


Abstract
In Escherichia coli, the endoribonuclease E (RNase E) can recruit several other ribonucleases and regulatory proteins via its noncatalytic domain to form an RNA degradosome that controls cellular RNA turnover. Similar RNA degradation complexes have been found in other bacteria; however, their compositions are varied among different bacterial species. In cyanobacteria, only the exoribonuclease PNPase was shown to bind to the noncatalytic domain of RNase E. Here, we showed that Alr1240, a member of the RNB family of exoribonucleases, could be co-isolated with RNase E from the lysate of the cyanobacterium Anabaena PCC 7120. Enzymatic analysis revealed that Alr1240 is an exoribonuclease II (RNase II), as it only degrades non-structured single-stranded RNA substrates. In contrast to known RNase E-interacting ribonucleases, which bind to the noncatalytic domain of RNase E, the Anabaena RNase II was shown to associate with the catalytic domain of RNase E. Using a strain in which RNase E and RNase II were tagged in situ with GFP and BFP, respectively, we showed that RNase E and RNase II form a compact complex in vivo by a fluorescence resonance energy transfer (FRET) assay. RNase E activity on several synthetic substrates was boosted in the presence of RNase II, suggesting that the activity of RNase E could be regulated by RNase II-RNase E interaction. To our knowledge, Anabaena RNase II is an unusual ribonuclease that interacts with the catalytic domain of RNase E, and it may represent a new type of RNA degradosome and a novel mechanism for regulating the activity of the RNA degradosome. As Anabaena RNase E interacts with RNase II and PNPase via different regions, it is very likely that the three ribonucleases form a large complex and cooperatively regulate RNA metabolism in the cell.
INTRODUCTION
In many bacteria, the initial cleavage of RNA degradation is mediated by a key endoribonuclease, RNase E (1–3). In the model organism Escherichia coli, RNase E consists of two functionally distinct domains, the N-terminal highly conserved catalytic domain and the C-terminal unstructured noncatalytic domain. Through its noncatalytic domain, RNase E functions as a molecular hub to recruit several interacting proteins and forms a multi-enzyme assembly known as the RNA degradosome (4).
The RNA degradosome controls the main pathway of mRNA degradation in E. coli (5–8); it also functions in the maturation of ribosomal RNAs and tRNAs (9,10). Recently, the RNA degradosome was found to participate in the degradation of hypomodified tRNAs in Vibrio cholerae, and thus could serve as a previously unrecognized bacterial tRNA quality control system (11). The major components of the RNA degradosome in E. coli are well characterized, and include the DEAD box RNA helicase RhlB, the exoribonuclease polynucleotide phosphorylase (PNPase) and the glycolytic enzyme, enolase (4,12–14). RNA degradation mediated by the RNA degradosome is a highly cooperative and efficient process, where the RNA helicase RhlB unwinds structured RNAs, the endoribonuclease RNase E cuts the substrates preferably at AU-rich sites, and the 3′-5′ exoribonuclease PNPase ultimately degrades the unwound or breakdown substrates into mononucleotides (6,15).
RNase E homologues have been found in various bacterial phyla, including Proteobacteria, Actinobacteria, Bacteroidetes, Chlamydiae, Cyanobacteria and Firmicutes, as well as in plant plastids (16). RNase E-based RNA degradosomes have been experimentally characterized in a few species, mostly in the Proteobacteria (16). These degradosomes are diverse in terms of their composition. For example, the RNA degradosome in Caulobacter crescentus is composed of RNase E, PNPase, the 3′-5′ exoribonuclease RNase D, a DEAD-box RNA helicase, and the Krebs cycle enzyme aconitase, whereas the RNA degradosome in the psychrotrophic bacterium, Pseudomonas syringae Lz4W contains RNase E, the 3′-5′ exoribonuclease RNase R and the DEAD-box helicase, RhlE (17–19). Machineries responsible for RNA degradation also exist in bacteria lacking RNase E. For example, in the Gram-positive bacterium, Bacillus subtilis, the endoribonuclease RNase Y recruits the exoribonucleases RNase J1/J2 (RNase J1 is also an endoribonuclease), the RNA helicase CshA, enolase and phosphofructokinase to form an RNA degradation complex that is compositionally different, but functionally equivalent to the E. coli RNA degradosome (20–22). In eukaryotes and archaea, an essential and conserved 3′-5′ exoribonuclease complex, called the RNA exosome, degrades or processes nearly every class of cellular RNA (23–26). Therefore, it is very likely that RNA degradosomes or similar assemblies are ubiquitous. However, how the different ribonucleases within these assemblies cooperate to degrade RNA remains largely unknown.
In addition to those ribonucleases found in the degradosome, other ribonucleases also play important roles in RNA turnover. For instance, RNase II, encoded by the rnb gene, was shown to be responsible for 90% of the hydrolytic activity in E. coli crude extracts (27). Escherichis coli RNase II (hereafter EcRnb) belongs to the RNB exoribonuclease family, whose members are present in all domains of life, and it efficiently hydrolyzes single-stranded RNAs from 3′ to 5′. RNase II has three functionally distinct regions: an N-terminal segment containing two cold-shock domains (CSD) involved in RNA binding, a C-terminal segment containing an S1 domain also involved in RNA binding, and a central RNB domain responsible for the catalytic activity (28,29). In addition to RNase II, E. coli genome encodes another RNB family exoribonuclease, RNase R (hereafter EcRnr). RNase R shares many structural and catalytic properties with RNase II; the key difference between the two enzymes is that RNase II only degrades RNAs without secondary structure, while RNase R is capable of degrading structured RNA, provided that a single-stranded 3′ end is initially available for binding (30–32).
Cyanobacteria is evolutionarily close to the chloroplasts of higher plants (33,34). Little is known about RNA metabolism in cyanobacteria. A single copy of an RNase E-encoding gene is present in the genome of each sequenced cyanobacterial strain (35). Cyanobacterial RNase E proteins share a domain architecture similar to that of E. coli RNase E, consisting of an N-terminal catalytic domain and a C-terminal noncatalytic domain. Although the noncatalytic region of cyanobacterial RNase E is much shorter than that of E. coli, it is also an intrinsically disordered region that might serve as the scaffold for a degradosome-like complex similar to E. coli RNase E. Indeed, we previously reported in Anabaena PCC 7120 that RNase E formed a complex with PNPase via a cyanobacterium-specific nonapeptide in the noncatalytic region (35). The remaining portion of the noncatalytic region in Anabaena RNase E could recruit other components. In this study, we attempted to identify such components by in vivo co-purification using the RNase E protein fused to an affinity purification tag. We successfully co-isolated a 3′-5′ exoribonuclease, RNase II, with RNase E from cell lysate. Surprisingly, we found that RNase II bound to the catalytic domain of RNase E. We then comprehensively characterized the interaction between RNase E and RNase II both in vitro and in vivo, and showed that their interaction could influence the catalytic activity of RNase E.
MATERIALS AND METHODS
Cyanobacterial strains and culture conditions
Anabaena strains were cultured in liquid BG11 medium under continuous illumination (30 μmol m−2 s−1) at 30°C in an incubator with orbital shaking at 150 rpm. When required, neomycin at a final concentration of 150 μg/ml, spectinomycin at a final concentration of 5 μg/ml, and streptomycin at a final concentration of 2.5 μg/ml, were added to the medium.
Plasmid construction
The plasmids used in this study are listed and described in Supplemental Table 1. The oligonucleotides used to construct the plasmids are listed in Supplemental Table 2. All constructed plasmids were verified by sequencing.
Construction of protein expression plasmids
The pET28a (Invitrogen)-derived vector pHSTag (GenBank accession number: MK948096) was used to express recombinant protein with a N-terminal His-tag and/or a C-terminal Strep-tag II (WSHPQFEK). The pHSTag was generated by inserting Strep-tag II-encoding duplex consisting of annealed complementary oligonucleotides of PCStagF into pET28a via the XhoI site. To construct plasmid for expressing recombinant protein AnaRnb-S (pHSAlr1240), the genomic region encoding Alr1240 was amplified with the primer pair PHSalr1240F1/PHSalr1240R2329 and cloned into pHSTag at the NdeI and XhoI sites. Other plasmids for the expression of recombinant proteins were similarly constructed. The plasmids for expressing the mutant proteins EcRnbR500A and AnaRnbR558A (pHSEcRnbR500A and pHSAnaRnbR558A, respectively) were constructed from pHSEcRnb and pHSAnaRnb by site-directed mutagenesis with the primer pairs PEcrnbR500AF/PEcrnbR500AR and Palr1240R558AF/Palr1240R558AR, respectively. Plasmids expressing His-AnaRne, His-AnaRneN, and His-AnaRneC were described previously (35).
