

Abstract
Bacterial genes are sometimes found to be inactivated by mutation. This inactivation may be observable simply because selection for function is intermittent or too weak to eliminate inactive alleles quickly. Here, I investigate cases in Salmonella enterica where inactivation is instead positively selected. These are identified by a rate of introduction of premature stop codons to a gene that is higher than expected under selective neutrality, as assessed by comparison to the rate of synonymous changes. I identify 84 genes that meet this criterion at a 10% false discovery rate. Many of these genes are involved in virulence, motility and chemotaxis, biofilm formation, and resistance to antibiotics or other toxic substances. It is hypothesized that most of these genes are subject to an ongoing process in which inactivation is favored under rare conditions, but the inactivated allele is deleterious under most other conditions and is subsequently driven to extinction by purifying selection.
Keywords: gene inactivation, positive selection, stop codons, bacterial evolution, pathogen evolution
Introduction
Bacteria are often found to contain genes that have been inactivated by mutation (Hall et al. 1983). Frequently, there is a single inactivating mutation: a premature stop codon, a frameshift mutation, a missense mutation, or the insertion of a transposable element. Although often referred to as pseudogenes, they usually lack the defining features of the mainly eukaryotic phenomenon. In most cases, they are simply presumptive null alleles.
Enteric bacteria provide the opportunity to study gene inactivation in exceptionally well-characterized organisms that are in many ways similar but exhibit important differences. In the first systematic search for inactivated genes in Escherichia coli genomes, Homma et al. (2002) identified 95 in the common laboratory strain K12. Ochman and Davalos (2006), who attempted to recognize additional types of gene disruptions, identified over 200 candidates in K12. McClelland et al. (2004) found a greater number of inactivated genes in Salmonella enterica serovars Typhi and Paratyphi A, which cause the severe disease typhoid and are restricted to human hosts, than in serovar Typhimurium, which is less pathogenic and has a broader host range. Several analogous comparisons have yielded similar results (Jin et al. 2002; Chain et al. 2004; Thomson et al. 2008; Feng et al. 2013). Whatever contribution relaxed selection may make to this systematic difference, positive selection in the course of adaptation to a new niche has been implicated in the inactivation or loss of particular genes (Day et al. 2001; Tong et al. 2005; Prosseda et al. 2012).
Selection-driven gene inactivation or loss can occur in response to ordinary laboratory conditions. Laboratory growth or storage can select for inactivation of rpoS in E. coli and Salmonella (Zambrano et al. 1993; Sutton et al. 2000; Spira et al. 2011; Snyder et al. 2012; Bleibtreu et al. 2014). In a long-term evolution experiment with E. coli B, ribose catabolism was consistently lost in parallel lines, and this loss was shown to confer a 1–2% selective advantage (Cooper et al. 2001). Parallel inactivations of other genes in multiple (though not all) lines, which are likely due to positive selection, were also observed in this experiment (Woods et al. 2006). Experiments designed to identify mutations subject to laboratory selection found parallel gene inactivations in E. coli K12 and in Salmonella Typhimurium LT2 (Knöppel et al. 2018).
Gene inactivation is often discussed as the first step in complete gene loss (Andersson JO and Andersson SG 2001; Dagan et al. 2006). Whatever may happen to any particular allele, this is the expected fate of the gene if selection is too weak to maintain gene function or if inactivation has a long-term net advantage. These are not, however, the only possibilities that can lead to the observation of an inactivated gene. In most populations, there will be recently arisen alleles that are so deleterious that they are likely doomed to eventual extinction. The more deleterious they are, the more recently they must have arisen (with high probability) in order to have persisted to the time of observation. This is a simple consequence of the fact that it takes time for selection to act. Related phenomena include disparities between the compositions of within-species polymorphism and between-species differences, which form the basis of the test introduced by McDonald and Kreitman (1991), and a higher apparent dN/dS in very close comparisons (Rocha et al. 2006).
Another possibility, actually a special case of deleterious mutation, is that inactivation of a gene is positively selected under some conditions but is deleterious in an average sense. Suppose, for example, that inactivation confers resistance to a rarely encountered substance, but also has an overriding disadvantage in the absence of the substance. Inactivated alleles might be short lived and remain rare in the population, despite the detectable effects of the condition-specific positive selection. This phenomenon might reflect fundamental tradeoffs faced by the organism, such as that between the advantages of importing useful substances and the associated risk of importing harmful substances.
Because such alleles are short lived, in a distant comparison, they will be rare relative to the evolutionary distance or to a proxy such as synonymous changes. Detection of positive selection will therefore require sufficiently close comparisons.
Here, I examine the occurrence of premature stop codons in S. enterica genes, identifying genes subject to selection-driven inactivation (SDI). The data set analyzed consists of over 100,000 genomes falling into a few thousand clusters of very closely related isolates. The timescale of comparison is very short because only within-cluster sequence changes are considered. A total of 84 genes were inferred to exhibit SDI based on a ratio of inactivation events to synonymous changes that exceeds the neutral expectation. Many of these genes are involved in pathogenesis, major “lifestyle” traits, or other processes of particular interest to researchers.
Materials and Methods
Data
Single-nucleotide polymorphism data and isolate information for S. enterica were obtained from the NCBI Pathogen detection project, build PDG0000002.1233 (archived at ftp://ftp.ncbi.nlm.nih.gov/pub/jcherry/sal_stop_codons, last accessed February 7, 2020), along with derived data and source code). Isolates with collection dates prior to 2012 were discarded, as were the few isolates of Salmonella bongori. Phylogenetic trees were built for each cluster with at least five remaining isolates by a variant of maximum compatibility (Cherry 2017) and midpoint rooted. Positions of single-nucleotide polymorphisms in coding sequences were determined from the annotation of the reference sequence for each cluster.
Inference of Sequence Changes
Sequence changes and their tree locations were inferred from a most parsimonious reconstruction under a “soft” interpretation of polytomies (Maddison 1989), which avoided inference of multiple parallel changes where a single change could explain the data. In cases where the reconstruction was ambiguous, usually due to ambiguities in the character state data, a single reconstruction was chosen. The reconstruction was chosen such that an ambiguous state for an isolate was reconstructed as ancestral rather than derived whenever possible, which is conservative with respect to inferring changes on internal branches. The details of handling ambiguities, and the choice of a “soft” rather than “hard” treatment of polytomies, were found not to affect the results substantially.
Determination of Presumptive LT2 Orthologs
Genes from different reference isolates were grouped based on presumptive orthology to LT2 genes. Orthology was inferred as follows. For speed, each protein was first compared with all LT2 proteins of identical length in a gapless alignment. A sequence identity of 70% or more was considered a match, and reciprocal best hits were classified as orthologs. The proteins remaining were then compared with all LT2 proteins by tetramer content, with a requirement of 10% of tetramers in common for a pair to be a candidate match. For this calculation, the denominator was taken to be the minimum of the two sequence lengths, and the numerator taken to be the sum of the minimum of the number of occurrences of the tetramer in the two sequences (usually 0 or 1, but possibly higher when a tetramer occurs multiple times in a single sequence). The candidate pairs were then aligned using BLAST, and a 70% identity threshold again applied, with gap positions counted as mismatches. It was also required that at least 80% of each protein in the pair was aligned in a nongap position. Reciprocal best hits were again classified as orthologs.
Inference of Selection for Inactivation
The ratio of the number of changes creating stop codons to the number of synonymous changes was compared with a neutral expectation for each gene (the ratio of the neutral expectations, as the expectation of the ratio is undefined). A one-sided binomial test for an elevated ratio was performed for each gene. The expectation for each gene was computed based on its codon composition and estimates of the relative rates of the 12 types of single-base substitution. First, all 4-fold-degenerate sites were analyzed and the frequencies of the different types of substitution determined. Then, for each of the 61 sense codons, relative rates of synonymous and nonsense changes were calculated. Finally, for each gene, the expected relative rate was determined from its codon composition. Estimates calculated from different isolates (which vary in codon composition) were found to correlate highly, so estimates based on the LT2 sequence were used.
Because small changes in stop codon location can occur without harm to protein function, the last 20 codons of each coding sequence were excluded from the analysis. Also, the test for elevated nonsense rates excluded changes expected to be accelerated by Dcm methylation and by M.SinI methylation in clusters containing the gene encoding that enzyme.
Correction for multiple testing (over 4,000 genes) was performed by a modified false discovery rate (FDR) procedure. The procedure was modified to account for the fact that the tested distribution (binomial) is discrete. The usual procedure estimates the expected number of tests with a P value not exceeding a threshold as the product of the P value and the number of tests. This is replaced with the sum of binomial probabilities that the P value is at least that small. Furthermore, this probability is based on an empirical distribution of relative rates that reflects the fact that nonsense rates for many genes are lower than the expected neutral rate, presumably due to selection. Details of this procedure are provided in the supplementary text, Supplementary Material online.
Results and Discussion
The analysis presented here is based on single-nucleotide variation data for clusters of closely related isolates of S. enterica. These clusters were formed by application of a clustering algorithm to pairwise sequence distances based on nucleotide k-mers. The size of the clusters varied from five isolates (smaller clusters were excluded from the analysis) to several thousand. Isolates within a cluster differ by fewer than 300 nucleotide substitutions among the millions of genomic positions compared. Thus, because the analysis considers only sequence changes inferred to occur within a cluster, the analyzed changes represent a relatively short evolutionary timescale. Coupled with the large numbers of isolates (over 100,000) and inferred sequence changes (over 400,000), this makes it possible to observe types of sequence change that, although driven by selection (perhaps under rare conditions), are rare in the population and unlikely to proliferate for long.
Reconstructions of sequence changes on inferred phylogenetic trees formed the basis for inferences about inactivation by nonsense mutations and, for comparison, synonymous changes. Figure 1 shows a phylogenetic tree for a small cluster with the reconstructed nonsense mutations indicated by gene names and branch coloring. A cluster with an atypically large number of inactivations for its size was chosen for the purpose of illustration. Supplementary figure S1, Supplementary Material online, shows the tree for a large cluster, with inactivation events for some frequently inactivated genes indicated.
