

ABSTRACT
We aimed to investigate the involvement of macroautophagy/autophagy in autoimmunity in rheumatoid arthritis (RA) through citrullination of VIM (vimentin) and its interaction with MHC class II in synovial fibroblasts (SFs). The cell surface expression of MHC class II and B7 costimulatory molecules on SFs was analyzed by flow cytometry after treatment with IFNG/IFN-γ (interferon gamma). Intracellular citrullinated autoantigens in SFs were analyzed by immunoblotting using serum from anti-citrullinated peptide antibodies (ACPA)-positive patient as a primary antibody. SFs were incubated in serum-free medium or treated with proteasome inhibitor MG132 to induce autophagy. An autophagy inhibitor 3-methyladenin (3-MA) was used. Intracellular citrullinated VIM (cVIM) was evaluated by immunoblotting and immunocytochemistry. The interaction between MHC class II and cVIM was evaluated with co-immunoprecipitation and proximity ligation assay (PLA). We demonstrated that MHC class II, CD274/B7-H1 and PDCD1LG2/B7-DC were expressed on SFs following treatment with IFNG whereas CD276/B7-H3 was detected on SFs regardless of the presence of IFNG. ACPA-positive sera recognized a 54 kDa protein in SFs. By immunoprecipitation, the 54 kDa protein recognized by RA sera was revealed to be cVIM. Following induction of autophagy, intracellular cVIM was increased in SFs but the effect was canceled by 3-MA. The interaction between MHC class II and cVIM was demonstrated by co-immunoprecipitation. Furthermore, PLA revealed the significant increase of MHC class II-cVIM interaction following induction of autophagy. Our findings suggest that SFs may contribute to the autoimmunity in RA through citrullination of VIM and its interaction with MHC class II promoted by autophagy.
Abbreviations: 3-MA: 3-methyladenine; ACPA: anti-citrullinated peptide antibodies; anti-CCP: anti-cyclic citrullinated peptide antibody; cVIM: citrullinated VIM; BECN1: beclin1; DAPI: 4ʹ,6-diamidino-2-phenylindole; FBS: fetal bovine serum; HLA: human leukocyte antigen; IFNG/IFN-γ: interferon gamma; IL6: interleukin 6; IP: immunoprecipitation; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MFI: mean fluorescence index; MHC: major histocompatibility complex; OA: osteoarthritis; PADI: peptidyl arginine deiminase; PepA: pepstatin A; PBS: phosphate-buffered saline; PtdIns3K: phosphatidylinositol 3-kinase; RA: rheumatoid arthritis; SFs: synovial fibroblasts; siRNA: small interfering RNA; VIM: vimentin
KEYWORDS: Autophagy, citrullination, MHC class II, rheumatoid arthritis, vimentin
Introduction
Rheumatoid arthritis (RA) is characterized by chronic joint inflammation with progressive bone and cartilage destruction caused by abnormal proliferation of synovial tissue. Anti-citrullinated peptide antibodies (ACPA) are disease specific autoantibodies included in 2010 American College of Rheumatology/European League Against Rheumatism RA classification criteria [1]. ACPA are frequently detected before onset of RA [2,3] and associated with high disease activity as well as joint destruction [4], suggesting ACPA as pathogenic autoantibodies. Although smoking [5] and carriage of human leukocyte antigen (HLA)-DRB1 shared epitope (SE) alleles [6,7] are proven to be associated with ACPA positivity, it still remains unclear what triggers the immune response to citrullinated peptides and how ACPA production is sustained.
Macroautophagy, hereinafter referred to as autophagy, is the major intracellular degradation system, characterized with delivery of cytoplasmic materials to the lysosome [8]. Autophagy mainly functions to adapt to starvation and other stresses but also regulates various cellular functions. Since autophagy is involved in antigen presentation with major histocompatibility complex (MHC) class II [9,10] and accelerates peptidylarginine deiminase (PAD) activity [11], autophagy is supposed to be involved in ACPA production through citrullination and antigen presentation.
Synovial fibroblasts (SFs) play a key role as effector cells in the pathogenesis of RA, releasing inflammatory cytokines, chemokines and matrix metalloproteinases [12]. We have previously reported a high autophagy activity in RASFs compared to those of osteoarthritis (OA) and autophagy’s protective effect against apoptotic cell death [13]. Although MHC class II molecules are mainly expressed on classical antigen-presenting cells including dendritic cells, macrophages and B cells, SFs function as antigen-presenting cells with MHC class II expression in the presence of IFNG/IFN-γ (interferon gamma) [14], leading to CD4+ T cell activation [15,16]. Moreover, SFs contain abundant amounts of VIM (vimentin) whose citrullinated form is a major antigen targeted by ACPA. In the present study, we investigated the role of autophagy on the citrullination of VIM and its presentation through MHC class II in SFs.
Results
Antigen-presenting potential of SFs
First, we evaluated the antigen-presenting potential of SFs by detecting cell surface expression of MHC class II and that of B7 costimulatory molecules and by comparing them with those of B cells, which were isolated from patients (Figure S1). HLA-DR, CD274 and PDCD1LG2 were expressed on SFs (Figure 1A) only when treated with IFNG with statistical significance (Figure 1B), whereas CD276 was expressed on SFs regardless of the presence of IFNG. Conversely, ICOSLG/B7-H2, VTCN1/B7-H4, CD80 and CD86 were not detected on SFs even after treatment with IFNG. On the other hand, HLA-DR, CD274, ICOSLG, VTCN1, PDCD1LG2 and CD86 were detected on B cells regardless of the presence of IFNG (Figure 1A,B), suggesting the difference in antigen-presenting potential between SFs and classical antigen-presenting cells (APCs). We also analyzed the effect of IL6 (interleukin 6), one of the major inflammatory cytokines involved in RA pathogenesis, on cell surface expression of HLA-DR and B7 molecules in SFs, however, no clear effect was observed (Figure 1C,D).
Figure 1.
