


Abstract
We have previously shown that the highly prevalent acute myeloid leukemia (AML) mutation, Arg882His, in DNMT3A disrupts its cooperative mechanism and leads to reduced enzymatic activity, thus explaining the genomic hypomethylation in AML cells. However, the underlying cause of the oncogenic effect of Arg882His in DNMT3A is not fully understood. Here, we discovered that DNMT3A WT enzyme under conditions that favor non-cooperative kinetic mechanism as well as DNMT3A Arg882His variant acquire CpG flanking sequence preference akin to that of DNMT3B, which is non-cooperative. We tested if DNMT3A Arg882His could preferably methylate DNMT3B-specific target sites in vivo. Rescue experiments in Dnmt3a/3b double knockout mouse embryonic stem cells show that the corresponding Arg878His mutation in mouse DNMT3A severely impairs its ability to methylate major satellite DNA, a DNMT3A-preferred target, but has no overt effect on the ability to methylate minor satellite DNA, a DNMT3B-preferred target. We also observed a previously unappreciated CpG flanking sequence bias in major and minor satellite repeats that is consistent with DNMT3A and DNMT3B specificity suggesting that DNA methylation patterns are guided by the sequence preference of these enzymes. We speculate that aberrant methylation of DNMT3B target sites could contribute to the oncogenic potential of DNMT3A AML variant.
INTRODUCTION
DNA methylation in mammals is critical for development and maintenance of the somatic cell state (1,2). DNA methylation has diverse functions including regulation of gene expression and silencing of repetitive elements (3–5). In mammals, CpG methylation is established and maintained by DNA methyltransferases (DNMTs) (6). DNMT1 largely functions as a maintenance methyltransferase by copying the methylation pattern from parent to daughter strand during DNA replication (7,8). The DNMT3 family includes two active homologs, DNMT3A and DNMT3B, and an inactive homolog DNMT3L (9,10). These enzymes perform de novo DNA methylation, which predominantly takes place during early embryogenesis and stem cell differentiation (9,11,12). Whereas DNMT3A is expressed ubiquitously, DNMT3B is highly expressed during early embryogenesis and largely silenced in somatic cell types (13–15). Despite having a high sequence similarity (>40%), DNMT3A and DNMT3B have distinct preferences for some target sites, although many sites can be methylated redundantly (16). This is represented by a preference of DNMT3A for major satellite repeats and a preference of DNMT3B for minor satellite repeats (9,11).
Mutations in DNMT3A and DNMT3B have been identified in several diseases. Germline transmitted mutations of DNMT3A and DNMT3B cause Tatton–Brown–Rahman syndrome and ICF (Immunodeficiency, Centromeric instability, and Facial anomalies) syndrome, respectively (9,17–20). Somatic mutations in DNMT3A are commonly found in patients with acute myeloid leukemia (AML) and other hematologic neoplasms (21,22). Detailed studies revealed that ∼20% of AML cases have heterozygous DNMT3A mutations, with the majority (∼60–70%) carrying the Arg882His mutation, which cause genome-wide hypomethylation (21,23–25). Given that genetic knockout of one copy of DNMT3A exhibits no obvious phenotype, the heterozygous DNMT3A Arg882His mutation was suggested to have a dominant negative effect (9,21). The observation that the expression of the murine DNMT3A Arg878His variant (equivalent to Arg882His in human DNMT3A) in mouse embryonic stem cells (mESCs) causes genome-wide loss of DNA methylation further supported the dominant negative activity of this variant (26,27). In contrast, DNMT3B mutations have not been identified in cancer. However, aberrant overexpression of DNMT3B is highly tumorigenic, including in AML (14,28–30). Overexpression of DNMT3B in AML leads to disease prognosis similar to that of patients with DNMT3A Arg882His mutation (29,30).
The effect of Arg882His on the activity of DNMT3A could be predicted from the crystal structure of the DNMT3A catalytic domain (DNMT3A-C), which shows that DNMT3A forms homodimers and tetramers through two interaction surfaces (31,32). Several AML-associated DNMT3A mutations, including Arg882His, are present at or close to the protein-protein interface and disrupt tetramer formation. This leads to reduced catalytic activity in vitro (21,27,33–36). Further, DNMT3A-C tetramers can oligomerize on DNA, forming nucleoprotein filaments (37,38). This oligomerization allows the enzyme to bind and methylate multiple CpG sites in a cooperative manner, thus exponentially increasing its catalytic activity (39). Our previous biochemical studies show that in DNMT3A-C, the Arg882His substitution results in loss of cooperativity, causing a significant decrease in its catalytic activity. Interestingly, we also reported that in DNMT3B, which is a non-cooperative enzyme, the homologous mutation, Arg829His, has no effect on its catalytic activity in vitro (40). Further, the DNMT3A-C Arg882His variant was shown to have a bias for G at N+3 position from CpG site (N = CG dinucleotide) (41). This is supported by the interaction of Arg882 with phosphate backbone at the N+3 position observed in co-crystal structure of the DNMT3A-C/DNA complex (42). However, it is unclear whether change in flanking sequence preference is directly due to the substitution of Arg with His or caused by loss of cooperative mechanism in the variant enzyme.
Our data show that all variants of Arg882 found in AML patients have low catalytic activity and lack a cooperative mechanism. We further show that in the absence of cooperative mechanism at low enzyme concentrations, the WT DNMT3A-C enzyme prefers G at the N+3 position, thus behaving like the AML variant, DNMT3A-C Arg882His. To test the role of cooperativity, we compared the flanking sequence preference of non-cooperative enzyme DNMT3B, with that of the DNMT3A-C WT and the Arg882His variant. DNA methylation of 124 CpGs in four different substrates was rank ordered to compute consensus sequence motifs that are preferred by these enzymes. Interestingly, our data show that the Arg882His variant and DNMT3B-C have a similar preference for nucleotides at N+1, 2 and 3 positions flanking the CpG site. These data strongly support that the gain of flanking sequence preference is due to loss of cooperative mechanism in DNMT3A-C Arg882His enzyme, and suggest that the variant could methylate DNMT3B-C preferred sites. We tested this ‘gain of function’ prediction by expressing WT mouse DNMT3A or the Arg878His (corresponding to Arg882His in human DNMT3A) in Dnmt3a/3b double knockout (DKO) mESCs. Our data show that whereas the DNMT3A Arg878His variant failed to rescue methylation at the major satellite repeats (DNMT3A preferred target sites), its ability to methylate the minor satellite repeats (DNMT3B preferred target sites) was comparable to that of DNMT3A WT enzyme. Further, our analysis of the CpG flanking sequences of the satellite repeats show that whereas major satellite repeats have an A or T at the N+3 position, the minor satellite repeats are enriched in G at N+3 position. This observation provides the previously unknown explanation for the substrate specificity of DNMT3A and DNMT3B for major and minor satellite repeats, respectively and suggets that the genome wide patterns of DNA methylation could be strongly influenced by the intrinsic substrate preference of DNMT3A and DNMT3B. Taken together, our data provide novel mechanistic insights into DNMT3A and DNMT3B substrate specificities which could infleunce the genomic pettern of DNA methylation and the oncogenic potential of these enzymes. We suggest that in leukemia, DNMT3A Arg882His substitution establishes a DNMT3B-like activity, the tumorigenic properties of which are exploited by cancer cells.
