


Abstract
Faithful translation of genetic information depends on the ability of the translational machinery to decode stop codons as termination signals. Although termination of protein synthesis is highly efficient, errors in decoding of stop codons may lead to the synthesis of C-terminally extended proteins. It was found that in eukaryotes such elongated proteins do not accumulate in cells. However, the mechanism for sequestration of C-terminally extended proteins is still unknown. Here we show that 3′-UTR-encoded polypeptides promote aggregation of the C-terminally extended proteins, and targeting to lysosomes. We demonstrate that 3′-UTR-encoded polypeptides can promote different levels of protein aggregation, similar to random sequences. We also show that aggregation of endogenous proteins can be induced by aminoglycoside antibiotics that promote stop codon read-through, by UAG suppressor tRNA, or by knokcdown of release factor 1. Furthermore, we find correlation between the fidelity of termination signals, and the predicted propensity of downstream 3′-UTR-encoded polypeptides to form intrinsically disordered regions. Our data highlight a new quality control mechanism for elimination of C-terminally elongated proteins.
INTRODUCTION
Efficient termination of messenger RNA (mRNA) translation ensures faithful translation of the correct coding sequence, by preventing the ribosomes from translating the 3′-untranslated region (3′-UTR) (1). Although termination of translation in eukaryotes is highly efficient, the translation process may not always terminate at the annotated stop codon, leading to translation of the 3′-UTR (stop codon read-through). Basal levels of stop codon read-through usually correlate with the intrinsic fidelity of the stop codon, where UAA is considered the most efficient termination codon, while UGA is less efficient than UAG. In addition, termination efficiency is affected by the nucleotides surrounding the stop codon. Particularly important, is the base at position +4, as stop codons followed by C are usually more efficient at promoting termination, than stop codons followed by A (2–5). Several studies suggested even more complicated context-dependent termination efficiencies, mediated by bases beyond position +4 (6–9).
Context-dependent stop codon read-through may enable the synthesis of more than one protein product from a single mRNA sequence; a process often referred to as programmed read-through. For example, in viruses, programmed read-through expands the coding capacity of the viral genome and serves as a regulatory mechanism for translation of essential genes (10,11). Programmed read-through was also documented in fungi and yeast (12–14). Bioinformatic analyses and experimental data show that stop codon read-through is relatively common in Drosophila melanogaster (15,16), and several genes were found to undergo translational read-through in mammals (8–9,17–19). While these examples suggest that context-dependent programmed read-through plays a regulatory role, erroneous (non-programmed) stop codon read-through can also be observed under normal physiological conditions; for example, following suppression by near cognate or mutant tRNAs (20,21). Furthermore, non-stop mutations as well as 3′-proximal frame-shift mutations can lead to elimination of the 3′ stop codon. Although such mutations do not promote stop codon read-through, their effect is practically similar; translation of the annotated 3′-UTR and expression of a C-terminally extended protein with potentially deleterious effects (22–25). Specifically, Shibata et al. identified over 400 read-through single nucleotide polymorphisms in humans, that lead to the expression of such C-terminally extended proteins, some with known hereditary disorders (25).
Eukaryotic ribosomes translating the 3′-UTR are expected to stall at the poly(A) tail at the 3′-end of the 3′-UTR. As an indication for faulty translation, stalled ribosomes can trigger several processes aimed at suppressing leaky termination, such as degradation of the C-terminally extended protein, to minimize the potentially deleterious effects of C-terminally extended proteins (26–28) [reviewed in Brandman and Hegde (29), and Inada T. (30)]. However, ribosomes translating the 3′-UTR are likely to encounter at least one in-frame termination codon before the poly(A) tail, where translation will terminate, with the possible release of a C-terminally extended protein. It was found that the expression of C-terminally extended proteins is downregulated following translation of the 3′-UTR (25,31–33). While post-translational elimination has been suggested as a possible mechanism, the exact quality control mechanism for elimination of C-terminally extended proteins is still elusive.
Currently, there are only a few examples for read-through-dependent protein depletion in mammalian cells, which limits our understanding of the corresponding mechanism. In an attempt to elucidate the effect of stop codon read-through on protein homeostasis at the cellular level, we decided to use UAG suppression machinery in order to increase stop codon read-through by co-translational incorporation of non-canonical amino acids. In particular, we were interested in understanding why 3′-UTR-encoded C-terminal extensions promote the elimination of extended proteins, and by which mechanism these proteins are depleted.
MATERIALS AND METHODS
General
Unless otherwise stated, chemicals and DNA oligomers were purchased from Sigma-Aldrich (Darmstadt, Germany) and used without further purification. Nε-tert-butyloxycarbonyl lysine (BocLys) was purchased from Chem-Impex International (Wood Dale, IL, USA). Enzymes and buffers for molecular biology were purchased from NEB (Ipswich, MA, USA) and used according to the manufacturer’s instructions. Escherichia coli DH10B cells (Life Technologies, Carlsbad, CA, USA) were used for molecular cloning and plasmid propagation. Plasmid DNA was purified using spin columns from Macherey Nagel (Düren, Germany). Bacteria were incubated in liquid LB media or plated on LB/agar plates supplemented with the required antibiotic (32 μg/mL zeocin, 50 μg/mL ampicillin). pEF.myc.ER-E2-Crimson plasmid (Addgene plasmid #38770) was a gift from Benjamin Glick (34). Plasmid directing the synthesis of shRNA against human release factor 1 and control plasmid (pEQ941 and pEQ936, respectively) were kindly provided by Adam P. Geballe (35). Anti-6×His (#G020) and anti-GFP (#G096) antibodies were purchased from abm (Richmond, ON, USA), anti-p53 (#ab90363) was purchased from Abcam (Cambridge, UK). Anti-Ser51-phosphorylated eIF2α (#3597), anti-PERK (#5683) and anti-ubiquitin (#3936) were purchased from cell signaling (Danvers, MA, USA). Anti-LC3 (#L7543) was purchased from Sigma-Aldrich (Darmstadt, Germany). Secondary horseradish peroxidase-conjugated anti-mouse IgG (#ab7068) and anti-rabbit IgG (#ab92080) were purchased from Abcam.
Plasmid construction
Unless otherwise stated, all plasmids used for transient transfection were based on the commercially available pBudCE4.1 vector (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA). The cDNA of the expressed protein of interest was cloned into the multiple cloning site downstream to the EF1α promoter, using KpnI, EcoRI, and XhoI restriction sites (Supplementary Figure S2).
mCherry-P2A-eGFP-3′-UTR extension
mCherry-P2A-eGFP cDNA was cloned between KpnI and XhoI sites downstream of the EF1α promoter (Supplementary Figure S2A). Indicated 3′-UTR sequences (Supplementary Table S1) were then cloned downstream of the mCherry-P2A-eGFP gene using EcoRI and XhoI restriction sites as shown in Supplementary Figure S2B. Random sequences of 13, 31 and 12 amino acids long (R1, R2 and R3, respectively) were generated using an in-house Python script.
mCherry-P2A-α-enolase-3′-UTR extension
The mCherry-P2A sequence was amplified from the mCherry-P2A-eGFP construct using primers 5′-GAAGGTACCATGGTGAGCAAGGGCG-3′ and 5′-GGCGAATTCGAGCTCAGGTCCAGGGTTCTCCTC-3′. α-enolase cDNA with N-terminal 6×His tag was amplified from plasmid HsCD00000030 (DNASU plasmid repository) using primers 5′-CTGAGCTCCACCACCACCATCATCACGTGCATCTGACTCCTG-3′ and 5′-GCGAATTCGTGATACTTGTGGGCCAGG-3′. The mCherry-P2A construct was cloned downstream of the EF1α promoter using KpnI and SacI restriction sites and 6×His-α-enolase was then cloned in frame using SacI and EcoRI cloning sites (Supplementary Figure S2C). Indicated 3′-UTRs were amplified from corresponding mCherry-P2A-eGFP-3′-UTR plasmids using primers 5′-GAAGGTACCATGGTGAGCAAGGGCG-3′ and 5′-ATCTCTCGAGTTAGTGATACTTGTGGGCCAGG-3′, and cloned in frame using the KpnI and XhoI restriction sites (Supplementary Figure S2D).
mRFP-GFP-3′-UTR
mRFP-GFP cDNA, without 3′-extension, was amplified from the ptfLC3 plasmid (gift from Tamotsu Yoshimori, Addgene plasmid #21074) (36) using primers 5′-GAAGGTACCATGGCCTTCTCCGAGGAC-3′ and 5′-TCTCTCGAGTTACTTGTACAGCTCGTCCATGC-3′. For the cloning of mRFP-GFP with 3′-UTR encoded C-terminal extension, mRFP-GFP cDNA was amplified using primers 5′-GAAGGTACCATGGCCTTCTCCGAGGAC-3′ and 5′-TGTGAATTCCTTGTACAGCTCGTCCATGC-3′. The polymerase chain reaction products of the control or C-terminally extended mRFP-GFP were cloned using KpnI and XhoI or KpnI and EcoRI restriction sites, respectively, downstream of the EF1α promoter (Supplementary Figures S2E and F).