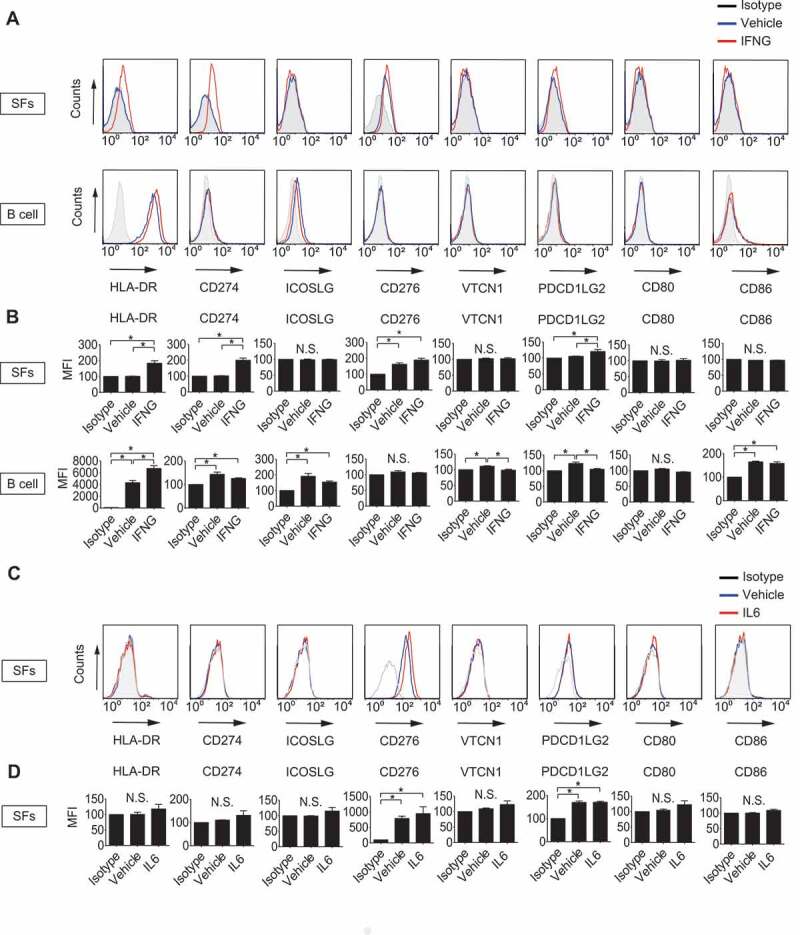
Determination of antigen-presenting potential of synovial fibroblasts (SFs) and B cells. SFs and B cells were left untreated or treated with 100 ng/mL IFNG for 72 h. Cell surface expression of human leukocyte antigen (HLA)-DR and that of B7 costimulatory molecules were analyzed by flow cytometry. Histograms are representative of 3 independent experiments using cells from 3 different patients (A). Cell surface expression levels of the molecules were quantified and presented as mean fluorescent intensity (MFI) compared to isotype controls (B). SFs were left untreated or treated with 100 ng/mL IL6 plus soluble IL6RA/IL-6R for 72 h. Cell surface expression of HLA-DR and that of B7 costimulatory molecules were analyzed by flow cytometry. Histograms are representative of 3 independent experiments using cells from 3 different patients (C). MFI of C (D). Values are presented as mean ± S.D. * = P < 0.05.
Detection of intracellular citrullinated antigens in SFs
Next, intracellular citrullinated autoantigens were analyzed by western blotting using ACPA positive or negative RA patient’s serum (ACPA-positive patient 1: 78-year-old female with anti-cyclic citrullinated peptide antibody [anti-CCP] of >300 IU/mL, patient 2: 72-year-old female with anti-CCP of 136.6 IU/mL and ACPA-negative patient 3: 69-year-old female with anti-CCP of <4.5 IU/mL) as a primary antibody. ACPA-positive serum, but not ACPA-negative serum, most strongly recognized a 54 kDa protein in the lysate of SFs (RA1: 74-year-old female with anti-CCP of <4.5 IU/mL, RA2: 67-year-old female with anti-CCP of 71.2 IU/mL), which was also recognized by anti-VIM, -citrulline and -citrullinated VIM (cVIM) antibodies (Figure 2A). By immunoprecipitation with anti-VIM antibodies, the 54-kDa protein recognized by ACPA-positive sera was revealed to be cVIM (Figure 2B).
Figure 2.
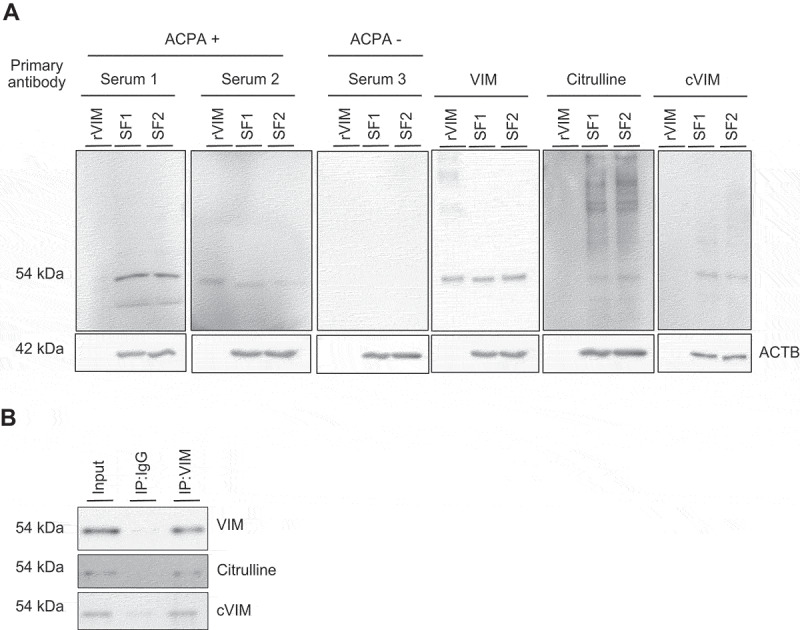
Detection of intracellular citrullinated antigens in synovial fibroblasts (SFs). Recombinant human VIM (rVIM) and SF lysates were immunoblotted using anti-citrullinated peptide antibodies (ACPA) positive or negative rheumatoid arthritis patient’s serum or anti-VIM, -citrulline or -citrullinated VIM (cVIM) antibody as a primary antibody (A). SF lysates were immunoprecipitated with anti-VIM antibody or normal goat IgG and blotted with anti-VIM, -citrulline or -cVIM antibody (B). IP, immunoprecipitation.