Construction of bacterial two-hybrid plasmids
The bacterial adenylate cyclase two-hybrid plasmids were constructed using the vectors pKT25 and pUT18C (36). For example, to make pKT25a-Alr1240 and pUT18Ca-Alr1240, the coding sequence of Alr1240 was amplified from the genome using primers Palr1240F1/Palr1240R2330, and then cloned into a linear vector fragment amplified from pKT25 using primers PKT25_F/PKT25_R and the fragment amplified from pUT18C using primers PUT18C_F/PUT18C_R via seamless cloning (Vazyme ClonExpress MultiS One Step Cloning Kit), to generate pKT25a-Alr1240 and pUT18Ca-Alr1240, respectively. The other two-hybrid plasmids were constructed in the same way.
Construction of plasmids for in vivo co-purification and fluorescent protein tagging
In the co-purification experiment, we used the inducible plasmid pCT-AnaRne, which is derived from pCT (GenBank accession number: MK948095), to express TwinStrep-tagged AnaRne. The vector pCT allows for expression of a gene in Anabaena under the control of the copper-inducible promoter PpetE and a theophylline-responsive riboswitch. To construct pCT-AnaRne, the ORF encoding AnaRne (gene id: alr4331) was amplified from the genome using primers Palr4331F5h and Palr4331R2061h and was inserted into SmaI-linearized pCT via seamless cloning (Vazyme ClonExpress MultiS One Step Cloning Kit). The plasmids pAlr1240GFP, pAlr1240BFP and pAlr4331GFP were used to construct fluorescent protein-labeled strains. To construct pAlr1240GFP, we first linearized the vector pRL271 with PstI and XhoI. Then, we excised the GFP-encoding fragment and the spectinomycin resistance gene from the synthetic plasmid pSfgfp-Sp (GenBank accession number: MK948098) using BamHI and SmaI. Subsequently, two alr1240 regions were amplified from the Anabaena chromosome using the primer pairs Palr1240F1471/Palr1240R2349 and Palr1240F2358/Palr1240R3240. Finally, the four fragments were assembled into pAlr1240GFP via seamless cloning (Vazyme ClonExpress MultiS One Step Cloning Kit). To construct pAlr1240BFP, the vector backbone together with the alr1240 fragments amplified from pAlr1240GFP using primers Palr1240BFP_F and Palr1240BFP_R, and the BFP-coding sequence and kanamycin resistance gene were amplified from the synthetic plasmid pMTagBFP2-npt (GenBank accession number: MK948097) using primers PV_16 and Pinsert2 and assembled via seamless cloning (Vazyme ClonExpress MultiS One Step Cloning Kit). The procedure used to construct pAlr4331GFP was same as that for pAlr1240GFP, except that the alr4331 regions were amplified with the primer pairs Palr4331F104/Palr4331R2061n and Palr4331F2065n/Palr4331R2986.
Construction of cyanobacterial strains
The cyanobacterial strains used in this study were constructed by transferring the relevant plasmids into the wild-type strain by conjugation, using a previously described procedure (37,38). The exconjugants were screened on BG11 plates containing 150 μg/ml neomycin when the plasmid carried a neomycin resistance gene, or 5 μg/mlL spectinomycin and 2.5 μg/ml streptomycin when it carried a spectinomycin/streptomycin resistance gene. The genotypes of all obtained strains were verified by PCR.
The strain used for co-purifying the proteins associated with RNase E from Anabaena was generated by transferring the plasmid pCT-AnaRne into the wild-type strain. The fluorescent protein-labeled strain containing AnaRnb-GFP was made by introducing the pAlr1240GFP plasmid into the wild-type strain. To construct the dual-labeled strain containing AnaRnb-BFP and AnaRne-GFP, pAlr4331GFP and pAlr1240BFP were sequentially introduced into the wild-type strain. The green fluorescent protein GFP has the major absorption spectrum of ∼440–508 nm (peaked at 488 nm) and the major emission spectrum of ∼493–532 nm (peaked at 510 nm); the blue fluorescent protein BFP has the major absorption spectrum of ∼370–425 nm (peaked at 402 nm) and the major emission spectrum of ∼439–498 nm (peaked at 457 nm). As the emission spectrum of BFP overlaps well with the excitation spectrum of GFP, the pair of fluorescent fusion proteins, AnaRnb-BFP and AnaRne-GFP, were used to study in vivo interaction/co-localization of AnaRnb and AnaRne by fluorescence resonance energy transfer (FRET).
Expression and purification of recombinant proteins
The proteins used in this study are listed and described in Supplemental Table 3. Escherichia coli BL21(DE3) cells transformed with the protein expression plasmids were grown in LB medium supplemented with 50 μg/ml kanamycin and 0.8% glucose at 37°C. When the cells reached an optical density at 600 nm (OD600) of 0.5, 0.5 mM IPTG was added to the culture. After 4 h of induction, the cells were pelleted by centrifugation and then resuspended in PBS buffer for lysis. After lysing with a high-pressure homogenizer, the cell lysate was loaded into a suitable affinity purification column. His-tag fusion proteins were purified using a Ni-NTA affinity column (Qiagen), and Strep-tag fusion proteins were purified using a Strep-Tactin XT affinity column (IBA). The purified proteins were dialyzed into protein storage buffer (20 mM Tris–HCl, 20 mM KCl, 100 mM NaCl and 25% glycerol, pH 7.5) and stored at −80°C.
Co-isolation of the proteins associated with RNase E in Anabaena cells
The procedure for the co-purification experiment was similar to that for the affinity purification of proteins associated with His-tagged AnaRneC, the noncatalytic region of AnaRne, from Anabaena (35). The strain containing pCT-AnaRne was used to overexpress the TwinStrep-tagged RNase E by adding the inducers of copper and theophylline. In a pilot experiment, we induced the expression of TwinStrep-tagged RNase E with different concentrations of the inducers for different time periods, and observed that when the concentration of the inducers were too high or the induction time was too long, Anabaena filaments would become severely fragmented and cells would die eventually (data not shown), indicating that high cellular level of RNase E is cytotoxic. To minimize the effect of overexpression on cell physiology, we here chose an induction condition under which the strain did not differ much from WT in growth (growing ∼10% slower than WT, no filament fragmentation). The strain containing pCT-AnaRne was grown in liquid BG11 medium to an OD750 of 0.5, then 1 μM CuSO4 and 2 mM theophylline were added to the medium. After 24 h of induction, AnaRne-TwinStrep was moderately induced (the induction level was examined by western blotting using an antibody against the Strep tag), and then the cells were collected by centrifugation and homogenized in lysis buffer (100 mM Tris–HCl, 150 mM NaCl, 0.1% Triton-X 100, 1× protease inhibitor cocktail, 50 μg/ml RNase A and 5 mM EDTA, pH 8.0) with FastPrep-24 (MP Biomedicals). After centrifugation at 14 000 g for 15 min at 4°C, the supernatant was transferred to a tube containing Strep-Tactin XT beads and incubated on a horizontal shaker at 4°C for 3 h. The beads were harvested by centrifugation and washed twice with buffer W (100 mM Tris–HCl, 150 mM NaCl, 0.1% Triton-X 100 and 5 mM EDTA, pH 8.0). The bound proteins were eluted into 500 μl of elution buffer (100 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 50 mM d-biotin, and 25% glycerol, pH 8.0). The proteins co-purified with AnaRne were then subjected to western blot analysis. A strain harboring the empty pCT vector was used as a negative control and was processed using the same procedure.