Cell culture
Cells were incubated at 37°C in a humidified chamber with 5% CO2. HEK293T, COS7, HeLa, MEF and MEFATG5−\− cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Biological Industries, Beit Haemek, Israel) and HCT116 cells were maintained in McCoy’s 5A medium (Biological Industries). All cell culture media were supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin G sodium, 0.1 mg/mL streptomycin sulfate, and 1.25 U/mL nystatin (Biological Industries). For Western blot analyses, 105 cells were seeded 24 h before transfection in a 24-well plate. For immunofluorescence and live cell imaging 3×104 cells were seeded 24 h before transfection in a 24-well plate with glass coverslips or in a 4-well μ-Slide ibiTreat plate (80426, Ibidi, Martinsried, Germany). HEK293T, MEF and MEFATG5−\− cells were transfected using LipoD293 reagent (Signagen Laboratories, Rockville, MD, USA), and COS7 and HCT116 cells were transfected using Lipofectamine 2000 (Invitrogen-Thermo Fisher Scientific). Transfections using LipoD293 were performed according to the manufacturer’s protocol, using 3:1 transfection reagent:DNA ratio. Transfections using Lipofectamine 2000 were performed at a 2:1 transfection reagent:DNA ratio, and culture media were exchanged 4 h post-transfection. For proteasomal inhibition, cells were transfected 24 h before lysis or imaging, and incubated with 1 μM of MG132 for 16 h. For lysosomal inhibition, cells were transfected 48 h before lysis or imaging, and incubated with indicated concentration of chloroquine for 14–40 h.
Stable transfection
The plasmid for stable transfection of mammalian cells with amber suppression machinery was kindly provided by Prof. Jason Chin (MRC-LMB, Cambridge). Stable transfection was based on the use of the PiggyBac transposase, and performed as described in Elsässer et al. (37), except that the pyrrolysine aminoacyl tRNA synthetase (PylRS) coding sequence in the original PiggyBac targeting plasmid was replaced with a chimeric version of the PylRS gene (38). 2.5×105 cells were plated in a 12-well plate and transfected with the PiggyBac targeting plasmid and Super PiggyBac Transposase (SBI, Palo Alto, CA, USA) at a ratio of 3:1. Twenty-four hours post-transfection, the cells were transferred to a 10 cm plate and cultured in DMEM supplemented with 5 μg/mL puromycin. Cells were incubated for at least 10 days under selection conditions, until polyclonal pools were established.
RNA interference
PERK knockdown was performed using three siRNA sequences (Invitrogen) as described in Oren et al. (39): 5′-GGCAGUGGAGUUUCUUCACAGUAAA-3′, 5′-CACCAGUAGCAAAUCUUCUUCUGAA-3′, and 5′-CAGAUGGAGAGAGUCAGGACCUUAA-3′. Non-specific control oligo (IDT) was used as control siRNA. HEK293T cells stably expressing UAG suppression machinery (at 15–20% confluency), were transfected using Oligofectamine (Invitrogen) following the manufacturer’s instructions. Twenty-four hours post-transfection, BocLys was added to the culture at 1 mM concentration. Cells were harvested 72 h post-transfection.
Label-free quantitative mass spectrometry
HEK293T cells stably transfected with UAG suppression machinery were seeded in 10 cm plates at ∼20% confluency. After 24 h, DMEM was replaced with fresh media supplemented with 5 μg/mL of puromycin and, where indicated, 1 mM BocLys for additional 48 h. Next, cells were collected (5 min, 500 g, 4°C) following incubation with trypsin, washed with 10 mL of sorting medium [phosphate buffered saline (PBS), 2 mM ethylenediaminetetraacetic acid (EDTA), 2% FBS], and resuspended in 500 μl of sorting medium supplemented with DAPI at a ratio of 1:1000. For label-free quantitative mass spectrometry (LFQMS) proteomic analysis, 1.5×106 cells were sorted and collected using FACSAria (BD Biosciences, San Jose, CA, USA). Sorting of cells cultured with BocLys was based on DAPI and Dendra2 fluorescence as an indication for active UAG suppression. Cells incubated in the absence of BocLys were sorted based on DAPI fluorescence only. Collected cells were then pelleted (10 min, 500 g, 4°C), flash-frozen in liquid nitrogen and stored at -80°C. Label-free quantitative mass spectrometry proteomic analysis was performed by The De Botton Protein Profiling institute of the Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science. For each condition, four biological replicates were analyzed. The samples were digested with trypsin using the S-trap method (Protifi, Huntington, NY, USA) (40). The resulting peptides were analyzed using nanoflow liquid chromatography (nanoAcquity) coupled to high-resolution mass spectrometry (Q Exactive HF, Thermo Fisher) (41). Each sample was analyzed on the instrument separately in a random order in discovery mode. Raw data were processed with MaxQuant v1.6.4.0 (42). The data were searched with the Andromeda search engine (43), against the Human proteome database (Swissprot Nov18) appended with common lab protein contaminants and the following modifications: fixed carbamidomethylation of cysteine, N-terminal protein acetylation, oxidation of methionine and deamidation of asparagine and glutamine. Quantification was based on the label-free quantification method (44). Data are available via ProteomeXchange with identifier PXD016728.
Fluorescence spectroscopy
Cells were lysed at indicated time points, with ice-cold RIPA lysis buffer [50 mM Tris buffer pH 8.0, 150 mM NaCl, 1% (v/v) Triton ×-100, 0.1% (w/v) sodium dodecyl sulphate (SDS)], supplemented with protease inhibitors (1.2 mg/mL leupeptin, 1 mM pepstatin A, 100 mM PMSF and 1 mg/mL aprotinin). Cell lysates were centrifuged in 14,000 g for 10 min at 4°C and the clear supernatants were collected for further analysis. Fifty microliters of lysate diluted in PBS (1:10 dilution) were analyzed in a 384-well dark plate. Fluorescence was measured with Spark plate reader using 485/535 nm (Ex/Em) for GFP fluorescence and 580/620 nm (Ex/Em) for mCherry fluorescence.
Western blotting
Total protein concentration in clear lysates was determined using a BCA assay kit (Thermo Fisher Scientific) and sample volumes were normalized accordingly. Clear lysates were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE) using gels supplemented with 1% 2,2,2-trichloroethanol (TCE). Following electrophoresis, proteins were visualized in gels by UV light to quantify total protein load (45). Proteins were transferred to a 0.2 μm nitrocellulose membrane using a semi-dry transfer apparatus (Trans-Blot Turbo, BioRad, Hercules, CA, USA). The Membrane was blocked with Tris-buffered saline containing 0.05% (v/v) Tween-20 (TBST) and 5% (w/v) non-fat dry milk, and incubated over night at 4°C with primary antibody diluted in 5% (w/v) bovine serum albumin in TBST. Following washing with TBST, the membrane was incubated with a secondary antibody for 1 h at room temperature. Proteins were then visualized using an ECL reagent (GE Healthcare). Gel bands were quantified using ImageJ (46). To separate between soluble and insoluble fractions, cells were incubated for 30 min with lysis buffer (PBS, 1% Triton ×-100) supplemented with protease inhibitors (1.2 mg/mL leupeptin, 1 mM pepstatin A, 100 mM PMSF, and 1 mg/L aprotinin). Cell lysates were centrifuged (250 g for 5 min at 4°C), and semi-cleared lysates were transferred to new tubes. Semi-cleared Lysates were centrifuged again (20,000 g for 10 min at 4°C), and supernatant and pellet were boiled separately in 1× and 2×sample buffer, respectively.