MATERIALS AND METHODS
Protein purification
Human DNMT3A-C WT, DNMT3A-C Arg882His, DNMT3A-C Arg882Cys, and DNMT3A-C Arg882Ser and mouse DNMT3B-C WT, DNMT3B-C Arg829His and DNMT3B-C Glu703Ala in pET28a(+) with a 6X His tag, were expressed and purified using affinity chromatography as described (40). Briefly, transformed BL21 (DE3) pLys cells were induced with 1 mM IPTG at OD600 0.3 and expressed for 2 h at 32°C. Harvested cells were washed with STE buffer (10 mM Tris–HCl (pH 8.0), 0.1 mM EDTA, 0.1 M NaCl), and resuspended in Buffer A (20 mM potassium phosphate (pH 7.5), 0.5 M NaCl, 10% (v/v) glycerol, 1 mM EDTA, 0.1 mM DTT, 40 mM imidazole). Cells were disrupted by sonication, followed by removal of cell debris by centrifugation. Clarified lysate was incubated with 0.4 ml Ni-NTA agarose for 3 h at 4°C. The protein bound slurry was packed in a 2 ml Biorad column and washed with 150 ml Buffer A. Protein was eluted using 200 mM imidazole in Buffer A at pH 7.5, then stored in 20 mM HEPES pH 7.5, 40 mM KCl, 1 mM EDTA, 0.2 mM DTT and 20% (v/v) glycerol at −80°C. The purity and integrity of recombinant proteins were checked by SDS-PAGE gel.
DNA methylation assays using radiolabeled AdoMet
Radioactive methylation assays to determine kinetic parameters of recombinant enzymes were performed using 3H-labeled S-adenosylmethionine (AdoMet) as a methyl group donor and biotinylated oligonucleotides of varying sizes bound on avidin-coated high-binding Elisa plates (Corning) as described (43). The DNA methylation reactions were carried out using either 250 nM 30-bp/32-bp DNA substrate in methylation buffer (20 mM HEPES pH 7.5, 50 mM KCl and 1 mM EDTA, 5 μg/ml BSA). The methylation reaction included 0.76 μM [methyl3H] AdoMet (PerkinElmer Life Sciences). Storage buffer was added to compensate for the different enzyme volumes in all reactions. Incorporated radioactivity was quantified by scintillation counting.
Processivity assay
Methylation kinetic analyses were performed using two enzyme concentrations and short oligonucleotide 30- and 32-bp substrates with 1 and 2 CpG sites, respectively. Low enzyme concentrations relative to DNA substrate concentrations were used to ensure that the reaction occurred under multiple turnover conditions. Each DNA substrate was used at 250 nM, and a 1:1 ratio of labelled and unlabeled AdoMet (final concentration 1.5 μM) was used.
Cooperativity assays
To examine cooperativity, increasing concentrations of enzyme were pre-incubated with DNA substrate for 10 min at room temperature prior to the addition of AdoMet to start the reaction. AdoMet was a mixture of unlabeled and 0.76 μM 3[H] labeled AdoMet to yield a final concentration of 2 μM. Methylation assays were performed using 100 ng of an unmethylated pUC19 plasmid purified from dam− /dcm−Escherichia coli strain (C2925I, NEB) or a 1-kb fragment containing 14 CpG sites amplified from the Meis1 enhancer were used as DNA substrates for filter binding assays. Briefly, 10 μl reaction mix was spotted on 0.5 in DE81 filter that was then washed five times in 0.2 M Ammonium Bicarbonate (NH4HCO3), washed 2–3 times with 100% ethanol, and air dried. Incorporated radioactivity was quantified by scintillation counting (44).
Flanking sequence preference
In vitro DNA methylation reactions were carried out using 100 ng of a 509-bp DNA fragment amplified from the SUHW1 promoter, 100 ng of a 1-kb DNA fragment amplified from the Meis1 enhancer region, 100 ng of a 721-bp DNA fragment amplified from the Sirt4 enhancer region, or 100 ng of a 1092-bp region of the pUC19 plasmid. Methylation reactions were carried out in methylation buffer (20 mM HEPES pH 7.5, 50 mM KCl, 1 mM EDTA and 0.05 mg/ml BSA) using varying concentrations of each enzyme at 37°C. Samples were taken at 10, 30 or 60 min and reaction was stopped by freeze/thaw. DNA methylation was analyzed by bisulfite sequencing as described below.
Flanking sequence preference was also measured using short oligonucleotides and DNA methylation assays using radiolabeled AdoMet as described above. Sixteen different 30-bp substrates were used with varying combinations of the second and third nucleotide around the CpG site on either side (Supplementary Table S3).
Bisulfite sequencing
Bisulfite conversion was performed using EpiTect Fast Bisulfite Conversion Kit (Qiagen, 59802) according to the manufacturer's protocol. PCR amplifications were performed with primers as described (39). The pooled samples were sequenced using NGS on Wide-Seq platform. The reads were assembled and analyzed by Bismark and Bowtie2. Methylated and unmethylated CpGs for each target were quantified, averaged, and presented as percent CpG methylation. Methylation assays were done at least 3 times using 3 different enzyme preparations and average percent methylation for each CpG was used to calculate preference. The standard error of the mean (SEM) for total percent methylation at all CpGs for each enzyme was also reported.
Rescue experiments in mESCs
WT (J1) and Dnmt3a/3b DKO mESCs were cultured on gelatin-coated petri dishes in mESC medium (DMEM supplemented with 15% fetal bovine serum, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 50 U/ml penicillin, 50 μg/ml streptomycin, and 103 U/ml leukemia inhibitory factor) (9). For the generation of stable clones expressing DNMT3A or DNMT3B proteins, mESCs were transfected with plasmid vectors using Lipofectamine 2000 (Invitrogen) and then seeded at low density on dishes coated with feeder cells, selected with 6 μg/ml of Blasticidin S HCl (Gibco) for 7–10 days, and individual colonies were picked. Southern blot analysis of DNA methylation at the major and minor satellite repeats were performed as previously described (11,26).
Data analysis
Comparative flanking sequence preference analysis
To compare the flanking sequence preference of different enzymes, we first calculated average percent methylation of all CpG sites in the substrate. The fractional variance (v) at each CpG site was calculated by dividing in the percent methylation of each site by average methylation. The preference for a site by an enzyme (B) over enzyme (A) was calculated as relative change (vB – vA)/vA, Positive values indicate a preference of enzyme B for a site and values greater than or equal to 1 were considered significant given the preference is >2-fold (Supplementary Table S1).
To determine the fractional distribution of nucleotides at preferred sites the occurrence of each nucleotide at N+1/2/3 positions was calculated as a fraction (N = CG dinucleotide). At each position, the number of times each nucleotide occurred was divided by the total number of preferred sites toc compute the fractional occurrence.
Individual flanking sequence preference analysis
Analysis of the bisulfite sequencing data was performed to determine optimal flanking sequence preferred by each enzyme. To consider the uneven distribution of nucleotides flanking the CpG site, the occurrence of each nucleotide at the N+1/2/3 positions (p1n, p2n and p3n) was calculated by dividing the number of times it occurred by the total number of CpG sites (s(p1n), s(p2n), s(p3n)). The expected occurrence of a trinucleotide set (O) was computed by multiplying the occurrence of three nucleotides as described by equation (1). This created an expected value, which would predict the probability at which this trinucleotide set would be methylated if there were no flanking sequence preference by the enzyme. With the data obtained from bisulfite sequencing, the fractional methylation (f) for each CpG site was calculated by dividing percent methylation at a site by sum of percent methylation at all sites as described by equation (2). Fractional methylation was sorted by nucleotides at position N+1/2/3 (f(p1n), f(p2n) and f(p3n)) and summed for each nucleotide at a position (Σf(p1n), Σf(p2n) and Σf(p3n)). The methylation for the flanking trinucleotide sets (M) was calculated then multiplying the summed value for a nucleotide at the three positions as described by equation (3). This gives us the value for observed methylation value at a CpG with a specific flanking trinucleotide. The preference for a flanking trinucleotide by an enzyme was calculated by determining the fold change between the observed and expected values as described by equation (4) (Supplementary Table S2).
