentre trial of 73 participants. Inclusion criteria included participants aged between two and 18 years of age: they must have had seizures for at least six months. All participants had a history of multiple seizure types with seizure onset during the first decade and had at least 40 clinically recognisable seizures every two weeks during the six weeks before study entry. Predominately generalised, slow, spike and wave discharges were demonstrated on an EEG during the three months before study entry. No individual was receiving more than three marketed antiepileptic drugs (AEDs) and none had previous exposure to cinromide. The trial consisted of a six‐week baseline period, following which participants were randomised to receive either cinromide or placebo for a period of 18 weeks. Study medication was initiated at 20 to 40 mg/kg/day, divided into four equal doses. Further increases (to a total daily maximum of 83 to 109 mg/kg) were prescribed at weekly visits, if each prior dose was tolerated and seizures continued. The method of randomisation is not stated. Both the recipients and assessors were blinded. There was loss to follow‐up and data were only analysed for 56 of the participants. Outcomes reported were complete cessation and a reduction in all seizure types.
Anonymous 1993
Anonymous 1993 was a randomised, double‐blind, placebo‐controlled multicentre trial of 73 participants. Individuals were included if they had a history of multiple types of seizures and a minimum of 90 atonic seizures or atypical absence seizures per month during an eight‐week pre‐study phase and were taking no more than two AEDs. The trial consisted of a 28‐day baseline period followed by a 14‐day titration phase and a 56‐day maintenance period. The initial dose of felbamate was 15 mg/kg/day, increased to 30 mg/kg after seven days and to either 14 mg/kg/day or 3600 mg/day (whichever was lower) after 14 days. The number of capsules taken by participants in the placebo group was based on body weight. Randomisation was by a computer‐generated schedule. Both recipients and assessors were blinded. Two participants withdrew from the study because of unacceptable adverse effects; there was no loss to follow‐up. Outcomes reported were cessation and/or reduction of atonic, tonic‐clonic and all seizures types.
Conry 2009
Conry 2009 was a randomised, double‐blind, dose ranging trial of 68 participants. Individuals were included if they had an EEG with slow spike and wave and multifocal spikes; had more than one type of generalised seizure for at least 6 months; were less than 11 years of age at the onset of LGS; weighed more than 12.5 kg; were on one to three anticonvulsant drugs and had at least two drop seizures per week. The trial consisted of a four‐week baseline period, a three‐week titration period and a four‐week maintenance period. Patients received either low‐dose clobazam (target dose of 25 mg/kg/day to a maximum of 10 mg/day) or high‐dose clobazam (target dose 1.0 mg/kg/day to a maximum of 40 mg/day). The method of randomisation was not stated. Both recipients and assessors were blinded. Seven patients were excluded from analysis because they did not have at least one measurement during the maintenance period. Nine patients withdrew as a result of unacceptable side effects. There were no deaths reported. Outcomes reported were percentage reductions in drop seizures and non‐drop seizures.
Conry 2010
Conry 2010 was a randomised, double‐blind, placebo‐controlled trial of 217 participants. Individuals were included between two and 60 years of age with LGS documented by both clinical and EEG criteria. Following a four‐week baseline phase, patients who had two drop seizures per week were randomised to placebo or one of three doses of clobazam (0.25, 0.5 and 1.0 mg/kg/day), up to a maximum dosage of 40 mg/day. Treatment included a three‐week titration phase and a 12‐week maintenance phase. The method of randomisation was not stated. Both participants and assessors were blinded. Forty patients did not complete the study. Outcomes reported were percentage reductions in drop seizures and non‐drop seizures.
Eriksson 1998
Eriksson 1998 was a randomised, double‐blind, placebo‐controlled trial of 30 participants (20 had LGS). All children and adolescents older than two years of age with refractory generalised epilepsy with more than two seizures per month were eligible for the trial. LGS was defined according to Gastaut's criteria. The trial consisted of six phases: an eight‐week baseline phase during which each child was observed on pre‐study medication; an open phase during which an attempt was made to find the optimal lamotrigine dose for each child; a double‐blind phase of two 12‐week periods during which, for each child, lamotrigine and placebo tablets were administered in random order. The treatment periods were separated by a three‐week washout phase. The method of randomisation was not stated. Both recipients and assessors were blinded. Results are given for 13 patients. Outcomes reported are reduction rates of all seizure types.
Glauser 2008
Glauser 2008 was a randomised, double‐blind, placebo‐controlled multicentre trial of 139 participants. Patients aged four to 30 years were eligible if they had a history of multiple seizure types, which had to include atypical absence seizures and drop attacks, a minimum of 90 seizures in the month prior to trial entry, an EEG demonstrating a pattern of slow spike and wave complexes, a body weight of at least 18 kg, a fixed‐dose regimen of one to three concomitant AEDs, and brain imaging confirming the absence of a progressive lesion. The trial consisted of a 28‐day baseline period at the end of which, patients continuing to meet the study criteria entered an 84‐day double‐blind treatment phase of either rufinamide or placebo. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period. Doses were titrated according to a recommended schedule based on body weight, up to a maximum dose of 45 mg/kg/day. The method of randomisation was not clear. Both recipients and assessors were blinded. Results were given for 123 participants. Outcomes reported are percentage reductions in seizure frequency for total seizure frequency, atonic‐tonic seizures, absence seizures, myoclonic seizures and partial‐onset seizures. Fifteen patients withdrew from the study because of unacceptable side effects. No deaths were reported
Glauser 2009 (Abstract only)
Glauser 2009 was a randomised, double‐blind, placebo‐controlled multicentre trial of 138 participants. Patients aged four to 30 years were eligible if they had a history of multiple seizure types including atypical absence seizures and drop attacks, a minimum of 90 seizures in the month prior to trial entry and had a recent history of a slow spike and wave pattern on EEG. The trial consisted of a 28‐day baseline period at the end of which, patients continuing to meet the study criteria entered an 84‐day double‐blind treatment phase. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period. Target dose of rufinamide was 45 mg/kg/day. The method of randomisation was not clear. Results were given for 129 participants. Outcomes reported were percentage reductions in seizure frequency for total seizure frequency and tonic‐atonic seizures. Nine patients were not included in the final analysis, the reasons for which were not given.
N.B. From the limited data available in this published abstract the study appears to use the same cohort of patients as Glauser 2008, but they report slightly different results and losses to follow‐up. For the purposes of this review the results of this paper have not been included in the meta‐analysis as the review authors were concerned that this might result in duplicate data entry giving rise to misleading figures. We will seek clarification from the study authors and will include the results given under the section effects of the interventions only.
Inanaga 1989
Inanaga 1989 was a randomised, open, dose‐finding multicentre trial of 190 participants of whom 98 had a diagnosis of LGS. Participants were either older than two years of age or weighed more than 15 kg and inclusion criteria included a stable condition during the pre‐treatment period with no excess sedation from other AEDs. The trial consisted of an eight‐week period where participants received either 0.4 mg/kg or 1.6 mg/kg of TRH DN‐1417 once a day orally. The method of randomisation was not stated and neither the recipients nor the assessors were blinded. Some participants were lost to follow‐up. Outcomes reported were reduction in absence, tonic and atonic seizures and all seizure types.