Effect of autophagy on citrullination of intracellular VIM in SFs
Given the acceleration of PAD activity by autophagy as shown in previous studies [11], we next evaluated the effect of autophagy on citrullination of VIM in SFs. cVIM was increased following induction of autophagy by 1–3 h of serum-free starvation (Figure 3A) with statistical significance (Figure 3B) in RASFs, which was parallel with the increase of MAP1LC3/LC3-II (microtubule associated protein 1 light chain 3), a marker for autophagy activity. Similarly, but to a lesser extent, citrullination of VIM and autophagy induction was upregulated by 1–2 h of serum-free starvation in OASFs (Figure 3C,D). The effect was canceled by the autophagy inhibitor 3-methyladenine (3-MA) (Figure 3E-H). To confirm the association between autophagy and citrullination of VIM, we induced autophagy by another means using a proteasome inhibitor MG132 [13] and lysosomal protease inhibitors pepstatin A (PepA) and E64d [17]. Proteasome inhibition with MG132 similarly promoted autophagy and cVIM in RASFs and in OASFs (Figure S2A-D). MG132 caused increase of LC3-II levels, and addition of PepA and E64d, which blocks later autolysosomal degradation, further increased the LC3-II levels and 3-MA also had inhibitory effect on autophagy and cVIM induced by proteasome inhibition (Figure S2E-H).
Figure 3.
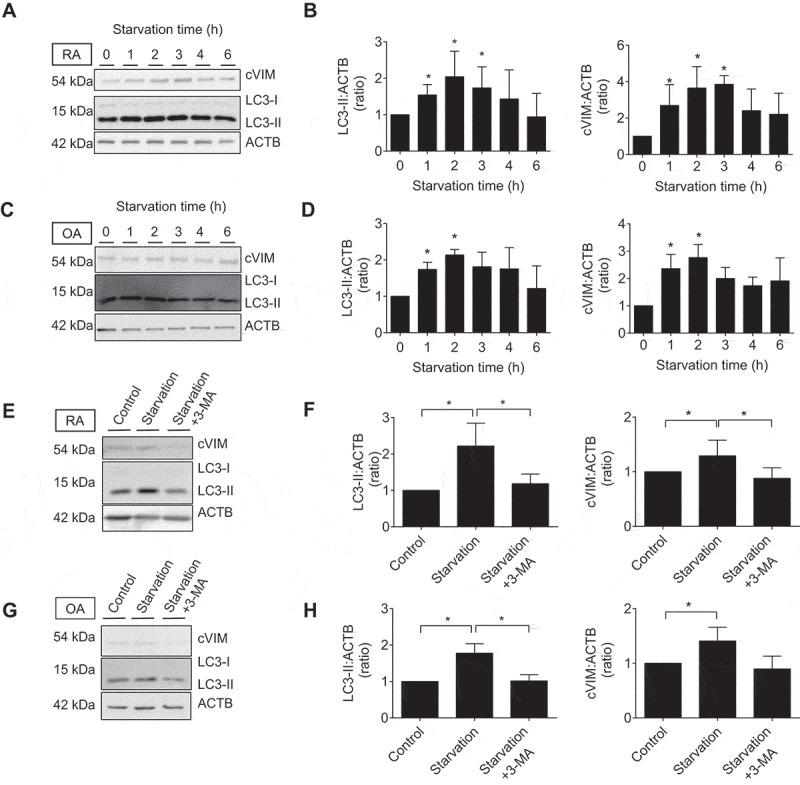
Effects of autophagy induction by serum-free starvation on citrullination of intracellular VIM in synovial fibroblasts (SFs). Rheumatoid arthritis (RA) and osteoarthritis (OA) SFs were normally cultured or incubated in serum-free medium for the indicated times and their lysates were immunoblotted. Blots are representative of 4 independent experiments, respectively (A and C). Densitometric analysis of A and C. Autophagy activity and citrullinated VIM (cVIM) were evaluated by LC3-II:ACTB/β-actin and cVIM:ACTB ratio, respectively (B and D). RASFs and OASFs were normally cultured or incubated in serum medium for 2 h in the presence or absence of 5 mM 3-methyladenine (3-MA), an autophagy inhibitor, and their lysates were immunoblotted. Blots are representative of 4 independent experiments (E and G). Densitometric analysis of E and G (F and H). Values are presented as mean ± S.D. * = P < 0.05.
Interaction between MHC class II and cVIM in SFs
Given the antigen-presenting potential of SFs and the promotive effect of autophagy on citrullination of VIM in SFs demonstrated by the previous experiments, we hypothesized the interaction between MHC class II and cVIM through autophagy. Prior to analysis, we evaluated the expression of intracellular cVIM and HLA-DR by immunocytochemistry. In RASFs, intracellular cVIM and HLA-DR were increased, compared to controls (Figure 4A,B, S3A,B), following treatment with serum-free starvation plus IFNG (Figure 4E,F) or with the combination of MG132 and IFNG (Figure S3E,F), again indicating the promotive effect of autophagy on citrullination of VIM in SFs. Moreover, as shown in merged images, HLA-DR colocalized with cVIM (Figure 4H; Figure S3H), suggesting the interaction between MHC class II and cVIM in SFs. The colocalization of HLA-DR and cVIM were also observed in OASFs (Figure 4I-P and Figure S3I-P). The interaction between MHC class II and cVIM in SFs was confirmed by co-immunoprecipitation with anti-HLA-DR antibodies (Figure 5A). Furthermore, proximity ligation assay revealed that MHC class II-cVIM interaction in SFs was significantly increased following treatment with serum-free starvation and IFNG (Figure 5B, 5Civ and Cix) or with MG132 and IFNG combination (Figure S4Aiv and Ax), and further increased with the addition of PepA and E64d (Figure S4Av and Axi), but the effect was canceled by 3-MA (Figure 5C-v and C-x; Figure S4A-vi and A-xii) with statistical significance (Figure 5D and S4B). Interestingly, treatment with either serum-free starvation, MG132 or IFNG did not significantly increase MHC class II-cVIM interaction, indicating that both autophagy induction and IFNG stimulation are necessary for MHC class II-cVIM interaction. As in RASFs, the proximity ligations of MHC class II and cVIM were also observed in OASFs, however, the results were less pronounced (Figure 5E and Figure S4C).