Equations
v = fractional variance
f = fractional methylation
n = any nucleotide
A, C, G, T = specific nucleotides
s = fraction (number of sites out/56)
p1 = nucleotide at position N+1
p2 = nucleotide at position N+2
p3 = nucleotide at position N+3
M = observed methylation based on fractional methylation
O = normalized occurrence of each site based on their frequency in the DNA substrate
Σ = sum
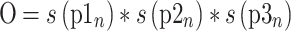 |
(1) |
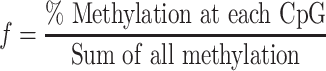 |
(2) |
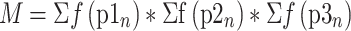 |
(3) |
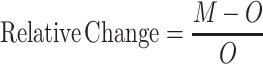 |
(4) |
Consensus sequence analysis of major and minor satellite repeats
From the mm9 genome, three regions, chr9:3000466–3028144, chr9:3033472–3037264 and chr2:98506820–98507474, were used for the sequences of the major satellite repeats. The minor satellite repeat sequences were obtained from chr2:98505036–98505275 and chr2:98506495–98506615. Additionally, more sequences were obtained from GenBank and used for analysis (accession no. X14462.1, X14463.1, X14464.1, X14465.1, X14466.1, X14468.1, X14469.1, X14470.1). Consensus sequences of the CpG sites and flanking regions in the major and minor satellite repeats were built using WebLogo (https://weblogo.berkeley.edu/logo.cgi).
Line and Bar graphs
Data were analyzed using the Prism software (GraphPad). For cooperativity graphs, data were fit with nonlinear, second order polynomial regression curves. Errors were calculated as standard error of the mean (SEM) for two to four independent experiments, as described in the figure legends. P-values for the relative change in flanking sequence preference was calculated with the following formula with Excel.
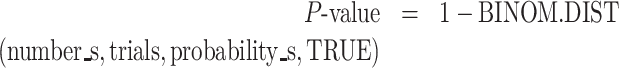 |
(5) |
RESULTS
The cooperative kinetic mechanism is absent in DNMT3A Arg882 variants
DNMT3A-C WT methylates neighboring CpGs on DNA by a cooperative kinetic mechanism, in which the DNA bound tetramer promotes successive binding events through protein-protein interaction (39). Our previous studies also show that the DNMT3A-C Arg882His variant fails to methylate DNA by a cooperative mechanism, which could be due to impaired tetramerization (26,33,40). It is not clear, however, whether this is due to the loss of Arg or gain of His at this position. Based on the observation that many AML patients have mutations that lead to substitution of Arg882 to Cys or Ser, we asked if these variant enzymes could methylate multiple CpGs on a single DNA molecule in a cooperative manner. His-tagged recombinant DNMT3A-C (catalytic domain) WT and Arg882 variants were produced in E. coli and purified using Ni-NTA affinity chromatography to ∼90–95% purity (Supplementary Figure S1A) (45). The catalytic activity of DNMT3A-C Arg882 variants was compared to the WT enzyme by performing DNA methylation assays using a 30-bp substrate containing one CpG site. Consistent with previous reports, a 60–80% loss of catalytic activity was observed in all variants compared to WT enzyme (Figure 1A) (27,33,35,36,40,41). We next assayed the cooperative kinetic mechanism of these enzymes. This is observed as an exponential relationship between the catalytic activity and enzyme concentrations as shown for DNMT3A-C WT enzyme. However, all Arg882 variant enzymes failed to methylate the substrate in a cooperative fashion (Figure 1B). From these data, we conclude that the Arg882 residue plays a key role in the cooperative mechanism of DNMT3A-C.
Figure 1.

Relative activity and kinetic mechanism of DNMT3A-C WT and Arg882 variants. (A) Methylation activity of 1 μM DNMT3A-C WT and Arg882 variants was measured for 5 and 10 min using 3[H] labelled S-Adenosylmethionine (AdoMet). The transfer of radiolabeled –CH3 group to DNA was measured as counts per minute (CPM) using the MicroBeta scintillation counter. (B) Methylation activity of DNMT3A-C WT and Arg882 variants was measured for 10 min using 100 ng of pUC19 plasmid as a substrate, at concentrations of enzymes varying from 0.25 to 1 μM. The enzymes were pre-incubated with DNA for 10 min at room temperature and the reaction was initiated by addition of AdoMet. Each data point is an average and standard error of the mean (n≥ 3 independent experiments).The data shows reduced activity and loss of cooperativity for all the variant enzymes.
Loss of cooperativity modulates flanking sequence preference of DNMT3A
Recent co-crystal structure of DNMT3A with DNA shows that the Arg882 residue interacts with the phosphate backbone of the nucleotide at N+3 position (N = CG dinucleotide) (42). It was also shown that Arg882His has a preference for G at the N+3 position compared to the WT enzyme by in vitro methylation assays where a 509-bp DNA fragment containing 56 CpG sites (SUHW1 promoter region) was used to compute relative preference of nucleotides at positions flanking the central CpG (41). Given that the Arg882 residue is also necessary for the cooperative kinetic mechanism of DNMT3A, we used the same experimental system and method of calculation to test if this preference was affected by cooperative kinetic mechanism of DNMT3A (Supplementary Table S1). We performed in vitro methylation of the SUHW1 DNA substrate using the WT and the Arg882His variant enzymes (Supplementary Figure S2A). DNA methylation was quantified by bisulfite conversion and high throughput sequencing. The preference of sites is represented by a relative change greater than 1. Our data confirmed the previously reported G preference at N+3 for Arg882His variant compared to the WT enzyme (Figure 2A). We next tested the relationship between loss of cooperative mechanism and flanking sequence preference. We have previously shown that at a low concentration (0.25 μM), DNMT3A-C does not multimerize on the SUHW1 promoter DNA substrate and therefore cannot methylate multiple CpGs using cooperative mechanism, which is observed at a higher concentration (1 μM) of the enzyme (40). DNA methylation assays were performed using 0.25 and 1 μM enzyme, and the flanking sequence preferences were compared. Strikingly, six out of seven sites preferred by DNMT3A-C at 0.25 μM have G at N+3 position, which matches the flanking sequence preference of the DNMT3A-C Arg882His variant (Figure 2B, Supplementary Figure S2B). Our data therefore suggest the role of cooperativity in modulating the interaction of the Arg882 residue with DNA.
Figure 2.

Effect of cooperativity on flanking sequence preference by DNMT3A-C. (A, B) DNA methylation of the 56 CpG sites in the SUHW1 promoter region (509-bp) substrate was analyzed using bisulfite sequencing. Methylation reaction was carried out using 1 μM DNMT3A-C WT, and 1 μM DNMT3A-C Arg882His in (A), and 0.25 μM and 1 μM DNMT3A-C WT in (B). Top panel shows relative preference calculated for each CpG site (1–56) by the DNMT3A-C Arg882His compared to DNMT3A-C WT in (A), and 0.25 μM compared to 1 μM of DNMT3A-C WT in (B). Relative preference of 1 is equal to a 2-fold change, and is represented by blue and light pink bars, respectively. Bottom panel shows the fractional distribution of nucleotides at the preferred sites. Each bar represents fractional distribution of nucleotides at positions N+1/2/3 respectively from the CpG site. Data presented are an average n = 3 independent experiments. The data show DNMT3A-C WT at low concentrations have flanking sequence preference similar to DNMT3A-C Arg882His variant.
