

OBJECTIVES:
Chronic pancreatitis (CP) is a serious condition whose pathogenic mechanism is unclear. Interactions of host genetic factors with gut microbiota have a role, but little is known, especially in children with CP (CCP), in which the external factors are less important. Our objective was to identify the main gut microbiota genera in CCP and to characterize the functional mutations of these patients.
METHODS:
We used 16S rRNA sequencing to compare the gut microbiota of healthy controls with patients who had CCP and different functional gene mutations.
RESULTS:
CCP is characterized by gut microbiota with remarkably reduced alpha diversity. Receiver operating characteristic curve analyses indicated that the abundances of 6 genera—Faecalibacterium, Subdoligranulum, Phascolarctobacterium, Bifidobacterium, Eubacerium, and Collinsella—were significantly decreased in CCP, with an area under curve (AUC) of 0.92 when considering all 6 genera together. Functional analysis of gut microbiota in CCP indicated reduced ribosomal activity, porphyrin and chlorophyll metabolism, starch and sucrose metabolism, and aminoacyl-tRNA biosynthesis, but an enrichment of phosphotransferase system pathways. The abundance of Butyricicoccus was significantly decreased in CCP in the presence of CFTR mutations when combined with mutations in CASR, CTSB, SPINK1, and/or PRSS1. The abundance of Ruminococcaceae was significantly increased in CCP when there were mutations in CASR, CTSB, SPINK1, and/or PRSS1. Patients with CCP but no gene mutations had greater abundances of Veillonella and reduced abundances of Phascolarctobacterium.
DISCUSSION:
CCP is associated with a depletion of probiotic gut microbiota, and CCP patients with different functional gene mutations have different gut microbiota.
INTRODUCTION
Chronic pancreatitis (CP) is a persistent fibroinflammatory process of the pancreas, which is associated with genetic, environmental, and other pathogenic factors, eventually leads to irreversible pancreas injury, and often causing pain and/or permanent loss of function. CP patients also have an increased risk of pancreatic cancer and impaired mental health (1,2). CP is especially harmful to children because malnutrition and malabsorption of food could potentially lead to a delay in their physical growth and development (3).
There is no effective treatment for CP and no simple and reliable diagnostic test for early CP (4). The annual incidence of CP ranges from 5 to 12 per 100,000, and the prevalence is about 50 per 100,000 (5). A survey in China reported that the prevalence of CP was 13.52 per 100,000 (6). On the other hand, based on the previous findings that most CP patients were 15–20 years old and a recent epidemiological study in France, which found an annual incidence of 7.8 per 100,000, the annual prevalence of CP may be as high as 120 to 143 per 100,000. The incidence and prevalence of CP are thus greatly underestimated, especially in China, and this is further compounded by the difficulty in diagnosing CP (4).
Clarification of the pathogenesis of CP is important for defining and treating this disease (1). Although there are well-established internal factors (genetic predispositions) and external factors (smoking, drinking alcohol) that increase the risk for CP, it remains a poorly understood disease (1,2,7,8).
Many researchers have examined the relationship of genomic variations of human hosts with the composition of the gut microbiome. There is strong evidence that an interaction of host genetics and gut microbiota underlie the onset and clinical presentation of many gastrointestinal diseases and that host genetics influence gut microbiota (9). Similarly, there is evidence that patients with CP and type 3c (pancreatogenic) diabetes mellitus (T3cDM) have higher levels of Bacteroidetes and lower levels of Faecalibacterium than patients with CP alone and that patients with T3cDM and PEI have lower levels of Bifidobacterium than those with T3cDM alone (10). Other studies that used gas liquid chromatography reported lower levels of Bifidobacterium and Lactobacillus and higher levels of Enterobacter, Proteus, Klebsiella, and Morganella in patients with CP than in healthy controls (HCs) (11,12). A study of oral microbiota reported that patients with CP had lower levels of Granulicatella adiacens than that of those with pancreatic ductal carcinoma (13). An analysis of the colonic microenvironment reported that patients with CP had lower levels of Lactobacillus and higher levels of Escherichia coli, Enterococcus faecalis, and Enterococcus faecium than that of HCs, but these 2 groups had no difference in the levels of Bifidobacterium (14). However, no studies have yet examined the link between gut microbiota and children with CP (CCP).