Confocal microscopy
Fluorescence imaging was performed using 3D Z-stacks of selected cells expressing indicated proteins. Images were collected using a fully incubated confocal spinning-disk microscope (Marianas; Intelligent Imaging, Denver, CO, USA) with a 63×N.A 1.4 oil immersion objective. Images were recorded on an electron-multiplying charge-coupled device camera (pixel size, 0.079 μm; Evolve; Photometrics, Tucson, AZ, USA). Image processing and analysis were done using SlideBook version 6 (Intelligent Imaging). Labeling of cellular aggregates in fixed cells (4% paraformaldehyde) was performed using Proteostat Aggresomes detection kit ENZ-51035-0025 (Enzo life sciences, Lausen Switzerland) according to the manufacturer’s protocol. As a positive control, cells were cultured in the presence of 5 μM MG132 for 16 h before fixation.
Bioinformatic analyses
Python scripts written in-house for random selection of 3′-UTR sequences downstream of UAG stop codons, with an in-frame stop codon at 30 and 150 bases (i.e. 10–50 amino acids) downstream of the annotated stop codon. To analyze the human 3′-UTR-encoded protein sequences, we used the Ensembl (release 99) human gene database (GRCh38.p13) (47) and generated two separated fasta files, one for the coding sequence of annotated proteins and one for their 3′-UTRs. The 3′-UTR sequences, between the annotated stop codon and the next in-frame stop codon, were translated using an in-house python code. The propensity of each amino acid to adopt an intrinsically disordered conformation was calculated using the the IUPred short algorithm as implemented in the IUPred2A server. The mean propensity was calculated for each polypeptide sequence, and all the sequences were then divided into six groups, according to the annotated upstream stop codon (UAA/UAG/UGA) and the first base of the coding 3′-UTR (A/C). Random polypeptide sequences with the same length distribution of the above six groups were created using an in-house python script, and analyzed in a similar way. Analyses of chicken, mouse, and zebrafish 3′-UTR sequences were performed as described above, using GRCg6a, GRCm38.p6, GRCz11 datasets, respectively. Standard packages in R were used for statistical analyses and generating boxplots.
RESULTS
3′-UTR-encoded C-terminal extensions promote post-translational depletion of extended soluble proteins
Amber suppression technology enables the encoding of non-canonical amino acids in response to in-frame stop codons, thereby provides a unique platform for studying cellular mechanisms related to stop codon read-through. For example, the pyrrolysine aminoacyl tRNA synthetase (PylRS) and its cognate UAG-suppressor tRNA (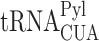 ) enable the co-translational incorporation of the non-canonical amino acid Nε-tert-butyloxycarbonyl lysine (BocLys) in response to the amber stop codon, UAG. In a preliminary quantitative mass spectrometry analysis, we found significant differences in protein expression levels between transiently transfected HEK293T cells incubated in the presence or absence of BocLys. That is, differences between cells with or without active UAG suppression, respectively. While providing the incentive for the current study, such measurements cannot provide definite conclusions regarding the expression levels of individual proteins, since the observed results may be attributed to secondary effects. For example, changes in signal transduction due to reduced cellular levels of a protein participating in a signaling pathway, or decrease in protein expression levels following degradation of transcription factors.
) enable the co-translational incorporation of the non-canonical amino acid Nε-tert-butyloxycarbonyl lysine (BocLys) in response to the amber stop codon, UAG. In a preliminary quantitative mass spectrometry analysis, we found significant differences in protein expression levels between transiently transfected HEK293T cells incubated in the presence or absence of BocLys. That is, differences between cells with or without active UAG suppression, respectively. While providing the incentive for the current study, such measurements cannot provide definite conclusions regarding the expression levels of individual proteins, since the observed results may be attributed to secondary effects. For example, changes in signal transduction due to reduced cellular levels of a protein participating in a signaling pathway, or decrease in protein expression levels following degradation of transcription factors.
To mechanistically study the effect of stop codon read-through on protein levels in a rational way, we randomly picked 3′-UTR sequences of 17 different human genes (the sequence between the natural UAG codon and the next in-frame stop codon; Supplementary Tables S1 and S2). The nucleotide sequence of each 3′-UTR was then cloned upstream to the stop codon of a gene encoding the expression of mCherry, followed by P2A sequence and eGFP (Figure 1A). Assuming mCherry and eGFP are translated at a 1:1 ratio, the ratio between mCherry and eGFP fluorescence can be used to quantify the post-translational depletion level of eGFP. For each C-terminally extended eGFP, the ratio of mCherry to eGFP fluorescence in soluble fractions of cell lysates was measured, and values were normalized to the ratio measured for the mCherry-P2A-eGFP construct without an extension.
Figure 1.

Relative levels of soluble C-terminally extended proteins. (A) mCherry-P2A-eGFP construct for measuring cellular levels of C-terminally extended eGFP. 3′-UTR sequences were added to the coding sequence of eGFP, while the use of a P2A sequence ensures translation of eGFP and mCherry at a 1:1 ratio. (B) Normalized levels of soluble eGFP with 3′-UTR-encoded C-terminal extension. The ratio between eGFP to mCherry fluorescence intensities is proportional to the level of post-translational elimination of eGFP from soluble fractions. (C) Elimination of soluble eGFP with selected 3′-UTR-encoded C-terminal extension, evaluated by quantification of immunoblot intensities. Values were normalized to total protein load using 1% TCE. (D) Post-translational elimination of soluble α-enolase. N’-6×His-labeled α-enolase variants with indicated 3′-UTR-encoded C-terminal extensions were expressed in HEK293T cells. Relative amounts of soluble α-enolase were evaluated from immunoblot intensities using anti-6×His antibody, as described in panel C. (E) The effect of variable C-terminal extensions on protein levels in soluble fractions. C-terminal extension with a random polypeptide sequence promoted the elimination of soluble eGFP at levels similar to those observed with 3′-UTR-encoded polypeptides (left). Shuffled and natural (un-shuffled) 3′-UTR-encoded polypeptides may promote reduction in soluble eGFP levels at different levels, suggesting the effect is sequence- and not composition-dependent (center). C-terminal extensions of polypeptides encoded by 3′-UTR sequences with frame-shift also promote post-translational elimination of soluble eGFP. Bars represent the mean ± S.D. of three independent measurements.
In good agreement with previous studies (25,31), we measured a decrease in levels of soluble eGFP following the addition of 3′-UTR-encoded C-terminal extensions (Figure 1B). The extent of eGFP depletion was dependent on the 3′-UTR-encoded C-terminal extension, with some polypeptides having small effect (e.g NES and TCEAL4) and others promoting almost complete depletion of soluble eGFP (e.g. CEP97 and SH3GLB2). Similar results were obtained when protein levels in soluble fractions of cell lysates were measured by quantification of anti-eGFP immunoblot intensities (Figure 1C). To verify that the observed C-terminal extension-dependent depletion is not protein specific, we replaced eGFP with human α-enolase (i.e., mCherry-P2A-α-enolase) and quantified from immunoblot intensities the cellular levels of soluble human α-enolase. We found that addition of 3′-UTR-encoded C-terminal extensions resulted in a decrease in cellular levels of soluble α-enolase, at levels similar to those measured for eGFP (Figure 1C versus D). In contrast, C-terminal extension encoded by the 3′-UTR of the mdh1 gene that is known to be expressed as an extended variant via programmed read-through (8), had no effect on soluble levels of eGFP (Supplementary Figure S1). Thus, our data confirm that mammalian 3′-UTR-encoded C-terminal extensions can promote the post-translational depletion of soluble proteins.