Figure 4.
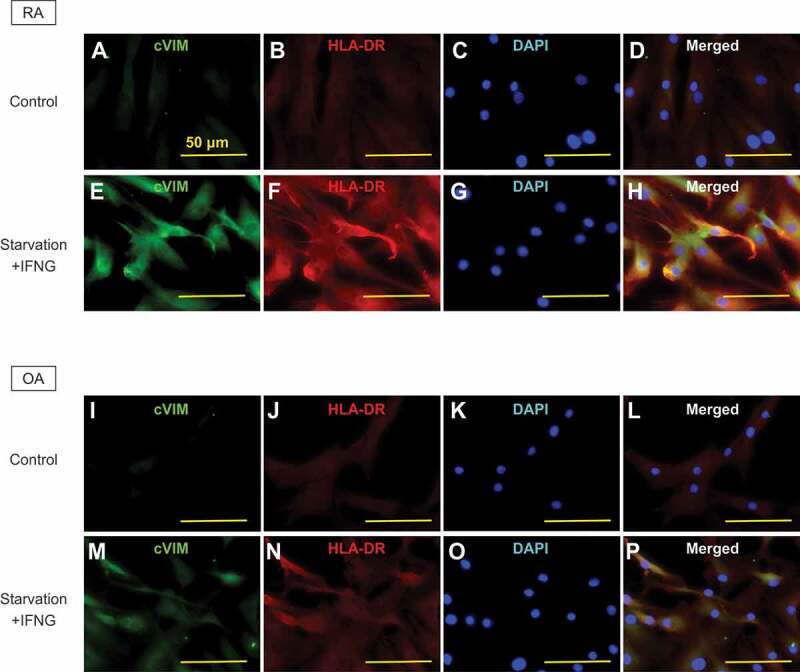
Immunocytochemical analysis of the co-expression of human leukocyte antigen (HLA)-DR-citrullinated VIM (cVIM) in synovial fibroblasts (SFs). cVIM (green), HLA-DR (red) were visualized and nuclei were stained with DAPI (blue). RASFs and OASFs were left untreated (control) or treated with 100 ng/mL IFNG for 72 h followed by serum-free starvation in the last 2 h (starvation + IFNG). Scale bar: 50 μm.
Figure 5.
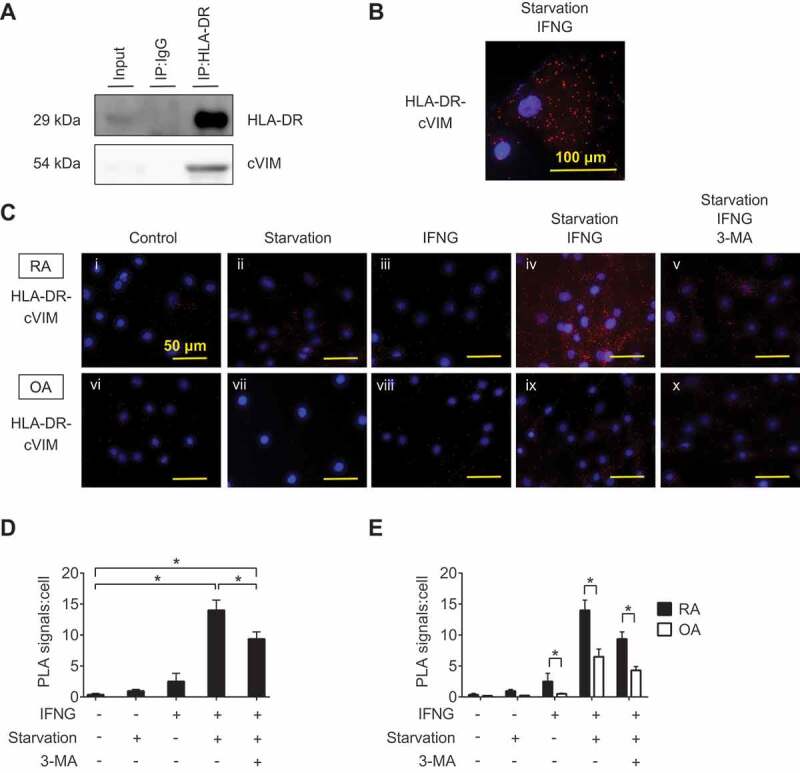
Interaction between HLA-DR and citrullinated VIM (cVIM) in synovial fibroblasts (SFs). RASFs were treated with IFNG for 72 h followed by serum-free starvation in the last 2 h and their lysates were co-immunoprecipitated with anti-HLA-DR antibodies and blotted with anti-HLA-DR or -cVIM antibodies (A). Proximity ligation assay to demonstrate the formation of HLA-DR-cVIM complexes. RA and OA SFs were left untreated, starved in serum-free medium, or treated with IFNG in the presence or absence of 3-MA. Figures are representative images of 3 independent experiments using cells from 3 different patients (C). Scale bar: 50 μm. Representative cell of Civ (B). Scale bar: 100 μm. Quantification of HLA-DR-cVIM PLA complexes in RASFs (D). Values are presented as mean PLA signal per cell ± S.D. * = P < 0.05. Comparison the quantification of HLA-DR-cVIM PLA complexes in RASFs and OASFs (E). Values are presented as mean PLA signal per cell ± S.D * = P < 0.05.
Discussion
We herein demonstrated the antigen-presenting potential of SFs in the presence of IFNG, the promotive effect of autophagy on citrullination of VIM in SFs, and the interaction between MHC class II and cVIM promoted by autophagy induction and IFNG stimulation in SFs, indicating a role of SFs, IFNG and autophagy in immune response to citrullinated peptides and ACPA production.