Flanking sequence preference of DNMT3B has similarity to that of DNMT3A Arg882His
DNMT3B is a homolog of DNMT3A that is frequently overexpressed in tumors, including AML (14,29). Similar to DNMT3A-C Arg882His variant, DNMT3B-C functions as a non-cooperative enzyme (40). Therefore, we asked if DNMT3B-C and the DNMT3A-C Arg882His variant have a similar flanking sequence preference. Although the nucleotide preference of DNMT3B is reported for N+1 position (G), the extended flanking sequence preference has not been thoroughly evaluated (46). Using recombinant DNMT3B-C WT and the Arg829His variant (homologous to DNMT3A Arg882His), we performed in vitro methylation assays using 4 different DNA substrate ranging in size from 500 to 1000 bp. As DNMT3B and DNMT3A Arg882His have lower activity compared to DNMT3A WT, we used catalytically dead DNMT3B Glu703Ala variant to detect the background of methylation analysis by bisulfite sequencing. The methylation level was significantly higher for all the enzymes compared to the control. The SEM for the percent methylation at all CpG sites was in the same range for all the active enzymes (Supplementary Figure S3A, B). Therefore, using the average percent methylation at each CpG site we computed the relative preference for each CpG site by DNMT3A-C WT, DNMT3A-C Arg882His and DNMT3B-C WT. The preferred sites of DNMT3A-C Arg882His compared to DNMT3A-C WT showed pronounced increase at 17 sites, of which nine had G at the N+3 position and 11 had G at N+1 position (Figure 3A–C). Simultaneously, there were 21 sites preferred by DNMT3B-C WT compared to DNMT3A-C WT, of which 10 had G at the N+3 position and 12 had G at N+1 position (Figure 3D–F). Therefore, we discovered a strong overlap in the fractional distribution of bases preferred by DNMT3B-C with those preferred by DNMT3A-C Arg882His over the DNMT3A-C enzyme. This was confirmed by a direct comparison of the preferred sites of DNMT3B-C WT and the DNMT3A-C Arg882His enzyme, which showed no significant preference for a specific nucleotide at examined CpG flanking positions at the 19 preferred sites (Figure 3G–I). These data suggest that the Arg882His substitution alters the specificity of DNMT3A to be like that of DNMT3B. Furthermore, a comparison between DNMT3B-C Arg829His variant and DNMT3B-C WT showed a very similar flanking sequence preference between the two enzymes, suggesting little or no effect of the Arg829 mutation on DNMT3B-C (Supplementary Figure S3C, D). DNMT3B methylates DNA using a processive kinetic mechanism (40), therefore, we assayed the kinetic mechanism of the DNMT3A-C Arg882His variant. Our data confirmed that the Arg882His variant, like the WT enzyme, is non-processive, suggesting a specific effect of Arg882His mutation on the flanking sequence preference of DNMT3A (Supplementary Figure S3E).
Figure 3.

Comparative analysis of the flanking sequence preferences of DNMT3A-C WT, DNMT3A-C Arg882His and DNMT3B-C WT. DNA methylation of 124 CpG sites in four substrates, SUHW1 promoter region (509-bp), Meis1 (1000-bp) and Sirt4 (721-bp) enhancer regions, and pUC 19 fragment (1092-bp), was analyzed using bisulfite sequencing. Methylation reaction was carried out using 1 μM enzyme for 10 minutes and relative preference was calculated for each CpG site represented on X axis from 1 to 124. A 2-fold or more change in substrate preference is represented by bars (colored) greater than 1. Preferred CpG sites are shown in pink for SUHW1, blue for Meis1, green for Sirt4, and purple for the pUC19 substrate. DNMT3A-C Arg882His compared to DNMT3A-C WT in (A), DNMT3B-C WT compared to DNMT3A-C WT in (D), and DNMT3B-C WT compared to DNMT3A-C Arg882His in (G). The distribution of nucleotides at positions N+1/2/3 respectively was plotted from 17 preferred CpG sites by DNMT3A-C Arg882His in (B), 21 preferred sites by DNMT3B-C WT in (E), and the 19 sites preferred by DNMT3B-C WT in (H). P-values of enrichment for each base at N+1/2/3, corresponding to B, E, and H, are shown in (C), (F) and (I) respectively. Data presented are an average n = 3 independent experiments.These data show that substrate preference of DNMT3A-C Arg882His variant is similar to that of DNMT3B-C WT.
The DNMT3A Arg882His variant acquires DNMT3B-like substrate preference
The methylation assays to determine the flanking sequence preference described above were performed for 10 min, which represents initial enzyme kinetics. To evaluate the substrate preference under multiple turnover conditions, the methylation assays were carried out for 30 and 60 min, allowing the enzyme kinetics to enter the steady state. Compared to the flanking sequence preference at 10 minutes, DNMT3A-C Arg882His and DNMT3B-C both showed a slight decrease in relative preference for G at the N+3 as well as at N+1 position (Figure 4A–D, Supplementary Figure S4A–C, E–G). This indicates that after methylating the preferred sites during the initial reaction, the enzyme methylates other sites under multiple turnover conditions. A similar comparison of site preference of DNMT3B-C WT over DNMT3A-C Arg882His variant showed no change in the preference during the steady state reaction that is decreased at the 30 and 60 min time points (Figure 4E, F, Supplementary Figure S4D, H). These data confirm the similarity between the DNMT3A-C Arg882His variant and DNMT3B-C WT both in the initial and steady state reaction conditions.
Figure 4.

Flanking sequence preference at steady state kinetics. Methylation reactions were carried out using 1 μM enzyme and four DNA substrates for 30 and 60 min. DNA methylation of the 124 CpG sites was analyzed using bisulfite sequencing. Top panels show the relative preference calculated for each CpG site (1–124) at 30 and 60 min by DNMT3A-C WT compared to the DNMT3A-C Arg882His variant in (A, B), DNMT3B-C WT compared to DNMT3A-C WT in (C, D) and DNMT3B-C WT compared to DNMT3A-C Arg882His in (E, F). A 2-fold or more change in substrate preference is represented by bars (colored) >1. Preferred CpG sites are shown in pink for SUWH1, blue for Meis1, green for Sirt4, and purple for the pUC19 substrate. Bottom panels show the fractional distribution of nucleotides at the preferred sites. Each bar represents nucleotides at positions N+1/2/3 respectively from the CpG site. Data presented are an average n = 3 independent experiments.The data show that the similarity in flanking sequence preference between DNMT3A-C Arg882His and DNMT3B-C is maintained during steady state kinetics.
Temporal change in flanking sequence preference by WT and variant DNMT3 enzymes
We extended the analysis of the flanking sequence preference to determine the preferred trinucleotides at the N+1/2/3 and N–1/2/3 positions by DNMT3A-C WT, DNMT3A-C Arg882His and DNMT3B-C WT. The bisulfite sequencing data of four DNA substrates was used for this analysis. Based on the occurrence of different sites, the methylation at a site was calculated as a ratio of observed to expected fractional methylation for the specific substrate (see Materials and Methods, Supplementary Table S2).