External pathogenic factors, such as tobacco and alcohol consumption, are generally irrelevant in studies of many children. Thus, the studies of children who had no smoking or drinking habits may simplify the identification of pathogenic factors associated with CP because there is no confounding by these factors. In this study, we used 16S rRNA sequencing and bioinformatics analysis to characterize the gut microbiota associated with CCP, the gut microbiota of hosts with different functional mutations, and the functional traits of hosts and metabolic pathways associated with the pathogenesis of CP.
MATERIAL AND METHODS
Inclusion and exclusion criteria
The diagnosis of CP was based on the criteria of the INternational Study Group of Pediatric Pancreatitis: In Search for a CuRE (INSPPIRE), as previously described (7). All cases and HCs were 4–18 years old and were examined from 2014 to 2017. Individuals who used an antibiotic or an oral probiotic drink within the previous 3 months were excluded.
Collection of samples
Samples were collected at Ruijin Hospital (Shanghai, China), which is affiliated with the Shanghai Jiao Tong University School of Medicine. All samples were stored at −80 °C within 2 hours after collection.
Isolation of fecal bacterial DNA
Fresh fecal specimens were collected in sterile containers and immediately subjected to DNA extraction using the OMEGA-soil DNA Kit (Omega Bio-Tek, Norcross, GA) following the instructions of the manufacturer. DNA concentration and purification were determined by using a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, DE). DNA quality was confirmed using 1% agarose gel electrophoresis, and isolated DNA was stored at −80 °C before sequencing.
16s rRNA amplicon sequencing of bacteria
PCR (GeneAmp 9700; Applied Biosystems, Foster City, CA) was used to amplify the V3-V4 hypervariable regions of the 16S rRNA gene with primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The PCR reactions (3 minutes of denaturation at 95 °C; 27 cycles of 30 seconds at 95 °C, 30 seconds for annealing at 55 °C, and 45 seconds for elongation at 72 °C; final extension at 72 °C for 10 minutes) were performed in triplicate in 20 μL mixtures (4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer [5 μM], 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA). DNA was extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA). Quantification was performed using QuantiFluor-ST (Promega, Madison, WI).
Illumina MiSeq sequencing
Purified amplicons were pooled in equimolar amounts and sent to Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China). Paired-end sequencing (2 × 300) was performed on an Illumina MiSeq platform (Illumina, San Diego, CA) using standard protocols.
Processing of sequencing data
Raw fastq files were subjected to read-quality filtering using Trimmomatic and merged by FLASH. First, reads were truncated at any site with an average quality score below 20 over a 50 bp sliding window. Second, sequences with overlap longer than 10 bp were merged so that the overlap with mismatch was no more than 2 bp. Third, sequences were separated by barcodes (exact matching) and primers (allowance for mismatch of 2 nucleotides); reads containing ambiguous bases were removed.
UPARSE (version 7.1, http://drive5.com/uparse/) was used to cluster the operational taxonomic units. A 97% similarity cutoff was implemented with a novel “greedy” algorithm that simultaneously removes chimeras and clusters operational taxonomic units. The identification of each 16S rRNA gene sequence was determined using the RDP Classifier algorithm (http://rdp.cme.msu.edu/) and a 16S rRNA database (Silva, SSU release 123), with a confidence threshold of 70%.
The α-diversity (within-sample diversity) was calculated from the gene profiles of each sample using the Shannon index, as described previously (15). The β diversity (between-sample diversity) was calculated by the principal coordinate analysis and nonmetric multidimensional scaling. The functional traits of each sample were assigned according to the known genomes and prediction using the KEGG orthology (KO) (16). The functional differences between groups were determined using Linear Discriminant Analysis Effect Size (LEfSe).