Citrullination is the post-translational modification of an arginine residue to citrulline in proteins catalyzed by PAD [18,19]. PAD converts positively charged arginine to uncharged citrulline, leading to changes in hydrogen bonding, ionic interactions and protein-protein interaction [20]. PADI4 gene, one of the isoforms of PAD coding regions, has been identified as one of the genetic susceptibility loci of RA, suggesting that disproportionate citrullination is associated with the pathogenesis of RA [21]. PAD4 also mediates autoantibody production against citrullinated antigens, promoting inflammatory arthritis [22].
Although ACPA recognize and bind to citrullinated epitopes present on numerous proteins including VIM, fibrin and enolase [19,23], anti-cVIM antibodies are particularly associated with joint destruction [24–26]. C-VIM is identified as a T cell epitope in SE containing RA-susceptible HLA-DRB alleles [6,27,28]. These findings suggest cVIM as a major autoantigen in RA.
Autophagy regulates various cellular functions and maintains homeostasis through preventing abnormal protein accumulation. Therefore, impairment of autophagy cause cellular damages and subsequent organ dysfunction, leading to neurodegenerative disease [29], diabetes mellitus [30] heart disease [31] and autoimmune diseases including systemic lupus erythematosus [32] and regional enteritis [33]. Meanwhile, some diseases are reported to have activated autophagy including cancer [34] and RA [13]. Moreover, autophagy promotes intracellular antigen presentation via MHC class II [9]. The term “Type 2 cross-presentation” has been recently proposed to define autophagy dependent intracellular antigen presentation via MHC class II [35], providing an evidence that cells with activated autophagy may be involved in antigen presentation. In our study, we have shown promotive effect of autophagy on citrullination of intracellular VIM in SFs. To analyze the effect of autophagy inhibition, we selected an inhibitor of phosphatidylinositol 3-kinases (PtdIns3K) 3-MA to inhibit autophagy. We also tried to inhibit autophagy via transient knockdown of BECN1, ATG5 and ATG7 by siRNAs. However, even after effective knockdown, there was little inhibition of basal autophagic flux shown by the accumulation of LC3-II. The siRNA approach to downregulate gene expression might be insufficient to block autophagic flux in SFs (Figure S5A,B), which is consistent with the published report [36,37]. Our results also demonstrated immunological diversity of ACPA. As shown in Figure 2, one ACPA-positive patient’s serum recognized not only cVIM but also uncitrullinated native VIM, meanwhile the other ACPA-positive serum recognized only cVIM. Autoimmunity against citrullinated autoantigen has been thought to be triggered by smoking [7] and by periodontal disease [38] through increased PAD activity and subsequent citrullination of various proteins in the lung or in the mouth, whereas the autoimmunity transition from citrullination to the joint remains unclear. To understand local autoimmunity in joints, possible pathophysiology of spondyloarthritis may provide a new clue on production of ACPA. Synovitis in spondyoarthritis has been proposed to be secondary synovitis due to cytokine release and growth factors from enthesitis [39]. Local production of ACPA in the joint has been suggested in patients with psoriatic arthritis, one of the subtypes of spondyloarthritis [40]. These various origin of ACPA may give us an explanation for the immunological diversity of ACPA. ACPA are associated with joint destruction and disease severity of psoriatic arthritis [41,42]. These findings support our result that citrullinated antigen presentation by SFs may contribute to the production and sustaining of ACPA.
Although our study demonstrated the antigen-presenting capacity of SFs, antigen-presenting potential may be different from classical professional APCs such as dendritic cells and B cells. MHC class II was expressed only following treatment with IFNG on SFs whereas it was expressed on B cells regardless of the presence of IFNG. We provide an evidence of cell surface expression of B7 costimulatory molecules on SFs. B7 superfamily is necessary for T cell activation and tolerance in addition to T cell receptor recognition of antigen [43]. CD276 was identified as a new B7 superfamily molecule in 2002, which was originally reported to have a costimulatory effect on T cell proliferation, enhancing cytotoxicity and IFNG production [44], however, its inhibitory effect was also reported [45]. In SFs, bidirectional CD276 signals to T cells have been reported [16]. CD276 has a costimulatory effect on activated T cells, while it has an inhibitory effect on resting T cells, suggesting that SFs may interact with T cells through CD276’s stimulatory effect and contribute to the progression of RA synovitis. CD274 (PD-L1) and PDCD1LG2 also belong to the B7 superfamily, which were identified as ligands for programmed death-1 [46]. Synovium related CD274 inhibits proinflammatory factors from local T cells [47]. The expression of PDCD1LG2 in rheumatoid synovium was also reported [48], indicating its involvement in the pathogenesis of RA, however, its contribution to RA is unclear. Further investigation is needed to determine the function of B7 superfamily on SFs.
Our study has shown the interaction between MHC class II and cVIM in the presence of IFNG and autophagy induction. Compared to classical APCs such as mature dendritic cells, MHC class II was shown to be less localized at the cell surface in non-classical APCs [49]. Consistent with the previous findings, our data showed that MHC class II was less expressed at the cell surface in SFs compared to B cells and that MHC class II-cVIM interaction was mainly localized in the cytoplasm in SFs. Therefore, the precise role of MHC class II-cVIM interaction on production and sustaining of ACPA remains to be determined. Further investigation is needed to demonstrate whether this interaction leads to T cell and B cell activation.
Materials and methods
Study approval
The protocol for the collection and the use of human sera and synovial samples was approved by the institutional review boards (IRBs) of Hokkaido University (008–0103). Written informed consent in accordance with the Declaration of Helsinki was obtained from all participants according to the relevant guidelines of the IRBs.