Fig. 1.
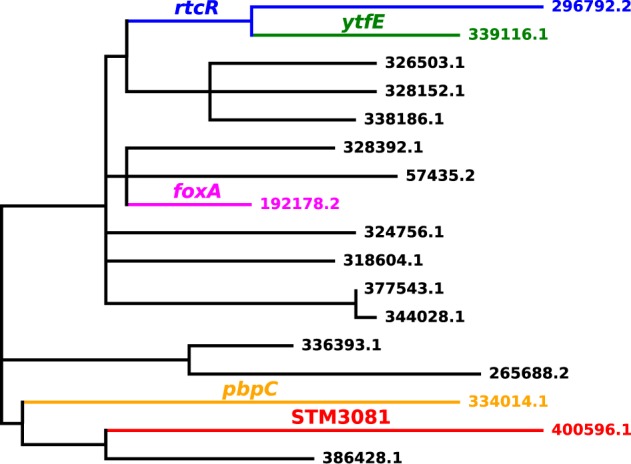
—The phylogenetic tree of a cluster showing inactivations of LT2 orthologs by stop codons. Every branch on which an inactivation event is inferred to occur is labeled with the gene name, and the branch and its descendants are colored. Note that the inactivation of rtcR affects two isolates, though its color is overridden on one isolate and a terminal branch because of the later inactivation of ytfE in a descendant.
After removal of Dcm mutation hotspots (see below) and sequence changes near the ends of coding sequences, there were a total of 11,456 nonsense mutations among presumptive orthologs of strain LT2 genes. The number of synonymous changes was 124,864.
Although synonymous changes are not strictly neutral in bacteria, selection is weak for most genes. On the short evolutionary timescale relevant here, the effect of selection on synonymous changes is likely negligible. In support of this claim, it can be noted that the ratio of nonsynonymous to synonymous changes in the data set is more than half of the neutral expectation for almost all genes. This contrasts with an average value of <0.1 for between-species differences (dN/dS), indicating the weakness of effects of selection on the observed sequence changes.
The observed ratio of nonsense to synonymous changes, 0.092, is 69% of what is expected in the absence of selection (0.133). It may seem surprising that so many nonsense mutations are observed, because they would be expected to be deleterious in most cases; otherwise, genes would not persist. However, because the clusters analyzed consist of such closely related isolates, the mutations represent recent events in evolutionary history. There has therefore not been much time for selection to act, and mutants subject to sufficiently weak purifying selection can still be observed. The nonsense mutations likely represent, for the most part, the mildly deleterious variants that can be found in any population. Their bearers would be examples of the “living dead” (Rice 1996): Individuals that are alive and viable, but doomed to likely eventual extinction due to purifying selection.
The present work concerns gene inactivation events that are driven by selection, even if the resulting mutants fail to persist because selection disfavors them in the long term. These two effects of selection are compatible: They can both apply if inactivation is favored under some conditions but disfavored more commonly or more strongly under others, such that it is disfavored in an average sense.
A total of 84 genes were inferred to exhibit SDI by premature stop codons. These are presented in table 1 and supplementary table S1, Supplementary Material online. The latter includes web links to the protein and gene entries at NCBI and additional information about the genes and their inactivation. Before addressing particular genes, I discuss several aspects of the analysis.
Table 1.
The 84 genes inferred to be subject to SDI at a 10% modified FDR. Genes are divided into functional categories. Within each category they are listed in descending order of the total (all branches) number of events producing premature stop codons. The categories are ordered by the highest number of events among the genes that they contain, except that “Other Genes” comes last.
| Category | Gene Name | Gene Alias | Protein (LT2) | Gene/Product Description | FDR | All Branches |
Internal Branches |
TBLI | TBLI P Values |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Obs/Exp | Stop | Syn. | Obs/Exp | Stop | Syn. | ≤0 versus >0 | ≥1 versus <1 | |||||||
| Stress response regulators | rpoS | STM2924 | NP_461845.1 | RNA polymerase sigma factor; master regulator of stress response | 2E-215 | 47.35 | 268 | 41 | 1.32 | 2 | 11 | 0.172 | 0.00703 | 1.01E-25 |
| iraP | STM0383 | NP_459378.3 | Antiadaptor protein; protects RpoS from degradation | 0.0003 | 9.27 | 9 | 5 | 5.15 | 2 | 2 | 0.0968 | 0.328 | 0.0464 | |
| crl | STM0319 | NP_459316.1 | Binds RpoS and increases its activity | 0.074 | 3.71 | 7 | 9 | 1.19 | 1 | 4 | −0.201 | 0.684 | 0.00119 | |
| cspC | STM1837 | NP_460793.1 | Increases production of RpoS | 0.055 | 19.32 | 3 | 1 | Inf | 0 | 0 | 0.102 | 0.315 | 0.122 | |
| Chemotaxis receptors | tsr | STM4533 | NP_463392.1 | Methyl-accepting chemotaxis protein | 9E-105 | 23.48 | 154 | 45 | 23.52 | 48 | 14 | 0.871 | 4.38E-13 | 0.163 |
| mcpC | STM3216 | NP_462130.1 | Methyl-accepting chemotaxis protein | 3E-07 | 4.01 | 28 | 46 | 3.76 | 12 | 21 | 1.2 | 0.000101 | 0.722 | |
| Outer membrane proteins | btuB | STM4130 | NP_463009.1 | TonB-dependent vitamin B12 receptor | 1E-75 | 18.64 | 126 | 39 | 13.6 | 33 | 14 | 0.771 | 4.13E-10 | 0.0469 |
| fhuA | STM0191 | NP_459196.1 | Ferrichrome porin | 3E-14 | 5.61 | 41 | 50 | 4.32 | 12 | 19 | 0.833 | 0.000264 | 0.267 | |
| nmpC | STM1572 | NP_460531.1 | Phosphoporin PhoE | 9E-09 | 4.52 | 29 | 51 | 3.61 | 5 | 11 | 1.21 | 4.76E-05 | 0.745 | |
| cirA | STM2199 | NP_461144.1 | TonB-dependent catecholate siderophore receptor | 4E-05 | 2.86 | 28 | 73 | 4.01 | 7 | 13 | 1.51 | 2.43E-06 | 0.927 | |
| ompC | STM2267 | NP_461210.1 | General porin | 7E-09 | 6.04 | 23 | 31 | 2.33 | 4 | 14 | 1.09 | 0.000374 | 0.607 | |
| yjbH | STM4225 | NP_463090.1 | Possibly involved in polysaccharide export | 0.0026 | 2.57 | 21 | 54 | 0.39 | 1 | 17 | 1.39 | 2.91E-06 | 0.879 | |
| Virulence regulators | hilD | STM2875 | NP_461796.1 | Regulator of virulence genes | 1E-56 | 22.65 | 89 | 23 | 5.85 | 11 | 11 | 0.238 | 0.0248 | 2.06E-07 |
| hilC | STM2867 | NP_461788.1 | Also sirC; regulator of virulence genes | 3E-21 | 8.47 | 50 | 32 | 14.72 | 19 | 7 | 0.491 | 0.0107 | 0.0187 | |
| UshA | ushA | STM0494 | NP_459489.1 | Phosphohydrolase; inactivated in LT2 | 1E-26 | 9.37 | 55 | 43 | 16.85 | 23 | 10 | 0.618 | 0.00108 | 0.0401 |
| Other cell surface related | fliB | STM1958 | NP_460911.1 | Flagellin lysine-N-methylase | 2E-27 | 10.54 | 55 | 34 | 4.34 | 8 | 12 | 0.446 | 0.00439 | 0.00272 |
| fimW | STM0552 | NP_459547.1 | Fimbriae regulatory protein | 0.0002 | 6.7 | 11 | 9 | 7.3 | 4 | 3 | 1.23 | 0.0197 | 0.647 | |
| Resistance to toxic compounds | ramR | STM0580 | NP_459572.1 | Repressor of efflux pump genes | 6E-40 | 28.57 | 55 | 14 | 39.99 | 11 | 2 | 0.562 | 0.000909 | 0.0104 |
| sbmA | STM0376 | NP_459371.1 | Peptide transporter | 8E-19 | 7.92 | 44 | 35 | 12.6 | 14 | 7 | 0.942 | 3.34E-05 | 0.412 | |
| glpT | STM2283 | NP_461225.1 | Glycerol-3-phosphate transporter; fosfidomycin resistance upon loss | 3E-05 | 3.67 | 21 | 43 | 8.34 | 10 | 9 | 1.3 | 0.000715 | 0.761 | |
| Uncharacterized polysaccharide | — | STM0724 | NP_459709.1 | Glycosyl transferase | 3E-10 | 3.7 | 46 | 69 | 3.96 | 10 | 14 | 0.878 | 7.19E-06 | 0.302 |
| — | STM0726 | NP_459711.1 | Glycosyl transferase | 4E-07 | 3.3 | 37 | 52 | 3.92 | 11 | 13 | 0.942 | 0.000082 | 0.416 | |
| — | STM0719 | NP_459704.1 | Putative UDP-galactose mutase | 3E-07 | 4.4 | 27 | 30 | 14.67 | 9 | 3 | 0.873 | 0.00245 | 0.369 | |
| — | STM0720 | NP_459705.1 | Glycosyl transferase | 2E-06 | 4.43 | 23 | 29 | 4.35 | 7 | 9 | 1.1 | 0.000427 | 0.635 | |
| — | STM0721 | NP_459706.1 | Glycosyl transferase | 8E-06 | 4.83 | 19 | 21 | 9.35 | 7 | 4 | 1.57 | 4.15E-05 | 0.913 | |
| — | STM0725 | NP_459710.1 | Glycosyl transferase | 0.082 | 2.13 | 15 | 36 | 2.27 | 4 | 9 | 0.795 | 0.0253 | 0.327 | |
| — | STM0717 | NP_459702.1 | Putative inner membrane protein | 0.02 | 20.75 | 4 | 1 | 10.37 | 2 | 1 | 0.0424 | 0.515 | 0.132 | |
| O-antigen/LPS | oafA | STM2232 | NP_461175.1 | Acetyltransferase; synthesizes determinant of O-antigen five | 2E-21 | 16.12 | 38 | 13 | 18.75 | 17 | 5 | 1.22 | 4.72E-06 | 0.792 |
| rfaL | STM3713 | NP_462613.1 | Also waaL; O-antigen ligase | 3E-09 | 4.9 | 31 | 35 | 0.31 | 1 | 18 | 0.879 | 0.000066 | 0.318 | |
| rfbP | STM2082 | NP_461027.1 | UDP-phosphate galactose phosphotransferase; LPS side chain defect | 0.056 | 2 | 21 | 42 | 0.5 | 1 | 8 | 0.493 | 0.0287 | 0.0545 | |
| wzzB | STM2079 | NP_461024.1 | O-antigen chain length determinant protein | 8E-05 | 4.19 | 17 | 26 | 2.14 | 3 | 9 | 0.245 | 0.218 | 0.0266 | |