Next, we quantified the effect of three random polypeptide sequences on post-translational depletion of C-terminally extended proteins (Figure 1E, left). Similar reduction in levels of soluble eGFP was measured upon addition of C-terminal random sequences, demonstrating that the effect of 3′-UTR-encoded protein sequences can be reproduced with random polypeptides. In addition, to understand if the observed protein depletion depends on the sequence of the C-terminal extension or its amino acid composition, we shuffled the amino acid sequence of four 3′-UTR-encoded polypeptides (Figure 1E, center). Some shuffled sequences promoted similar levels of protein depletion as the un-shuffled sequences (e.g. MYO5C and CEP97), while shuffled TCEAL4 sequence had much stronger effect, compared to the natural sequence. Finally, we changed the composition of 3′-UTR-encoded polypeptides, by adding part of the upstream coding sequence to the 3′-UTR, thereby simulating a frame-shift in the ‘reading frame’ of the 3′-UTR (Figure 1E, right). The measured ratios between eGFP and mCherry fluorescence showed that such polypeptides can also promote the elimination of C-terminally extended soluble proteins. Taken together, these data show that the post-translational depletion of proteins with 3′-UTR-encoded C-terminal extension is not dependent on a specific amino acid composition or a consensus sequence. It is rather dependent on the order of variable amino acid compositions, as may be found in random protein sequences.
Elimination of soluble C-terminally extended proteins is independent of the ubiquitin-proteasome pathway or macroautophagy
To gain more insight into the mechanism by which 3′-UTR-encoded C-terminal extensions promote post-translational reduction in levels of soluble proteins, we first inhibited the ubiquitin-dependent proteasomal degradation pathway using MG132. As expected, incubation of cells with MG132 resulted in an increase in cellular levels of ubiquitinated proteins such as p53 (Supplementary Figures S3A and B). However, eGFP levels in soluble fractions of cell lysates were not increased following incubation with MG132, but rather decreased (Figure 2A and Supplementary Figure S3C). These data show that proteasomal degradation is not the default pathway for sequestration of soluble proteins with a 3′-UTR-encoded C-terminal extension.
Figure 2.

3′-UTR-encoded C-terminal extensions promote aggregation of extended proteins. (A and B) Effect of proteasome inhibition and chloroquine on cellular levels of soluble eGFP with 3′-UTR-encoded C-terminal extension. Transfected HEK293T cells were cultured for 16 h in the presence of MG132 (A) or 40 h in the presence of chloroquine (B). Cellular levels of indicated C-terminally extended eGFP variants were determined from the ratio between fluorescence intensities of soluble eGFP and mCherry. (C) Total levels of eGFP with 3′-UTR-encoded C-terminal extension. HEK293T cells expressing mCherry-P2A-eGFP with indicated C-terminal extension were boiled in 2×sample buffer 48 h post-transfection and clear samples were analyzed by SDS-PAGE. Bars represent immunoblot intensities (mean ± S.D. of three independent measurements) normalized to total protein load (using TCE). (D) Distribution of eGFP between soluble and insoluble fractions. Soluble (Sup) and insoluble (Pellet) fractions of HEK293T lysates prepared with Triton×-100, were boiled in 2×SDS sample buffer before analysis. Depletion of eGFP from soluble fractions was accompanied by increased levels in insoluble fractions. TCE fluorescence of soluble fraction was used as a loading control. (E) Spatial distribution of eGFP with 3′-UTR-encoded C-terminal extension. COS7 cells co-expressing mCherry-P2A-eGFP with indicated C-terminal extensions and ER-E2-Crimson for ER labeling (red channel) were visualized by fluorescence confocal microscopy 48 h post-transfection. Soluble eGFP (control, NES) displayed a diffusive phenotype, while eGFP with 3′-UTR-encoded C-terminal extensions that promoted depletion from soluble fractions, formed green fluorescent puncta. No overlap was found between eGFP puncta and ER (red). Scale bar = 10 μm.
A decrease in cellular levels of a given protein upon inhibition of proteasomal degradation may indicate that the protein undergoes lysosomal degradation, which is upregulated to compensate for inhibition of the proteasomal pathway (48). To examine if C-terminally extended soluble proteins are targeted to lysosomes, HEK293T cells expressing mCherry-P2A-eGFP with 3′-UTR-encoded C-terminal extensions were treated with chloroquine, that inhibits the fusion of autophagosomes with lysosomes by neutralizing the intrinsic lysosomal acidic pH. As expected, LC3-II levels increased in response to treatment with chloroquine (Supplementary Figure S3D). However, levels of C-terminally extended eGFP in soluble fractions were not affected by incubation with chloroquine (Figure 2B). In addition, we checked if C-terminally extended soluble eGFP is eliminated via autophagy-related gene 5 (ATG5)-dependent macroautophagy, using ATG5 knockout mouse embryonic fibrolplasts (MEFATG5−\−). We found that compared to proteins expressed in MEFATG5+\+, the ratio of eGFP to mCherry fluorescence was not higher in MEFATG5−\− expressing mCherry-P2A-eGFP with different 3′-UTR-encoded C-terminal extensions (Supplementary Figure S4). Thus, ATG5 is not essential for the elimination of soluble proteins with a 3′-UTR-encoded C-terminal extension.
Proteins with a 3′-UTR-encoded C-terminal extension are prone to aggregation and targeted to lysosomes
One mechanism that can explain the observed depletion of C-terminally extended proteins from soluble fractions is aggregation. Following this hypothesis, we first quantified the total amount of expressed eGFP in whole cell lysates (i.e. soluble and insoluble fractions). HEK293T cells expressing mCherry-P2A-eGFP with selected 3′-UTR-encoded C-terminal extensions were boiled in 2×SDS sample buffer, and proteins were separated by SDS-PAGE (Figure 2C). Quantification of immunoblot intensities revealed that total levels of eGFP were not as sensitive to 3′-UTR-encoded C-terminal extension, as levels of soluble eGFP (Figure 2C versus 1B). Furthermore, when cells were lysed using 1% Triton ×-100 and soluble and insoluble fractions were separated and evaluated independently, we found an inverse correlation between the levels of eGFP in soluble fractions, and its levels in insoluble fractions (Figure 2D). These results suggest that 3′-UTR-encoded C-terminal extensions can promote the accumulation of the extended proteins in insoluble states, such as aggregates.
To verify the formation of aggregates, we monitored the spatial distribution of eGFP with selected 3′-UTR-encoded polypeptide extensions in COS7 cells co-expressing mCherry-P2A-eGFP and ER-E2-Crimson for ER labeling (34). According to confocal fluorescence microscopy, 3′-UTR-encoded C-terminal extensions that promoted weak depletion (NES) had no effect on the distribution of expressed eGFP, which presented a diffusive phenotype. In contrast, C-terminal extensions that reduced eGFP levels in soluble fractions, also promoted the formation of green fluorescent puncta, that were not localized to the ER (Figure 2E). These data further support our observation that 3′-UTR-encoded C-terminal extensions can derive the depletion of extended proteins from soluble fractions, in the form of aggregates.
One of the cellular mechanisms for segregation of aggregates is through lysosomal targeting (49). To check if C-terminally extended proteins are targeted to lysosomes, we expressed the RFP-GFP fusion protein with and without C-terminal extensions and visualized the expressed chimeric proteins in living cells incubated with or without chloroquine. As GFP fluorescence is quenched at acidic pH, expression of RFP-GFP with C-terminally fused LC3 that targets proteins to lysosomes, resulted in formation of red-fluorescent puncta, indicating that the chimeric protein is within the acidic environment of lysosomes and not fully degraded (Figure 3). A similar phenotype was observed when RFP-GFP’s C-terminal was extended by 3′-UTR-encoded polypeptides, while RFP-GFP without C-terminal extension was mainly diffusive (Supplementary Figure S5). Importantly, treatment of cells with the lysosomal inhibitor chloroquine, resulted in a pronounced increase in GFP fluorescence, observed as GFP- and RFP-fluorescent puncta. These data indicate that aggregates of the C-terminally extended proteins were targeted to lysosomes. Taken together, we found that 3′-UTR-encoded C-terminal extensions can promote aggregation of the extended proteins, that are then targeted to lysosomes.