The top 10 preferred sites were used in WebLogo application (47) to determine the consensus sequence logo flanking the CpG site for each enzyme. The analysis was performed for methylation data collected at the 10, 30 and 60 min time points to monitor the temporal order of site preference by these enzymes (Figure 5A, B). As expected, the data again show a dramatic difference in flanking sequence preference between DNMT3A-C and DNMT3B-C. Whereas DNMT3A-C WT shows a preference for C or T at the N+1/2/3 positions, DNMT3B-C WT prefers G and A. The preferred sequence of the DNMT3A-C Arg882His variant strikingly resembles that of DNMT3B-C, particularly at N+1, where it loses preference for T and gains a preference for G, and at N+2, where it gains a preference for C. Consistent with previous observations (46), the sites with T at the N+1 position are most preferred by DNMT3A-C WT, which is evident by its preference at 10 min (Figure 5B). DNMT3A-C WT enzyme has the least preference for sites with G at the N+1 position, evident by its appearance at 60 min. Comparatively, the preference pattern at the flanking positions is similar for DNMT3B-C and DNMT3A-C Arg882His, but different from that of DNMT3A-C WT. Both DNMT3B-C and DNMT3A-C Arg882His enzymes prefer G at the N+1 position and C at N+2, and disfavor sites with T at the N+1 position. The preference of all three enzymes for nucleotides at position N–1/2/3 showed a weak or no preference (Figure 5B). This is in agreement with crystal structure data showing fewer interactions between DNMT3A and the nucleotides upstream of CpG site (42).
Figure 5.

Trinucleotide sequence flanking CpG preferred by WT and variant DNMT3 enzymes. (A) Heat-map showing the preference of different trinucleotide sets by DNMT3A-C WT (3AWT), DNMT3A-C Arg882His (3ARH), DNMT3B-C WT (3BWT) at 10, 30 and 60 min. Sequences with values greater or equal to 1 are considered as preferred. Upper panels in blue represent the flanking sequence preference at the N+1/2/3 positions. Lower panels in purple represent the flanking sequence preference at the N–1/2/3 positions. (B) Consensus sequence generated by WebLogo from top ten preferred sequences by each enzyme as methylation reaction proceeds from 10 to 60 min. (C) Consensus flanking sequence for methylated sites with a G at the N+3 position. The data show that for the DNMT3A-C Arg882His variant the nucleotide preference at N+1 and N+3 matches that of DNMT3B-C. Similarly, at methylated sites with G at the N+3 position, inner flanking sequence are similar between DNMT3A-C Arg882His and DNMT3B-C, whereas it is different for DNMT3A-C WT enzyme.
We next selected CpG sites that had G at the N+3 position and generated the preferred flanking sequence logo using the WebLogo application (47). A comparison between the consensus sequences again show a striking similarity between DNMT3A-C Arg882His and DNMT3B-C WT enzymes. Whereas G at the N+1 position is the most preferred nucleotide by these enzymes, it is the least preferred by the DNMT3A-C WT enzyme. The consensus flanking sequence of DNMT3A-C WT has a T at the N+1 position. These observations uncover the importance of the nucleotide at the N+1 position, which can affect the interaction of DNMT3 enzymes with DNA (Figure 5C). We therefore tested if T at the N+1 position affects the preference for G at the N+3 position by DNMT3B-C and DNMT3A-C Arg882His enzymes. Methylation assays were performed using 30-bp oligonucleotide substrates, which contain a central CpG site, and varying combinations of nucleotides at N+2/3 or N-2/3 positions. The positions N+1 and N–1 were held constant with T and A, respectively (Supplementary Table S3). Radiolabeled AdoMet was used in enzyme assays and total methylation at 10 minutes was measured. Our data show that for these substrates, the preference of DNMT3B-C and the DNMT3A-C Arg882 variants for G at the N+3 position is lost (Supplementary Figure S5A–E). DNMT3A-C and its variants show rather strong preference for sites with A at the N+2 position, whereas DNMT3B-C shows a weak preference for sites with A or C occupying the N+2 position (Supplementary Figure S5E). Interestingly, DNMT3B-C Arg829His shows reduced activity when compared to the WT enzyme, indicating an adverse effect of T at N+1 position on its activity (Supplementary Figure S5F). These data confirm our previous observations that the interaction of DNMT3A-C Arg882His at the N+3 position is strongly influenced by the nucleotide at the N+1 position.
DNMT3A-C Arg882His and DNMT3B-C preferably methylate the same CpG site in the Meis1 enhancer
We next tested if the change in sequence preference of DNMT3A-C Arg882His could affect methylation of the regions that are known to be spuriously hypomethylated in AML patients (48). The Meis1 gene is expressed during development and, by promoting the self-renewal of progenitor-like cells, it regulates leukemogenesis and hematopoiesis (49,50). The enhancer of Meis1 is methylated by DNMT3A during normal hematopoietic stem cell (HSC) differentiation, whereas in AML patients expressing the DNMT3A Arg882His variant, this region is largely hypomethylated (48,51). A 1-kb region of the Meis enhancer was used as a substrate for methylation reactions (Supplementary Figure S3A). The average methylation by DNMT3A-C Arg882His and DNMT3B-C WT was significantly lower than by DNMT3A-C WT (Figure 6A). Examination of the flanking sequence preference showed that DNMT3B methylated with a strong bias for one site (#7), which had G at N+1 and A at the N+3 position (Figure 6B, Supplementary Figure S3A). Interestingly, this site was one of the two sites preferred by DNMT3A-C Arg882His (Figure 6C). A comparison between DNMT3B-C and DNMT3A-C Arg882His confirmed that DNMT3B-C has a higher preference for this site (Figure 6D). Although there are 3 sites in this substrate with G at N+3 position, these sites have either T, C or A at the N+1 position, which are weakly preferred by DNMT3B-C as well as by the DNMT3A-C Arg882His variant. The data again confirms the effect of the nucleotide at the N+1 position on the flanking sequence preference of DNMT3 enzymes and the effect of DNMT3A-C Arg882His on target site specificity.
Figure 6.

Relative activity and site preference of DNMT3 WT and mutant enzymes on Meis1 enhancer substrate. A 1-kb Meis1 enhancer region was used as substrate for in vitro methylation reactions by DNMT3A-C WT, the Arg882His variant, and DNMT3B-C WT. (A) Methylation activity of DNMT3A-C WT, DNMT3A-C Arg882His, DNMT3B-C WT, and catalytically inactive DNMT3B-C Glu703Ala on the Meis1 substrate at 10 minutes, analyzed by bisulfite sequencing. Relative preference for each of the 14 CpG sites was calculated for DNMT3B-C compared to DNMT3A-C in (B), DNMT3A-C Arg882His compared to DNMT3A-C WT in (C), and DNMT3B-C WT compared to DNMT3A-C Arg882His in (D). Relative preference of 1 which is equal to a 2-fold change, is represented by green, blue and pink bars, respectively. Data presented are an average n = 3 independent experiments. DNMT3A-C Arg882His and DNMT3B-C prefer to methylate same site in this substrate, however DNMT3B-C shows a very strong preference compared to the DNMT3A-C Arg882His variant.