| rfaY | STM3716 | NP_462616.1 | Also waaY; phosphorylation of heptose region of the LPS core | 0.086 | 2.02 | 16 | 46 | 2.9 | 2 | 4 | 0.433 | 0.0637 | 0.0436 | |
| rfaJ | STM3717 | NP_462617.1 | Lipopolysaccharide 1,2-glucosyltransferase | 0.095 | 2.01 | 16 | 39 | 1.09 | 2 | 9 | 0.741 | 0.0039 | 0.177 | |
| rfaB | STM3719 | NP_462619.1 | Lipopolysaccharide 1,6-galactosyltransferase | 0.045 | 2.48 | 14 | 27 | 0 | 0 | 5 | 1.21 | 0.00128 | 0.699 | |
| pmrA | STM4292 | NP_463157.1 | Also basR; regulates LPS modification and O-antigen chain length | 0.06 | 4.89 | 5 | 9 | 8.8 | 3 | 3 | 1.75 | 0.0357 | 0.781 | |
| Environmental sensors | barA | STM2958 | NP_461879.1 | Two-component system histidine kinase | 1E-08 | 3.77 | 34 | 83 | 0.35 | 1 | 26 | 0.31 | 0.0483 | 0.00145 |
| rcsD | STM2269 | NP_461211.1 | Phosphotransferase | 0.061 | 1.82 | 23 | 90 | 0.61 | 3 | 35 | 0.607 | 0.0193 | 0.147 | |
| uvrY | STM1947 | NP_460900.1 | Also sirA; two-component system response regulator | 3E-09 | 10.17 | 18 | 13 | 1.22 | 1 | 6 | −0.105 | 0.652 | 0.000116 | |
| rcsB | STM2270 | NP_461212.1 | DNA-binding response regulator | 2E-05 | 5.71 | 13 | 26 | 1.63 | 1 | 7 | 0.0452 | 0.405 | 0.00549 | |
| ompR | STM3502 | NP_462405.1 | Two-component system response regulator | 0.09 | 3.36 | 6 | 19 | 0 | 0 | 8 | 0.268 | 0.226 | 0.0532 | |
| Regulators of motility | ydiV | STM1344 | NP_460310.1 | Repressor of motility under starvation conditions | 6E-17 | 11.42 | 32 | 19 | 14.7 | 13 | 6 | 0.489 | 0.028 | 0.0516 |
| fliZ | STM1955 | NP_460908.1 | Positive regulator of motility | 0.077 | 2.71 | 10 | 19 | 0.64 | 1 | 8 | 0.552 | 0.0741 | 0.132 | |
| lrhA | STM2330 | NP_461272.1 | Repressor of flhDC, and hence of motility and chemotaxis | 0.086 | 2.65 | 9 | 26 | 2.87 | 3 | 8 | 1.91 | 0.0036 | 0.896 | |
| Acid resistance | cadC | STM2557 | NP_461492.1 | Transcriptional activator of cad (cadaverine) operon | 1E-07 | 4.3 | 28 | 40 | 3.51 | 8 | 14 | 1.04 | 0.000387 | 0.559 |
| adiY | STM4295 | NP_463160.1 | Transcriptional activator of arginine decarboxylase | 4E-07 | 6.13 | 19 | 19 | 5.26 | 6 | 7 | 0.879 | 0.015 | 0.402 | |
| Repressors of sugar utilization | uxuR | STM4507 | NP_463366.1 | Repressor of hexuronate catabolism | 3E-07 | 6.02 | 20 | 19 | 7.15 | 5 | 4 | 0.469 | 0.0503 | 0.0644 |
| galS | STM2191 | NP_461136.1 | Repressor of galactose catabolism | 0.0002 | 4.68 | 13 | 26 | 2.34 | 3 | 12 | 0.927 | 0.0231 | 0.463 | |
| mlc | STM1488 | NP_460448.1 | Global repressor of carbohydrate metabolism | 0.03 | 2.9 | 11 | 30 | 2.63 | 2 | 6 | 1.46 | 0.0034 | 0.789 | |
| rbsR | STM3886 | NP_462785.1 | Repressor of ribose catabolism | 0.091 | 2.59 | 9 | 27 | 0 | 0 | 8 | 0.471 | 0.124 | 0.157 | |
| Biofilm formation | bcsA | STM3619 | NP_462520.1 | Cellulose synthase catalytic subunit | 0.09 | 1.85 | 19 | 68 | 1.65 | 5 | 20 | 0.789 | 0.00447 | 0.252 |
| bcsB | STM3618 | NP_462519.1 | Cellulose synthase regulatory subunit | 0.03 | 2.35 | 15 | 63 | 4.93 | 7 | 14 | 1.03 | 0.00449 | 0.531 | |
| bcsE | STM3622 | NP_462523.1 | Cellulose synthesis c-di-GMP-binding protein | 0.074 | 2.13 | 15 | 43 | 2.04 | 4 | 12 | 1.18 | 0.00107 | 0.681 | |
| pdeH | STM3611 | NP_462512.1 | Cyclic-di-GMP phosphodiesterase | 0.0024 | 3.76 | 13 | 20 | 2.32 | 4 | 10 | 0.613 | 0.0738 | 0.18 | |
| Misc. regulatory proteins | proQ | STM1846 | NP_460802.1 | RNA chaperone | 4E-07 | 7.06 | 17 | 16 | 3.32 | 2 | 4 | 0.721 | 0.00819 | 0.208 |
| ptsP | STM3003 | NP_461920.1 | Nitrogen phosphotransferase enzyme EI(Ntr) | 0.043 | 2.2 | 16 | 60 | 1.84 | 4 | 18 | 1.26 | 0.000777 | 0.727 | |
| icc | STM3183 | NP_462098.1 | Called cpdA in E. coli; 3′,5′-cyclic-AMP phosphodiesterase | 0.066 | 2.47 | 12 | 27 | 0.43 | 1 | 13 | 0.446 | 0.115 | 0.0968 | |
| Structural virulence proteins | sseK1 | STM4157 | NP_463026.1 | SPI-2 T3SS; inhibits NF-κB signaling and macrophage death | 0.07 | 2.18 | 15 | 37 | 1.54 | 2 | 7 | 1.12 | 0.00081 | 0.633 |
| vrgS | STM0289 | NP_459287.1 | SPI-6-encoded Type VI secretion protein; vgrG homolog | 0.045 | 2.56 | 12 | 34 | 2.72 | 3 | 8 | 0.988 | 0.00657 | 0.495 | |
| sseI | STM1051 | NP_460026.1 | SPI-2 T3SS; inhibits host cell migration; long-term systemic infection | 0.05 | 3.26 | 9 | 14 | 0 | 0 | 3 | 0.624 | 0.0628 | 0.213 | |
| Nitroaromatic reductases | mdaA | STM0874 | NP_459851.1 | nfsA in E. coli; nitroaromatic reductase A; NADPH dependent | 1E-05 | 5.76 | 15 | 19 | 9.12 | 5 | 4 | 0.774 | 0.0226 | 0.273 |
| nfnB | STM0578 | NP_459570.1 | nfsB in E. coli; nitroaromatic reductase B; NAD(P)H dependent | 2E-06 | 14.37 | 10 | 6 | 30.19 | 7 | 2 | 1.61 | 0.025 | 0.801 | |
| Other transporter proteins | nupC | STM2409 | NP_461350.1 | Nucleoside permease | 0.013 | 3.37 | 10 | 37 | 0.89 | 1 | 14 | 1.51 | 0.000119 | 0.879 |
| — | STM3333 | NP_462243.1 | Cytosine permease | 0.056 | 3.05 | 8 | 33 | 5.04 | 4 | 10 | 0.365 | 0.268 | 0.21 | |
| ydiE | STM1346 | NP_460312.1 | Possible hemin transport protein | 0.028 | 17.99 | 3 | 2 | Inf | 1 | 0 | 0.818 | 0.249 | 0.462 | |
| Metabolic genes | fhlA | STM2859 | NP_461780.1 | Formate hydrogenlyase transcriptional activator | 0.045 | 2.18 | 16 | 60 | 0.39 | 1 | 21 | 0.827 | 0.00361 | 0.3 |
| dcuA | STM4325 | NP_463189.1 | Anaerobic C4-dicarboxylate transporter | 0.0008 | 4.8 | 10 | 31 | 1.06 | 1 | 14 | 1.18 | 0.0279 | 0.623 | |
| melR | STM4297 | NP_463162.1 | Positive regulator of melibiose operon | 0.09 | 2.69 | 9 | 20 | 0 | 0 | 8 | 0.43 | 0.105 | 0.0596 | |
| Sulfur assimilation | cutR | STM0459 | NP_459455.1 | Positive regulator of cysteine catabolism | 7E-12 | 17.85 | 19 | 7 | 1.32 | 1 | 5 | 0.166 | 0.268 | 0.00556 |
| cyuP | STM3239 | NP_462153.1 | Anaerobic cysteine transport protein | 0.035 | 2.78 | 11 | 36 | 1.14 | 2 | 16 | 0.664 | 0.0371 | 0.232 | |
| cysK | STM2430 | NP_461365.1 | Cysteine synthase A | 0.083 | 3.11 | 7 | 23 | 0 | 0 | 6 | 0.141 | 0.366 | 0.0856 | |
| Other genes | pepT | STM1227 | NP_460197.1 | Aminotripetidase (peptidase T) | 0.0004 | 3.39 | 17 | 44 | 1.75 | 3 | 15 | 0.557 | 0.0418 | 0.125 |
| ligB | STM3739 | NP_462639.1 | DNA ligase B | 0.029 | 2.44 | 17 | 28 | 3.12 | 7 | 9 | 0.888 | 0.00948 | 0.398 | |
| speC | STM3114 | NP_462030.1 | Ornithine decarboxylase; catalyzes step in polyamine biosynthesis | 0.086 | 2.13 | 13 | 57 | 0.93 | 2 | 20 | 0.816 | 0.0149 | 0.331 | |
| ybhL | STM0807 | NP_459785.1 | Hypothetical protein | 2E-05 | 6.34 | 13 | 17 | 0 | 0 | 3 | 1.02 | 0.00356 | 0.519 | |
| clsC | STM1148 | NP_460119.3 | Cardiolipin synthase C | 0.097 | 2.18 | 12 | 43 | 2.44 | 5 | 16 | 0.782 | 0.118 | 0.365 | |
| ddlB | STM0130 | NP_459135.1 | d-Alanine–d-alanine ligase | 0.095 | 2.34 | 11 | 29 | 1.54 | 2 | 8 | 1.13 | 0.00376 | 0.622 | |
| — | STM2238 | NP_461181.1 | Predicted P-loop NTPase | 0.012 | 4.47 | 9 | 10 | 0 | 0 | 4 | 0.351 | 0.139 | 0.0463 | |
| rlmF | STM0826 | NP_459803.1 | 23S rRNA (adenine[1618]-N[6])-methyltransferase | 0.086 | 2.91 | 8 | 19 | 2.59 | 3 | 8 | 2.32 | 0.000219 | 0.975 | |
| yhaH | STM3234 | NP_462148.1 | Hypothetical protein; similar to E. coli putative cytochrome | 0.06 | 3.56 | 8 | 10 | 6.67 | 3 | 2 | 1.6 | 0.0131 | 0.789 | |
| — | STM3026 | NP_461943.1 | Hypothetical protein | 0.095 | 3.18 | 7 | 13 | 3.54 | 3 | 5 | 0.857 | 0.0689 | 0.461 | |
| yaeQ | STM0239 | NP_459244.1 | Hypothetical protein | 0.06 | 3.89 | 7 | 9 | 2.5 | 1 | 2 | 1.55 | 0.00298 | 0.825 | |
| — | STM0839 | NP_459816.1 | Putative inner membrane protein | 0.097 | 3.24 | 7 | 11 | 5.09 | 1 | 1 | 0.982 | 0.0078 | 0.475 | |
| — | STM2742 | NP_461669.1 | Putative cytoplasmic protein | 0.083 | 7.12 | 4 | 3 | 8 | 3 | 2 | 1.07 | 0.175 | 0.571 | |
| — | STM0295 | NP_459293.1 | Putative cytoplasmic protein | 0.078 | 6.63 | 4 | 4 | 0 | 0 | 1 | 1.28 | 0.0333 | 0.656 | |
Inferring Selection for Inactivation