Patient samples and cell preparation
SFs were derived from synovial tissue specimens obtained from RA and OA patients during joint replacement surgery (Department of Orthopedic Surgery, Hokkaido University, Sapporo, Japan). All RA patients fulfilled the American College of Rheumatology/European League Against Rheumatism criteria for classification of RA [1]. SFs were cultured as described elsewhere [50], synovial tissue samples were digested with collagenase type I (Sigma-Aldrich, C0130) and cultured in Iscove’s modified Dulbecco’s medium (Sigma-Aldrich, I6529) supplemented with 10% fetal bovine serum (FBS; Cosmo Bio, CCP-FBS-BR-500), 100 IU/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, P4333) and used between passages 4–8 for all experiments. Peripheral blood mononuclear cells (PBMCs) were isolated from patients’ peripheral blood using Ficoll®-paque PLUS (GE healthcare, 17144002). B cells were enriched by negative selection using the EasySepTM Human B cell isolation kit (StemcellTM Technologies, 17954) as per the manufacturer’s instructions. Isolated cells were cultured in RPMI-1640 medium (Sigma-Aldrich, 8758) supplemented with 10% FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin. Purity of isolated B cells was assessed by flow cytometry using phycoerythrin-conjugated antibodies against CD19 (Biolegend, 152,407).
Flow cytometry
To analyze antigen-presenting potential, SFs and B cells were labeled with phycoerythrin-conjugated antibodies against HLA-DR (Biolegend, 307605) or B7 costimulatory molecules including CD274/B7-H1 (Biolegend, 329705), ICOSLG/ICOSL/B7-H2 (Biolegend, 309403), CD276/B7-H3 (Biolegend, 351003), VTCN1/B7-H4 (Biolegend, 358103), PDCD1LG2/B7-DC (Biolegend, 329605), CD80 (Biolegend, 305207) and CD86 (Biolegend, 305405) for 15 min in the dark. Stained cells were applied to BD FACSCaliburTM (BD Biosciences, U.S.A) and analyzed using the FlowJo software (BD Biosciences, U.S.A).
Treatment of cells
To induce autophagy, SFs were starved using serum-free medium for 0–6 h or treated with 0–10 μM of the proteasome inhibitor MG132 (Sigma-Aldrich, M7449) for 24 h in the presence or absence of 5 mM of the autophagy inhibitor 3-MA (Cayman Chemical, 5142-23-4). To inhibit lysosomal function, 10 μM of pepA (Sigma-Aldrich, P5318) and 10 μM of E64d (Sigma-Aldrich, E8640) were used. To induce MHC class II, SFs and B cells were treated with 100 ng/mL IFNG (Biolegend, 570204) or with combination of 100 ng/mL IL6 (Biolegend, 570804) and 100 ng/mL of soluble IL6RA/IL-6R (Biolegend, 751504) for 72 h. Control samples were treated with matched amount of phosphate-buffered saline (PBS; Sigma-Aldrich, D8537) or dimethyl sulfoxide (DMSO; Dojindo, 349–01025).
Small interfering RNA (siRNA) knockdown
SFs were seeded to 12 well plates and incubated in Iscove’s modified Dulbecco’s medium containing 10% FBS to 70–80 confluency. After incubating for 48 h, transient transfection experiments were conducted using Lipofectamine® RNAiMAX (Invitrogen, 13778100) according to the manufacturers’ protocols. A set of small interfering RNA (siRNA) of Control (Cell Signaling Technology, 6568) BECN1 (Cell Signaling Technology, 6222) ATG5 (Cell Signaling Technology, 6345) and ATG7 (Cell Signaling Technology, 6604) were purchased from Cell Signaling Technology. After 24 h incubation at 37°C, the treated cells were collected, and total RNA was isolated.
Quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR)
Total RNA was obtained using TRIzol RNA Reagent (Invitrogen, 15596–018) according to the manufacturer’s instructions. The RNA samples were reverse transcribed using SuperScript VILO (Invitrogen, 11755250). Complementary DNA was amplified using SYBR Green Master Mix (Applied biosystems, 4385610). The level of the BECN1, ATG5 and ATG7 transcript was normalized to that of the GAPDH transcript with an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, U.S.A) and custom primers for BECN1, ATG5, ATG7 and GAPDH (Hokkaido system science, Japan); primer sequences are listed in Table 1. Relative quantification was performed using the ΔΔCt method.
Table 1.
Primers used in this study.
| Gene name | Primer |
|---|---|
| GADPH Forward | 5ʹ-GGGAAGCTTGTCATCAATGGA-3’ |
| GADPH Reverse | 5ʹ-TCTGGCTCCTGGAAGATGGT-3’ |
| BECN Forward | 5ʹ-GCTACCGGGAAGTCGCTGAAGACAG-3’ |
| BECN Reverse | 5ʹ-CGTCTTGACCCTTCCATCCCTCGG-3’ |
| ATG5 Forward | 5ʹ-GATGTGTGGTTTGGACGAATTCC-3’ |
| ATG5 Reverse | 5ʹ-GTATGGTTCTGCTTCCCTTTCAG-3’ |
| ATG7 Forward | 5ʹ-AACCTCTCTTGGGCTTGTGCCTCAC-3’ |
| ATG7 Reverse | 5ʹ-CATCGCTCATGTCCCAGATCTCAGC-3’ |
BECN1, beclin 1
Western blotting
Cells were lysed in Laemmli buffer (62.5 mM Tris HCl, pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 0.1% bromophenol blue, 5 mM β-mercaptoethanol). Whole cell lysates and recombinant human VIM (R&D systems, 2105-VI-100) were separated on 10-15% polyacrylamide gels and electroblotted onto polyvinylidene difluoride (PVDF) membranes (Merck, IPVH304F0). Membranes were blocked for 1 h in 5% nonfat milk in TBS-T (20 mM Tris base, 137 mM sodium chloride, 0.1% Tween 20 [Nacalai tesque, 23926–35], pH 7.6). After blocking, the membranes were probed with antibodies against MAP1LC3B (Cell Signaling Technology, 2775), VIM (Abcam, ab45939), citrulline (Merck, 07–377), cVIM (Cayman Chemical, 22054), HLA-DR (Abcam, ab226820), BECN1 (Cell Signaling Technology, 3738), ATG5 (Cell Signaling Technology, 12994), ATG7 (Cell Signaling Technology, 2631), or ACTB/β-actin (Sigma-Aldrich, A3854) as an endogenous control. To detect intracellular citrullinated antigens, ACPA-positive or negative RA patient’s serum was used as a primary antibody. As secondary antibodies, horseradish peroxidase-conjugated goat anti-rabbit (Sigma-Aldrich, A4914), goat anti-mouse antibodies (Sigma-Aldrich, A8642) and goat anti-human antibodies (Sigma-Aldrich, A0170) were used. Signals were detected using enhanced chemiluminescence western blot detection reagents (GE healthcare, RPN2109) and LAS-4000 imaging system (FUJIFILM, Japan). For quantification of protein expression, densitometric analysis was performed using ImageJ software (National Institutes of Health, U.S.A).