Far-western blotting
Far-western blotting was performed as previously described (39), with minor modifications. In the far-western blot, 5 ng of Strep-tagged Alr1240 (as the positive control) and 2 μg each of AnaRne, AnaRneN, and AnaRneC were separated in parallel on two 13% SDS-PAGE gels and electrotransferred onto two PVDF membranes. Then, the membranes were blocked with 5% skimmed milk in PBS-T (PBS containing 0.1% Tween-20) for 1 h, and then incubated in PBS-T containing 1% skim milk for 1.5 h. One membrane was incubated with 0.2 mg of Strep-tagged Alr1240 and 100 μg RNase A (to exclude the effect of RNA on the interaction), while the other was not. Subsequently, the membranes were sequentially incubated with anti-Strep tag polyclonal antibodies (1:1000) for 1 h and with HRP-conjugated goat-anti-rabbit IgG antibodies (1:5000), as in conventional western blot protocols. The immunoblotting signals were developed using ECL reagent.
Bacterial two-hybrid assay
The bacterial two-hybrid system, which is based on reconstitution of the Bordetella pertussis adenylate cyclase CyaA (36), was used to evaluate the interaction between Anabaena RNase E and RNase II. This two-hybrid system exploits the two complementary fragments of the catalytic domain of CyaA, T25 and T18, that are not active when physically separated. To test the interaction between two proteins of interest, say A and B, the fusions of T25-A and T18-B (or T18-A and T25-B) can be co-expressed in an adenylate cyclase deficient (cya−) strain of E. coli. The interaction between A and B brings T25 and T18 together so that CyaA is reconstituted and become active in cAMP synthesis. The synthesized cAMP further activates the catabolite activator protein CAP to turn on the expression of several metabolic enzymes, including β-galactosidase. Thus, on the medium containing, X-gal, the chromogenic substrate for β-galactosidase, the interaction between A and B will be indicated by the blue color of the colonies. In this study, the pKT25- and pUT18C-derived plasmids, which respectively express the T25 and T18 fusions of the proteins of interest (i.e. the full-length or sub-regions of AnaRne and AnaRnb), were co-transformed into the cya− strain BTH101. The transformants were checked after grown in the dark at 30°C for 1–3 days on LB agar plates containing 50 μg/ml kanamycin, 100 μg/ml ampicillin, 0.5 mM IPTG, and 40 μg/ml X-gal. A strain co-transformed with pKT25-zip and pUT18C-zip was used as the positive control, and a strain co-transformed with the empty vectors pKT25a and pUT18Ca was used as the negative control.
ITC assay
Isothermal titration calorimetry (ITC) assays were performed at 25°C in a Nano ITC Low Volume isothermal calorimeter (TA Instruments, New Castle, DE, USA). Prior to the assay, all proteins were dialyzed into the same buffer (100 mM Tris–HCl and 10% glycerol, pH 8.0) and then centrifuged and degassed. Twenty-five 2-μl aliquots of Alr1240 (250 μM) were titrated into 190 μl of AnaRneN (25 μM) at 200-s intervals and a stirring rate of 250 rpm. The measured heat data, after subtracting the dilution heat obtained by titrating buffer into AnaRneN, was analyzed using the NanoAnalyze Software provided by the manufacturer. The titration curves were fitted to the independent-site binding model. Images were generated by R ggplot2.
Ribonuclease activity assay
Ribonucleolytic activity was assayed using synthetic RNA substrates. Two 5′ FAM-labeled single-stranded RNA substrates of 16-mer (5′-FAM-CCCGACACCAACCACU-3′) and 30-mer (5′-FAM-CCCGACACCAACCACUAAAAAAAAAAAAAA-3′) were synthesized first (31,40). They were then hybridized to the complementary unlabeled 16-mer oligodeoxyribonucleotide (5′-AGTGGTTGGTGTCGGG-3′) to obtain the double-stranded substrates 30–16ds and 16–16ds, respectively. These single- and double-stranded substrates were used to assess the activities of the exoribonucleases. The activity of the endonuclease RNase E was determined using three 5′-monophosphorylated and 3′-FAM-labelled RNA substrates: LU13 (5′-p-GAGACAGUAUUUG-FAM-3′), CRISPR3 (5′-p-GUCUCCACUCGU↓AGGAGAAAUUAAUUGAUUGGAAAC-FAM-3′), and RNA62 (5′-p-GGUUAUAAAUCAACACAUUGAUUUAUAAGGUCAAAUAAUAAAGCCCGUCUCCUACGAUGGGC-FAM-3′) (41–43). All the oligoribonucleotides were synthesized by GenScript.
The ribonucleolytic reactions were performed in a final volume of 10 μl containing 20 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 100 mM NaCl, 5% glycerol, 0.1% Triton X-100 and 0.1 mM DTT. The reactions were started by the addition of enzyme. After incubation at 30°C for the indicated time periods, the reactions were stopped by adding RNA loading Dye (New England Biolabs B0363S) and denatured for 5 min at 95°C prior to electrophoresis. The degradation products were resolved in 15% or 20% polyacrylamide gels containing 8 M urea and photographed with a FUJIFILM FLA-5100 fluorescent image analyzer.
FRET assay
The FRET experiment to analyze the interaction between AnaRnb-BFP and AnaRne-GFP was carried out with a fv1000mp two-photon laser scanning confocal microscope (OLYMPUS). AnaRnb-BFP was excited by light at 405 nm, AnaRne-GFP was excited by light at 488 nm. FRET between the donor (AnaRnb-BFP) and the acceptor (AnaRne-GFP) was measured using the acceptor photobleaching method. Fluorescence intensity of the donor was measured before and after photobleaching the acceptor fluorescence, and the FRET efficiency (E) was calculated as follows: E = 1 – (prebleaching/postbleaching). The distance (r) between the donor and acceptor was calculated as follows: r = Ro[(1/E) – 1] 1/6, where Ro is the Forster distance of BFP-GFP (4.14 nm).
RESULTS
Alr1240 binds to the catalytic domain of Anabaena RNase E
We previously reported that RNase E forms a complex with PNPase in cyanobacteria via a C-terminal motif in the noncatalytic region (35). To determine if RNase E recruits other ribonucleases to form a large RNA degradosome in cyanobacteria, we attempted to isolate RNase E-associating proteins via co-purification. We constructed an Anabaena RNase E-overexpressing strain, which expressed the RNase E protein with a TwinStrep tag fused at the C-terminus from a replicative plasmid. The TwinStrep-tagged RNase E, together with the proteins that bound to it, was then isolated from the cell lysate of this strain with a streptavidin column. Subsequently, the co-isolated proteins were checked for the presence of other ribonucleases by western blotting using the available antibodies in our laboratory. The results showed that one ribonuclease II/R family protein, Alr1240, was co-isolated with RNase E (Figure 1A).
Figure 1.