Selection for inactivation was inferred when a gene’s rate of inactivation by nonsense mutations, relative to the synonymous rate, exceeded the neutral expectation, provided that this excess was statistically significant. Under neutrality, we would expect many more synonymous changes than acquisitions of stop codons because mutations are more likely to be synonymous than to create a stop codon. Of the 61 × 9 = 549 possible single-base changes to sense codons, 134 are synonymous but only 23 create stop codons. Not all types of mutations occur at the same rate, and sense codons occur at different frequencies. Analysis of changes at 4-fold-degenerate sites yielded estimates of the relative rates of different types of mutations. Each gene’s codon composition was then used to obtain a gene-specific neutral expectation of the nonsense:synonymous ratio. This ratio was on average 0.126 but varied significantly among genes (standard deviation 0.049).
Correction for multiple testing (over 4000 genes) was performed by a modified FDR procedure. The procedure was modified in two ways. The standard FDR procedure assumes that P values are uniformly distributed between 0 and 1 if the null hypothesis is true, so that the probability of a P value less than or equal to a threshold p* is equal to p*. This assumption is correct for continuous distributions, but not for discrete distributions such as the binomial that is relevant here: The probability that the P value is at least as small as p* may be smaller than p* (even 0), making the FDR too conservative. The modified procedure sums the actual probabilities that the P values are at least as small as the threshold. Another reason that the standard FDR is too conservative is that nonsense rates for many genes are much lower than the expected neutral rate, presumably due to selection, making them much less likely to produce small P values by chance. The modified procedure estimates the distribution of nonsense rates relative to the neutral rate for genes not selected for inactivation and uses it for computing the probabilities of P values under the null hypothesis. This distribution is used only for correction for multiple tests; P values are computed under the neutral expectation. Details of this procedure are provided in the supplementary text, Supplementary Material online.
Estimation of this distribution introduces some uncertainty into the procedure. However, simulations indicate that it estimates FDR reasonably well, with actual FDRs at a nominal rate of 10% equal to 10.4–11.1% depending on the assumptions (supplementary text S1, Supplementary Material online). These simulations also confirm that the standard FDR is far too conservative for this data set, with an actual FDR of only 0.5% at a nominal 10% FDR.
Supplementary table S1, Supplementary Material online, reports the P values and the standard FDR values for each gene along with the modified FDR. At a standard FDR of 10%, 42 genes show a significant effect. With a very conservative Bonferroni correction, 33 genes are significant at a 5% P value cutoff.
The inferred distribution of relative nonsense rates has a substantial peak at 81–82% of the calculated neutral value, but negligible probability mass near the neutral value itself (supplementary fig. S2, Supplementary Material online). Simulations suggest that this is unlikely to be an artifact of the estimation procedure. Because inactivation of many genes may be close to neutral on a short timescale, this may indicate that the calculated nonsense mutation rates are too large, perhaps due to context effects on mutation rate. If so, the calculated P values are too conservative. If the actual rate is assumed to be 82% of the calculated rate, a total of 135 genes show a significant effect. Many of the additional genes fall into the categories in table 1.
Dcm Methylation Sites and Other Hotspots of Mutation
Cytosines methylated by the Dcm methyltransferase (CCWGG, W = A or T) are hotspots for transition mutations in the laboratory (Coulondre et al. 1978) and in nature (Cherry 2018). The high-frequency transitions can produce stop codons: a CCAGG → CTAGG transition, which is equivalent to a CCTGG → CCTAG transition on the opposite strand, produces a TAG stop codon in two of the six possible reading frames. This phenomenon is quite significant: 28% of the observed stop codons are due to transitions at Dcm sites.
It would be possible to incorporate Dcm hotspots into the model used to compute expected rates under neutrality. However, there appears to be significant variation in mutation rate among Dcm sites, and there are in most cases only a few of them in each gene, so the reliability of the calculation would be questionable. Furthermore, the inclusion of mutational hotspots makes it more likely that independent parallel sequence changes will appear to be a single change. For these reasons, Dcm sites are excluded from the analysis presented in table 1.
For most genes, Dcm sites contribute modestly to inactivation by stop codons, typically adding only a fraction of the total due to other sites. The notable exception is ydiV, for which a single Dcm site is responsible for 401 inactivations. This site apparently has an extraordinarily high mutation rate, which may be an evolved feature. This phenomenon is discussed further in the section on ydiV.
Also excluded were transitions at M.SinI methylation sites (GGWCC) in clusters containing that enzyme. These have a much smaller total effect than Dcm sites, but a few M.SinI hotspots would contribute significantly to some of the nonsense counts in table 1 without this measure.
The possibility that other mutational hotspots create the appearance of selection can be addressed by examining the distributions of the positions of the nonsense mutations in the identified genes. As can be seen from supplementary figure S3, Supplementary Material online, for all genes but one (lrhA), stops are distributed across multiple positions, none of which dominate. This indicates that hotspots of mutation are not responsible for the results.
The lrhA gene may not be a genuine exception. Although eight of the nine observed nonsense mutations occurred within a single codon, they are split 3:5 between the second and third positions, either of which can give rise to a stop codon by a transition. These adjacent nucleotides might both have unusually high mutation rates. However, the observation would also be explained by selection that is specific for a stop codon late in the gene because this only partially eliminates function.
Tree-Based Tests for Laboratory Artifacts
A possible cause of gene inactivation is inadvertent selection during growth and storage in the laboratory. Indeed, the gene in table 1 with the largest number of inactivations, rpoS, is known to be subject to such selection (Zambrano et al. 1993; Sutton et al. 2000; Spira et al. 2011; Snyder et al. 2012; Bleibtreu et al. 2014). This laboratory phenomenon is not without interest, but it should be distinguished from SDI in the wild.
In experiments designed to identify mutations subject to selection during laboratory growth of a derivative of S. enterica Typhimurium LT2, Knöppel et al. (2018) observed mutations in only two of the genes in table 1 (barA and nmpC) during growth in lysogeny broth, the usual medium for growth of S. enterica. This result supports the view that the results presented here identify mostly natural SDI. However, although several genes or gene categories were affected in all four replicates, these experiments cannot be assumed to have identified all genes subject to selection in the laboratory. Furthermore, what is true of the particular strain used in these experiments need not hold for all of S. enterica. It is noteworthy that mutations in rpoS were not observed during growth in lysogeny broth, although one was observed in a minimal medium.
Two tests can be applied to a case of SDI to provide evidence that it is a natural phenomenon rather than a laboratory artifact. These tests are illustrated in figure 2. Both make use of the phylogenetic trees and reconstructions. Neither can distinguish between laboratory-selected inactivation and strong effects of long-term negative selection in the wild, which are difficult or impossible to distinguish with the data at hand. Culture-free sequencing might eventually resolve the question in all cases.
Fig. 2.