Figure 3.

Fluorescence imaging of cells expressing C-terminally extended RFP-GFP. An increase in the number of green fluorescent puncta following treatment with chloroquine indicates localization of GFP in acidic compartments. Imaging of living COS7 cells was performed 48 h post-transfection, following 14 h incubation with 10 μM chloroquine. RFP-GFP with C-terminal LC3 was used as a positive control. Scale bar=10 μm.
Stop codon read-through affects cellular proteostasis by promoting aggregation of endogenous proteins
Following our experiments with model proteins, we asked if 3′-UTR-encoded C-terminal extension-dependent aggregation is a general phenomenon that also applies to endogenous proteins. To answer this question, we decided to increase the expression levels of C-terminally extended proteins by promoting stop codon read-through and monitor any subsequent formation of cellular-protein aggregates. To this end, we created a polyclonal HEK293T cell line, stably expressing the orthogonal PylRS/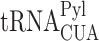 couple, for the co-translational incorporation of BocLys in response to the UAG stop codon (37). In these cells, the encoded wild-type PylRS gene is terminated with a UAG stop codon, followed by a P2A sequence and the fluorescent protein Dendra2. Therefore, Dendra2 is expressed only if the UAG stop codon is suppressed by the incorporation of BocLys into the expressed protein. The polyclonal HEK293T cells were cultured for 48 h in the presence or absence of BocLys, and equal numbers of cells were isolated by fluorescence activated cell sorting, using Dendra2 fluorescence as an indication for functional UAG suppression machinery in cells incubated in the presence of BocLys (Supplementary Figure S6).
couple, for the co-translational incorporation of BocLys in response to the UAG stop codon (37). In these cells, the encoded wild-type PylRS gene is terminated with a UAG stop codon, followed by a P2A sequence and the fluorescent protein Dendra2. Therefore, Dendra2 is expressed only if the UAG stop codon is suppressed by the incorporation of BocLys into the expressed protein. The polyclonal HEK293T cells were cultured for 48 h in the presence or absence of BocLys, and equal numbers of cells were isolated by fluorescence activated cell sorting, using Dendra2 fluorescence as an indication for functional UAG suppression machinery in cells incubated in the presence of BocLys (Supplementary Figure S6).
Label-free quantitative high-resolution mass spectrometry was then used in order to compare between the proteome of cells with active UAG stop codon read-through (RT) and the proteome of control cells cultured in the absence of BocLys (Cont). For each condition, we measured four independent samples, and identified 5275 proteins in total (Supplementary File 1). Statistical analysis based on Student’s t-test with false discovery rate of q < 0.05, identified 423 proteins with significantly altered cellular levels. The abundance of 251 proteins decreased and of 172 proteins increased in cells with UAG suppression compared to control (Figure 4A and B). Functional annotation using the PANTHER classification system found that most changes in protein levels were observed in proteins related to metabolic processes, and that in all identified functional categories, protein levels both increased and decreased in response to UAG suppression (Figure 4C). However, this observation may be related to the natural high abundance of proteins in these categories. Although the measured differences may be attributed to secondary effects and therefore cannot be directly related to UAG suppression, the data clearly show that UAG read-through significantly affected cellular proteostasis.
Figure 4.

Read-through-dependent aggregation of endogenous proteins. (A) Volcano plot displaying the significance and fold change of all 5275 proteins identified by LFQMS. Significantly altered proteins are marked in red. Positive Log2 fold change values mean lower cellular levels in cells with UAG read-through (RT) and negative values mean higher levels, relative to control (Cont). (B) Clustering of 423 significantly altered proteins in cells with UAG read-through and control. Red and blue colors mark higher and lower cellular levels of proteins, respectively. (C) PANTHR functional classification of significantly altered protein levels in read-through and control cells (69). Bars show the number of proteins in each functional category, whose abundance was lower (blue bars) or higher (red bars) in RT samples, relative to control. (D) Protein aggregation in response to UAG-suppression. MEF cells stably transfected with PylRS and 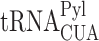 genes that enable UAG suppression-dependent expression of Dendra2 were cultured in the presence (+) or absence (−) of BocLys for 48 h. To detect the formation of aggregates, cells were labeled with proteostat (red channel). UAG suppression promoted the formation of protein aggregates, as also observed in cells incubated with the proteasome inhibitor MG132. (E) Gentamicin-dependent aggregation of endogenous proteins. MEF cells were cultured for 24 or 48 h in the presence of 200 μg/mL gentamicin, and stained as described in panel D. Images show the formation of aggregates following 48 h incubation with gentamicin. (F) Naïve HEK293T cells were transfected with a plasmid encoding shRNA against RF1 or control shRNA (35), and stained with Hoechst and proteostat 48 h post-transfection. Images show increase in protein aggregation following RF1-knockdown and subsequent enhanced read-through. Scale bar = 10 μm.
genes that enable UAG suppression-dependent expression of Dendra2 were cultured in the presence (+) or absence (−) of BocLys for 48 h. To detect the formation of aggregates, cells were labeled with proteostat (red channel). UAG suppression promoted the formation of protein aggregates, as also observed in cells incubated with the proteasome inhibitor MG132. (E) Gentamicin-dependent aggregation of endogenous proteins. MEF cells were cultured for 24 or 48 h in the presence of 200 μg/mL gentamicin, and stained as described in panel D. Images show the formation of aggregates following 48 h incubation with gentamicin. (F) Naïve HEK293T cells were transfected with a plasmid encoding shRNA against RF1 or control shRNA (35), and stained with Hoechst and proteostat 48 h post-transfection. Images show increase in protein aggregation following RF1-knockdown and subsequent enhanced read-through. Scale bar = 10 μm.
Next, to verify the formation of aggregates in response to stop codon read-through, we created polyclonal MEF cells, stably transfected with the above described 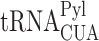 and PylRS for UAG suppression-dependent expression of Dendra2. The cells were cultured for 48 h in the presence or absence of BocLys, as well as with the proteasome inhibitor MG132, as a positive control. Cells were then stained with proteostat, for specific fluorescent labeling of aggregates. As seen in Figure 4D, aggregates were found in cells with active UAG suppression machinery (+BocLys, Dendra2 positive), as well as in cells cultured in the presence of MG132, but not in cells without active UAG suppression (-BocLys) or in naïve cells cultured in the presence of BocLys (Supplementary Figure S7A). UAG suppression-dependent aggregation of endogenous proteins was also observed in stably transfected human HCT116 cell line (Supplementary Figure S7B). In addition, aggregation of endogenous proteins was detected in naïve MEF cells treated with 200 μg/mL of the aminoglycoside antibiotic gentamicin, which is known to promote stop codon read-through (Figure 4E) (50,51). Aggregation was time-dependent, and was clearly observed after 48 h. Aminoglycoside-dependent aggregation was also detected in MEF cells treated with G418 as well as in naïve HeLa and HEK293T cells (Supplementary Figure S7C and D). As expected, aggregation of endogenous proteins was not found in MEF cells cultured in the presence of Ataluren (PTC124) that promotes read-through of premature termination codons (Supplementary Figure S7E). Moreover, we found high levels of aggregation following knockdown of release factor 1 (RF1) that promotes stop codon read-through (Figure 4F) (35). Finally, we expected the observed protein aggregation to trigger stress responses, such as the unfolded protein response (UPR) that is characterized by Ser51-phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α). Indeed, in stably transfected HEK293T cells we measured a two-fold increase in eIF2α Ser51-phosphorylation upon addition of BocLys, indicating the activation of cellular stress responses in response to UAG read-through (Supplementary Figure S8). Knockdown of protein kinase R-like endoplasmic reticulum kinase (PERK) significantly reduced eIF2α phosphorylation, suggesting that read-through-dependent aggregation activates the PERK-eIF2α UPR branch. Together, these results further support our observations of protein aggregation that is dependent on stop codon read-through and expression of proteins with 3′-UTR-encoded C-terminal extension.