Motte 1997
Motte 1997 was a randomised, double‐blind, placebo‐controlled multicentre trial of 179 participants. Participants aged three to 25 years were included if they had more than one type of predominantly generalised seizure for at least one year, were younger than 11 years of age at the onset of seizures and had seizures at least every other day; and they had intellectual impairment or deterioration and a recent EEG demonstrating an abnormal background and a pattern of slow‐wave complexes. The trial consisted of a four‐week baseline period during which all recipients received placebo. Participants were then randomised to receive either lamotrigine or placebo for a 16‐week treatment period. Participants were assigned to one of four dosing regimens according to concomitant valproate use and body weight based on paediatric dosing recommendations at that time. The method of randomisation was not stated. Both recipients and assessors were blinded. Results were given for 169 participants. Outcomes reported were overall reduction in absence seizures, drop attacks, tonic‐clonic seizures and all seizure types.
Sachdeo 1999
Sachdeo 1999 was a randomised, double‐blind, placebo‐controlled multicentre trial of 112 participants. Participants aged one to 30 years were eligible if they had an EEG showing a slow spike and wave pattern and seizure types including drop attacks and atypical absence seizures, with a frequency of at least 60 seizures during the month prior to the baseline phase while being maintained on one or two standard AEDs. The trial consisted of a baseline phase of four weeks and an 11‐week treatment phase. The participants were titrated up to a dose of 6 mg/kg/day or their maximal tolerated dosage of either topiramate or placebo over the first three weeks of the treatment period. Randomisation was by a computer‐generated schedule and there was concealment of allocation. Both recipients and assessors were blinded. Results were given for 97 participants. Outcomes reported were cessation and reduction in drop attacks and overall reduction of all seizure types.
Risk of bias in included studies
All but one of the nine studies were double‐blind placebo‐controlled trials, but only two gave the method of randomisation, of which only one stated that concealment of allocation had been performed. Eight had loss of participants to follow‐up. The ninth study was an open dose‐finding study that did not state the method of randomisation and had loss of patients to follow‐up (see Table 6).
1. Methodological quality of included studies.
| Study ID | Concealment | Assessors blinded | Recipients blinded | Loss to follow‐up |
| Anonymous 1989 | Not clear | Yes | Yes | Yes |
| Anonymous 1993 | Not clear | Yes | Yes | No |
| Inanaga 1989 | Not clear | No | No | Yes |
| Motte 1997 | Not clear | Yes | Yes | Yes |
| Glauser 2008 | Not clear | Yes | Yes | Yes |
| Glauser 2009 | Not clear | Yes | Yes | Yes |
| Eriksson 1998 | Not clear | Yes | Yes | Yes |
| Sachdeo 1999 | Yes | Yes | Yes | Yes |
| Conry 2009 | Not clear | Yes | Yes | Yes |
| Conry 2010 | Not clear | Yes | Yes | Yes |
We had planned to look at characteristics of the population that may affect outcome regardless of treatment (see Table 7); unfortunately not only was there great heterogeneity among the populations studied, but the information was not always presented. The male:female ratio given for six studies was 1.5:1.0 and the age at trial entry given for eight studies ranged from two to 60 years of age. The age at diagnosis of LGS was not given for any of the studies other than one study that stated age of onset was before 11 years of age. It is not possible, therefore, to comment on any effect that these characteristics might have had on final outcome.
2. Population characteristics.
| Study ID | Treatment | Participant numbers | Male:female | Age at Dx of LGS | Age at trial entry |
| Glauser 2008 (Glauser 2009) | Rufinamide | 74 | 46:28 | Not given | 13 (4 to 35) years |
| Placebo | 64 | 40:24 | Not given | 10.5 (4 to 37) years | |
| Eriksson 1998 | Lamotrigine | 13 | Not given | Not given | 10 (4.6 to 16.9) years |
| Placebo | 13 | Not given | Not given | 10 (4.6 to 16.9) years | |
| Anonymous 1989 | Cinromide | 26 | 13:13 | Not given | 7.38 ± 3.65 (2 to 17) years |
| Placebo | 30 | 21:9 | Not given | 7.93 ± 4.87 (2 to 18) years | |
| Anonymous 1993 | Felbamate | 37 | 27:10 | Not given | 12 (4 to 24) years |
| Placebo | 36 | 24:12 | Not given | 14 (4 to 36) years | |
| Inanaga 1989 | Low‐dose TRH | 48 | Not given | Not given | Not given |
| High‐dose TRH | 50 | Not given | Not given | Not given | |
| Motte 1997 | Lamotrigine | 79 | 54:25 | Not given | 9.6 ± 5.2 years |
| Placebo | 90 | 45:45 | Not given | 10.9 ± 5.9 years | |
| Sachdeo 1999 | Topiramate | 48 | 28:20 | Not given | 11.2 ± 6.2 (2 to 29) years |
| Placebo | 50 | 25:25 | Not given | 11.2 ± 7.7 (2 to 42) years | |
| Conry 2009 | Low‐dose clobazam | 32 | 42:26 | Not given | 7.4 (2 to 26) years |
| High‐dose clobazam | 36 | Not given | |||
| Conry 2010 | Clobazam (3 doses) | 160 | 60.5% male | Not given | 12.4 years |
| Placebo | 57 | Not given |
Dx: diagnosis; LGS: Lennox‐Gastaut syndrome; TRH: thyrotrophin‐releasing hormone.
Effects of interventions
Cinromide versus placebo
Anonymous 1989: two outcome measures were considered in this study. The effect on cessation of all seizure types: number of participants in either the cinromide or placebo group that had complete cessation of their seizures and the reduction in number of all seizure types. In the cinromide group, three participants had 75% to 100% reduction in total seizures, four participants had 50% to 74% reduction in total seizures, five participants had 25% to 49% reduction in total seizures, six participants had 0% to 24% reduction in seizures and eight participants had an increase in their total number of seizures. In the placebo group, no participant had 75% to 100% reduction in total seizures, seven participants had 50% to 74% reduction in total seizures, six participants had 2% to 49% reduction in total seizures, six participants had 0% to 24% reduction in seizures and 11 participants had an increase in their total number of seizures. None of these had a significant risk ratio (RR) (Analysis 1.1; Analysis 1.2; Analysis 1.3; Analysis 1.4; Analysis 1.5).
1.1. Analysis.
Comparison 1 Cinromide versus placebo, Outcome 1 Number of participants in whom there was complete cessation of seizures.
1.2. Analysis.
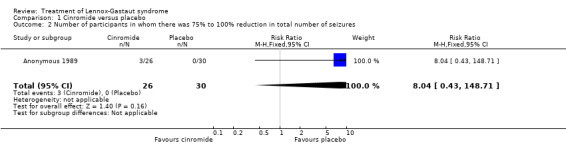
Comparison 1 Cinromide versus placebo, Outcome 2 Number of participants in whom there was 75% to 100% reduction in total number of seizures.