—Distinguishing an effect in nature from a laboratory phenomenon. Branches on which sequence changes occur are marked with a slash, and they and their descendants are colored. Upper panels: Laboratory mutations occur on terminal branches (magenta, upper left panel). Natural mutations can occur on internal branches as well (green, upper right panel). An inflated nonsense:synonymous ratio on internal branches is evidence for a natural phenomenon. Lower panels: A laboratory mutation is just as likely to occur on a short terminal branch (red) as a long one (blue), as illustrated in the lower left panel. Naturally occurring mutations are more likely to occur on longer terminal branches (lower right panel). The TBLI quantifies the extent to which the latter expectation is realized.
The first test is to consider only sequence changes that are found on internal, as opposed to terminal, branches of the tree. A mutation that occurs in the laboratory affects a single isolate and occurs on a terminal branch, and the reconstruction will, with rare exceptions, reflect this fact. If the ratio of nonsense to synonymous changes on internal branches exceeds the neutral expectation, this phenomenon cannot be attributed to events in the laboratory.
A diminished nonsense:synonymous ratio on internal branches may reflect purifying selection in nature rather than positive selection in the laboratory. Long-term selection against inactivated alleles would mean that they are less likely to survive from deep in the tree to be observed and also less likely to be found in multiple isolates (and hence on internal branches). Thus, this test can provide evidence for a natural phenomenon but otherwise is ambiguous rather than demonstrating conclusively that frequent inactivation is a laboratory phenomenon.
The second test is based on the lengths of the terminal branches on which observed nonsense mutations arise, with any contribution of the nonsense mutation itself removed. It is assumed that the other sequence changes occurred in the wild. One justification for this is that they are enriched for synonymous changes, which are presumably unlikely to be selected strongly in the laboratory.
The probability that a nonsense mutation is observed because of selection in the laboratory is expected to be unrelated to the length of the terminal branch leading to the isolate. A naturally occurring nonsense mutation, on the other hand, is more likely on a longer branch, which on average corresponds to a longer evolutionary time. If the possibility of long-term selection is neglected, this probability is proportional to the true length of the branch, which is estimated by the observed length. The expected values of terminal branch length are computed for each nonsense mutation under both assumptions, based on the observed terminal branch lengths of the tree on which it occurs. The length of the branch on which it occurs is scaled according to these expectations such that a value of 0 corresponds to the expectation for a laboratory mutant and a value of 1 corresponds to the expectation for a mutation that occurred in the wild. These are averaged for the nonsense mutations observed for a gene to yield a measure that I call the terminal branch length index (TBLI). Values of this measure are given in table 1, along with P values for the null hypotheses that it is ≤0 and that it is ≥1. Details of these calculations are given in the supplementary text, Supplementary Material online. This test, too, can fail to provide evidence for a genuine natural phenomenon due to long-term purifying selection, which may restrict observed nonsense mutations to the very tips of terminal branches, which are not longer on branches beyond a certain length.
The best evidence for a natural effect occurs when the TBLI is close to 1 or higher and statistics reject a value of 0 or less but not a value of 1 or more. However, an intermediate value of the TBLI can also provide evidence for a natural phenomenon, provided that the ratio of observed to expected nonsense mutations remains convincingly high after correction. For example, the TBLI for fliB, 0.446, is well below 1, but it is statistically distinguishable from 0. Even if only 44.6% of the 47 terminal-branch nonsense mutations are natural, combined with the eight internal nonsense mutations they give approximately a 29:34 nonsense:synonymous ratio, yielding an observed:expected ratio of 5.6, indicating a strong natural effect. In this case, the ratio on internal branches (4.84) agrees.
An important difference between the two tests is that the internal branch ratio is computed using all trees, whereas the TBLI utilizes only those on which nonsense mutations occur for a gene. They may also be affected differently by selection because occurrence of a mutation on an internal branch requires its presence in multiple isolates, whereas the TBLI depends only on evolutionary time.
For about 83% of the genes in table 1, at least one of the tests provides evidence that SDI occurs in nature. Where evidence is lacking, laboratory selection is a possible explanation, but purifying selection in the wild or insufficient power due to small numbers might be responsible. Most of the isolates included in the study were grown only for the purpose of molecular characterization rather than being cultivated extensively, which should minimize laboratory selection. Most of the inactivated alleles would be expected to experience long-term negative selection in the wild, because most of the genes have been maintained by purifying selection for millions of years.
Genes Exhibiting SDI
A few genes exhibiting SDI are discussed below. A more comprehensive discussion, which considers most of the genes in table 1, can be found in the supplementary text, Supplementary Material online.
Sigma Factor RpoS
The gene with the largest number of inactivations, by a factor of 2.5 if methylation hotspots are excluded, is rpoS. This gene encodes a sigma subunit of RNA polymerase that activates transcription of a variety of genes in response to starvation and other stresses (reviewed by Battesti et al. 2011 and Landini et al. 2014), including virulence genes (Fang et al. 1992; Velásquez et al. 2016).
Inactivated rpoS genes have frequently been observed in S. enterica and E. coli, due in part to selection during storage or growth in the laboratory (Zambrano et al. 1993; Sutton et al. 2000; Spira et al. 2011; Snyder et al. 2012; Bleibtreu et al. 2014). However, rpoS is frequently inactivated in fresh isolates of S. enterica serovar Typhi, though not of Typhimurium (Robbe-Saule et al. 2003).
There is a severe relative deficiency of nonsense mutations on the internal as compared with terminal branches. Furthermore, the TBLI is small and clearly distinguishable from 1. It is reassuring that both tests indicate the possibility of laboratory artifacts in this known case of laboratory selection for inactivation.
It is nonetheless possible that SDI occurs in nature, as suggested by the observations of Robbe-Saule et al. (2003). Both tests might be affected by long-term selection for RpoS function in nature, and the counts on internal branches are statistically compatible with a 2-fold or greater enhancement of the inactivation rate by selection. Furthermore, the TBLI (0.172) is statistically distinguishable from 0 or less. Taking it as an estimate of the fraction of terminal-branch inactivations that are natural implies that a total of about 48 inactivations have occurred in the wild. This figure should not be regarded as a precise estimate. However, if it is qualitatively correct then rpoS is among the genes most frequently inactivated in nature and its inactivation rate is several times the neutral expectation.
SDI of rpoS in the wild might be driven by the same forces that drive it in the laboratory. However, some of the genes regulated by RpoS are most relevant in a host. Among them are virulence genes (Fang et al. 1992; Chen et al. 1995; Velásquez et al. 2016) and genes involved in biofilm formation (Davidson et al. 2008), functions that are shared with many other genes that exhibit SDI (discussed below). Ferenci (2003) discusses possible reasons for rpoS inactivation.
Chemotaxis Receptor Tsr
The second most frequently inactivated gene (neglecting methylation hotspots) is tsr, which encodes a methyl-accepting chemotaxis protein (MCP). MCPs serve as receptors for chemotaxis. The loss of an MCP can be viewed as a partial loss of motility and chemotaxis. It may cause the bacteria to remain stationary when they otherwise would have moved along a concentration gradient. Loss of one MCP might also allow the cells to follow a weaker concentration gradient sensed by another MCP.
Tsr-mediated taxis toward alternative electron acceptors (“energy taxis”) is important for one route to invasion of host cells (Rivera-Chávez et al. 2013). Selection for tsr inactivation may be related to this process, either because inactivation permits invasion by another route or because it prevents invasion.
Because this is the first such gene to be discussed, it is worth pointing out that there is strong evidence that SDI of tsr occurs in nature. The observed:expected ratio is high on internal branches—nearly as high as the overall ratio—and is based on a large enough number of sequence changes that there is no great uncertainty in its estimate. Also, the TBLI is close to 1, statistically indistinguishable from 1, and easily distinguishable from 0. Inactivations of tsr that occur on internal branches can be seen in supplementary figure S1, Supplementary Material online.
Virulence Regulators HilD and HilC
Salmonella enterica virulence determinants are regulated by a complex network of proteins and small RNAs (Hölzer et al. 2009). The hilC and hilD genes are part of SPI-1 and encode transcriptional activators that activate virulence genes indirectly and directly (Ellermeier et al. 2005). There is also mutual activation between HilC, HilD, and RtsA. Both HilC and HilD activate SPI-1 genes, but only HilD can activate SPI-2 genes.
It is notable that there are no nonsense mutations, but 52 synonymous changes, in hilA. The derepression of hilA by hilC and hilD is the main means by which they activate virulence genes. Inactivation of hilA may give a more severe phenotype that is not favored. Also, HilC and HilD can activate a subset of SPI-1 genes independently of HilA (Akbar et al. 2003), which may be important to selection for their loss.
Phosphohydrolase UshA
UshA is a periplasmic phosphohydrolase. It has long been known that ushA is inactivated by a missense mutation in many Typhimurium isolates, including the laboratory strain LT2 (Burns and Beacham 1986), and by a different mutation in isolates of the serovars Gallinarum and Pullorum (Edwards et al. 1993; Innes et al. 2001)., which are close relatives. The present results indicate that inactivation of ushA is driven by positive selection.
In many clusters, ushA is inactivated in all isolates. In addition, as table 1 indicates, many inactivations are observed within clusters. More so than with other genes, these sometimes occur fairly deep in the tree and affect many isolates.
UshA is active toward a wide range of substrates (Neu 1967; Alves-Pereira et al. 2008). Selection for inactivation of ushA may be related to the intracellular lifestyle: Because of its periplasmic location, UshA might hydrolyze important compounds of the host cell, causing it unnecessary harm. Expression of ushA is increased when S. enterica enters a nongrowing state that is induced by acid (Núñez-Hernández et al. 2013), so selection for inactivation may be related to this state or to its abandonment.
The four Typhimurium isolates studied by Innes et al. (2001) all contained the same inactivating missense mutation, suggesting that inactivation in Typhimurium was due to a single mutational event. Although several Typhimurium clusters are completely affected by the know missense mutation, in other clusters several inactivations by nonsense mutations are observed. Thus, even within Typhimurium, inactivation is due to multiple events, and inactivation is an ongoing process.
Notably, no stop mutations are observed in ushB, which encodes another extracytoplasmic phosphohydrolase and is inactivated in E. coli (Edwards et al. 1993). The notion that inactivation of ushA and ushB are essentially equivalent losses of redundant functions is not supported by these results.