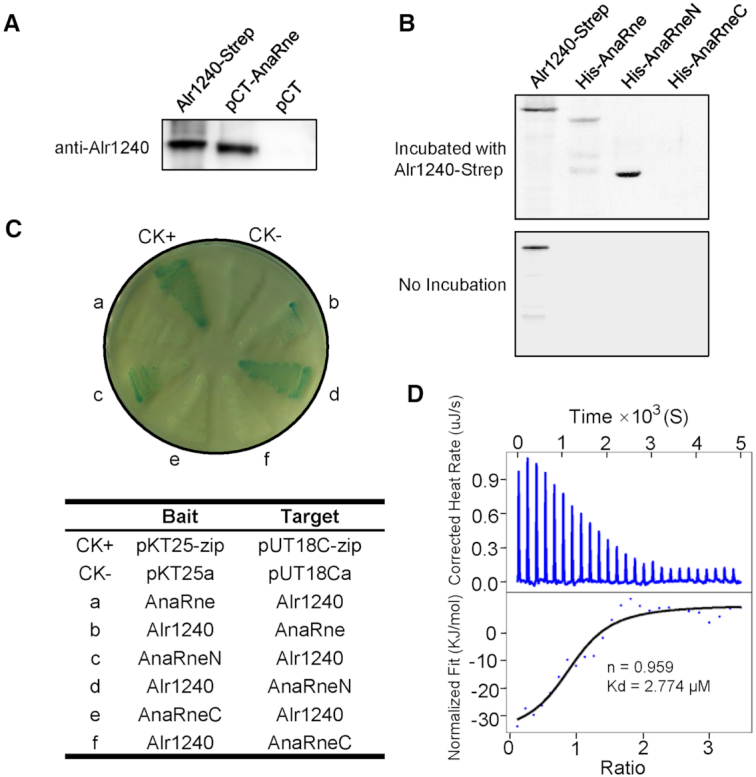
Identifying the interaction between Alr1240 and AnaRne. (A) Alr1240 was co-purified with TwinStrep-tagged AnaRne expressed in Anabaena cells using Strep-Tactin Sepharose. The eluted protein samples from cells carrying the TwinStrep-tagged AnaRne-expressing plasmid (pCT-AnaRne) and the empty vector (pCT) were evaluated using antibodies against Alr1240. The recombinant protein Alr1240-Strep (Alr1240 with a C-terminal Strep tag fusion) served as the positive control. (B) Far-western blot assay showing that Alr1240 interacts with the catalytic domain of AnaRne. Duplicate samples of the full-length, N-terminal half, and C-terminal half of the AnaRne protein, together with recombinant Alr1240-Strep (the positive control), were separated on 10% SDS PAGE gels and transferred onto nitrocellulose membranes. One membrane was incubated with Alr1240-Strep, and the other was not. Subsequently, the signals on both membranes were developed with an antibody against the Strep tag (see Materials and methods for details). (C) Investigating the interaction between Alr1240 and AnaRne by the bacterial adenylate cyclase two-hybrid assay. Escherichia coli BTH101 cells were co-transformed with the indicated two-hybrid plasmid pairs and incubated on plates containing X-gal, IPTG, and the appropriate antibiotics (Kan and Amp) at 30°C for 3 days. Cells co-transformed with pKT25-zip and pUT18C-zip were used as the positive control (CK+), and cells co-transformed with the empty vectors pKT25a and pUT18Ca were used as the negative control (CK–). (D) ITC assay evaluating the interaction between Alr1240 and AnaRneN. The original titration data and integrated heat measurements are shown in the upper and lower plots, respectively.
To understand whether RNase E and Alr1240 form a complex directly, we checked their interaction by two different methods, far-western blotting and bacterial two-hybrid assay. Anabaena RNase E (AnaRne) consists of an N-terminal catalytic domain (AnaRneN) and a C-terminal noncatalytic domain (AnaRneC) (35). In the far-western blotting assay, we examined the interactions between Alr1240 and full-length AnaRne or different truncations of AnaRne (AnaRneN and AnaRneC) using recombinant proteins purified from E. coli. The results showed that both AnaRne and AnaRneN were able to interact with Alr1240, while AnaRneC was not (Figure 1B).
In the bacterial two-hybrid assay, we confirmed that AnaRneN could interact with Alr1240 but AnaRneC could not (Figure 1C). It should be noted that when AnaRne was used in the assay, the interaction signal became much weaker (a and b in Figure 1C). One reason could be that the full-length RNase E had much lower expression in the host strain probably due to stronger cellular toxicity, consequently resulting in weaker interaction signal (we also found that when AnaRne and AnaRneN were individually expressed in E. coli from the same vector for protein purification, AnaRne always had much lower yield than AnaRneN). Nevertheless, combining the far-western blotting result and the bacterial two-hybrid assay result, we can conclude that Alr1240 interacts with AnaRne by binding to its catalytic domain. This is in contrast to PNPase, which interacts with the noncatalytic domain of AnaRne (35).
We further measured the binding stoichiometry and strength of the interaction by an isothermal titration calorimetry (ITC) assay. A titration curve was obtained by titrating drops of AnaRneN into a solution of Alr1240. From this curve, we calculated that the binding stoichiometry between the two proteins was 1:1, and the dissociation constant (Kd) was 2.774 μM (Figure 1D).
The C-terminal and N-terminal extensions of Alr1240 interact with AnaRneN
The primary sequence of Alr1240 is close to the E. coli RNB family proteins EcRnr and EcRnb (see the sequence alignment in Supplementary Figure S1). BLASTP analysis showed that Alr1240 matched EcRnr with 49% similarities and 27% identities over 745 aa, and it matched EcRnb with 41% similarities and 20% identities over 576 aa. Domain analysis further showed that Alr1240 has an architecture typical of RNB family proteins, which features two cold-shock domains (CSD) within the N-terminus, an exoribonuclease domain (RNB) in the center, and an S1 domain within the C-terminus (Figure 2A and Supplementary Figure S1). Using the crystal structure of EcRnr (PDB ID: 5XGU) as the template, we were also able to build a good-quality 3D model for Alr1240 using SWISS-MODEL (Supplementary Figure S2). These analyses indicate that Alr1240 is a member of the RNB family proteins.
Figure 2.
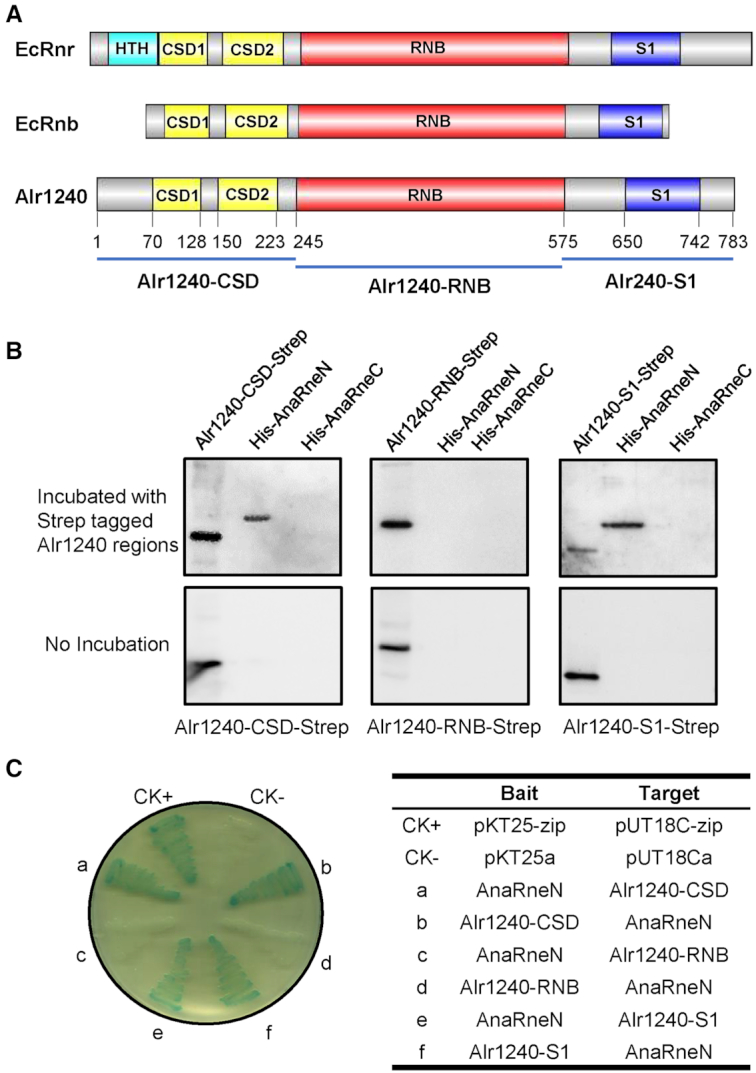
Determining the Alr1240 regions responsible for the interaction between AnaRne and Alr1240. (A) Schematic diagram of the domains in E. coli RNase R (EcRnr), E. coli RNase II (EcRnb) and Alr1240. All these proteins contain two cold shock domains (CSD), one exoribonuclease domain (RNB) and one S1 RNA-binding domain (S1). EcRnr also contains one helix-turn-helix domain (HTH) at the N-terminus. Based on the domain architecture, three regions of Alr1240 (Alr1240-CSD, Alr1240-RNB and Alr1240-S1) were chosen for the interaction assays. (B) Far-western blot assay assessing the interactions between AnaRneN/AnaRneC and different regions of Alr1240. The experiment procedure was same as that in Figure 1B. (C) Bacterial adenylate cyclase two-hybrid assay investigating the interactions between AnaRneN and different regions of Alr1240. The experiment procedure was same as that in Figure 1C.
