

ABSTRACT
Damaged mitochondria are selectively removed from the cell in a process termed mitophagy. This mitochondrial quality control mechanism is important for neuronal homeostasis, and mutations in pathway components are causative for Parkinson disease and amyotrophic lateral sclerosis (ALS). Here, we discuss our recent work using a novel mild induction paradigm to investigate the spatiotemporal dynamics of mitophagy in primary neurons. Using live-cell imaging, we find that mitophagy-associated proteins translocate to depolarized mitochondrial fragments. These mitophagic events were primarily localized to somatodendritic compartments, suggesting neuronal mitophagy is primarily a somal quality control mechanism. Damaged mitochondria were efficiently sequestered within autophagosomes, but lysosomal fusion or acidification was significantly delayed. Surprisingly, engulfed mitochondria persisted in non-acidified vesicular compartments for hours to days after initial damage. Expression of an ALS-associated mutation disrupted the membrane potential of the mitochondrial network, and oxidative stress exacerbated this effect. Importantly, our results highlight the slow kinetics of mitophagy and suggest that slow turnover of damaged mitochondria may increase neuronal susceptibility to neurodegeneration.
KEYWORDS: Autophagy, mitophagy, mitochondria, lysosome, neurodegenerative diseases
Dysfunctional mitochondria are specifically removed from the cell through the selective autophagy mechanism known as mitophagy, where autophagy receptors bind ubiquitinated mitochondria and recruit phagophores, precursors to autophagosomes, to sequester and eliminate damaged organelles. Mitophagy initiates with the stabilization of PINK1 (PTEN induced putative kinase 1) on the surface of damaged mitochondria where it recruits PRKN/Parkin, an E3 ubiquitin ligase, leading to phospho-ubiquitination of outer mitochondrial membrane proteins. The autophagy receptor OPTN (optineurin) translocates to ubiquitinated mitochondria and is stabilized on the surface via phosphorylation by TBK1 (TANK-binding kinase 1), a serine/threonine kinase. Subsequently, OPTN binds to MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), facilitating the engulfment of damaged organelles into phagophores, and ultimate degradation by a conserved lysosomal degradation pathway. Mutations in pathway components are linked to both Parkinson disease and ALS.
In neurons, the regulated removal and replenishment of mitochondria to support local energy demands at fusion sites is important to maintain homeostasis. As a result of the unique spatial and temporal constraints of neurons, multiple mitochondrial quality control mechanisms may work collectively to maintain network health. Investigation of mitophagy in neurons, both in vivo and primary cultures, has resulted in conflicting views regarding the spatiotemporal dynamics of mitophagy and the relative significance of PINK1- and PRKN-dependent mitophagy as a predominant mitochondrial quality control mechanism. These controversies are heightened by the use of a variety of harsh chemical uncouplers that are toxic to neurons. Here, we used a novel mitophagy induction protocol to investigate the dynamics of neuronal mitophagy [1]. Antioxidant deprivation increases intracellular reactive oxygen species resulting in low levels of mitochondrial damage, but maintains the morphology and dynamics of the mitochondrial network and neuronal integrity.
Live-cell imaging in primary hippocampal neurons indicates that mitophagy-associated proteins, including PRKN, TBK1, OPTN, and LC3, translocate to ubiquitinated mitochondria within an hour of antioxidant removal. Sequestered mitochondria are negative for TMRE, a vital dye used to monitor mitochondrial membrane potential and health. Thus, mild oxidative stress through antioxidant removal induces mitochondrial depolarization and initiates neuronal mitophagy, where OPTN specifically translocates to damaged mitochondrial fragments downstream of PRKN recruitment.
The temporal dynamics of lysosome fusion and subsequent autophagosome acidification to degrade damaged mitochondria was visualized using live-cell imaging. In control conditions the bulk of OPTN-positive mitochondria fuse with lysosomes (marked by LAMP1 [lysosomal-associated membrane protein 1]), but this fusion is significantly reduced in stressed conditions, possibly because antioxidant removal overwhelms the system and stalls lysosomal fusion. Analysis of lysosomal acidification (visualized using LysoTracker) revealed that nearly all mitophagic events in either basal or induced conditions have yet to acidify 6 h after treatment, suggesting this step in mitophagy is slow. These findings were confirmed using dual labeled fluorescent probes targeted to mitochondria or lysosomes, which allow for simultaneous monitoring of localization and acidification, indicating damaged organelles stay in non-acidified vesicular compartments for hours. It is possible that the antioxidant removal unexpectedly neutralizes the lumen of cellular autolysosomes. As a result, the acidification state of both autophagosomes and lysosomes was examined, and neither organelle display disrupted acidification resulting from antioxidant deprivation. Thus, the delay in degradation is specific to these mitophagic events. Additionally, we compared the slow kinetics of neuronal mitophagy to that of HeLa cells overexpressing exogenous PRKN and determined that >80% of mitophagic events in HeLa cells had undergone lysosomal fusion and acidification within an hour of treatment. Surprisingly, the temporal dynamics of neuronal mitophagy are much slower, with lysosome fusion and acidification to degrade damaged mitochondria occurring hours after initial damage (Figure 1).
Figure 1.