SbmA
The sbmA gene encodes a periplasmic transport protein. In E. coli, this protein is involved in entry of some bacteriophages and bacteriocins into the cell, and its inactivation results in resistance to them. SbmA is involved in the import of PR-39, a porcine antimicrobial peptide, and its inactivation confers resistance to this peptide (Pranting et al. 2008).
The isolate information points to PR-39 as a driving force for inactivation. Of the 30 cases of terminal-branch inactivation, five, or 16.7%, are from pigs or pork products. By comparison, only 3.6% of all the isolates are from porcine sources. This difference is statistically significant (P = 0.0045, two-sided Fisher’s exact test). If the total terminal branch length is considered (4.5% porcine), the enrichment is slightly smaller but remains statistically significant (P = 0.011, two-sided binomial test).
Of the nonporcine isolates, eight, or 26.7% of the total, are from cattle or beef products, whereas only 3.1% of all isolates are from these sources and they correspond to only 4.0% of the terminal branch length (differences statistically significant: P = 2.2E-6, two-sided Fisher’s exact test, and P = 1.6E-5, two-sided binomial test). This suggests that cattle produce a peptide similar to PR-39 that also is imported by SbmA and selects for sbmA inactivation. Analysis of bovine sequences identifies a candidate peptide (the mature peptide component of NP_777251.1). Like PR-39, it is rich in proline, arginine, and phenylalanine, and it has similar length.
Although humans produce a PR-39 counterpart of sorts, FALL-39 (Agerberth et al. 1995), sequence similarity is limited to the portion of the precursor protein that is excised; the mature peptides bear little resemblance. The six identifiable human cases may represent infections from pork and beef products, as the phylogenies strongly suggest in four cases (they are uninformative in the other two).
Biosynthesis of an Uncharacterized Polysaccharide
Strong evidence for SDI exists for seven genes that are part of a cluster apparently involved in the synthesis of a polysaccharide. These include five glycosyltransferases, a UDP-sugar (probably galactose) mutase, and an inner membrane protein (possibly involved in polysaccharide or precursor export).
This polysaccharide might, like cellulose, be involved in biofilm formation. In addition to cellulose, another polysaccharide is present in the extracellular matrix in Typhimurium biofilms, and it contains galactose (de Rezende et al. 2005). This substance is different from an additional polysaccharide present in S. enterica Enteritidis biofilms (White et al. 2003), and this gene cluster is present in Typhimurium but absent from most Enteritidis genomes. In any case, the frequent inactivation of these genes suggests that this polysaccharide has an undiscovered importance.
Four of these genes were identified by Barquist et al. (2013) as being “required” in Typhimurium, but not in Typhi. Many of the inactivations by stop codons, however, occur in Typhimurium. This seeming contradiction might be explained by the operational definition of “required” used by Barquist et al., which is compatible with the viability of an inactivated mutant. Barquist et al. also argue, based in part on differing “requirements” for these genes, that the cell surface is more important for Typhimurium than for Typhi. However, the evidence does not support this conclusion. These two points are discussed in greater detail in the supplementary text, Supplementary Material online.
O-Antigen Ligase RfaL
The rfaL gene (also called waaL) encodes O-antigen ligase. Its inactivation leads to lack of attachment of O-antigen to the LPS core, a major change in the cell surface.
Inactivated rfaL is found disproportionately in isolates from urine. Of the 30 cases of inactivation restricted to a single isolate, 8 (26.7%) are from urine and for 15 the relevant information is not available. Only 1.2% of all isolates are marked as isolated from urine, so this is an enrichment by more than a factor of 22 (P = 1.4E-9, two-sided Fisher’s exact test), or ∼18 compared with the fraction of terminal branch length (P = 1.2E-8, two-sided binomial test). In addition, one isolate is from porcine kidney. Selection for loss of this gene presumably operates most strongly in the urinary tract and is perhaps related to adhesion. Adhesion to host cells is known to be increased in rfaL mutants (Hölzer et al. 2009).
In addition to a high TBLI, the overrepresentation of isolates from urine is evidence that SDI occurred in the wild. On internal branches, however, the nonsense:synonymous ratio is <1/3 the neutral expectation. This appears to be a case where a lack of apparent SDI on internal branches is caused by long-term selection against inactivated alleles and does not indicate a laboratory artifact. Because of the second form of evidence that SDI is natural, this gene provides a particularly good example of this phenomenon.
Motility Regulator YdiV
Salmonella enterica is motile (and chemotactic) under some conditions and nonmotile under others. Expression of flagellar genes is under tight regulation and responds to a variety of inputs. The choice between motility and nonmotility is a major “lifestyle” decision that can involve tradeoffs. Even when motility would otherwise be advantageous, it comes with costs: Expression of flagella consumes resources and results in a 2% decrease in growth rate (Macnab 1996), and flagella may be a target of antibodies, are recognized by the immune system as pathogen-associated molecular patterns (Miao et al. 2007), and serve as attachment sites for some bacteriophages (Choi et al. 2013; Hendrix et al. 2015). Mutations that eliminate flagella confer advantages under some conditions and disadvantages under others (Weinstein et al. 1984; Allen-Vercoe et al. 1999; Van Asten et al. 2000; Kilroy et al. 2017).
YdiV represses expression of flagella under conditions of poor nutrient availability. Salmonella enterica is unusual in that it exhibits motility and chemotaxis only when nutrients are plentiful (Koirala et al. 2014). In most other bacteria, including E. coli, just the opposite is true: Motility and chemotaxis are exhibited only when nutrients are rare. It may be that most bacteria use chemotaxis for foraging, whereas S. enterica uses it for host colonization (Koirala et al. 2014). Loss of YdiV might make S. enterica adopt the use of chemotaxis for foraging like most bacteria. It might also be selected because it prevents biofilm formation, or because it allows the use of chemotaxis for host invasion under conditions that normally prevent it.
A notable feature of ydiV is that 401 nonsense inactivations occur at a single Dcm-methylated position, nucleotide 367 of the LT2 coding sequence. This is more than ten times the total at other positions in the gene, and by itself gives ydiV a higher rate of these events than any other gene if Dcm sites are included. This phenomenon is not accounted for by the typical effect of Dcm methylation, which increases the transition rate by a factor of ∼8 (Cherry 2018). Assuming that selection for a stop codon is not significantly stronger at this position than at others in the gene, its transition rate must be higher than average for a Dcm site by more than an order of magnitude.
Although there is significant rate heterogeneity among Dcm sites, this site appears to be exceptional. The coding sequences contain just one other with a comparable transition rate, and only three more that come within a factor of 2. This analysis accounts for the ∼11-fold effect of selection for ydiV inactivation: the fastest Dcm position elsewhere in the genome exhibits 31 transitions, which is lower than the 401/11.34 = 35 expected for the ydiV site without selection.
The methylated position on the opposite strand of this Dcm site does not share the extraordinary transition rate: No changes are observed there, even though a transition would be synonymous. Another Dcm site lies just upstream of this one, with six base pairs separating the two, but such closely spaced Dcm sites are not rare in the Salmonella genome. The second site is apparently not highly mutable for a Dcm site either: Only three transitions are observed there. Transitions at both methylated positions of the second site are nonsynonymous, but if they are strongly disruptive their frequency should be increased by SDI.
It is tempting to speculate that the presence of this extraordinarily mutable site in a gene subject to SDI is an evolved mechanism for generating inactivated alleles. However, the straightforward version of this hypothesis faces two difficulties. First, there is the likelihood that inactivated alleles are “dead ends” due to long-term purifying selection. Second, no allele that destroys itself will be favored by the success of the derived allele, which does not increase the frequency of its highly mutable parent. Both objections could be overcome by sufficiently frequent reversion, but there are only two candidate reversion events at this position of ydiV, both of which are suspect because an equally parsimonious reconstruction eliminates them.
A plausible alternative is that a small fraction of cells with inactivated alleles contribute to the success of the parental genotype, in which case a high rate of inactivation could be favored due to kin selection. However, in this case, it is the intact parental allele that enjoys a fitness advantage, so SDI does not follow, though the two phenomena might be related.
Gene Presence and Absence among Isolates
If inactivated alleles are not subject to negative selection in the long term, the usual outcome is expected to be complete loss of the gene. Sufficiently weak purifying selection would also diminish the rate of acquisition of a gene, and borderline selection in some lineages may correlate with weak selection in others.
Figure 3 shows the cumulative distribution of the frequency of occurrence of the genes that display SDI in a set of Salmonella genomes (blue), including alleles with stop codons or frame-changing insertions and deletions but requiring a match over at least 80% of the coding sequence. Also shown (gray) is the distribution for the remaining LT2 orthologs. The fractions for the individual genes are given in supplementary table S1, Supplementary Material online. With the goal of obtaining a more representative sampling of diversity, only one genome (the reference) was included from each cluster of isolates, and the analysis included all clusters, not just those containing at least 5 isolates, for a total of 9,594 genomes.
Fig. 3.