Mouse DNMT3A Arg878His retains its activity for minor satellite DNA in mESCs
While DNMT3A and DNMT3B redundantly methylate many genomic regions in cells, they also have preferred and specific targets (9,11,52). For example, in murine cells, DNMT3A preferentially methylates the major satellite repeats in pericentric regions, whereas DNMT3B preferentially methylates the minor satellite repeats in centromeric regions (9,11). Based on our observations, we predicted that DNMT3A Arg882His would prefer DNMT3B-specific targets. To test the idea, we carried out rescue experiments in late-passage Dnmt3a/3b DKO mESCs, which show severe loss of global DNA methylation, including at the major and minor satellite repeats (11). mESCs express two major DNMT3A isoforms, DNMT3A1 (full length) and DNMT3A2 (a shorter form that lacks the N terminus of DNMT3A1), with both showing identical activity (11,26,53). We transfected Dnmt3a/3b DKO mESCs with plasmid vectors and generated stable lines expressing mouse DNMT3A1 WT, DNMT3A1 Arg878His, DNMT3B1 WT, or DNMT3B1 Arg829His (catalytically inactive DNMT3A1 and DNMT3B1, with their PC motif in the catalytic center being mutated (54), were included as negative controls) (Figure 7A). The genomic DNA from these cell lines was harvested, and DNA methylation at the major and minor satellite repeats was analyzed by digestion with methylation-sensitive restriction enzymes followed by Southern blot. Consistent with our previous results (26), the ability of DNMT3A1 to rescue methylation at the major satellite repeats is severely impaired with the Arg878His substitution (Figure 7B). However, at the minor satellite repeats, which are largely methylated by DNMT3B, DNMT3A Arg878His rescues DNA methylation comparable to the DNMT3A WT enzyme (Figure 7C). These data demonstrate that the DNMT3A AML mutant specifically retains its ability to methylate DNMT3B-preferred target sites, while losing preference for sites methylated by DNMT3A.
Figure 7.

Rescue of DNA methylation at the major and minor satellite repeats in Dnmt3a/3b DKO mESCs. (A) Dnmt3a/3b DKO mESCs were transfected with plasmids encoding mouse DNMT3A1 WT, DNMT3A1 Arg878His (3A1:RH), DNMT3A Pro705Val/Cys706Asp (3A1:PC), DNMT3B1 WT, DNMT3B1 Arg829His (3B1:RH), or DNMT3B1 Pro656Gly/Cys657Thr (3B1:PC), and stable clones were derived. Total cell lysates were used to analyze the expression of DNMT3A or DNMT3B proteins by western blot with anti-DNMT3A, anti-DNMT3B, and anti-β-Actin antibodies. A long exposure of the DNMT3A blot is included to show endogenous DNMT3A1 in WT (J1) mESCs. Note that stable clones showing similar expression levels to those of endogenous DNMT3A or DNMT3B were used for the experiments. (B, C) DNA methylation was analyzed by Southern blot. Genomic DNA was digested with MaeII (major satellite repeats) or HpaII (minor satellite repeats), and probed for the major (B) or minor (C) satellite repeats. J1 (WT) and untransfected DKO mESCs were used as controls. The numbers on the top indicate clone#. Complete digestion due to low or no DNA methylation is indicated by low molecular weight bands as seen in untransfected DKOs, and high molecular weight bands as seen in J1 indicate high DNA methylation and protection from digestion. Comparing the activity of DNMT3A1 clones 19/22 with 3A1:RH clones 15/16 at major and minor satellite repeats shows that 15/16 methylate minor repeats similar to 19/22 whereas at major satellite repeats the activity of 15/16 is severely impaired. (D) Consensus sequence of the nucleotides flanking the CpG site in either the major or minor satellite repeats, created using WebLogo shows high prevalence G at N+1 and N+3 positions at minor repeats.
To test if the preference of DNMT3A and DNMT3B was driven by a potential sequence bias in the major or minor satellite repeats, we computed the consensus sequence logo for +3 and −3 flanking nucleotides around the CpG sites using the WebLogo application (52) (Figure 7D). The analysis shows that the major satellite repeats are enriched with CpG sites carrying T at N+1 and A at N+3, and are depleted in CpG sites carrying G at the N+1 and N+3 positions. However, the minor satellite repeats have a high percentage of sites with G at the N+1 and N+3 positions, which are highly preferred by DNMT3B as well as DNMT3A Arg882His. These data confirm that DNMT3A Arg878His acquires catalytic properties analogous to DNMT3B, which may allow it to target DNMT3B-specific sites in somatic cells and contribute to cancer development.
Our data also show that unlike the WT enzyme, the DNMT3B Arg829His variant was unable to rescue methylation of the major satellite repeats and only partially rescued methylation of the minor satellite repeats (Figure 7B, C). This is explained by the observation that substrates with T at the N+1 position are strongly disfavored by the DNMT3B-C Arg829His variant (Supplementary Figure S5F), and, about half of the CpG sites in both the major and minor satellite repeats have T at the N+1 position.
Taken together, we propose a model in which substrate specificity and kinetic mechanism of DNMT3A and DNMT3B regulate the DNA methylation of various genomic regions. At repetitive elements where CpG content is intermediate to high, DNMT3A WT enzyme acts cooperatively on multiple CpGs where it prefers A and T at N+1 and N+3 positions, respectively. Loss of cooperativity in the DNMT3A WT or DNMT3A Arg882His variant modifies the specificity to G at both N+1 and N+3 analogous to DNMT3B (Figure 8). Whereas DNMT3B methylates its targets in a processive manner, DNMT3A WT and Arg882His methylate these sites distributively explaining the lower activity of these enzymes at these sites. Given that the DNA methylation at repetitive elements represents the bulk of DNA methylation in mammalian genomes, these data indicate that besides being targeted by protein-protein interaction and chromatin modifications, DNA methylation by DNMT3A and DNMT3B is largely guided by their intrinsic sequence preference.
Figure 8.
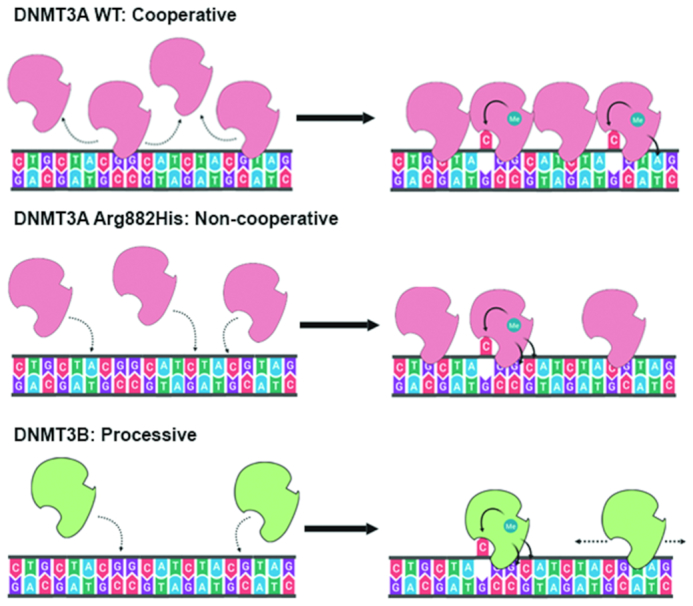
Model showing the different kinetic mechanisms of DNMT3A and DNMT3B and their influence their flanking sequence preferences. At high concentrations, DNMT3A methylates multiple CpG sites rapidly by using cooperative mechanism, and disfavours G at N+1 and N+3, and prefers T at N+2. However, in absence of cooperative kinetic mechanism DNMT3A WT enzyme, the DNMT3A Arg882His variant, and DNMT3B, the flanking sequence preference switches to G at the N+1. Both the DNMT3A Arg882His variant and DNMT3B disfavour T at N+1 position.