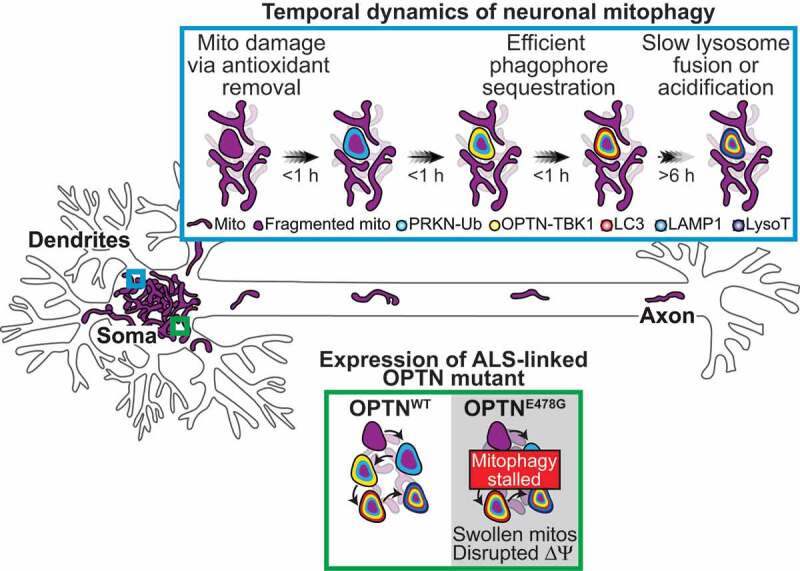
Model of the spatiotemporal regulation of neuronal mitophagy following mild oxidative stress. Mitochondrial damage was induced via antioxidant removal, which increases intracellular reactive oxygen species and initiates mitophagy, yet maintains mitochondrial network health. Upon damage, PINK1-recruited PRKN phospho-ubiquitinates outer mitochondrial membrane proteins. The abundant ubiquitin chains on the damaged organelles recruit OPTN and TBK1, which leads to autophagosome formation and sequestration. These steps in the pathway occur within an hour of initial damage. Interestingly, only a minor fraction of mitophagic events are acidified in either basal or induced conditions 6 h after treatment. Thus, autophagosome formation is efficient, but lysosomal fusion and acidification to degrade mitochondria is rate-limiting. Furthermore, overexpression of an ALS-linked OPTN mutant disrupts the mitochondrial network membrane potential, and stress to the system via antioxidant removal exacerbates this phenotype increasing neuronal susceptibility to damage and neurodegeneration. ∆ψ, mitochondrial membrane potential; Ub, ubiquitin; LysoT, LysoTracker.
Pulse-chase experiments to observe long-term sequestration in autolysosomes illustrated that engulfed mitochondrial fragments can be visualized 24 h post-treatment. These events persist in the soma of basal and induced conditions, implying acidification is a rate-limiting step in neuronal mitophagy. Slow mitochondrial turnover may expose the neuron or mitochondrial network to damage, as dysfunctional organelles could build up over time. This hypothesis was tested by expressing an ALS-associated OPTNE478 G mutant that fails to bind ubiquitin. Compared to wild type, mutant expression disrupts the mitochondrial membrane potential. Importantly, the health of the mitochondrial network worsens with stress to the system via antioxidant deprivation, providing a mechanism by which mutations to pathway components can disrupt mitophagy and cause neurodegeneration.
In summary, our recent work uses a novel mild mitochondrial damaging paradigm to demonstrate the spatial and temporal dynamics of OPTN-mediated mitophagy in primary hippocampal neurons. We provide additional evidence that OPTN acts downstream of PINK1 and PRKN, where it specifically translocates to damaged mitochondria and recruits phagophore engulfment and degradation via lysosomal fusion. Interestingly, we find that the time course of lysosomal fusion and removal of damaged mitochondria is remarkably slow in neurons, occurring hours to days after initial damage. In an OPTN mutant background, mitochondrial health is altered, and this decline is exacerbated by a second hit to the system, such as antioxidant removal to stress the mitochondrial network.
These findings support a model where phagophore sequestration of damaged mitochondria is efficient, but lysosomal acidification to fully degrade mitochondria is slow and rate-limiting. Phagophore engulfment may prevent dissemination of damaged mitochondrial components and potential poisoning of the mitochondrial network, which could be detrimental in terminally differentiated neurons. Furthermore, the slow kinetics of mitophagy could increase neuronal sensitivity to age-related factors or perturbations to the system, ultimately leading to neurodegeneration. Additionally, our conditions of mild oxidative stress to mimic highly metabolic neurons, induces low levels of mitochondrial damage and mitophagy. It is likely that additional mitochondrial quality control mechanisms are contributing to maintaining mitochondrial and neuronal health.
Funding Statement
This work was supported by a grant from NIH NINDS [R37 NS060698] to E.L.F.H. C.S.E. was supported by the Howard Hughes Medical Institute Hanna H. Gray Fellowship
Disclosure statement
The authors declare no competing financial interests.
Reference
- [1].Evans CS, Holzbaur EL.. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife. 2020;9. DOI: 10.7554/eLife.50260 [DOI] [PMC free article] [PubMed] [Google Scholar]