1.3. Analysis.
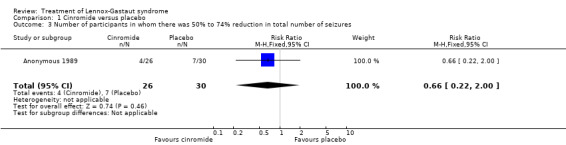
Comparison 1 Cinromide versus placebo, Outcome 3 Number of participants in whom there was 50% to 74% reduction in total number of seizures.
1.4. Analysis.
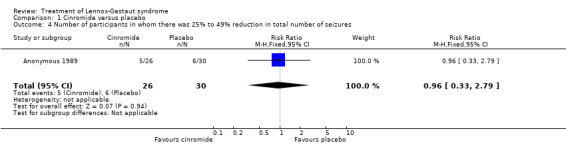
Comparison 1 Cinromide versus placebo, Outcome 4 Number of participants in whom there was 25% to 49% reduction in total number of seizures.
1.5. Analysis.
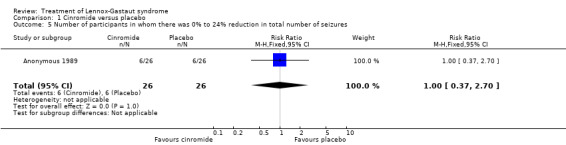
Comparison 1 Cinromide versus placebo, Outcome 5 Number of participants in whom there was 0% to 24% reduction in total number of seizures.
This study did not report the effect of treatment on either stopping or reducing the number of absence, tonic, atonic, myoclonic, tonic‐clonic or partial seizures, or drop attacks.
No participants were reported as having the treatment stopped due to adverse effects and no deaths were reported.
Felbamate versus placebo
Anonymous 1993: this study reported that five out of 28 participants treated with felbamate had total cessation of atonic seizures as compared with none out of 22 participants treated with placebo, giving an RR with 95% confidence intervals (CIs) of 5.7 (95% CI 0.5 to 149.80). The reduction in number of atonic seizures was given as an overall reduction of 44% in the 28 participants treated with felbamate compared with an overall reduction of 9% in the 22 participants treated with placebo (P = 0.02 using analysis of variance). They also reported that seven out of 16 participants treated with felbamate had total cessation of tonic‐clonic seizures as compared with one out of 13 participants treated with placebo, giving an RR of 5.7 (95% CI 0.8 to 40.5), it was unclear how many participants had a reduction in the number of tonic‐clonic seizures. Overall, for all seizure types, four out of 37 participants treated with felbamate had total cessation of all seizure types as compared with one out of 36 participants treated with placebo, giving an RR of 3.9 (95% CI 0.5 to 33.2) and the reduction in number of all seizure types was given as an overall reduction of 19% in the 37 participants treated with felbamate compared with an overall increase of 4% in the 36 participants treated with placebo (P = 0.002 using analysis of variance) (Analysis 2.1; Analysis 2.2; Analysis 2.3).
2.1. Analysis.
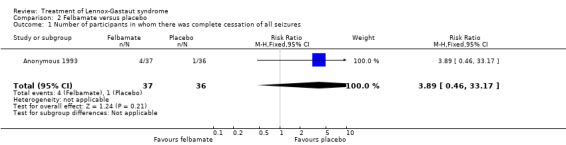
Comparison 2 Felbamate versus placebo, Outcome 1 Number of participants in whom there was complete cessation of all seizures.
2.2. Analysis.
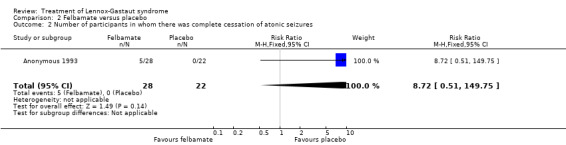
Comparison 2 Felbamate versus placebo, Outcome 2 Number of participants in whom there was complete cessation of atonic seizures.
2.3. Analysis.
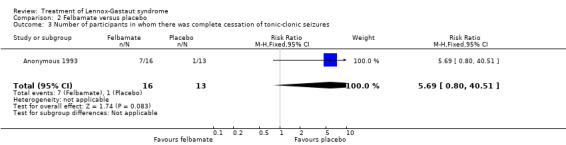
Comparison 2 Felbamate versus placebo, Outcome 3 Number of participants in whom there was complete cessation of tonic‐clonic seizures.
This study did not report the effect of treatment on either stopping or reducing the number of absence, tonic or myoclonic seizures or drop attacks. One participant had treatment stopped because of somnolence and ataxia in the felbamate group and one because of pancreatitis in the placebo group. There were no deaths reported.
Low‐dose versus high‐dose TRH DN 1417
Inanaga 1989: this group reported that in the low‐dose group, one person had 75% to 100% reduction in absence seizures, none had 50% to 74% reduction in absence seizures, two people had 25% to 49% reduction in absence seizures, nine people had 0% to 24% reduction in absence seizures and nine people had an increase in their total number of absence seizures. In the high‐dose group, two people had 75% to 100% reduction in absence seizures, three individuals had 50% to 74% reduction in absence seizures, six individuals had 25% to 49% reduction in absence seizures and 15 individuals had 0% to 24% reduction in absence seizures. None of these had a significant RR (Analysis 3.1; Analysis 3.2; Analysis 3.3; Analysis 3.4).
3.1. Analysis.
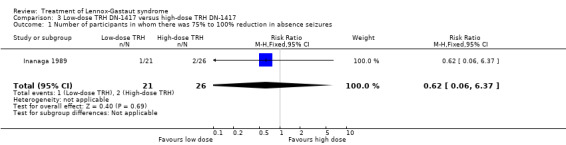
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 1 Number of participants in whom there was 75% to 100% reduction in absence seizures.
3.2. Analysis.
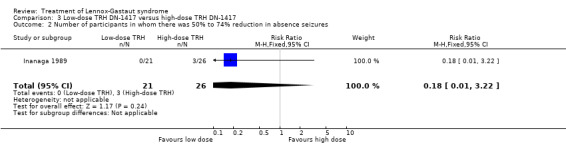
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 2 Number of participants in whom there was 50% to 74% reduction in absence seizures.
3.3. Analysis.
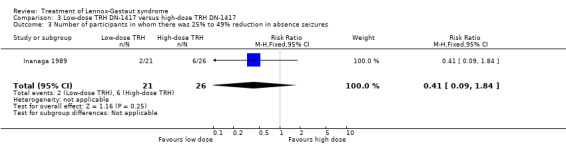
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 3 Number of participants in whom there was 25% to 49% reduction in absence seizures.
3.4. Analysis.
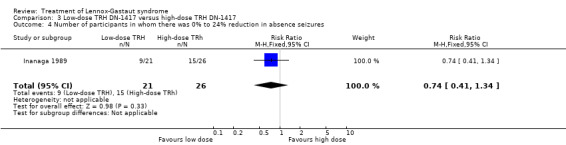
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 4 Number of participants in whom there was 0% to 24% reduction in absence seizures.