We then identified the sub-regions in Alr1240 that are responsible for interaction with AnaRneN by far-western blotting. The protein sequence of Alr1240 was split into three parts: the N-terminal region that contains two CSD domains (Alr1240-CSD), the central catalytic RNB domain (Alr1240-RNB), and the C-terminal region that contains the S1 domain (Alr1240-S1) (Figure 2A). Far-western blotting data showed that the two noncatalytic regions, Alr1240-CSD and Alr1240-S1, could interact with AnaRneN, while the catalytic region (Alr1240-RNB) could not (Figure 2B). This result was further confirmed by a bacterial two-hybrid assay (Figure 2C). We additionally tested the specificity of such interaction by checking the interaction between AnaRneN and the S1 domains from EcRnb, EcRnr and E. coli translation initiation factor IF-1 (EcIF1) with a bacterial two-hybrid assay, and found that AnaRneN could interact with none of them (Supplementary Figure S3). Based on these results, we concluded that the noncatalytic domains of Alr1240 are responsible for its interaction with AnaRneN.
Alr1240 is an RNB family RNase II
Members of RNB family include the well-characterized E. coli exoribonucleases RNase II and RNase R. Both RNase II and RNase R catalyze 3′ to 5′ RNA degradation. A major difference between these two exoribonucleases is that RNase R can degrade structured RNA substrates, while RNase II cannot (28,32). As to our knowledge, the enzymatic properties of Alr1240 have not been investigated, we tested the activity of Alr1240 on several synthetic oligoribonucleotides that have been previously used as substrates for other exoribonucleases (31,40). Towards this, 16-mer and 30-mer RNAs with an amine derivative of fluorescein (FAM fluorophore) fused at their 5′ terminus, were used in combination with a complementary 16-mer DNA to generate the following four RNA substrates: 16ss, 30ss, 16-16ds and 30-16ds. Using these substrates, the exoribonucleolytic activities of Alr1240, E. coli RNase II and E. coli RNase R were characterized and compared.
As expected, all three RNB-family proteins efficiently degraded the 16ss and 30ss substrates (Figure 3A and B), and none of them digested the blunt-ended double stranded substrate 16-16ds (Figure 3C). However, the three proteins showed varied activities with respect to the 30-16ds substrate. EcRnb and Alr1240 rapidly degraded the 3′-single stranded region and generated a final product of ∼20 nt in length, indicating that the enzymes were stalled by the double-stranded region (Figure 3D). In contrast, EcRnr degraded the entire RNA chain and produced short oligoribonucleotides as products, supporting the requirement of a 3′-single-stranded extension for degradation of structured substrates by RNase R (30,31). The results of the enzymatic assay indicate that alr1240 encodes an RNase II rather than an RNase R, although the Alr1240 is more similar to EcRnr in primary sequence. Hence, we named Alr1240 AnaRnb (Anabaena RNase II, to distinguish it from its E. coli counterpart EcRnb).
Figure 3.
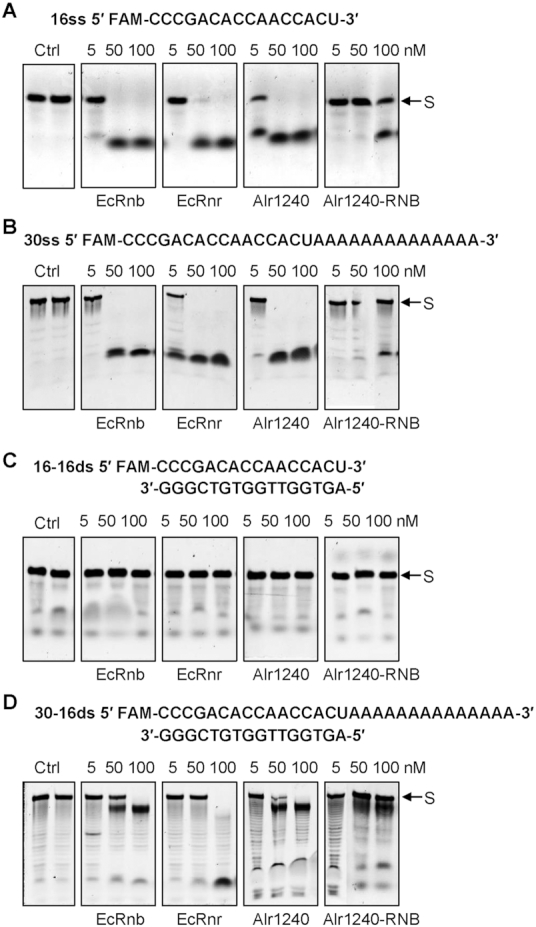
Comparing the catalytic property of Alr1240 with those of the Escherichia coli exoribonucleases RNase II (EcRnb) and RNase R (EcRnr). Activity assays were performed using 16ss (A), 30ss (B), 16-mer and its complement (16-16ds) (C) or a 30-mer hybridized to the complement 16-mer (30-16ds) (D) as substrates at 30°C (see Materials and methods for details). In each reaction, the same amount of RNA substrate (50 nM) and different amounts of purified proteins (shown at the top of each lane) were used. The 16-mer and 30-mer RNA strands were labeled the 5′-end with the FAM fluorescein-based dye. The control reactions, which contained only the RNA substrates (Ctrl) and the reaction buffer, were incubated for either 0 min (left lane) or 30 min (right lane), while all the other reactions were incubated for 30 min. The FAM-labeled substrate strands are indicated by arrows (S).
To illustrate the contribution of the noncatalytic regions of AnaRnb to its catalytic activity, the catalytic region (Alr1240-RNB) was evaluated in an exoribonucleolytic activity assay using the 16ss, 30ss, 16-16ds and 30-16ds substrates. The results showed that Alr1240-RNB had exonuclease activity similar to—but lower than—that of full-length Alr1240 (Figure 3). We also tested the activities of the noncatalytic regions of Alr1240-CSD and Alr1240-S1, and as expected, they could not cleave the substrates (Supplementary Figure S4). This result is in accordance with previous studies that showed that the RNB domain is involved in substrate degradation and the CSD and S1 domains are involved in substrate binding (28,29).
In E. coli RNase II, several conserved residues within the RNB domain are essential for the catalytic activity, which include the residues for metal coordination (D201, D207, D209, D210) and those for substrate interaction (Y253, Y313, F358, E390, R500, E542) (28,44,45). Two of these residues—aspartic acid 209 (D209) and arginine 500 (R500)—are absolutely required for enzymatic activity, as mutating either of them (D209N and R500A) completely inactivates the enzyme (44–46). These two residues are also present in EcRnr (corresponding to D280 and R571, respectively), suggesting that they may be critical for the exoribonuclease activity of RNB-family proteins. However, sequence alignment showed that only one of the two residues is conserved in AnaRnb (R558, corresponding to R500 in EcRnb, Supplementary Figure S1). We then checked if R558 was required for the activity of AnaRnb. In contrast to the EcRnb R500A mutant, which shows no activity, the AnaRnb R558A mutant was still capable of degrading RNA substrates, although its activity was decreased (Supplementary Figure S5). We additionally checked the presence of the other key residues, and found that except for Y253 and E542, the other key residues in EcRnb are not conserved in AnaRnb (Supplementary Figure S1). These data together indicate that AnaRnb and EcRnb likely use a different set of key residues for catalytic activity.
RNase II and RNase E co-localize in Anabaena cytoplasm
Although AnaRne and AnaRnb were co-isolated from Anabaena cell lysate and their interaction was confirmed by in vitro assays, the compartment where AnaRne and AnaRnb co-localize, and the extent of interaction, in vivo was unknown. Here, we used an Anabaena strain that expresses RNase E tagged with GFP at the C-terminus (AnaRne-GFP) and RNase II tagged with BFP at the C-terminus (AnaRnb-BFP) in situ to characterize their interaction and subcellular localization. Although the cells of the dual-labelled strain appeared slightly longer than the WT strain (Figure 4A and B), its growth rate was similar to that of WT (data not shown), implying that fusing fluorescent proteins to RNase E and RNase II does not interfere with their functionality. Confocal microscopy showed that fluorescent signals for AnaRne-GFP and AnaRnb-BFP were both localized to the cytoplasm and were well overlapped (Figure 4A), indicating that the majority of AnaRne and AnaRnb are co-localized in vivo. It is worth noting that cytoplasmic localization of AnaRne is distinct from the membrane localization of EcRne (47,48).