and PylRS for UAG suppression-dependent expression of Dendra2. The cells were cultured for 48 h in the presence or absence of BocLys, as well as with the proteasome inhibitor MG132, as a positive control. Cells were then stained with proteostat, for specific fluorescent labeling of aggregates. As seen in Figure 4D, aggregates were found in cells with active UAG suppression machinery (+BocLys, Dendra2 positive), as well as in cells cultured in the presence of MG132, but not in cells without active UAG suppression (-BocLys) or in naïve cells cultured in the presence of BocLys (Supplementary Figure S7A). UAG suppression-dependent aggregation of endogenous proteins was also observed in stably transfected human HCT116 cell line (Supplementary Figure S7B). In addition, aggregation of endogenous proteins was detected in naïve MEF cells treated with 200 μg/mL of the aminoglycoside antibiotic gentamicin, which is known to promote stop codon read-through (Figure 4E) (50,51). Aggregation was time-dependent, and was clearly observed after 48 h. Aminoglycoside-dependent aggregation was also detected in MEF cells treated with G418 as well as in naïve HeLa and HEK293T cells (Supplementary Figure S7C and D). As expected, aggregation of endogenous proteins was not found in MEF cells cultured in the presence of Ataluren (PTC124) that promotes read-through of premature termination codons (Supplementary Figure S7E). Moreover, we found high levels of aggregation following knockdown of release factor 1 (RF1) that promotes stop codon read-through (Figure 4F) (35). Finally, we expected the observed protein aggregation to trigger stress responses, such as the unfolded protein response (UPR) that is characterized by Ser51-phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α). Indeed, in stably transfected HEK293T cells we measured a two-fold increase in eIF2α Ser51-phosphorylation upon addition of BocLys, indicating the activation of cellular stress responses in response to UAG read-through (Supplementary Figure S8). Knockdown of protein kinase R-like endoplasmic reticulum kinase (PERK) significantly reduced eIF2α phosphorylation, suggesting that read-through-dependent aggregation activates the PERK-eIF2α UPR branch. Together, these results further support our observations of protein aggregation that is dependent on stop codon read-through and expression of proteins with 3′-UTR-encoded C-terminal extension.
3′-UTR-encoded protein sequences may have high aggregation propensity
In line with the observed effect of 3′-UTR-encoded polypeptide extensions on protein aggregation, we asked if there is a correlation between the measured depletion levels of extended proteins (Figure 1B), and the propensity of the extensions to form intrinsically unfolded regions or aggregates. To answer this question, we used the IUPred short algorithm as implemented in the IUPred2A server (52), and calculated the predicted propensity of the 3′-UTR-encoded polypeptides studied here (Figure 1B and Supplementary Table S1), to form intrinsically disordered regions. Plotting of cellular levels in soluble fractions (taken from Figure 1B), as a function of the propensity of each 3′-UTR-encoded polypeptide to form intrinsically disordered region, revealed that on average, disordered C-terminal polypeptide extensions have a lower tendency to promote protein depletion, compared to structured polypeptides extensions (Figure 5A). We also estimated the tendency of each 3′-UTR-encoded polypeptide to form aggregates using the TANGO algorithm (53,54). The plot of measured protein levels in soluble fractions as a function of predicted aggregation propensities, demonstrates that 3′-UTR-encoded polypeptides with high aggregation propensity also promoted depletion of proteins from soluble fractions, and viceversa (Figure 5B). In comparison, the correlation between depletion levels and length of C-terminal extension was less explicit (Supplementary Figure S9). Taken together, the experimental data and the predicted disorder and aggregation propensities of 3′-UTR-encoded polypeptides, suggest that proteins extended by sequences with a lower tendency to form intrinsically disordered regions and a higher tendency to form aggregates, are sequestered more efficiently from soluble fractions.
Figure 5.

Predicted disorder and aggregation propensities. (A) Correlation between measured cellular levels of C-terminally extended soluble eGFP (as described in Figure 1B) and the calculated tendency of C-terminal extensions to form intrinsically disordered regions. Straight line and shading represent the fitting of data to a linear model and 95% prediction intervals, respectively. (B) Aggregation propensities of 3′-UTR-derived protein sequences. Relative levels of soluble C-terminally extended eGFP (measured as described in Figure 1B) were plotted against the averaged aggregation propensity of the C-terminal extension, calculated using the TANGO server. (C) Average disorder propensity of 3′-UTR-encoded protein sequences, displayed as a function of the upstream termination signal. Median values are presented within each box; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. n = 1126, 656, 909, 973, 1854, 1468 sample points. P-values were calculated using the unpaired two sample Mann–Whitney–Wilcoxon test as implemented in R. Horizontal red line marks the average disorder propensity of random protein sequences of the same length distribution (0.41).
Next, we asked if a similar correlation can also be found between the fidelity of termination sequences (stop codon and the base at position +4), and protein sequences encoded by the 3′-UTR downstream of the termination sequence. To answer this question, we chose 3′-UTR sequences in which there are 75–150 bases (i.e. 25–50 amino acids) between the initial stop codon and the next in-frame stop codon (total of 6986 sequences), and calculated the averaged propensity of the corresponding protein sequence to form intrinsically disordered regions using the IUPred short algorithm. The averaged values were then plotted according to the expected fidelity of the annotated termination codon (UAA>UAG>UGA), while comparing between stop codons followed by C at the +4 position (more leaky) and stop codons followed by A at the +4 position (Figure 5C) (2–5). As control, we used random protein sequences of the same length distribution. As seen in Figure 5C, we found a statistically significant correlation between the fidelity of the annotated termination sequence (four bases) and the predicted disorder propensity of the downstream 3′-encoded protein sequence. On average, polypeptides derived from 3′-UTR sequences downstream of more leaky C at the +4 position are predicted to be more disordered, compared to sequences downstream of the less leaky A at the +4 position of the same termination codon (for alternative representation of the data see Supplementary Figure S10). In contrast, no significant correlation was found between the length of the same 3′-encoded polypeptides and the fidelity of the annotated upstream termination signal (Supplementary Figure S11A). Similarly, we found no statistically significant differences between random polypeptide sequences of the same length distribution (Supplementary Figure S10). Moreover, the averaged median disorder propensities of random polypeptide sequences (0.41, red horizontal line in Figure 5C), was lower than the median value of the most leaky termination sequences (UAGC and UGAC, 0.47 and 0.46, respectively) and higher than the median value of the most efficient termination sequence (UAAA, 0.32). A similar correlation between the identities of termination sequences and averaged disorder propensities was found in 10–24 amino acids long 3′-UTR-encoded polypeptides, and in genomes of other organisms (Supplementary Figures S11B and S12). Hence, our calculations suggest that on average, 3′-UTR-encoded polypeptides following leaky termination sequences have a higher propensity to form intrinsically disordered regions, compared to polypeptides downstream of more efficient termination sequences.
DISCUSSION
The immediate consequence of failed termination of translation at annotated 3′ stop codons, is translation of the 3′-UTR, followed by termination at the following in-frame stop codon or stalling at the poly(A) sequence. In the case of the former, a C-terminally extended protein with potentially deleterious effects may be synthesized. It was previously suggested that such proteins are eliminated, but the mechanism, as well as the properties of 3′-UTR-encoded polypeptides that promote their segregation are still elusive. Here, we found that such proteins can form aggregates that are then targeted to lysosomes. Our results suggest a new quality control mechanism for the elimination of read-through protein products, that is driven by the intrinsic property of some 3′-UTR-encoded polypeptides to form aggregates.