In the low‐dose group, one participant had 75% to 100% reduction in tonic seizures, three participants had 50% to 74% reduction in tonic seizures, five participants had 25% to 49% reduction in tonic seizures, 19 participants had 0% to 24% reduction in tonic seizures and five participants had an increase in their total number of tonic seizures. In the high‐dose group, one participant had 75% to 100% reduction in tonic seizures, four participants had 50% to 74% reduction in tonic seizures, seven participants had 25% to 49% reduction in tonic seizures, 10 participants had 0% to 24% reduction in tonic seizures, and two had an increase in their number of tonic seizures. Only the reduction of 0% to 24% had a small significant RR of 2.0 (95% CI 0.09 to 46.09) (Analysis 3.5; Analysis 3.6; Analysis 3.7; Analysis 3.8).
3.5. Analysis.
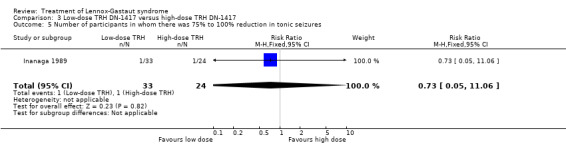
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 5 Number of participants in whom there was 75% to 100% reduction in tonic seizures.
3.6. Analysis.
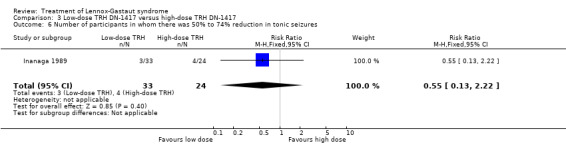
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 6 Number of participants in whom there was 50% to 74% reduction in tonic seizures.
3.7. Analysis.
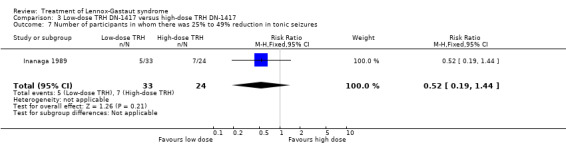
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 7 Number of participants in whom there was 25% to 49% reduction in tonic seizures.
3.8. Analysis.
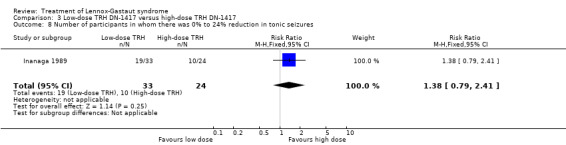
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 8 Number of participants in whom there was 0% to 24% reduction in tonic seizures.
In the low‐dose group, one participant had 75% to 100% reduction in atonic seizures, four participants had 50% to 74% reduction in atonic seizures, one participant had 25% to 49% reduction in atonic seizures, 16 participants had 0% to 24% reduction in atonic seizures and one participant had an increase in their total number of atonic seizures. In the high‐dose group, none had 75% to 100% reduction in absence seizures, three participants had 50% to 74% reduction in absence seizures, four participants had 25% to 49% reduction in absence seizures, seven participants had 0% to 24% reduction in absence seizures and one participant had an increase in their total number of atonic seizures. None of these had a significant RR (Analysis 3.9; Analysis 3.10; Analysis 3.11; Analysis 3.12.
3.9. Analysis.
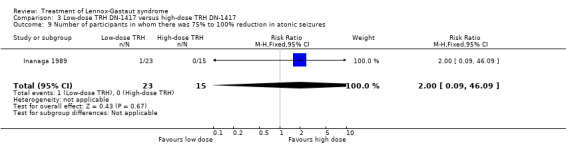
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 9 Number of participants in whom there was 75% to 100% reduction in atonic seizures.
3.10. Analysis.
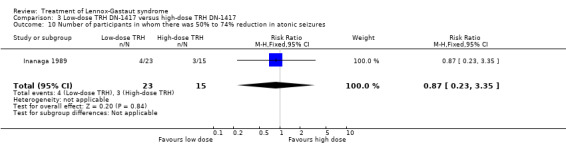
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 10 Number of participants in whom there was 50% to 74% reduction in atonic seizures.
3.11. Analysis.
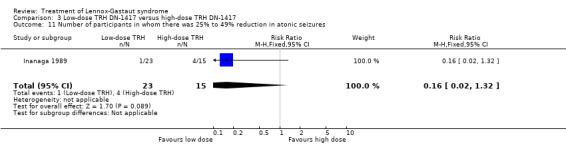
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 11 Number of participants in whom there was 25% to 49% reduction in atonic seizures.
3.12. Analysis.
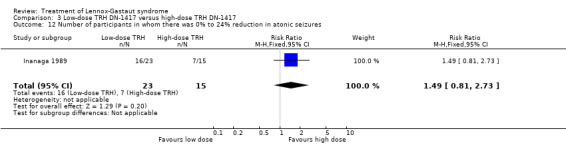
Comparison 3 Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417, Outcome 12 Number of participants in whom there was 0% to 24% reduction in atonic seizures.
This study did not report the effects of treatment on stopping absence, tonic, atonic, myoclonic, tonic‐clonic or partial‐onset seizures or drop attacks. It did not report the effects of treatment on reducing the number of myoclonic, tonic‐clonic or partial‐onset seizures or drop attacks.
No participants were reported as stopping treatment because of adverse effects and no deaths were reported.
Lamotrigine versus placebo
There were two studies that looked at the effect of lamotrigine versus placebo (Eriksson 1998; Motte 1997).
Motte 1997 reported that overall 47 participants treated with lamotrigine had a 13% decrease in the number of absence seizures compared with 44 participants treated with placebo who had an overall reduction of 38%. Eriksson 1998 did not report reduction in the number of absence seizures as an outcome.
Motte 1997 reported that in the lamotrigine group 75 participants had a 34% reduction in the number of drop attacks compared with 89 participants treated with placebo who had an overall reduction of 9%. In the lamotrigine group 28 out of 75 participants had a 50% to 100% reduction in drop attacks compared with 20 out of the 89 participants treated with placebo, giving an RR of 1.66 (95% CI 1.0 to 2.7) (Analysis 4.1). In the lamotrigine group 19 out of 75 participants had a 26% to 49% reduction in drop attacks compared with 14 out of the 89 participants treated with placebo, giving an RR of 1.61 (95% CI 0.87 to 2.99) (Analysis 4.2). In the lamotrigine group 28 out of 75 participants had either a reduction of less than 25% or an increase in their number of drop attacks compared with 55 out of the 89 participants treated with placebo, giving an RR of 0.60 (95% CI 0.43 to 0.85) (Analysis 4.3). Eriksson 1998 did not report reduction in the number of drop attacks as an outcome.
4.1. Analysis.
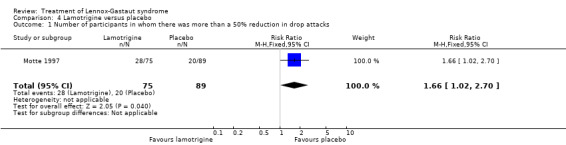
Comparison 4 Lamotrigine versus placebo, Outcome 1 Number of participants in whom there was more than a 50% reduction in drop attacks.