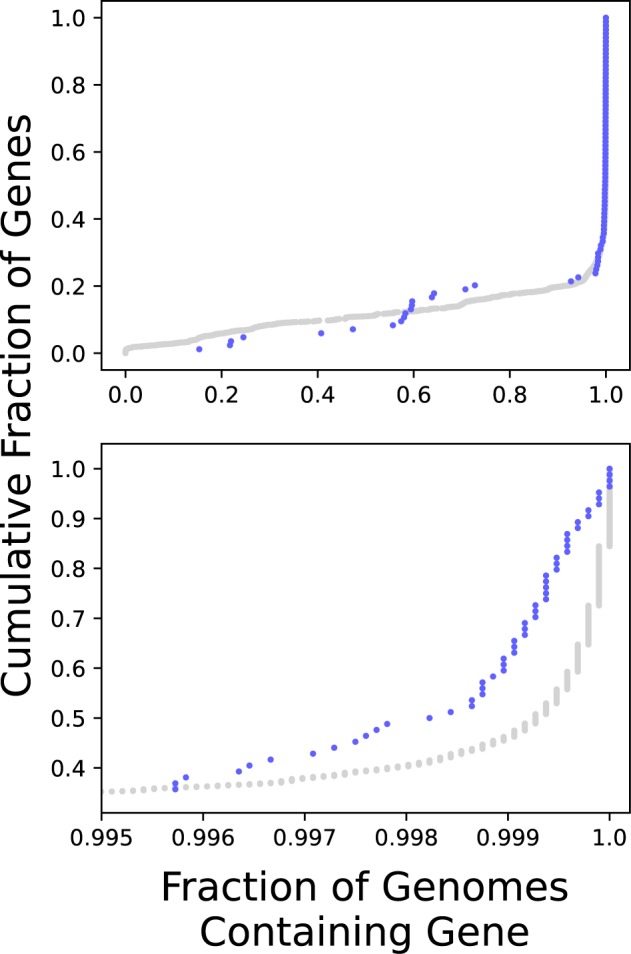
—The distribution among genes of the fraction of reference genomes in which they are found. Cumulative distributions are shown for the 84 genes for which SDI is apparent (blue) and for the remainder of the genes (gray). Each point corresponds to a gene. The lower panel shows just the part of the distribution between 99.5% and 100% occurrence.
The distribution for the genes exhibiting SDI is similar to that for other genes. Perhaps surprisingly, there is no deficit of genes present nearly universally. Of the 84 genes, 55, or 65%, are present in more than 99.5% of the genomes, which is slightly larger than the fraction among other genes. Thus, SDI is not associated with one indication of weak selection for function or an accessory or lineage-specific role. Furthermore, this result supports the view that the selectively inactivated genes enjoy only a short-term advantage and are usually eliminated by negative selection in the long term.
Above 99.5%, the two distributions diverge (fig. 3, lower panel), with generally lower fractions of presence among the 84 genes showing SDI. This is to be expected. The fact that any inactivated alleles are observed indicates that a gene is not absolutely required for survival. Furthermore, selection for inactivation is expected to increase the rate of gene loss, both because deletion would be positively selected under some circumstances and a gene inactivated in some other way is not subject to selection against deletion.
General Discussion
It was found that 84 S. enterica genes exhibit SDI. For the majority of them, this could be shown to have occurred in nature. There may be many others for which this phenomenon could not be detected due to insufficient power or the effects of purifying selection on inactivated mutants.
In many cases, several genes with related functions, or whose inactivation would produce related phenotypes, were found to be subject to SDI. This commonality is partially reflected in the categories in table 1, but several recurring themes transcend the categories. Many of the genes affect processes that are connected by a vast regulatory network: virulence, motility and chemotaxis, biofilm formation, and the stress response. A large number of the genes affect the cell surface, which forms the cell’s interface with the outside world, both abiotic and biotic. It is likely that inactivation of many of the genes is selected, at least in some instances, because it provides resistance to toxic substances. Only three genes were designated as being inactivated due to selection by specific substances, but resistance is a suspected reason for inactivation of many other genes. Most of the genes affecting the cell surface fall into this category, especially if “toxic substances” is extended to include bacteriophages.
The results provide evidence for several hypotheses that do not concern selection and may merit empirical exploration. One is that ydiV contains a site with an extraordinarily high mutation rate. If this phenomenon can be observed in the laboratory, the sequence requirements and mechanism could be investigated. Another is that cattle, like swine, produce an antimicrobial peptide that is active against Salmonella, and that enters the cell through the product of the sbmA gene, the inactivation of which confers resistance to the peptide. Finally, the results suggest the unrecognized importance of an exopolysaccharide produced by the products of a gene cluster found in Typhimurium and some isolates of other serovars. The presence of the polysaccharide might be easily observable by microscopy (de Rezende et al. 2005), and any effects on host colonization and pathogenesis would be of interest.
Condition-Specific Advantage and Eventual Extinction
Although inactivated alleles of a few genes, such as ushA and oafA, may be establishing themselves, it seems unlikely that a large fraction of the genes in table 1 are in the process of being lost. This is especially so for genes with orthologs in E. coli and other enteric bacteria. It seems more plausible that these genes are regularly subject to occasional condition-specific selection that drives inactivation, but that the mutants are eventually eliminated by the same purifying selection that has maintained the genes for millions of years. The fact that the majority of these genes are present in almost all isolates analyzed (fig. 3) supports this view.
A straightforward example of this phenomenon that involved resistance to a harmful substance was given in the Introduction section. In other cases, the selective force that favors inactivation might be less obvious and more complex. Salmonella enterica encounters a wide variety of conditions. It can live inside of host cells of various types (most notably epithelial cells and macrophages), and outside of cells in various host compartments and extrahost environments. Hosts vary in such aspects as species, genotype, nutrition, immune state, and resident microbiota. Uncommon conditions, or combinations of conditions, may select for loss of a gene’s function, allowing inactivated mutants to out-compete the parent strain. This would, however, be a Pyrrhic victory in that the mutation would doom its bearer to likely extinction.
A different possibility is that a short-term advantage under common, ordinary conditions is offset by a long-term disadvantage. A mutant might be capable of out-competing its parent within any locale, but less effective at spreading to new locales. Mutations of this sort can be compared with somatic mutations that lead to increased cell growth rate and cancer, in that they enjoy only a temporary advantage and they harm the parental genotype.
Nontyphoidal human infections (the source of the vast majority of the human isolates) continually derive from nonhuman reservoirs, mainly poultry, cattle, and swine. If selection for inactivation occurred only upon such a host switch, the combination of positive selection and nonpersistence might be explained by source-sink dynamics (Sokurenko et al. 2006). However, human isolates are not overrepresented among isolates singly affected by SDI events, and the major reservoir species are found among them at representative frequencies (supplementary table S1, Supplementary Material online, rightmost columns). Thus, although source-sink dynamics due to zoonosis might contribute to the phenomenon, it is apparently not the main explanation for it.
Why Not Regulate?
If it is sometimes advantageous to eliminate a gene product, why has regulation not evolved to repress gene expression under the relevant conditions? This would allow exploitation of those conditions without eliminating future gene expression under other conditions where it is favorable.
The rarity of some conditions may explain the absence of seemingly advantageous regulation. If the conditions are sufficiently rare, selection will be too weak to establish and maintain regulation. Selection may even disfavor such regulation because the cost of the necessary additional regulatory apparatus outweighs the small gain in fitness.
Some of the conditions that favor gene inactivation may be sufficiently novel that there has not been sufficient time for complete adaptation to them. Although most conditions will have been encountered before in the history of S. enterica, their occurrence at high frequencies may be phenomena of the modern world. Possible examples include the presence of truly novel antimicrobial agents and the crowded conditions of modern methods of rearing poultry.
Another possibility is that the mutant phenotype locally out-competes the parent under ordinary conditions yet is harmful to the success of the pathogen. Conflicts likely exist between individual growth rate and the success of a clone that infects a host; fast growth might reduce transmission by, for example, reducing achievable density. As suggested earlier, such a mutant is akin to a cancer on the parental strain. More abstractly, the mutant phenotype amounts to defection in the cooperative effort of the clone. Repression of the relevant gene would be harmful, much like repression of a tumor suppressor gene, at least when a single clone infects a host.
In some cases, the necessary regulation might be difficult or impossible, at least with the usual mechanisms of bacterial regulation. Several negative regulators of sugar utilization are among the genes displaying SDI. These regulators seemingly serve to allow gene expression when it is appropriate (e.g., when the sugar is available) and prevent wasteful expression otherwise, so why would their inactivation ever be favorable? It might be advantageous under conditions of intermittent sugar availability because constitutive expression allows avoidance of phenotypic lag, which overcomes the cost involved. The impossibility of predicting the future precludes a simple regulatory solution. In principle, a regulatory “decision” to express the catabolic genes continuously could be made based on the history of availability of this sugar and other carbon sources. However, this would require some sort of long-term memory and a somewhat sophisticated integration of historical information.
An inactivating mutant might have occasional success due only to chance. The mutant phenotype would represent a “decision” that is usually detrimental but, unpredictably, advantageous on occasion, much as probabilistically incorrect play in a game of chance sometimes beats correct play. If the outcome is for practical purposes random, always making the probabilistically correct play—that is, never repressing the gene—is the best strategy.
Conclusion and Perspectives
Selection-driven gene inactivation is a notable phenomenon in S. enterica, and presumably in other bacteria as well. It is likely that most selective inactivation events enjoy only temporary success and are ultimately eliminated by purifying selection. This may be an example of a more general phenomenon that is not limited to bacteria or to inactivating mutations but occurs in most species and involves nonsynonymous and regulatory mutations as well. This could complicate efforts to infer distributions of selection coefficients for various types of mutations but might provide opportunities for other types of studies.
The identities of genes subject to selection for inactivation provide a window into the selective tradeoffs faced by the organism and may point to testable nonevolutionary hypotheses concerning extant bacteria. Furthermore, because the genes affected are enriched for genes of high interest to researchers, they may identify other genes of unrecognized importance that deserve further study. This phenomenon may be especially important for other bacteria that have been less intensively studied than S. enterica.