DISCUSSION
Despite numerous studies addressing the biological roles of DNMT3A and DNMT3B in development and diseases, the differences and similarities in their kinetic mechanisms remain poorly understood. Germline mutations in DNMT3A and DNMT3B have deleterious effects and are associated with congenital diseases (17,18). In AML and other types of leukemia, the majority of somatic mutations in DNMT3A affects Arg882, mostly leading to an Arg-to-His substitution (21). Because of its high prevalence (∼20% in AML) and early occurrence during disease development, the Arg882His variant is considered a founder mutation (55). Therefore, the variant enzyme has been the subject of many studies. Through these studies, the Arg882His substitution was shown to alter DNA binding properties, attenuate tetramerization, disrupt cooperativity, and change the flanking sequence preference of the DNMT3A enzyme (22,27,33,40,41). In this study, we show that the altered sequence preference of the AML variant can potentially cause aberrant methylation of DNMT3B target sites, thus contributing to its oncogenic potential.
We show that disruption of cooperativity alters the flanking sequence preference of DNMT3A-C WT to match the sequence preferred by DNMT3A-C Arg882His variant, demonstrating the effect of kinetic mechanism on substrate specificity. Using the DNA methylation analysis of 124 CpG sites, we systematically computed the temporal order of site preference for the DNMT3A-C WT, DNMT3A-C Arg882His, and DNMT3B-C WT enzymes. The most important finding in this study is the discovery that the altered flanking sequence preference of the DNMT3A-C Arg882His variant is nearly identical to the preferred substrate sequence of DNMT3B-C. Consequently, we predicted that the DNMT3A Arg882His variant would potentially methylate DNMT3B-specific targets. Indeed, DNA methylation of major and minor satellite repeats rescued by the WT or mutant DNMTs in Dnmt3a/3b DKO mESCs provide strong evidence supporting this prediction. The data revealed that, compared to the DNMT3A WT, the mouse DNMT3A Arg878His (equivalent to human DNMT3A Arg882His) variant has little effect in rescuing DNA methylation at major satellite repeats. However, its ability to methylate the minor satellite repeats is almost fully retained. These data demonstrated that target site specificity of DNMT3A and DNMT3B in vivo could be strongly influenced by their respective CpG flanking sequence preference. Consistent with this, we observed that at the minor satellite repeats CpG flanking sequence showed an overrepresentation of G at N+1 and N+3 positions that is preferred by DNMT3B and the DNMT3A Arg882His variant. The major satellite repeats have high percentages of T and A at the N+1 and N+3 positions, respectively, which are preferred by DNMT3A WT enzyme and disfavored by DNMT3B WT and the Arg829His variant.
Given that under conditions favoring non-cooperative mechanism, the flanking sequence preference of DNMT3A WT is modified to match that of DNMT3A Arg878His variant, we propose that DNMT3A methylates minor satellite repeats using a non-cooperative kinetic mechanism. This prediction also explains the rationale behind the lower activity of DNMT3A at minor satellite repeats compared to that of DNMT3B, which methylates its target sites using processive kinetic mechanism. Similarly, although DNMT3A Arg878His variant prefers DNMT3B sites, it methylates these sites using a non-cooperative distributive mechanism, explaining an incomplete rescue by the variant enzyme compared to DNMT3B. A high activity of DNMT3A at the major satellite repeats could be justified by the cooperative mechanism used to methylate multiple CpG sites. Therefore, these data also provide evidence for the role of distinct kinetic properties and substrate specificity of DNMT3A and DNMT3B in selection of their genomic target sites.
These observations also suggest that at genomic regions with sparse or dispersed CpG sites, the absence of cooperativity would favor methylation of sequences with Gs at N+1,3 by both DNMT3A and DNMT3B at common target sites. However, the distinct tissue specific expression of DNMT3A and DNMT3B ensures that these enzymes methylate their specific targets, thus regulating their biological activity. The importance of this regulation is highlighted by aberrant expression of DNMT3B in various types of cancer, including AML (14,28–30). The effect of DNMT3B overexpression in AML is similar to that of DNMT3A Arg882His mutation, leading to an increase in stemness, downregulation in apoptotic genes, and poor patient prognosis (27,29). Our data here reveal a mechanism by which the DNMT3A Arg882His variant acts like the DNMT3B enzyme, thus providing a mechanistic explanation to the aforementioned observations in AML patients. We speculate that the oncogenic potential of DNMT3A Arg882His variant may not be only due to its lower activity causing DNA hypomethylation in AML cells, but also due to the gain of DNMT3B-like activity generating aberrant patterns of DNA methylation. While the DNMT3A Arg882 mutation may have other effects, these data suggest that changes of substrate specificity contribute to leukemogenesis caused by the DNMT3A Arg882His mutation. Thus, our observations provide novel insights into consequences of cancer-causing mutations on the enzymatic activity of DNMT3 enzymes.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to Gowher lab members for discussions.
Author contributions: A.B.N. and H.G. conceived the project and designed the experiments. A.B.N., L.A., A.R.M. and N.E.F. purified recombinant proteins and performed radiolabeled methylation assays. A.B.N. and Y.T. performed in vitro methylation assays and bisulfite conversion. A.B.N., H.G. and T. C. analyzed the data, generated figures, and wrote the manuscript. B.L. performed the rescue experiments in mESCs, and T.C. supervised the tissue culture work.