4.2. Analysis.
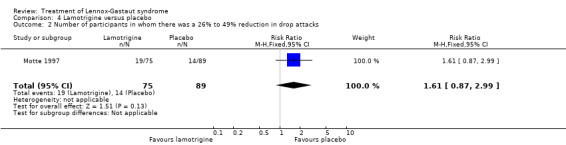
Comparison 4 Lamotrigine versus placebo, Outcome 2 Number of participants in whom there was a 26% to 49% reduction in drop attacks.
4.3. Analysis.
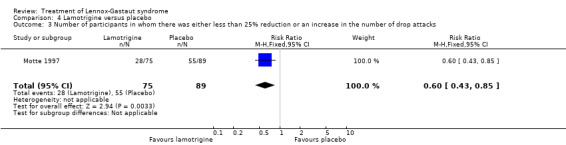
Comparison 4 Lamotrigine versus placebo, Outcome 3 Number of participants in whom there was either less than 25% reduction or an increase in the number of drop attacks.
Motte 1997 reported that in the lamotrigine group 60 participants had a 36% reduction in the number of tonic‐clonic seizures compared with 64 participants treated with placebo who had an overall increase of 10%. In the lamotrigine group 26 out of 60 participants had a 50% to 100% reduction in tonic‐clonic seizures compared with 13 out of the 64 participants treated with placebo, giving an RR of 2.13 (95% CI 1.21 to 3.75) (Analysis 4.4). In the lamotrigine group six out of 60 participants had a 26% to 49% reduction in tonic‐clonic seizures compared with 14 out of the 64 participants treated with placebo, giving an RR of 0.46 (95% CI 0.19 to 1.11) (Analysis 4.5). In the lamotrigine group 28 out of 60 participants had either a reduction of less than 25% or an increase in their number of tonic‐clonic seizures compared with 44 out of the 64 participants treated with placebo, giving an RR of 0.68 (95% CI 0.49 to 0.93) (Analysis 4.6). Eriksson 1998 did not report reduction in the number of tonic‐clonic seizures as an outcome.
4.4. Analysis.
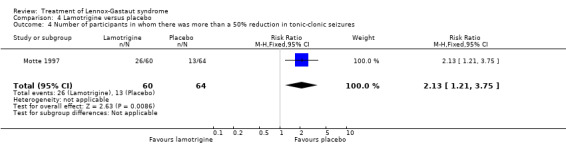
Comparison 4 Lamotrigine versus placebo, Outcome 4 Number of participants in whom there was more than a 50% reduction in tonic‐clonic seizures.
4.5. Analysis.
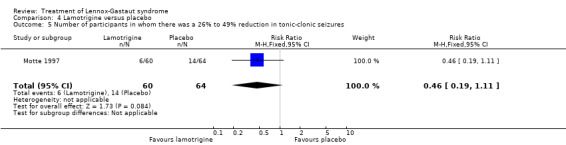
Comparison 4 Lamotrigine versus placebo, Outcome 5 Number of participants in whom there was a 26% to 49% reduction in tonic‐clonic seizures.
4.6. Analysis.
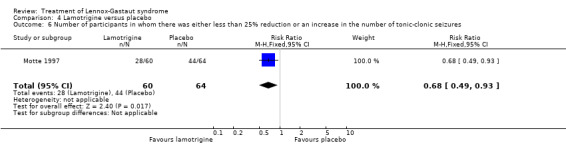
Comparison 4 Lamotrigine versus placebo, Outcome 6 Number of participants in whom there was either less than 25% reduction or an increase in the number of tonic‐clonic seizures.
Motte 1997 reported that in the lamotrigine group 78 participants had a 32% reduction in all seizures compared with 89 participants treated with placebo who had an overall increase of 9%. In the lamotrigine group 26 out of 78 participants had a 50% to 100% reduction in all seizures compared with 14 out of the 89 participants treated with placebo giving an RR of 2.12 (95% CI 1.19 to 3.76) (Analysis 4.7). Motte 1997 reported in the lamotrigine group 21 out of 78 participants had a 26% to 49% reduction in all seizures compared with 17 out of the 89 participants treated with placebo giving an RR of 1.41 (95% CI 0.80 to 2.47) (Analysis 4.8). Motte 1997 reported in the lamotrigine group 31 out of 78 participants had either a reduction of less than 25% or an increase in their number of all seizures compared with 58 out of the 89 participants treated with placebo giving an RR of 0.61 (95% CI 0.45 to 0.83) (Analysis 4.9).
4.7. Analysis.
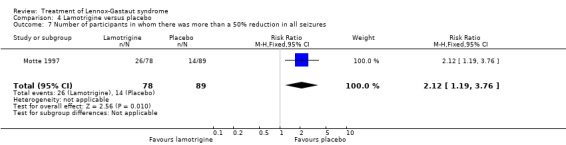
Comparison 4 Lamotrigine versus placebo, Outcome 7 Number of participants in whom there was more than a 50% reduction in all seizures.
4.8. Analysis.
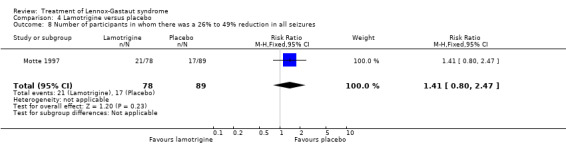
Comparison 4 Lamotrigine versus placebo, Outcome 8 Number of participants in whom there was a 26% to 49% reduction in all seizures.
4.9. Analysis.
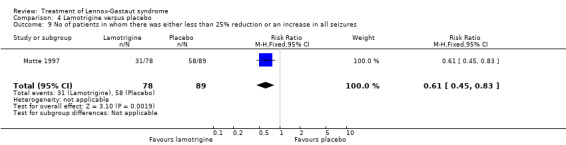
Comparison 4 Lamotrigine versus placebo, Outcome 9 No of patients in whom there was either less than 25% reduction or an increase in all seizures.
Eriksson 1998 reported that seven out of 13 children with LGS entered into the double‐blind phase of the trial showed improvement in the lamotrigine phase compared with the placebo phase, with one child showing a 100% reduction in their seizures.
The effect of treatment on the reduction in number of tonic, atonic, myoclonic and partial seizures was not reported in these two studies.
Three participants on lamotrigine had treatment withdrawn, one had deterioration of seizure control, the other two developed a rash and seven participants receiving placebo had it withdrawn, six because of deterioration in seizure control and one other developed a rash. There were no deaths reported.
Topiramate versus placebo
Sachdeo 1999 study reported the effect of treatment on drop attacks and the reduction of overall seizures. One out of 46 participants treated with topiramate had complete cessation of their drop attacks compared with none of the 50 participants treated with placebo; this had a significant RR of 3.26 (95% CI 0.14 to 77.97) (Analysis 5.1). Overall the 46 participants treated with topiramate had a 14.8% reduction in drop attacks compared with the 50 participants treated with placebo who had an overall increase of 5.1% in their drop attacks (P = 0.041 using the Bonferroni method of analysis). Eight participants treated with topiramate and three participants treated with placebo had a 75% to 100% reduction, giving an RR of 2.90 (95% CI 0.82 to 10.27) (Analysis 5.2). Thirteen participants treated with topiramate and seven treated with placebo had a 50% to 74% reduction, giving an RR of 2.02 (95% CI 0.88 to 4.62) (Analysis 5.3). Overall the 48 participants treated with topiramate had a 20.6% reduction in total seizures compared with the 50 participants treated with placebo who had 8.8% reduction in total seizures (P = 0.037 using the Bonferroni method of analysis).