Figure 4.
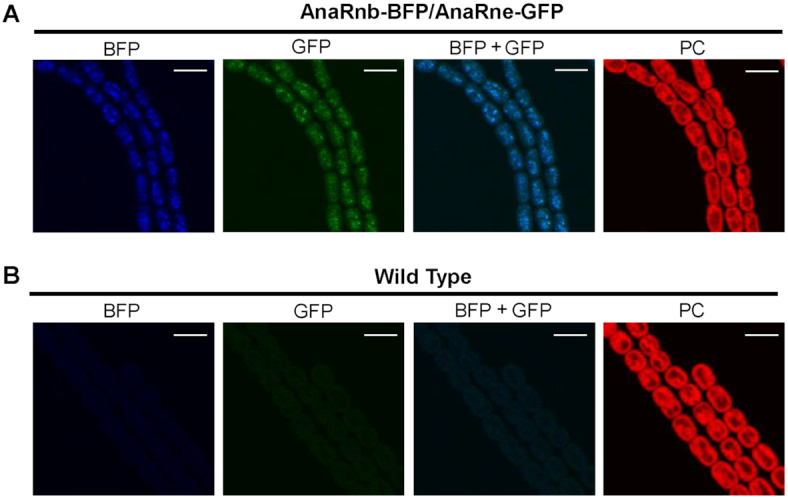
Subcellular localization of AnaRnb and AnaRne. The filaments of a dual-labeled strain with AnaRnb-BFP and AnaRne-GFP (A) and the filaments of the wild-type (WT) strain (B) were observed under a confocal microscope. The fluorescent signals in the same field were recorded in the channels for green fluorescence (GFP), blue fluorescence (BFP) and photosynthetic pigment fluorescence (PC). Merged images of blue and green fluorescence are also shown (GFP+BFP). Scale bar: 5 μm.
Using the dual-labeled strain, we observed the fluorescence resonance energy transfer (FRET) between AnaRne-GFP and AnaRnb-BFP using the sensitized emission method (Figure 5A). To do so, the fluorescence of filaments of the dual-labeled strain and a control strain that only expresses AnaRnb-GFP were observed in the BFP and GFP channels, respectively, using the excitation light of 405 nm (for BFP) or a combination of 405 nm (for BFP) and 488 nm (for GFP). Filaments of the control strain showed bright green fluorescence after excitation at 405/488 nm only. In contrast, the dual-labeled strain showed dim blue fluorescence and bright green fluorescence under both excitation conditions, indicating FRET between AnaRnb-BFP (the donor) and AnaRne-GFP (the acceptor).
Figure 5.

Examining the intracellular interaction between AnaRnb and AnaRne by FRET. (A) Checking the FRET between AnaRne-GFP and AnaRnb-BFP using the sensitized emission method. For the dual-labelled strain (left), GFP fluorescence could be observed at the BFP excitation wavelength (405 nm), indicating energy transfer from AnaRne-BFP to AnaRne-GFP; while for the GFP-labeled control strain (right), GFP fluorescence was emitted only at the GFP excitation wavelength (488 nm). Scale bar: 5 μm. (B) Measurement of the FRET efficiency between AnaRnb-BFP and AnaRne-GFP by the acceptor photobleaching method. The filaments of the dual-labelled cells were imaged in the GFP and BFP channels before and after the acceptor AnaRne-GFP was photobleached (left panel). Q and DQ are the fluorescence signals for the quenched and dequenched state of the donor protein. Pr and Po are the fluorescence signals for the acceptor before and after photobleaching. The middle and right panels are the color intensity maps showing the FRET efficiency and distance between donor and acceptor calculated from the ratio of the donor fluorescence signals measured before and after photobleaching of the acceptor. Scale bar: 10 μm.
We then quantified the FRET efficiency and distance between AnaRnb-BFP and AnaRne-GFP using the acceptor photobleaching method (Figure 5B). To accomplish this, the fluorescence of the cells needed to be imaged before and after the acceptor is photobleached by light at its excitation wavelength. Before being photobleached, the acceptor protein, AnaRne-GFP, received energy from the donor protein AnaRnb-BFP, resulting in weak donor fluorescence (donor quenched) and bright acceptor fluorescence. After photobleaching of AnaRne-GFP, the donor, AnaRnb-BFP, showed increased fluorescence, as its energy could no longer be transferred to AnaRne-GFP (donor dequenched). By measuring the increase in AnaRnb-BFP fluorescence before and after photobleaching of AnaRne-GFP, the FRET efficiency and distance between the two proteins were calculated, showing that AnaRnb-BFP and AnaRne-GFP had a high FRET efficiency of ∼0.7, and a distance of ∼5 nm.
Anabaena RNase II enhances the endonuclease efficiency of RNase E
To investigate the effect of the interaction between AnaRnb and AnaRne on their catalytic activities, we tested the degradation of different RNA substrates by AnaRnb and AnaRne, either alone or in combination. Five RNA substrates were selected for the assay: two 5′-FAM-labeled RNAs used above (16ss and 30ss), and three newly synthesized 5′-monophosphorylated and 3′-FAM-labeled RNA substrates (LU13, CRISPR3 and RNA62) (41–43). The 5′-FAM-modified 16ss RNA was degraded only by the 3′-5′ exoribonuclease AnaRnb, not by RNase E. We observed that it was equally degraded by AnaRnb in the presence or absence of AnaRne (Figure 6A). Similarly, the AnaRnb activity on the 5′-FAM-modified 30ss was not influenced by the presence of AnaRne either (Supplementary Figure S6). Thus, with the tested substrates, the activity of AnaRnb was not influenced by the presence of AnaRne. We further checked if the activity of AnaRne was affected by the presence of AnaRnb. The 5′-monophosphorylated and 3′-FAM-modified LU13, CRISPR3 and RNA62 were cleaved only by AnaRne or AnaRneN, not by AnaRnb, and their cleavage by either AnaRneN or AnaRne was significantly enhanced in the presence of AnaRnb (Figure 6B). To quantify the enhancing effect, the percentage of cleaved substrates in the presence or absence of AnaRnb was measured and compared (Figure 6B). It showed that AnaRnb had greater enhancing effect on AnaRne activity when the reaction time is short. After 5 min of incubation, 16% of LU13 was cleaved by AnaRne alone, whereas 38% was cleaved when AnaRnb was present, showing 1.5-fold enhancement of cleavage. Similarly, 34% of CRISPR3 was cleaved by AnaRne alone and 65% cleaved in the presence of AnaRnb in 5 min, showing 0.91-fold enhancement. For RNA62, the enhancing effect could not be accurately estimated, since the substrate was already efficiently cleaved by AnaRne alone (83%), and it was almost completely cleaved (98%) in the presence of AnaRnb in 5 minutes. With longer reaction time, cleavage of these substrates by AnaRne was also stimulated by AnaRnb, but to a lesser extent (e.g. the cleavage of LU13 and CRISPR3 by AnaRne was enhanced in the presence of AnaRnb by 0.71-fold and 0.16-fold, respectively), probably due to the decrease of substrate concentration and reduced enzyme activity. The enhancing effect of AnaRnb on the activity AnaRneN was similar (Figure 6B). These data indicate that the activity of AnaRne can be regulated by its interaction with AnaRnb.
Figure 6.

The effect of AnaRnb and AnaRne interaction on their ribonucleolytic activities. (A) Testing the influence of AnaRne on the exoribonuclease activity of AnaRnb on the 5′ FAM-labeled 16-base single-stranded RNA 16ss. AnaRne or BSA at 10 nM was included in the indicated reactions. (B) Testing the influence of AnaRnb on the endoribonuclease activity of AnaRne on the 5′-monophosphorylated and 3′ FAM-labeled substrates of LU13, CRISPR3 and RNA62. The major cleavage sites in the substrates are indicated by rightwards arrows. The positions of the bands corresponding to intact substrates (S) and the degradation/cleavage products (P) are indicated on the right. The numbers below the gel lanes are the percentages of degraded/cleaved products in the corresponding reactions, which were calculated based on the intensity of the gel bands quantified using ImageJ (https://imagej.nih.gov/ij). All the reactions were performed at 30°C for the indicated time periods. AnaRne at 10 nM or AnaRneN at 100 nM was added in the indicated reactions, and BSA at the same concentrations was included as the control reactions. Each reaction mixture contained 50 nM RNA substrate.