Immunoprecipitation
SFs were lysed with CelLytic M Cell lysis reagent (Sigma-Aldrich, C2978). VIM and HLA-DR were precipitated with anti-VIM antibody (R&D systems, AF2105) or -HLA-DR antibody (Abcam, ab136320) and protein G-sepharose (GE healthcare, 71-7083-00). Normal goat IgG (Santa Cruz Biotechnology, sc-2028) or normal mouse IgG (Abcam, ab188776) was used as a control. The immunoprecipitates were eluted by boiling with Laemmli buffer and analyzed by western blotting as described above.
Immunocytochemistry
Cells were cultured in chamber slides (Lab-Tec, 177399PK), fixed with 4% paraformaldehyde (FUJIFILM Wako Pure Chemical Corporation, 163-20145), and permeabilized with PBS containing 0.1% Triton X-100 (Nacalai tesque, 35501-15). Nonspecific binding was blocked with 1% bovine serum albumin (Sigma-Aldrich, A6003) for 1 h. Slides were incubated overnight at 4°C with primary antibodies. After washing, slides were incubated for 3 h with Alexa Fluor® 488- (Thermo Scientific, 11029) or Alexa Fluor® 594-conjugated (Thermo Scientific, 21207) secondary antibodies and nuclei were stained with 4ʹ,6-diamidino-2-phenylindole (DAPI; Biolegend, 422801). Slides were covered with fluorescent mounting medium (Diagnostic Biosystems, K024) and analyzed with a fluorescence microscope BZ-X800 (Keyence, U.K.).
In situ proximity ligation assays
Cells were cultured in 96-well plates, fixed with 4% paraformaldehyde, and permeabilized with PBS containing 0.1% Triton X-100. Nonspecific binding was blocked with Duolink® Blocking Solution (Sigma-Aldrich) for 1 h at 37°C following overnight incubation with primary antibodies against HLA-DR and cVIM at 4°C. After washing, plates were incubated with the PLA probes (Anti-Rabbit Plus and Anti-Mouse Minus; Sigma-Aldrich, DUO92002, DUO92004) for 1 h at 37°C followed by chromogenic substrate development and nuclear staining with DAPI. Plates were covered with PBS and analyzed with a fluorescence microscope. Fluorescence spots were quantified using Image J software.
Statistical analysis
Statistical analysis was performed by Student’s t-test, One-way ANOVA with post-hoc Tukey’s test. P values less than 0.05 were considered significant. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, U.S.A.).
Supplementary Material
Funding Statement
This work was supported by the Japanese Ministry of Education, Culture, Sports, Science, and Technology; [KAKENHI-16K19591].
Acknowledgments
The authors would like to thank all the members and laboratory assistants of Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University for their technical support and valuable discussion.
Disclosure statement
Dr. Atsumi has received research support from Astellas, Takeda, Mitsubishi Tanabe, Chugai, Pfizer, Daiichi Sankyo and Otsuka (more than $10, 000 each) and speaking fees from Mitsubishi Tanabe, Chugai, Pfizer, Eisai, AbbVie (less than $10, 000 each), Astellas and Daiichi Sankyo (more than $10,000 each).
Dr. Yasuda has received research support from Chugai Pharmaceutical, Novartis Pharmaceutical, MSD (less than $10,000 each) and Bristol Myers Squibb (more than $10,000), speaking fees from Chugai Pharmaceutical, Bristol Myers Squibb and Tanabe-Mitsubishi Pharmaceutical and Pfizer and consulting fees from Glakso Smith Kline (less than $10,000 each).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].Aletaha D, Neogi T, Silman AJ, et al. Rheumatoid arthritis classification criteria: an American College of Rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–2581. [DOI] [PubMed] [Google Scholar]
- [2].van Venrooij WJ, van Beers JJ, Pruijn GJ.. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol. 2011;7(7):391–398. [DOI] [PubMed] [Google Scholar]
- [3].Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–2749. [DOI] [PubMed] [Google Scholar]
- [4].van den Broek M, Dirven L, Klarenbeek NB, et al. The association of treatment response and joint damage with ACPA-status in recent-onset RA: a subanalysis of the 8-year follow-up of the BeSt study. Ann Rheum Dis. 2012;71(2):245–248. [DOI] [PubMed] [Google Scholar]
- [5].Catrina AI, Svensson CI, Malmstrom V, et al. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat Rev Rheumatol. 2016;13(2):79–86. [DOI] [PubMed] [Google Scholar]
- [6].Scally SW, Petersen J, Law SC, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210(12):2569–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Klareskog L, Ronnelid J, Lundberg K, et al. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol. 2008;26:651–675. [DOI] [PubMed] [Google Scholar]
- [8].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [9].Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102(22):7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schmid D, Munz C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ. 2005;12 Suppl 2(Suppl 2):1519–1527. [DOI] [PubMed] [Google Scholar]
- [11].Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. 2011;208(13):2625–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kato M, Ospelt C, Gay RE, et al. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014;66(1):40–48. [DOI] [PubMed] [Google Scholar]
- [14].Navarrete Santos A, Riemann D, Thiele K, et al. Constitutive expression of HLA class II mRNA in synovial fibroblast-like cells from patients with rheumatoid arthritis. Immunol Lett. 1997;58(1):53–58. [DOI] [PubMed] [Google Scholar]
- [15].Boots AM, Wimmers-Bertens AJ, Rijnders AW. Antigen-presenting capacity of rheumatoid synovial fibroblasts. Immunology. 1994;82(2):268–274. [PMC free article] [PubMed] [Google Scholar]
- [16].Tran CN, Thacker SG, Louie DM, et al. Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol. 2008;180(5):2989–2998. [DOI] [PubMed] [Google Scholar]