5.1. Analysis.
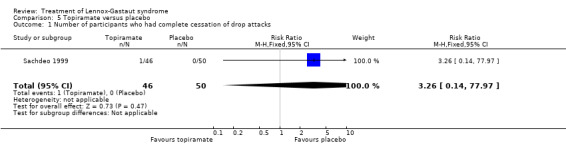
Comparison 5 Topiramate versus placebo, Outcome 1 Number of participants who had complete cessation of drop attacks.
5.2. Analysis.
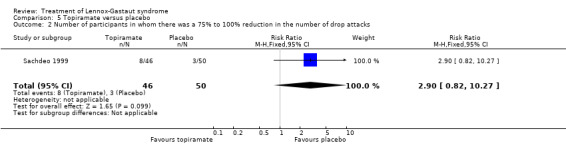
Comparison 5 Topiramate versus placebo, Outcome 2 Number of participants in whom there was a 75% to 100% reduction in the number of drop attacks.
5.3. Analysis.
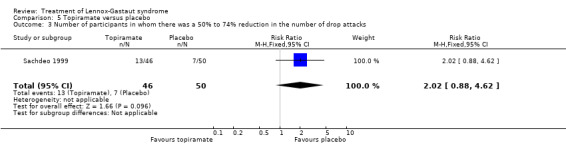
Comparison 5 Topiramate versus placebo, Outcome 3 Number of participants in whom there was a 50% to 74% reduction in the number of drop attacks.
This study did not report the effect of treatment on stopping or reducing the number of absence, tonic, clonic, myoclonic, tonic‐clonic or partial seizures or the effect on stopping all seizure types.
No participant was reported as having had treatment stopped due to adverse effects and no deaths were reported.
Rufinamide versus placebo
Glauser 2008: this study reported the effect of treatment as an overall percentage change in number of seizures for each seizure type reported. There was a 50.6% reduction in the number of absence seizures experienced by the rufinamide group compared with a 30% reduction in the placebo group. There was a 28% reduction in the number of tonic seizures experienced by the rufinamide group compared with a 1.6% reduction in the placebo group. There was a 45% reduction in the number of atonic seizures experienced by the rufinamide group compared with a 21% increase in the placebo group. There was a 42.5% reduction in the number of atonic‐clonic seizures experienced by the rufinamide group compared with a 1.4% reduction in the placebo group. There was a 50.6% reduction in the number of absence seizures experienced by the rufinamide group compared with a 30% reduction in the placebo group. There was a 30% reduction in the number of myoclonic seizures experienced by the rufinamide group compared with a 14% reduction in the placebo group. There was a 46% reduction in the number of tonic‐clonic seizures experienced by the rufinamide group compared with an 18% reduction in the placebo group. There was a 70% reduction in the number of partial‐onset seizures experienced by the rufinamide group compared with an 11% reduction in the placebo group. Overall there was a 33% reduction in the number of all seizures experienced by the rufinamide group compared with an 11.7% increase in the placebo group.
this study reported the effect of treatment as an overall percentage change in number of total seizures and tonic‐atonic seizures at weeks 1, 2, 4, 8 and 12 post‐randomisation.
At week 12 there was a 50.3% reduction in the number of tonic‐atonic seizures experienced by the rufinamide group as compared with an increase of 1.0% in the placebo group.
At week 12 there was a 43.1% reduction in the number of all seizures experienced by the rufinamide group as compared with a 1.5% reduction in the placebo group.
Low‐dose clobazam versus high‐dose clobazam
this study looked at the effect of low‐dose clobazam compared with high‐dose clobazam on the effect of drop attacks only.
In the low‐dose group 56% of patients had a reduction of at least 25% in the number of drop attacks compared with 89% in the high‐dose group.
In the low‐dose group 38% of patients had a reduction of at least 50% in the number of drop attacks compared with 83% in the high‐dose group.
In the low‐dose group 25% of patients had a reduction of at least 75% in the number of drop attacks compared with 67% in the high‐dose group.
In the low‐dose group 6% of patients had a reduction of 100% in the number of drop attacks compared with 22% in the high‐dose group.
Seven patients were not included in the follow‐up because they did not have at least on measurement of the number of drop attacks experienced during the maintenance period.
Ten patients discontinued medication due to side effects: somnolence (2 people), convulsion (1 person), aggression (1 person), reduced oral intake (1 person), chorea (1 person), behaviour (1 person), encephalopathy (1 person) and sedation (1 person) (1 person) at carers request, no further details given. No deaths were reported.
Clobazam versus placebo
this study looked at the effects of three dosages of clobazam as compared with placebo on the reduction in the number of drop seizures.
There was no statistical decrease with low‐dose clobazam (0.25 mg/kg/day) as compared with placebo.
In the 0.5 mg/kg/day dose 59% had a reduction in the number of drop seizures of 50% as compared with 32% of those treated with placebo.
In the 0.5 mg/kg/day dose 38% had a reduction in the number of drop seizures of 75% as compared with 11% of those treated with placebo.
In the 0.5 mg/kg/day dose 78% had a reduction in the number of drop seizures of 50% as compared with 32% of those treated with placebo.
In the 0.5 mg/kg/day dose 63% had a reduction in the number of drop seizures of 75% as compared with 11% of those treated with placebo.
Forty patients were excluded from the results, the reasons for loss to follow‐up are not given.
Patients discontinuing medication due to adverse effects are not given and no deaths were reported.
Discussion
LGS is a complex epileptic encephalopathy that encompasses different seizure types that often vary in their frequency over a period of time. It is a syndrome that has historically been difficult to treat, with many people receiving polypharmacy without seizure control. The aim of our review was to try and determine the evidence base for the optimal treatment of this syndrome (on a single therapy basis, though it must be remembered that many individuals will have been receiving polytherapy at the time of entry into a trial). This has not been possible from current data available for the reasons discussed below.
LGS is not rare; it is thought to account for up to 10% of all childhood epilepsies, yet despite an extensive review of the literature we found only nine RCTs looking at the pharmaceutical treatment of this syndrome. There are several possible explanations for this lack of information. LGS is primarily a syndrome of childhood; although long‐term prognosis is poor with regard to cognitive outcome, seizures become less troublesome into adulthood. Treatments require evaluation early in its course. There has traditionally been reluctance to set up trials in the paediatric age group as it is both difficult and expensive, and continues to be so, despite the early exclusivity clause set out by EU Regulations (European Union 2006). In addition, although LGS is not rare, its prevalence would require a multicentre collaboration to enrol the numbers of individuals required for sufficient power. This would require agreement on selection of individuals, drug therapy and outcome measures (Arzimanoglou 2009). LGS is usually diagnosed after a period of time and by definition the individual must suffer from several different seizure types with specific EEG changes, so that by the time the diagnosis is certain many individuals will have already received several AEDs. Assessment of the efficacy of any add‐on or single therapy is then difficult. The natural history of the syndrome shows that the frequency and type of seizures often fluctuate with time so that improvement/deterioration seen might not be due to the drug therapy being studied. Perhaps it is not surprising that no monotherapy (to date) has been shown to be highly effective in this syndrome.