DISCUSSION
In the E. coli RNA degradosome, several protein components are recruited by the endoribonuclease RNase E. Because AnaRne and EcRne share a similar domain architecture, it is likely that AnaRne may also recruit other components to form a large, E. coli-like RNA degradosome. Our previous studies have shown that RNase E forms a complex with PNPase in cyanobacteria via interaction with a C-terminal motif in the noncatalytic region (35). PNPase is the major exoribonuclease in all RNase E-based degradosomes, and it binds to the C-terminal end of the noncatalytic domain of RNase E (18,49,50). In addition to PNPase, RNB family exoribonucleases are also found in the RNA degradosome of some species. For example, RNase R is a component of the RNA degradosome in the psychrotrophic bacterium, Pseudomonas syringae Lz4W (17), and RNase II might be also present in the E. coli RNA degradosome (51). However, how RNase R and RNase II interact with RNase E within the degradosome, and how they regulate the activities of the degradosome and RNA metabolism are unknown. In this study, we determined that RNase E formed a complex with RNase II in Anabaena. We found that AnaRnb, through its S1 and CSD domains, bound to the catalytic domain of AnaRne, and that AnaRne cleaved the synthetic substrates of LU13, CRISPR3 and RNA62 significantly faster in the presence of AnaRnb, implying that the binding of AnaRnb to the active core of AnaRne regulates its catalytic activity. Furthermore, we demonstrated that AnaRnb and AnaRne co-localized in the cytoplasm by FRET assay. It is very likely that the exonuclease AnaRnb and the endonuclease AnaRne cooperatively degrade RNA substrates in vivo. It should be noted that to our knowledge, AnaRnb is the first ribonuclease enzyme shown to interact with the catalytic domain of RNase E. The interaction between AnaRne and AnaRnb in Anabaena PCC 7120 not only provides another example of the physical link between RNB family exoribonucleases and the essential endonuclease RNase E, but also represents a new regulatory mechanism for RNase E activity. As RNase II and PNPase bind to the catalytic and noncatalytic regions of AnaRne, respectively, it is very likely that these three ribonucleases form a large RNA degradation complex in vivo.
The RNB family enzymes RNase II and RNase R, as ubiquitous 3′-5′ exoribonucleases, are important for the regulation of gene expression and are involved in virulence in prokaryotes (52–54). Using short artificial substrates, we determined that AnaRnb only degraded single-stranded substrates, a catalytic property very similar to that of E. coli RNase II. However, only a small portion of key residues in E. coli RNase II (i.e. Y253, R500 and E542) are also conserved in AnaRnb. EcRnb with the mutation of R500A completely loses the activity, while mutation of the corresponding residues in AnaRnb (R558A) only decreased but did not eliminate enzyme activity (Supplementary Figure S5). EcRnb uses four acidic residues (D201, D207, D209, D210) for metal ion coordination at the catalytic center, but the counterpart region in AnaRnb contains only two acidic residues (D262 and E267) (Supplementary Figure S1). Thus, AnaRnb and EcRnb likely use a different set of key residues for catalytic activity. We further compared the predicted structure of AnaRnb and the experimentally determined structure of EcRnb (28). The two proteins have a similar overall structure, but the space between the CSD1 domain and the RNB domain looks smaller in AnaRnb than in EcRnb (Supplementary Figure S2). The differences in key residues and structure imply that they may have different substrate preference in their hosts.
In E. coli, RNase II, together with PNPase, performs the major mRNA degradation activities (55); it also plays a critical role in A-site mRNA cleavage (56). In addition, RNase II regulates RNase PH levels and is essential for cell survival during starvation and prolonged stationary phase (57). Besides E. coli, the biological and physiological functions of RNase II are much less understood. In cyanobacteria, RNase II has only been investigated in two unicellular strains, Synechocystis PCC 6803 and Synechococcus PCC 7002. In both strains, RNase II is dispensable. In Synechocystis, mutation of RNase II led to resistance to the carbonic anhydrase inhibitor acetazolamide (58), and in Synechococcus PCC 7002, the RNase II mutant grew slower and had reduced phycocyanin content (59). However, the mechanisms underlying the functions of RNase II in cyanobacteria remain unclear. The interaction between RNase II and RNase E in Anabaena revealed in this study implies that RNase II may regulate RNA metabolism and cellular activities mainly through its cooperation with RNase E in this cyanobacterium.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Shaoran Zhang for the help in ITC assay and Dr Zhe Hu for the help in the use of two-photon laser scanning confocal microscope.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31570048]; National Key Research and Development Program of China [2018YFE0105600]; Featured Institute Service Project from the Institute of Hydrobiology, the Chinese Academy of Sciences [Y65Z021501]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Callaghan A.J., Marcaida M.J., Stead J.A., McDowall K.J., Scott W.G., Luisi B.F.. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005; 437:1187–1191. [DOI] [PubMed] [Google Scholar]
- 2. Carpousis A.J., Luisi B.F., McDowall K.J.. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 2009; 85:91–135. [DOI] [PubMed] [Google Scholar]
- 3. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 4. Vanzo N.F., Li Y.S., Py B., Blum E., Higgins C.F., Raynal L.C., Krisch H.M., Carpousis A.J.. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998; 12:2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Py B., Causton H., Mudd E.A., Higgins C.F.. A protein complex mediating mRNA degradation in Escherichia coli. Mol. Microbiol. 1994; 14:717–729. [DOI] [PubMed] [Google Scholar]
- 6. Carpousis A.J. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007; 61:71–87. [DOI] [PubMed] [Google Scholar]
- 7. Bandyra K.J., Bouvier M., Carpousis A.J., Luisi B.F.. The social fabric of the RNA degradosome. Biochim. Biophys. Acta. 2013; 1829:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chao Y., Li L., Girodat D., Förstner K.U., Said N., Corcoran C., Śmiga M., Papenfort K., Reinhardt R., Wieden H.J. et al.. In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell. 2017; 65:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bessarab D.A., Kaberdin V.R., Wei C.L., Liou G.G., Lin-Chao S.. RNA components of Escherichia coli degradosome: evidence for rRNA decay. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:3157–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ow M.C., Kushner S.R.. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 2002; 16:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kimura S., Waldor M.K.. The RNA degradosome promotes tRNA quality control through clearance of hypomodified tRNA. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carpousis A.J., Van Houwe G., Ehretsmann C., Krisch H.M.. Copurification of E. coli RNase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell. 1994; 76:889–900. [DOI] [PubMed] [Google Scholar]
- 13. Miczak A., Kaberdin V.R., Wei C.L., Lin-Chao S.. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Py B., Higgins C.F., Krisch H.M., Carpousis A.J.. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996; 381:169–172. [DOI] [PubMed] [Google Scholar]
- 15. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aït-Bara S., Carpousis A.J.. RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol. Microbiol. 2015; 97:1021–1135. [DOI] [PubMed] [Google Scholar]
- 17. Purusharth R.I., Klein F., Sulthana S., Jäger S., Jagannadham M.V., Evguenieva-Hackenberg E., Ray M.K., Klug G.. Exoribonuclease R interacts with endoribonuclease E and an RNA helicase in the psychrotrophic bacterium Pseudomonas syringae Lz4W. J. Biol. Chem. 2005; 280:14572–14578. [DOI] [PubMed] [Google Scholar]