Notes
Present address: Lama AlAbdi, Department of Zoology, King Saud University, Riyadh, Saudi Arabia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01GM118654-01 to H.G. and R01AI12140301A1 to T.C.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Reik W., Dean W., Walter J.. Epigenetic reprogramming in mammalian development. Science. 2001; 293:1089–1093. [DOI] [PubMed] [Google Scholar]
- 2. Smith Z.D., Meissner A.. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013; 14:204–220. [DOI] [PubMed] [Google Scholar]
- 3. Jaenisch R., Jahner D.. Methylation, expression and chromosomal position of genes in mammals. Biochim. Biophys. Acta. 1984; 782:1–9. [DOI] [PubMed] [Google Scholar]
- 4. Surani M.A. Imprinting and the initiation of gene silencing in the germ line. Cell. 1998; 93:309–312. [DOI] [PubMed] [Google Scholar]
- 5. Ng H.H., Bird A.. DNA methylation and chromatin modification. Curr. Opin. Genet. Dev. 1999; 9:158–163. [DOI] [PubMed] [Google Scholar]
- 6. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018; 19:81–92. [DOI] [PubMed] [Google Scholar]
- 7. Bestor T., Laudano A., Mattaliano R., Ingram V.. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988; 203:971–983. [DOI] [PubMed] [Google Scholar]
- 8. Yoder J.A., Soman N.S., Verdine G.L., Bestor T.H.. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol. 1997; 270:385–395. [DOI] [PubMed] [Google Scholar]
- 9. Okano M., Bell D.W., Haber D.A., Li E.. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. [DOI] [PubMed] [Google Scholar]
- 10. Aapola U., Kawasaki K., Scott H.S., Ollila J., Vihinen M., Heino M., Shintani A., Kawasaki K., Minoshima S., Krohn K. et al.. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics. 2000; 65:293–298. [DOI] [PubMed] [Google Scholar]
- 11. Chen T., Ueda Y., Dodge J.E., Wang Z., Li E.. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell Biol. 2003; 23:5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gujar H., Weisenberger D.J., Liang G.. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes. 2019; 10:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watanabe D., Suetake I., Tada T., Tajima S.. Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis. Mech. Dev. 2002; 118:187–190. [DOI] [PubMed] [Google Scholar]
- 14. Linhart H.G., Lin H., Yamada Y., Moran E., Steine E.J., Gokhale S., Lo G., Cantu E., Ehrich M., He T. et al.. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007; 21:3110–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen B.F., Chan W.Y.. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics. 2014; 9:669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schubeler D. Function and information content of DNA methylation. Nature. 2015; 517:321–326. [DOI] [PubMed] [Google Scholar]
- 17. Norvil A.B., Saha D., Saleem Dar M., Gowher H.. Effect of disease-associated germline mutations on structure function relationship of DNA methyltransferases. Genes. 2019; 10:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ehrlich M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin. Immunol. 2003; 109:17–28. [DOI] [PubMed] [Google Scholar]
- 19. Xu G.L., Bestor T.H., Bourc’his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E.. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999; 402:187–191. [DOI] [PubMed] [Google Scholar]
- 20. Hansen R.S., Wijmenga C., Luo P., Stanek A.M., Canfield T.K., Weemaes C.M., Gartler S.M.. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. PNAS. 1999; 96:14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ley T.J., Ding L., Walter M.J., McLellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J. et al.. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010; 363:2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brunetti L., Gundry M.C., Goodell M.A.. DNMT3A in leukemia. Cold Spring Harb. Perspect. Med. 2017; 7: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research, N. Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A., Hoadley K., Triche T.J. Jr., Laird P.W. et al.. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu Y., Lennartsson A., Gaidzik V.I., Deneberg S., Karimi M., Bengtzen S., Hoglund M., Bullinger L., Dohner K., Lehmann S.. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014; 9:1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L., Rodriguez B., Mayle A., Park H.J., Lin X., Luo M., Jeong M., Curry C.V., Kim S.B., Ruau D. et al.. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell. 2016; 29:922–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S.J., Zhao H., Hardikar S., Singh A.K., Goodell M.A., Chen T.. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013; 122:4086–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Russler-Germain D.A., Spencer D.H., Young M.A., Lamprecht T.L., Miller C.A., Fulton R., Meyer M.R., Erdmann-Gilmore P., Townsend R.R., Wilson R.K. et al.. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014; 25:442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gagliardi M., Strazzullo M., Matarazzo M.R.. DNMT3B functions: novel insights from human disease. Front. Cell Dev. Biol. 2018; 6:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niederwieser C., Kohlschmidt J., Volinia S., Whitman S.P., Metzeler K.H., Eisfeld A.K., Maharry K., Yan P., Frankhouser D., Becker H. et al.. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acute myeloid leukemia. Leukemia. 2015; 29:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hayette S., Thomas X., Jallades L., Chabane K., Charlot C., Tigaud I., Gazzo S., Morisset S., Cornillet-Lefebvre P., Plesa A. et al.. High DNA methyltransferase DNMT3B levels: a poor prognostic marker in acute myeloid leukemia. PLoS One. 2012; 7:e51527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jia D., Jurkowska R.Z., Zhang X., Jeltsch A., Cheng X.. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007; 449:248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo X., Wang L., Li J., Ding Z., Xiao J., Yin X., He S., Shi P., Dong L., Li G. et al.. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2015; 517:640–644. [DOI] [PubMed] [Google Scholar]
- 33. Holz-Schietinger C., Matje D.M., Reich N.O.. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012; 287:30941–30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sandoval J.E., Huang Y.H., Muise A., Goodell M.A., Reich N.O.. Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 2019; 294:4898–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yan X.J., Xu J., Gu Z.H., Pan C.M., Lu G., Shen Y., Shi J.Y., Zhu Y.M., Tang L., Zhang X.W. et al.. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011; 43:309–315. [DOI] [PubMed] [Google Scholar]
- 36. Yamashita Y., Yuan J., Suetake I., Suzuki H., Ishikawa Y., Choi Y.L., Ueno T., Soda M., Hamada T., Haruta H. et al.. Array-based genomic resequencing of human leukemia. Oncogene. 2010; 29:3723–3731. [DOI] [PubMed] [Google Scholar]
- 37. Jurkowska R.Z., Anspach N., Urbanke C., Jia D., Reinhardt R., Nellen W., Cheng X., Jeltsch A.. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008; 36:6656–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rajavelu A., Jurkowska R.Z., Fritz J., Jeltsch A.. Function and disruption of DNA methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res. 2012; 40:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emperle M., Rajavelu A., Reinhardt R., Jurkowska R.Z., Jeltsch A.. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 2014; 289:29602–29613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Norvil A.B., Petell C.J., Alabdi L., Wu L., Rossie S., Gowher H.. Dnmt3b methylates DNA by a noncooperative mechanism, and its activity is unaffected by manipulations at the predicted dimer interface. Biochemistry. 2018; 57:4312–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emperle M., Rajavelu A., Kunert S., Arimondo P.B., Reinhardt R., Jurkowska R.Z., Jeltsch A.. The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res. 2018; 46:3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Z.M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S.B., Wang Y. et al.. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018; 554:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roth M., Jeltsch A.. Biotin-avidin microplate assay for the quantitative analysis of enzymatic methylation of DNA by DNA methyltransferases. Biol. Chem. 2000; 381:269–272. [DOI] [PubMed] [Google Scholar]
- 44. Hemeon I., Gutierrez J.A., Ho M.C., Schramm V.L.. Characterizing DNA methyltransferases with an ultrasensitive luciferase-linked continuous assay. Anal. Chem. 2011; 83:4996–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gowher H., Jeltsch A.. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 2002; 277:20409–20414. [DOI] [PubMed] [Google Scholar]
- 46. Handa V., Jeltsch A.. Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J. Mol. Biol. 2005; 348:1103–1112. [DOI] [PubMed] [Google Scholar]
- 47. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu R., Wang P., Parton T., Zhou Y., Chrysovergis K., Rockowitz S., Chen W.Y., Abdel-Wahab O., Wade P.A., Zheng D. et al.. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell. 2016; 30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mercader N., Leonardo E., Azpiazu N., Serrano A., Morata G., Martinez C., Torres M.. Conserved regulation of proximodistal limb axis development by Meis1/Hth. Nature. 1999; 402:425–429. [DOI] [PubMed] [Google Scholar]
- 50. Cai M., Langer E.M., Gill J.G., Satpathy A.T., Albring J.C., Kc W., Murphy T.L., Murphy K.M.. Dual actions of Meis1 inhibit erythroid progenitor development and sustain general hematopoietic cell proliferation. Blood. 2012; 120:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ferreira H.J., Heyn H., Vizoso M., Moutinho C., Vidal E., Gomez A., Martinez-Cardus A., Simo-Riudalbas L., Moran S., Jost E. et al.. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene. 2016; 35:3079–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Challen G.A., Sun D., Mayle A., Jeong M., Luo M., Rodriguez B., Mallaney C., Celik H., Yang L., Xia Z. et al.. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014; 15:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen T., Ueda Y., Xie S., Li E.. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 2002; 277:38746–38754. [DOI] [PubMed] [Google Scholar]
- 54. Chen T., Tsujimoto N., Li E.. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell Biol. 2004; 24:9048–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mayle A., Yang L., Rodriguez B., Zhou T., Chang E., Curry C.V., Challen G.A., Li W., Wheeler D., Rebel V.I. et al.. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015; 125:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.