Although we did find nine RCTs, we were unable to perform any sort of useful meta‐analysis, primarily because all but two trials studied a different therapy. However, even in the two trials that had considered the same therapy, overall numbers were small and the methodology of the two studies differed significantly, making it difficult to combine the results. We could have assumed that the participants enrolled in the seven studies were all from the same population (i.e. did all suffer LGS), but it was clear that the studies had used different entry criteria and definitions (summarised under Description of studies) leading to heterogeneity between the groups. In addition, the studies all used different outcome measures; for example Anonymous 1989 only considered cessation or reduction of all seizure types, while Inanaga 1989 considered a reduction in the number of absence, tonic and atonic seizures and Motte 1997 reported a reduction in drop attacks, tonic‐clonic seizures and all seizure types. Even when studies did report the same outcomes, the results were often presented in different ways; for example Anonymous 1989 gave the reduction in all seizure types as the percentage reduction in number of seizures for each participant, while Sachdeo 1999 gave an overall reduction for all the participants combined.
Finally, and it might be argued most importantly, no trials have been undertaken to examine the effects of treatment on behaviour or cognitive outcome, even though these are frequently cited by the families as being the most difficult features of the syndrome to accept and manage.
Authors' conclusions
Implications for practice.
The optimum treatment for LGS remains uncertain and no study to date has shown any one drug to be highly efficacious; lamotrigine, rufinamide, topiramate and felbamate may be helpful as add‐on therapy. Until further research has been undertaken, clinicians will need to continue to consider each patient individually, taking into account the potential benefit of each therapy weighed against the risk of adverse effects.
Implications for research.
Technical limitations, logistical challenges and ethical constraints should not provide excuses for not conducting well‐designed drug trials in children. It is clear that consideration should be given to trial of new medications early in the development of new medications, particularly if they show broad‐spectrum efficacy. It is recommended that the key considerations proposed by the consensus group (Arzimanoglou 2009) are borne in mind by future researchers.
What's new
| Date | Event | Description |
|---|---|---|
| 18 October 2012 | New citation required but conclusions have not changed | Three new studies have been included (Conry 2009; Conry 2010; Glauser 2009). Conclusions remain unchanged. |
| 18 October 2012 | New search has been performed | Searches updated 18 October 2012. |
Acknowledgements
None.
Appendices
Appendix 1. MEDLINE search strategy
For the most recent update of this review, we used the following search strategy. It is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials published in Lefebvre 2009.
1. randomized controlled trial.pt.
2. controlled clinical trial.pt.
3. randomized.ab.
4. placebo.ab.
5. clinical trials as topic.sh.
6. randomly.ab.
7. trial.ti.
8. 7 or 5 or 2 or 6 or 1 or 4 or 3
9. exp animals/ not humans.sh.
10. 8 not 9
11. Lennox Gastaut.tw.
12. 10 and 11
For earlier versions of this review, we used the following search strategy.
1. Lennox Gastaut.tw.
2. randomized controlled trial.pt.
3. controlled clinical trial.pt.
4. exp Randomized Controlled Trials/
5. exp Random Allocation/
6. exp Double‐Blind Method/
7. exp Single‐Blind Method/
8. 2 or 3 or 4 or 5 or 6 or 7
9. (animals not human).sh.
10. 8 not 9
11. clinical trial.pt.
12. Clinical Trial/
13. (clin$ adj trial$).ab,ti.
14. ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ab,ti.
15. exp PLACEBOS/
16. placebo$.ab,ti.
17. random$.ab,ti.
18. exp Research Design/
19. 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18
20. (animals not human).sh.
21. 19 not 20
22. 10 or 21
23. 1 and 22
Data and analyses
Comparison 1. Cinromide versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants in whom there was complete cessation of seizures | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Number of participants in whom there was 75% to 100% reduction in total number of seizures | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.04 [0.43, 148.71] |
| 3 Number of participants in whom there was 50% to 74% reduction in total number of seizures | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.22, 2.00] |
| 4 Number of participants in whom there was 25% to 49% reduction in total number of seizures | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.33, 2.79] |
| 5 Number of participants in whom there was 0% to 24% reduction in total number of seizures | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.37, 2.70] |
Comparison 2. Felbamate versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants in whom there was complete cessation of all seizures | 1 | 73 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.89 [0.46, 33.17] |
| 2 Number of participants in whom there was complete cessation of atonic seizures | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.72 [0.51, 149.75] |
| 3 Number of participants in whom there was complete cessation of tonic‐clonic seizures | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.69 [0.80, 40.51] |
Comparison 3. Low‐dose TRH DN‐1417 versus high‐dose TRH DN‐1417.
Comparison 4. Lamotrigine versus placebo.
Comparison 5. Topiramate versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants who had complete cessation of drop attacks | 1 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [0.14, 77.97] |
| 2 Number of participants in whom there was a 75% to 100% reduction in the number of drop attacks | 1 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [0.82, 10.27] |
| 3 Number of participants in whom there was a 50% to 74% reduction in the number of drop attacks | 1 | 96 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.02 [0.88, 4.62] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anonymous 1989.
| Methods | RCT | |
| Participants | 73 participants | |
| Interventions | Cinromide versus placebo | |
| Outcomes | Complete cessation and reduction in all seizure types | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Unclear concealment of allocation |
Anonymous 1993.
| Methods | RCT | |
| Participants | 73 participants | |
| Interventions | Felbamate versus placebo | |
| Outcomes | Cessation and/or reduction in atonic, tonic‐clonic and all seizure types | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Unclear concealment of allocation |
Conry 2009.
| Methods | RCT | |
| Participants | 68 participants | |
| Interventions | Low‐dose clobazam versus high‐dose clobazam | |
| Outcomes | Reduction in drop seizures | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Unclear concealment of allocation |
Conry 2010.
| Methods | RCT | |
| Participants | 217 participants | |
| Interventions | Clobazam versus placebo | |
| Outcomes | Reduction in drop seizures | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Unclear concealment of allocation |
Eriksson 1998.
| Methods | RCT | |
| Participants | 30 participants | |
| Interventions | Lamotrigine versus placebo | |
| Outcomes | Reduction rates of all seizures | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation unclear |
Glauser 2008.
| Methods | RCT | |
| Participants | 139 participants | |
| Interventions | Rufinamide versus placebo | |
| Outcomes | Percentage reductions in: total seizure frequency, atonic‐tonic seizures, absence seizures, myoclonic seizures and partial‐onset seizures | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation unclear |
Glauser 2009.
| Methods | RCT | |
| Participants | 138 participants | |
| Interventions | Rufinamide versus placebo | |
| Outcomes | Percentage reductions in total seizure frequency and tonic‐atonic seizure frequency | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation un clear |
Inanaga 1989.
| Methods | RCT | |
| Participants | 98 participants | |
| Interventions | Low‐dose versus high‐dose TRH DN 117 | |
| Outcomes | Reduction in absence, tonic, atonic and all seizure types | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation unclear |
Motte 1997.
| Methods | RCT | |
| Participants | 179 participants | |
| Interventions | Lamotrigine versus placebo | |
| Outcomes | Reduction in absence, tonic‐clonic, drops and all seizure types | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | High risk | No blinding of recipients or assessors. Concealment of allocation unclear |
Sachdeo 1999.
| Methods | RCT | |
| Participants | 112 participants | |
| Interventions | Topiramate versus placebo | |
| Outcomes | Cessation and reduction in drops and reduction in all seizure types | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
RCT: randomised controlled trial.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Jensen 1994 | It is possible that this is the same cohort of participants reported by Anonymous in 1993 |
| Vajda 1985 | Only some participants had LGS but the results were not given independently for this group |
| Vassella 1978 | This paper is written in German, following translation the results remained unclear |
LGS: Lennox‐Gastaut syndrome.