- [17].Vomero M, Manganelli V, Barbati C, et al. Reduction of autophagy and increase in apoptosis correlates with a favorable clinical outcome in patients with rheumatoid arthritis treated with anti-TNF drugs. Arthritis Res Ther. 2019;21(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fuhrmann J, Thompson PR. Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem Biol. 2016;11(3):654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vossenaar ER, Radstake TR, van der Heijden A, et al. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis. 2004;63(4):373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fuhrmann J, Clancy KW, Thompson PR. Chemical biology of protein arginine modifications in epigenetic regulation. Chem Rev. 2015;115(11):5413–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Suzuki A, Yamada R, Chang X, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34(4):395–402. [DOI] [PubMed] [Google Scholar]
- [22].Shelef MA, Sokolove J, Lahey LJ, et al. Peptidylarginine deiminase 4 contributes to tumor necrosis factor alpha-induced inflammatory arthritis. Arthritis Rheumatol. 2014;66(6):1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wegner N, Lundberg K, Kinloch A, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233(1):34–54. [DOI] [PubMed] [Google Scholar]
- [24].Syversen SW, Goll GL, van der Heijde D, et al. Prediction of radiographic progression in rheumatoid arthritis and the role of antibodies against mutated citrullinated vimentin: results from a 10-year prospective study. Ann Rheum Dis. 2010;69(2):345–351. [DOI] [PubMed] [Google Scholar]
- [25].Mathsson L, Mullazehi M, Wick MC, et al. Antibodies against citrullinated vimentin in rheumatoid arthritis: higher sensitivity and extended prognostic value concerning future radiographic progression as compared with antibodies against cyclic citrullinated peptides. Arthritis Rheum. 2008;58(1):36–45. [DOI] [PubMed] [Google Scholar]
- [26].Bang H, Egerer K, Gauliard A, et al. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum. 2007;56(8):2503–2511. [DOI] [PubMed] [Google Scholar]
- [27].Feitsma AL, van der Voort EI, Franken KL, et al. Identification of citrullinated vimentin peptides as T cell epitopes in HLA-DR4-positive patients with rheumatoid arthritis. Arthritis Rheum. 2010;62(1):117–125. [DOI] [PubMed] [Google Scholar]
- [28].Snir O, Rieck M, Gebe JA, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. 2011;63(10):2873–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345–357. [DOI] [PubMed] [Google Scholar]
- [30].Kaniuk NA, Kiraly M, Bates H, et al. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes. 2007;56(4):930–939. [DOI] [PubMed] [Google Scholar]
- [31].Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619. [DOI] [PubMed] [Google Scholar]
- [32].Pierdominici M, Vomero M, Barbati C, et al. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. Faseb J. 2012;26(4):1400–1412. [DOI] [PubMed] [Google Scholar]
- [33].Lees CW, Barrett JC, Parkes M, et al. New IBD genetics: common pathways with other diseases. Gut. 2011;60(12):1739–1753. [DOI] [PubMed] [Google Scholar]
- [34].White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Valecka J, Almeida CR, Su B, et al. Autophagy and MHC-restricted antigen presentation. Mol Immunol. 2018;99:163–170. [DOI] [PubMed] [Google Scholar]
- [36].Staskiewicz L, Thorburn J, Morgan MJ, et al. Inhibiting autophagy by shRNA knockdown: cautions and recommendations. Autophagy. 2013;9(10):1449–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lundberg K, Kinloch A, Fisher BA, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58(10):3009–3019. [DOI] [PubMed] [Google Scholar]
- [39].McGonagle D, Gibbon W, Emery P. Classification of inflammatory arthritis by enthesitis. Lancet. 1998;352(9134):1137–1140. [DOI] [PubMed] [Google Scholar]
- [40].Spadaro A, Riccieri V, Scrivo R, et al. Anti-cyclic citrullinated peptide antibody determination in synovial fluid of psoriatic arthritis. Clin Exp Rheumatol. 2007;25(4):599–604. [PubMed] [Google Scholar]
- [41].Dalmády S, Kiss M, Képíró L, et al. Higher levels of autoantibodies targeting mutated citrullinated vimentin in patients with psoriatic arthritis than in patients with psoriasis vulgaris. Clin Dev Immunol. 2013;2013:474028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Korendowych E, Owen P, Ravindran J, et al. The clinical and genetic associations of anti-cyclic citrullinated peptide antibodies in psoriatic arthritis. Rheumatology. 2005;44(8):1056–1060. [DOI] [PubMed] [Google Scholar]
- [43].Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2(2):116–126. [DOI] [PubMed] [Google Scholar]
- [44].Chapoval AI, Ni J, Lau JS, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2(3):269–274. [DOI] [PubMed] [Google Scholar]
- [45].Castriconi R, Dondero A, Augugliaro R, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci U S A. 2004;101(34):12640–12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bardhan K, Anagnostou T, Boussiotis VA. The PD1: PD-L1/2Pathway from discovery to clinical implementation. Front Immunol. 2016;7:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Raptopoulou AP, Bertsias G, Makrygiannakis D, et al. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. 2010;62(7):1870–1880. [DOI] [PubMed] [Google Scholar]
- [48].Guo G, Shang Y, Zhu G, et al. The expression and distribution of immunomodulatory proteins B7-H1, B7-DC, B7-H3, and B7-H4 in rheumatoid synovium. Clin Rheumatol. 2012;31(2):271–281. [DOI] [PubMed] [Google Scholar]
- [49].Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26(1):79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kono M, Yasuda S, Stevens RL, et al. Ras guanine nucleotide-releasing protein 4 is aberrantly expressed in the fibroblast-like synoviocytes of patients with rheumatoid arthritis and controls their proliferation. Arthritis Rheumatol. 2015;67(2):396–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.