One possible cellular mechanism that can explain our observations is aggrephagy—the selective sequestration of protein aggregates by autophagy (55,56). The two main pathways for protein degradation in mammalian cells are ubiquitination-dependent proteasomal degradation and autophagy [reviewed in (57–59)]. Individual misfolded or unfolded proteins that are not cleared efficiently by the ubiquitin-proteasome system, or cannot properly refold by molecular chaperons, may form aggregates that accumulate in the form of compact structures, namely aggresomes (60–63). To advance the clearance of potentially harmful aggregates, aggresomes trigger autophagy and are targeted to lysosomes (60). In agreement with this mechanism, we found that sequestered C-terminally extended proteins form aggregates that accumulate in typical perinuclear structures and are targeted to lysosomes. It is not clear though if aggregation is necessary for the elimination of C-terminally extended proteins or whether other mechanisms exist. An aggregation-independent degradation process, if exists, would probably take place when extended proteins are expressed at low levels. In addition, as accumulation of single aggresomal particles in aggresomes is not necessarily ubiquitination-dependent (63), it is yet to be determined if aggregated C-terminally extended proteins in aggresomes must be ubiquitinated prior to aggregation, accumulation in aggresomes, or lysosomal degradation.
We also found a negative correlation between the calculated propensity of 3′-UTR-encoded polypeptide sequences to form intrinsically disordered regions, and their tendency to form aggregates in cells. In addition, we found that polypeptides encoded by 3′-UTR sequences positioned downstream of leaky termination signals (stop codon and the following base), have a higher tendency to form intrinsically disordered structures. Assuming that 3′-UTR sequences are random, the polypeptides encoded by these sequences should have random propensities to form intrinsically disordered regions or aggregates. Therefore, a strong termination signal can provide an evolutionary advantage, if positioned upstream to 3′-UTR sequence encoding a polypeptide with high aggregation propensity. Following this logic, it would be interesting to find if the identity and distribution of termination sequences were shaped, among other factors, by the structural properties of the protein sequences encoded by the downstream 3′-UTR. Specifically, one may suggest that the population of genes with 3′-UTR sequences that encode for aggregation-prone polypeptides, was enriched by high-fidelity termination sequences.
In a study based on the Human Gene Mutation Database (HGMD November 2007; www.hgmd.org), Mort et al. found that nonsense mutations account for ∼11% of mutations that cause human inherited diseases (64). Therefore, therapeutic strategies aimed at suppressing translation termination at premature stop codons offer an attractive route for treatment of a wide variety of human diseases (65). One approach is based on the use of small molecule drugs to promote read-through of premature stop codons. For example, aminoglycoside antibiotics that bind the decoding center of the ribosome and promote misincorporation at both sense and termination codons. Despite their higher affinity to bacterial ribosomes, several aminoglycosides can bind the eukaryotic ribosome and promote stop codon read-through by misincorporation of amino acids in response to premature termination codons. One of the commonly used aminoglycoside in stop codon suppression studies, is gentamicin. For instance, the ability of gentamicin to restore physiologically relevant levels of functional proteins was studied in the context of cystic fibrosis and Duchenne muscular dystrophy (50,51). However, chronic use of gentamicin is prohibited by the risk of renal toxicity and ototoxicity (66,67). Our data, showing read-through-dependent protein aggregation, stress the need to rule out protein aggregation in studies of therapies based on read-through of stop codons. It also highlights the importance of identifying small molecules that promote read-through of premature, and not 3′-encoded, termination codons.
An important mechanism of the adaptive immune system, is the recognition of proteolytic peptide fragments presented to CD8+ T cells on major histocompatibility complex (MHC) molecules. Following our observation of read-through-dependent protein aggregation and targeting to lysosomes, one may suggest that proteolytic peptides derived from 3′-UTR-encoded C-terminal extensions can be presented on MHC molecules. Since 3′-UTR sequences are usually not translated, the derived polypeptide sequences can be recognized as foreign sequences and trigger autoimmune response. Indeed, treatment with gentamicin can promote MHC class I presentation of cryptic peptides or a model peptide encoded downstream of an in-frame premature termination codon (68). These results not only support our observations, but also provoke the question whether read-through-dependent aggregation is one of the mechanisms leading to autoimmune responses in specific human pathologies.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016728. All python scripts are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Uzi Hadad, from the Flow Cytometry and Cellular Imaging Lab at the Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University, for his technical support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council (ERC) under the European Union Horizon 2020 Research and Innovation Programme [678461 to E.A.] and [639313]; Israel Science Foundation [807/15 to E.A.]. Funding for open access charge: ERC under the European Union Horizon 2020 Research and Innovation Programme [678461 to E.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jackson R.J., Hellen C.U.T., Pestova T.V.. Termination and post-termination events in eukaryotic translation. Adv. Protein Chem. Struct. Biol. 2012; 86:45–93. [DOI] [PubMed] [Google Scholar]
- 2. Phillips-Jones M.K., Hill L.S., Atkinson J., Martin R.. Context effects on misreading and suppression at UAG codons in human cells. Mol. Cell Biol. 1995; 15:6593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCaughan K.K., Brown C.M., Dalphin M.E., Berry M.J., Tate W.P.. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:5431–5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cridge A.G., Crowe-McAuliffe C., Mathew S.F., Tate W.P.. Eukaryotic translational termination efficiency is influenced by the 3′ nucleotides within the ribosomal mRNA channel. Nucleic Acids Res. 2018; 46:1927–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anzalone A.V., Zairis S., Lin A.J., Rabadan R., Cornish V.W.. Interrogation of Eukaryotic stop codon readthrough signals by in vitro RNA selection. Biochemistry. 2019; 58:1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harrell L., Melcher U., Atkins J.F.. Predominance of six different hexanucleotide recoding signals 3′ of read-through stop codons. Nucleic Acids Res. 2002; 30:2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Namy O., Hatin I., Rousset J.-P.. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001; 2:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loughran G., Chou M.-Y., Ivanov I.P., Jungreis I., Kellis M., Kiran A.M., Baranov P.V., Atkins J.F.. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 2014; 42:8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schueren F., Lingner T., George R., Hofhuis J., Dickel C., Gärtner J., Thoms S.. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. Elife. 2014; 3:e03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Firth A.E., Brierley I.. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012; 93:1385–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pelham H.R. Leaky UAG termination codon in tobacco mosaic virus RNA. Nature. 1978; 272:469–471. [DOI] [PubMed] [Google Scholar]
- 12. True H.L., Lindquist S.L.. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000; 407:477–483. [DOI] [PubMed] [Google Scholar]
- 13. Freitag J., Ast J., Bölker M.. Cryptic peroxisomal targeting via alternative splicing and stop codon read-through in fungi. Nature. 2012; 485:522–525. [DOI] [PubMed] [Google Scholar]
- 14. Stiebler A.C., Freitag J., Schink K.O., Stehlik T., Tillmann B.A.M., Ast J., Bölker M.. Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in fungi and animals. PLoS Genet. 2014; 10:e1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jungreis I., Lin M.F., Spokony R., Chan C.S., Negre N., Victorsen A., White K.P., Kellis M.. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011; 21:2096–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunn J.G., Foo C.K., Belletier N.G., Gavis E.R., Weissman J.S.. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife. 2013; 2:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamaguchi Y., Hayashi A., Campagnoni C.W., Kimura A., Inuzuka T., Baba H.. L-MPZ, a novel isoform of Myelin P0, is produced by stop codon readthrough. J. Biol. Chem. 2012; 287:17765–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geller A.I., Rich A.. A UGA termination suppression tRNATrp active in rabbit reticulocytes. Nature. 1980; 283:41–46. [DOI] [PubMed] [Google Scholar]