Supplementary Material
Acknowledgment
This research was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Literature Cited
- Agerberth B, et al. 1995. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U S A. 92(1):195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar S, Schechter LM, Lostroh CP, Lee CA.. 2003. AraC/XylS family members, HilD and HilC, directly activate virulence gene expression independently of HilA in Salmonella typhimurium. Mol Microbiol. 47(3):715–728. [DOI] [PubMed] [Google Scholar]
- Allen-Vercoe E, Sayers AR, Woodward MJ.. 1999. Virulence of Salmonella enterica serotype Enteritidis aflagellate and afimbriate mutants in a day-old chick model. Epidemiol Infect. 122(3):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves-Pereira I, et al. 2008. CDP-alcohol hydrolase, a very efficient activity of the 5′-nucleotidase/UDP-sugar hydrolase encoded by the ushA gene of Yersinia intermedia and Escherichia coli. J Bacteriol. 190(18):6153–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson JO, Andersson SG.. 2001. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol Biol Evol. 18(5):829–839. [DOI] [PubMed] [Google Scholar]
- Barquist L. et al. 2013. A comparison of dense transposon insertion libraries in the Salmonella serovars Typhi and Typhimurium. Nucleic Acids Res. 41(8):4549–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, Gottesman S.. 2011. The RpoS-Mediated General Stress Response in Escherichia coli Annu Rev Microbiol. 65(1):189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleibtreu A, et al. 2014. The rpoS gene is predominantly inactivated during laboratory storage and undergoes source-sink evolution in Escherichia coli species. J Bacteriol. 196(24):4276–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DM, Beacham IR.. 1986. Identification and sequence analysis of a silent gene (ushA0) in Salmonella typhimurium J Mol Biol. 192(2):163–175. [DOI] [PubMed] [Google Scholar]
- Chain PSG, et al. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 101(38):13826–13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, et al. 1995. Central regulatory role for the RpoS sigma factor in expression of Salmonella dublin plasmid virulence genes. J Bacteriol. 177(18):5303–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JL. 2017. A practical exact maximum compatibility algorithm for reconstruction of recent evolutionary history. BMC Bioinformatics 18(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JL. 2018. Methylation-induced hypermutation in natural populations of bacteria. J Bacteriol. 200(24):pii: e00371–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Shin H, Lee J-H, Ryu S.. 2013. Identification and characterization of a novel flagellum-dependent Salmonella-infecting bacteriophage, iEPS5. Appl Environ Microbiol. 79(16):4829–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper VS, Schneider D, Blot M, Lenski RE.. 2001. Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J Bacteriol. 183(9):2834–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W.. 1978. Molecular basis of base substitution hotspots in Escherichia coli. Nature 274(5673):775–780. [DOI] [PubMed] [Google Scholar]
- Dagan T, Blekhman R, Graur D.. 2006. The ‘domino theory’ of gene death: gradual and mass gene extinction events in three lineages of obligate symbiotic bacterial pathogens. Mol Biol Evol. 23(2):310–316. [DOI] [PubMed] [Google Scholar]
- Davidson CJ, White AP, Surette MG.. 2008. Evolutionary loss of the rdar morphotype in Salmonella as a result of high mutation rates during laboratory passage. ISME J. 2(3):293–307. [DOI] [PubMed] [Google Scholar]
- Day WA, Fernández RE, Maurelli AT.. 2001. Pathoadaptive mutations that enhance virulence: genetic organization of the cadA regions of Shigella spp. Infect Immun. 69(12):7471–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rezende CE, Anriany Y, Carr LE, Joseph SW, Weiner RM.. 2005. Capsular polysaccharide surrounds smooth and rugose types of Salmonella enterica serovar Typhimurium DT104. Appl Environ Microbiol. 71(11):7345–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CJ, Innes DJ, Burns DM, Beacham IR.. 1993. UDP-sugar hydrolase isozymes in Salmonella enterica and Escherichia coli: silent alleles of ushA in related strains of group I Salmonella isolates, and of ushB in wild-type and K12 strains of E. coli, indicate recent and early silencing events, respectively. FEMS Microbiol Lett. 114:293–298. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Ellermeier JR, Slauch JM.. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol. 57(3):691–705. [DOI] [PubMed] [Google Scholar]
- Fang FC, et al. 1992. The alternative sigma factor KatF (RpoS) regulates Salmonella virulence. Proc Natl Acad Sci U S A. 89(24):11978–11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Johnston RN, Liu G-R, Liu S-L.. 2013. Genomic comparison between Salmonella Gallinarum and Pullorum: differential pseudogene formation under common host restriction. PLoS One 8(3):e59427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenci T. 2003. What is driving the acquisition of mutS and rpoS polymorphisms in Escherichia coli?. Trends Microbiol. 11(10):457–461. [DOI] [PubMed] [Google Scholar]
- Hall BG, Yokoyama S, Calhoun DH.. 1983. Role of cryptic genes in microbial evolution. Mol Biol Evol. 1(1):109–124. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, et al. 2015. Genome sequence of Salmonella Phage χ. Genome Announc. 3(1):e01229–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölzer SU, Schlumberger MC, Jäckel D, Hensel M.. 2009. Effect of the O-antigen length of lipopolysaccharide on the functions of Type III secretion systems in Salmonella enterica. Infect Immun. 77:5458–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Fukuchi S, Kawabata T, Ota M, Nishikawa K.. 2002. A systematic investigation identifies a significant number of probable pseudogenes in the Escherichia coli genome. Gene 294(1-2):25–33. [DOI] [PubMed] [Google Scholar]
- Innes D, et al. 2001. The cryptic ushA gene (ushA(c)) in natural isolates of Salmonella enterica (serotype Typhimurium) has been inactivated by a single missense mutation. Microbiology 147(7):1887–1896. [DOI] [PubMed] [Google Scholar]
- Jin Q, et al. 2002. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 30(20):4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilroy S, et al. 2017. Salmonella Enteritidis flagellar mutants have a colonization benefit in the chicken oviduct. Comp Immunol Microbiol Infect Dis. 50:23–28. [DOI] [PubMed] [Google Scholar]
- Knöppel A, et al. 2018. Genetic adaptation to growth under laboratory conditions in Escherichia coli and Salmonella enterica. Front Microbiol. 9:756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, et al. 2014. A nutrient-tunable bistable switch controls motility in Salmonella enterica serovar Typhimurium. mBio 5(5):e01611–01614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landini P, Egli T, Wolf J, Lacour S.. 2014. sigmaS, a major player in the response to environmental stresses in Escherichia coli: role, regulation and mechanisms of promoter recognition. Environ Microbiol Rep. 6(1):1–13. [DOI] [PubMed] [Google Scholar]
- Macnab RM. 1996. Flagella and motility. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. FC Neidhardt. Washington (DC: ): ASM Press. [Google Scholar]
- Maddison W. 1989. Reconstructing character evolution on polytomous cladograms. Cladistics 5(4):365–377. [DOI] [PubMed] [Google Scholar]
- McClelland M, et al. 2004. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet. 36(12):1268–1274. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M.. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351(6328):652–654. [DOI] [PubMed] [Google Scholar]
- Miao EA, Andersen-Nissen E, Warren SE, Aderem A.. 2007. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 29(3):275–288. [DOI] [PubMed] [Google Scholar]
- Neu HC. 1967. The 5′-nucleotidase of Escherichia coli. I. Purification and properties. J Biol Chem. 242(17):3896–3904. [PubMed] [Google Scholar]
- Núñez-Hernández C, et al. 2013. Genome expression analysis of nonproliferating intracellular Salmonella enterica serovar Typhimurium unravels an acid pH-dependent PhoP-PhoQ response essential for dormancy. Infect Immun. 81(1):154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Davalos LM.. 2006. The nature and dynamics of bacterial genomes. Science 311(5768):1730–1733. [DOI] [PubMed] [Google Scholar]
- Pränting M, Negrea A, Rhen M, Andersson DI.. 2008. Mechanism and fitness costs of PR-39 resistance in Salmonella enterica serovar Typhimurium LT2. Antimicrob Agents Chemother. 52(8):2734–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosseda G, et al. 2012. Shedding of genes that interfere with the pathogenic lifestyle: the Shigella model. Res Microbiol. 163(6–7):399–406. [DOI] [PubMed] [Google Scholar]
- Rice WR. 1996. Evolution of the Y sex chromosome in animals. BioScience 46(5):331–343. [Google Scholar]
- Rivera-Chávez F, et al. 2013. Salmonella uses energy taxis to benefit from intestinal inflammation. PLoS Pathog. 9(4):e1003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe-Saule V, Algorta G, Rouilhac I, Norel F.. 2003. Characterization of the RpoS status of clinical isolates of Salmonella enterica. Appl Environ Microbiol. 69(8):4352–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC, et al. 2006. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol. 239(2):226–235. [DOI] [PubMed] [Google Scholar]
- Snyder E, Gordon DM, Stoebel DM.. 2012. Escherichia coli lacking RpoS are rare in natural populations of non-pathogens. G3 (Bethesda) 2:1341–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokurenko EV, Gomulkiewicz R, Dykhuizen DE.. 2006. Source-sink dynamics of virulence evolution. Nat Rev Microbiol. 4(7):548–555. [DOI] [PubMed] [Google Scholar]
- Spira B, de Almeida Toledo R, Maharjan RP, Ferenci T.. 2011. The uncertain consequences of transferring bacterial strains between laboratories—rpoS instability as an example. BMC Microbiol. 11(1):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton A, Buencamino R, Eisenstark A.. 2000. rpoS mutants in archival cultures of Salmonella enterica serovar Typhimurium. J Bacteriol. 182(16):4375–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson NR, et al. 2008. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 18(10):1624–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z, et al. 2005. Pseudogene accumulation might promote the adaptive microevolution of Yersinia pestis. J Med Microbiol. 54(3):259–268. [DOI] [PubMed] [Google Scholar]
- Van Asten FJ, Hendriks HG, Koninkx JF, Van der ZB, Gaastra W.. 2000. Inactivation of the flagellin gene of Salmonella enterica serotype Enteritidis strongly reduces invasion into differentiated Caco-2 cells. FEMS Microbiol Lett. 185(2):175–179. [DOI] [PubMed] [Google Scholar]
- Velásquez JC, et al. 2016. SPI-9 of Salmonella enterica serovar Typhi is constituted by an operon positively regulated by RpoS and contributes to adherence to epithelial cells in culture. Microbiology 162(8):1367–1378. [DOI] [PubMed] [Google Scholar]
- Weinstein DL, Carsiotis M, Lissner CR, O’Brien AD.. 1984. Flagella help Salmonella typhimurium survive within murine macrophages. Infect Immun. 46(3):819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AP, Gibson DL, Collinson SK, Banser PA, Kay WW.. 2003. Extracellular polysaccharides associated with thin aggregative fimbriae of Salmonella enterica serovar Enteritidis. J Bacteriol. 185(18):5398–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods R, Schneider D, Winkworth CL, Riley MA, Lenski RE.. 2006. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 103(24):9107–9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano M, Siegele D, Almiron M, Tormo A, Kolter R.. 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259(5102):1757–1760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.