- 18. Hardwick S.W., Chan V.S., Broadhurst R.W., Luisi B.F.. An RNA degradosome assembly in Caulobacter crescentus. Nucleic Acids Res. 2011; 39:1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Voss J.E., Luisi B.F., Hardwick S.W.. Molecular recognition of RhlB and RNase D in the Caulobacter crescentus RNA degradosome. Nucleic Acids Res. 2014; 42:13294–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Even S., Pellegrini O., Zig L., Labas V., Vinh J., Bréchemmier-Baey D., Putzer H.. Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005; 33:2141–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shahbabian K., Jamalli A., Zig L., Putzer H.. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009; 28:3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lehnik-Habrink M., Lewis R.J., Mäder U., Stülke J.. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol. Microbiol. 2012; 84:1005–1017. [DOI] [PubMed] [Google Scholar]
- 23. Liu Q., Greimann J.C., Lima C.D.. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006; 127:1223–1237. [DOI] [PubMed] [Google Scholar]
- 24. Evguenieva-Hackenberg E., Klug G.. RNA degradation in Archaea and Gram-negative bacteria different from Escherichia coli. Prog. Mol. Biol. Transl. Sci. 2009; 85:275–317. [DOI] [PubMed] [Google Scholar]
- 25. Januszyk K., Lima C.D.. Structural components and architectures of RNA exosomes. Adv. Exp. Med. Biol. 2010; 702:9–28. [PMC free article] [PubMed] [Google Scholar]
- 26. Zinder J.C., Wasmuth E.V., Lima C.D.. Nuclear RNA exosome at 3.1 Å reveals substrate specificities, RNA paths, and allosteric inhibition of Rrp44/Dis3. Mol. Cell. 2016; 64:734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deutscher M.P., Reuven N.B.. Enzymatic basis for hydrolytic versus phosphorolytic mRNA degradation in Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:3277–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frazão C., McVey C.E., Amblar M., Barbas A., Vonrhein C., Arraiano C.M., Carrondo M.A.. Unravelling the dynamics of RNA degradation by ribonuclease II and its RNA-bound complex. Nature. 2006; 443:110–114. [DOI] [PubMed] [Google Scholar]
- 29. Amblar M., Barbas A., Fialho A.M., Arraiano C.M.. Characterization of the functional domains of Escherichia coli RNase II. J. Mol. Biol. 2006; 360:921–933. [DOI] [PubMed] [Google Scholar]
- 30. Vincent H.A., Deutscher M.P.. Substrate recognition and catalysis by the exoribonuclease RNase R. J. Biol. Chem. 2006; 281:29769–29775. [DOI] [PubMed] [Google Scholar]
- 31. Matos R.G., Barbas A., Arraiano C.M.. RNase R mutants elucidate the catalysis of structured RNA: RNA-binding domains select the RNAs targeted for degradation. Biochem. J. 2009; 423:291–301. [DOI] [PubMed] [Google Scholar]
- 32. Vincent H.A., Deutscher M.P.. Insights into how RNase R degrades structured RNA: analysis of the nuclease domain. J. Mol. Biol. 2009; 387:570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin W., Rujan T., Richly E., Hansen A., Cornelsen S., Lins T., Leister D., Stoebe B., Hasegawa M., Penny D.. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:12246–12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jensen P.E., Leister D.. Chloroplast evolution, structure and functions. F1000Prime Rep. 2014; 6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J.Y., Deng X.M., Li F.P., Wang L., Huang Q.Y., Zhang C.C., Chen W.L.. RNase E forms a complex with polynucleotide phosphorylase in cyanobacteria via a cyanobacterial-specific nonapeptide in the noncatalytic region. RNA. 2014; 20:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karimova G., Pidoux J., Ullmann A., Ladant D.. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cai Y.P., Wolk C.P.. Use of a conditionally lethal gene in Anabaena sp. strain PCC 7120 to select for double recombinants and to entrap insertion sequences. J. Bacteriol. 1990; 172:3138–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elhai J., Vepritskiy A., Muro-Pastor A.M., Flores E., Wolk C.P.. Reduction of conjugal transfer efficiency by three restriction activities of Anabaena sp. strain PCC 7120. J. Bacteriol. 1997; 179:1998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu Y., Li Q., Chen X.Z.. Detecting protein-protein interactions by Far western blotting. Nat. Protoc. 2007; 2:3278–3284. [DOI] [PubMed] [Google Scholar]
- 40. Matos R.G., Fialho A.M., Giloh M., Schuster G., Arraiano C.M.. The rnb gene of Synechocystis PCC6803 encodes a RNA hydrolase displaying RNase II and not RNase R enzymatic properties. PLoS. One. 2012; 7:e32690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kime L., Jourdan S.S., Stead J.A., Hidalgo-Sastre A., McDowall K.J.. Rapid cleavage of RNA by RNase E in the absence of 5′ monophosphate stimulation. Mol. Microbiol. 2010; 76:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Behler J., Sharma K., Reimann V., Wilde A., Urlaub H., Hess W.R.. The host-encoded RNase E endonuclease as the crRNA maturation enzyme in a CRISPR-Cas subtype III-Bv system. Nat. Microbiol. 2018; 3:367–377. [DOI] [PubMed] [Google Scholar]
- 43. Bandyra K.J., Wandzik J.M., Luisi B.F.. Substrate recognition and autoinhibition in the central ribonuclease RNase E. Mol. Cell. 2018; 72:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amblar M., Arraiano C.M.. A single mutation in Escherichia coli ribonuclease II inactivates the enzyme without affecting RNA binding. FEBS J. 2005; 272:363–374. [DOI] [PubMed] [Google Scholar]
- 45. Barbas A., Matos R.G., Amblar M., López-Viñas E., Gomez-Puertas P., Arraiano C.M.. Determination of key residues for catalysis and RNA cleavage specificity: one mutation turns RNase II into a “SUPER-ENZYME”. J. Biol. Chem. 2009; 284:20486–20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barbas A., Matos R.G., Amblar M., López-Viñas E., Gomez-Puertas P., Arraiano C.M.. New insights into the mechanism of RNA degradation by ribonuclease II: identification of the residue responsible for setting the RNase II end product. J. Biol. Chem. 2008; 283:13070–13076. [DOI] [PubMed] [Google Scholar]
- 47. Khemici V., Poljak L., Luisi B.F., Carpousis A.J.. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 2008; 70:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murashko O.N., Kaberdin V.R., Lin-Chao S.. Membrane binding of Escherichia coli RNase E catalytic domain stabilizes protein structure and increases RNA substrate affinity. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:7019–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erce M.A., Low J.K., Wilkins M.R.. Analysis of the RNA degradosome complex in Vibrio angustum S14. FEBS J. 2010; 277:5161–5173. [DOI] [PubMed] [Google Scholar]
- 50. Płociński P., Macios M., Houghton J., Niemiec E., Płocińska R., Brzostek A., Słomka M., Dziadek J., Young D., Dziembowski A.. Proteomic and transcriptomic experiments reveal an essential role of RNA degradosome complexes in shaping the transcriptome of Mycobacterium tuberculosis. Nucleic Acids Res. 2019; 47:5892–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu F., Taghbalout A.. The Escherichia coli major exoribonuclease RNase II is a component of the RNA degradosome. Biosci. Rep. 2014; 34:e00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arraiano C.M., Matos R.G., Barbas A.. RNase II: the finer details of the Modus operandi of a molecular killer. RNA Biol. 2010; 7:276–281. [DOI] [PubMed] [Google Scholar]
- 53. Arraiano C.M., Andrade J.M., Domingues S., Guinote I.B., Malecki M., Matos R.G., Moreira R.N., Pobre V., Reis F.P., Saramago M. et al.. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010; 34:883–923. [DOI] [PubMed] [Google Scholar]
- 54. Matos R.G., Bárria C., Moreira R.N., Barahona S., Domingues S., Arraiano C.M.. The importance of proteins of the RNase II/RNB-family in pathogenic bacteria. Front. Cell Infect. Microbiol. 2014; 4:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Donovan W.P., Kushner S.R.. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Garza-Sánchez F., Shoji S., Fredrick K., Hayes C.S.. RNase II is important for A-site mRNA cleavage during ribosome pausing. Mol. Microbiol. 2009; 73:882–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sulthana S., Quesada E., Deutscher M.P.. RNase II regulates RNase PH and is essential for cell survival during starvation and stationary phase. RNA. 2017; 23:1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beuf L., Bédu S., Cami B., Joset F.. A protein is involved in accessibility of the inhibitor acetazolamide to the carbonic anhydrase(s) in the cyanobacterium Synechocystis PCC 6803. Plant Mol. Biol. 1995; 27:779–788. [DOI] [PubMed] [Google Scholar]
- 59. Cameron J.C., Gordon G.C., Pfleger B.F.. Genetic and genomic analysis of RNases in model cyanobacteria. Photosynth. Res. 2015; 126:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.