- 19. Eswarappa S.M., Potdar A.A., Koch W.J., Fan Y., Vasu K., Lindner D., Willard B., Graham L.M., DiCorleto P.E., Fox P.L.. Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell. 2014; 157:1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dabrowski M., Bukowy-Bieryllo Z., Zietkiewicz E.. Translational readthrough potential of natural termination codons in eucaryotes The impact of RNA sequence. RNA Biol. 2015; 12:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Namy O., Rousset J.-P.. Atkins JF, Gesteland RF. Specification of standard Amino Acids by stop codons. Nucleic Acids and Molecular Biology. 2010; NY: Springer; 79–100. [Google Scholar]
- 22. Hollingsworth T.J., Gross A.K.. The severe autosomal dominant retinitis pigmentosa rhodopsin mutant Ter349Glu mislocalizes and induces rapid rod cell death. J. Biol. Chem. 2013; 288:29047–29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vidal R., Frangione B., Rostagno A., Mead S., Révész T., Plant G., Ghiso J.. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999; 399:776–781. [DOI] [PubMed] [Google Scholar]
- 24. Pang S., Wang W., Rich B., David R., Chang Y.T., Carbunaru G., Myers S.E., Howie A.F., Smillie K.J., Mason J.I.. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3β-Hydroxysteroid Dehydrogenase (3β-HSD) gene causing, respectively, nonclassic and classic 3β-HSD deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2002; 87:2556–2563. [DOI] [PubMed] [Google Scholar]
- 25. Shibata N., Ohoka N., Sugaki Y., Onodera C., Inoue M., Sakuraba Y., Takakura D., Hashii N., Kawasaki N., Gondo Y. et al.. Degradation of stop codon Read-through mutant proteins via the Ubiquitin-Proteasome system causes hereditary disorders. J. Biol. Chem. 2015; 290:28428–28437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frischmeyer P.A. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002; 295:2258–2261. [DOI] [PubMed] [Google Scholar]
- 27. Doma M.K., Parker R.. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006; 440:561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sitron C.S., Brandman O.. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 2019; 26:450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brandman O., Hegde R.S.. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016; 23:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Inada T. The ribosome as a platform for mRNA and nascent polypeptide quality control. Trends Biochem. Sci. 2017; 42:5–15. [DOI] [PubMed] [Google Scholar]
- 31. Arribere J.A., Cenik E.S., Jain N., Hess G.T., Lee C.H., Bassik M.C., Fire A.Z.. Translation readthrough mitigation. Nature. 2016; 534:719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hashimoto S., Nobuta R., Izawa T., Inada T.. Translation arrest as a protein quality control system for aberrant translation of the 3′UTR in mammalian cells. FEBS Lett. 2019; 593:777–787. [DOI] [PubMed] [Google Scholar]
- 33. Yordanova M.M., Loughran G., Zhdanov A.V., Mariotti M., Kiniry S.J., O’Connor P.B.F., Andreev D.E., Tzani I., Saffert P., Michel A.M. et al.. AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nature. 2018; 553:356–360. [DOI] [PubMed] [Google Scholar]
- 34. Strack R.L., Hein B., Bhattacharyya D., Hell S.W., Keenan R.J., Glick B.S.. A rapidly maturing Far-Red derivative of DsRed-Express2 for whole-cell labeling. Biochemistry. 2009; 48:8279–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Janzen D.M., Geballe A.P.. The effect of eukaryotic release factor depletion on translation termination in human cell lines. Nucleic Acids Res. 2004; 32:4491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kimura S., Noda T., Yoshimori T.. Dissection of the autophagosome maturation process by a novel reporter protein, tandem Fluorescent-Tagged LC3. Autophagy. 2007; 3:452–460. [DOI] [PubMed] [Google Scholar]
- 37. Elsässer S.J., Ernst R.J., Walker O.S., Chin J.W.. Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat. Methods. 2016; 13:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bryson D.I., Fan C., Guo L.-T., Miller C., Söll D., Liu D.R.. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017; 13:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oren Y.S., McClure M.L., Rowe S.M., Sorscher E.J., Bester A.C., Manor M., Kerem E., Rivlin J., Zahdeh F., Mann M. et al.. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol. Med. 2014; 6:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zougman A., Selby P.J., Banks R.E.. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 2014; 14:1006–1000. [DOI] [PubMed] [Google Scholar]
- 41. Scheltema R.A., Hauschild J.-P., Lange O., Hornburg D., Denisov E., Damoc E., Kuehn A., Makarov A., Mann M.. The Q Exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field orbitrap analyzer. Mol. Cell Proteomics. 2014; 13:3698–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tyanova S., Temu T., Cox J.. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016; 11:2301–2319. [DOI] [PubMed] [Google Scholar]
- 43. Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M.. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011; 10:1794–1805. [DOI] [PubMed] [Google Scholar]
- 44. Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M.. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014; 13:2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ladner C.L., Yang J., Turner R.J., Edwards R.A.. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004; 326:13–20. [DOI] [PubMed] [Google Scholar]
- 46. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S. et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O. et al.. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007; 447:860–864. [DOI] [PubMed] [Google Scholar]
- 49. Leeman D.S., Hebestreit K., Ruetz T., Webb A.E., McKay A., Pollina E.A., Dulken B.W., Zhao X., Yeo R.W., Ho T.T. et al.. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018; 359:1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wilschanski M., Yahav Y., Yaacov Y., Blau H., Bentur L., Rivlin J., Aviram M., Bdolah-Abram T., Bebok Z., Shushi L. et al.. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl. J. Med. 2003; 349:1433–1441. [DOI] [PubMed] [Google Scholar]
- 51. Malik V., Rodino-Klapac L.R., Viollet L., Wall C., King W., Al-Dahhak R., Lewis S., Shilling C.J., Kota J., Serrano-Munuera C. et al.. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010; 67:771–80. [DOI] [PubMed] [Google Scholar]
- 52. Mészáros B., Erdős G., Dosztányi Z.. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018; 46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Linding R., Schymkowitz J., Rousseau F., Diella F., Serrano L.. A comparative study of the relationship between protein structure and β-Aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004; 342:345–353. [DOI] [PubMed] [Google Scholar]
- 54. Fernandez-Escamilla A.-M., Rousseau F., Schymkowitz J., Serrano L.. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004; 22:1302–1306. [DOI] [PubMed] [Google Scholar]
- 55. Øverbye A., Brinchmann M.F., Seglen P.O.. Proteomic analysis of Membrane-Associated proteins from rat liver autophagosomes. Autophagy. 2007; 3:300–322. [DOI] [PubMed] [Google Scholar]
- 56. Tan S., Wong E.. Kinetics of protein aggregates disposal by aggrephagy. Methods Enzymol. 2017; 588:245–281. [DOI] [PubMed] [Google Scholar]
- 57. Finley D. Recognition and processing of Ubiquitin-Protein conjugates by the proteasome. Ann. Rev. Biochem. 2009; 78:477–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kroemer G., Mariño G., Levine B.. Autophagy and the integrated stress response. Mol. Cell. 2010; 40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mizushima N., Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011; 147:728–741. [DOI] [PubMed] [Google Scholar]
- 60. Johnston J.A., Ward C.L., Kopito R.R.. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 1998; 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Burnett B.G., Pittman R.N.. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:4330–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Garcia-Mata R., Gao Y.-S., Sztul E.. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002; 3:388–396. [DOI] [PubMed] [Google Scholar]
- 63. García-Mata R., Bebök Z., Sorscher E.J., Sztul E.S.. Characterization and dynamics of aggresome formation by a cytosolic Gfp-Chimera. J. Cell Biol. 1999; 146:1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A.. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008; 29:1037–1047. [DOI] [PubMed] [Google Scholar]
- 65. Keeling K.M., Xue X., Gunn G., Bedwell D.M.. Therapeutics based on stop codon readthrough. Ann. Rev. Genomics Hum. Genet. 2014; 15:371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lopez-Novoa J.M., Quiros Y., Vicente L., Morales A.I., Lopez-Hernandez F.J.. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 2011; 79:33–45. [DOI] [PubMed] [Google Scholar]
- 67. Huth M.E., Ricci A.J., Cheng A.G.. Mechanisms of Aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011; 2011:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goodenough E., Robinson T.M., Zook M.B., Flanigan K.M., Atkins J.F., Howard M.T., Eisenlohr L.C.. Cryptic MHC class I-binding peptides are revealed by aminoglycoside-induced stop codon read-through into the 3′ UTR. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:5670–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mi H., Muruganujan A., Ebert D., Huang X., Thomas P.D.. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019; 47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016728. All python scripts are available upon request.