Contributions of authors
Eleanor Hancock and Helen Cross were involved with all aspects of this review including: protocol design; data collection and analysis. Eleanor Hancock was primarily responsible for the writing of the review.
Sources of support
Internal sources
St Piers, St Piers Lane, Lingfield, Surrey RH7 6PW, UK.
Great Ormond Street Hospital, Great Ormond Street, London, UK.
External sources
No sources of support supplied
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Anonymous 1989 {published data only}
- Anonymous. Double‐blind, placebo‐controlled evaluation of cinromide in patients with the Lennox‐Gastaut syndrome. The Group for the Evaluation of Cinromide in the Lennox‐Gastaut syndrome. Epilepsia 1989;30(4):422‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Anonymous 1993 {published data only}
- Anonymous. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox‐Gastaut syndrome). The Felbamate Study Group in Lennox Gastaut syndrome. New England Journal of Medicine 1993;328(1):29‐33. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Conry 2009 {published data only}
- Conry J, Ng Y, Paolicchi JM, Kernitsky L, Mitchell WG, Ritter FJ, et al. Clobazam in the treatment of Lennox Gastaut syndrome. Epilepsia 2009;50(5):1158‐66. [DOI] [PubMed] [Google Scholar]
Conry 2010 {published data only}
- Conry J, Ng Y, Drummond R, Stolle J, Sagar S. Efficacy and safety of clobazam in the treatment of seizures associated with Lennox Gastaut syndrome: results of a phase III trial (abstract no: 1.283). Proceedings of the 64th Annual Meeting of the American Epilepsy Society. December 3‐7, 2010, San Antonio, TX. www.aesnet.org/go/publications/aes‐abstracts/abstract‐search/mode/display/st/clobazam/sy/2010/sb/All/id/12483 (accessed 28 April 2012).
Eriksson 1998 {published data only}
- Eriksson AS, Nergardh A, Hoppu K. The efficacy of lamotrigine in children and adolescents with refractory generalised epilepsy: a randomised, double blind, crossover study. Epilepsia 1998;39(5):495‐501. [DOI] [PubMed] [Google Scholar]
Glauser 2008 {published and unpublished data}
- Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox‐Gastaut syndrome. Neurology 2008;70(21):1950‐8. [DOI] [PubMed] [Google Scholar]
Glauser 2009 {published data only}
- Glauser T, Santana B V, Wang W, Narurkar M. Early and sustained response to rufinamide as adjunctive therapy for seizures associated with Lennox Gastaut syndrome. Epilepsia 2009;50 (suppl 11):261. [Google Scholar]
Inanaga 1989 {published data only}
- Inanaga K, Kumashiro H, Fukyama Y, Ohtahara S, Shirouzu M. Clinical study of oral administration of DN‐1417, a TRH analog, in patients with intractable epilepsy. Epilepsia 1989;30(4):438‐45. [MEDLINE: ; 1989194763] [DOI] [PubMed] [Google Scholar]
Motte 1997 {published data only}
- Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalised seizures associated with the Lennox‐Gastaut syndrome. Lamictal Lennox‐Gastaut Study Group. New England Journal of Medicine 1997;337(25):1807‐12. [MEDLINE: ; 1998002039] [DOI] [PubMed] [Google Scholar]
Sachdeo 1999 {published data only}
- Sachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double‐blind, randomized trial of topiramate in Kennox‐Gastaut syndrome. Topiramate YL Study Group. Neurology 1999;52(9):1882‐7. [MEDLINE: ; 1999198021] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Jensen 1994 {published data only}
- Jensen PK. Felbamate in the treatment of Lennox‐Gastaut syndrome. Epilepsia 1994;35 (suppl)(5):S54‐7. [MEDLINE: ; 1994265566] [DOI] [PubMed] [Google Scholar]
Vajda 1985 {published data only}
- Vajda FJ, Bladin PF, Parsons BJ. Clinical experience with clobazam: a new 1,5 benzodiazepine in the treatment of refractory epilepsy. Clinical and Experimental Neurology 1985;21:177‐82. [MEDLINE: ] [PubMed] [Google Scholar]
Vassella 1978 {published data only}
- Vassella F, Rudeberg A, Silva V, Pavlincova E. Double‐blind study on the anticonvulsive effect of phenobarbital and valproate in the Lennox syndrome [Doppletblind‐Untersuchung uber die antikonvulsive Wirkung von Phenobarbital und Valproat beim Lennox‐Syndrom]. Schweizerische Medizinische Wochenschrift 1978;108(19):713‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Additional references
Aicardi 1996
- Aicardi J. Lennox‐Gastaut syndrome. In: Wallace editor(s). Epilepsy in Children. First Edition. London: Chapman & Hall Medical, 1996:249‐62. [Google Scholar]
Arzimanoglou 2009
- Arzimanoglou A, French J, Blume WT, Cross J, Ernst JP, Feucht M, et al. Lennox Gastaut Syndrome: a consensus approach on diagnosis, assessment, management and trial methodology. Lancet Neurology 2009;8(1):82‐93. [DOI] [PubMed] [Google Scholar]
Beaumanoir 2005
- Beaumanoir A, Blume W. The Lennox‐Gastaut syndrome. In: Roger J, Bureau M, Dravet C, Tassinari C, Wolf P editor(s). Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th Edition. London: John Libby Eurotext, 2005:125‐48. [Google Scholar]
Berg 2011
- Berg A, Scheffer I. New concepts in classification of the epilepsies: entering the 21st century. Epilepsia 2011;52(6):1058‐61. [DOI] [PubMed] [Google Scholar]
European Union 2006
- The European Parliament and the Council of the European Union. Regulation (EC) No 1901/2006 of the European Council and of the Council of 12th December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. www.emea.europa.eu/htms/human/paediatrics/regulation.htm 2006.
Lefebvre 2009
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 (updated September 2009). The Cochrane Collaboration, 2009. Available from www.cochrane‐handbook.org.
Patwardhan 2000
- Patwardhan RV, Strong B, Bebin EM, Mathisen J, Grabb PA. Efficacy of vagal nerve stimulation in children with medically refractory epilepsy. Neurosurgery 2000;47:1353‐7. [PubMed] [Google Scholar]