The abundances of different gut genera in CP patients and HCs were compared by AUROC curve analysis to determine their performance in the diagnosis of CP.
Genetic analysis and grouping
Genomic DNA was extracted from peripheral blood leukocytes of patients using the FlexiGene DNA Extraction Kit (Qiagen, Hilden, Germany). PRSS1, SPINK1, CFTR, CTRC, CASR, CTSB, KRT8, CLDN2, CPA1, and ATP8B1 were sequenced and analyzed, as previously described (6). The patients were classified into 3 groups, according to the sequencing results, and the abundance of gut bacteria in these groups were compared. Patients in group 1 had heterozygous or homozygous mutations in CFTR that were responsible for abnormal secretions in the pancreatic duct (17,18) and also had or had no heterozygous or homozygous mutations in CASR, CTSB, SPINK1, and/or PRSS1 that were responsible for the abnormal activation of trypsinogen (19). Patients in group 2 only had mutations in CASR, CTSB, SPINK1, and/or PRSS1. Patients in group 3 had no mutations in any of these genes.
Clinical trial registry
This study was approved by the Institutional Review Board (IRB) of Shanghai Jiao Tong University, and the protocols were approved by the Committee of Human Subjects Protection of Ruijin Hospital. Informed consent was obtained from the parents of all recruited children. The clinical trial registry number is NCT03809247 (https://register.clinicaltrials.gov/).
Statistical analysis
Quantitative data are presented as means ± SDs and analyzed using the t-test (for normal distributions), the Wilcoxon signed-rank test (for non-normal distributions), or one-way analysis of variance (for multiple samples). Categorical data are presented as frequencies and analyzed by the χ2 tests (with or without the Yates correction for continuity) or Fisher exact tests, where appropriate. SPSS 17.7 software for Windows (SPSS, Chicago, IL) was used for statistical analyses. A P value below 0.05 was considered significant.
AUROC curve calculations were performed using R software, as previously described (20). In these calculations, the relative abundance of each genus was subjected to arcsine square-root transformation before the analysis. To evaluate the diagnostic performance of the fitted logistic regression models, the area under the curve (AUC) values were calculated. The significance of differences between different AUCs was determined using the DeLong method.
RESULTS
Basic clinical data
We compared the gut microbiota of 30 CCP patients with 35 HCs (Table 1, see Table 1, Supplementary Digital Content 1, http://links.lww.com/CTG/A229). These 2 groups had no significant differences in age (7.2 ± 0.5 vs 8.3 ± 0.7 years, P = 0.224) or sex ratio (53.3% vs 65.7% men, P = 0.122).
Table 1.
Basic clinical data of children with chronic pancreatitis


We collected the peripheral blood samples from 21 CCP patients (Tables1–3). The 3 groups of CCP patients had no statistically significant differences in age (8.9 ± 4.0 vs 10.0 ± 5.0 vs 7.5 ± 3.0 years, P = 0.515) or sex ratio (57.1% vs 33.3% vs 62.5% men, P = 0.646).
Table 3.
Molecular characteristics of mutated genes in 13 patients

Table 2.
Mutations present in the different groups

Alterations of gut microbial diversity in CCP
CCP was associated with a significantly reduced alpha diversity of gut microbiota (P < 0.01) (Figure 1a,b). Analysis of beta diversity showed that the abundance of gut microbial genera had distinct clustering in CCP patients and in HCs (Figure 1c,d). In particular, the KEGG analysis indicated that CCP patients had gut microbiota that were enriched in the phosphotransferase system and depleted in ribosomal activity, porphyrin and chlorophyll metabolism, starch and sucrose metabolism, and aminoacyl-tRNA biosynthesis (Figure 1e).
Figure 1.
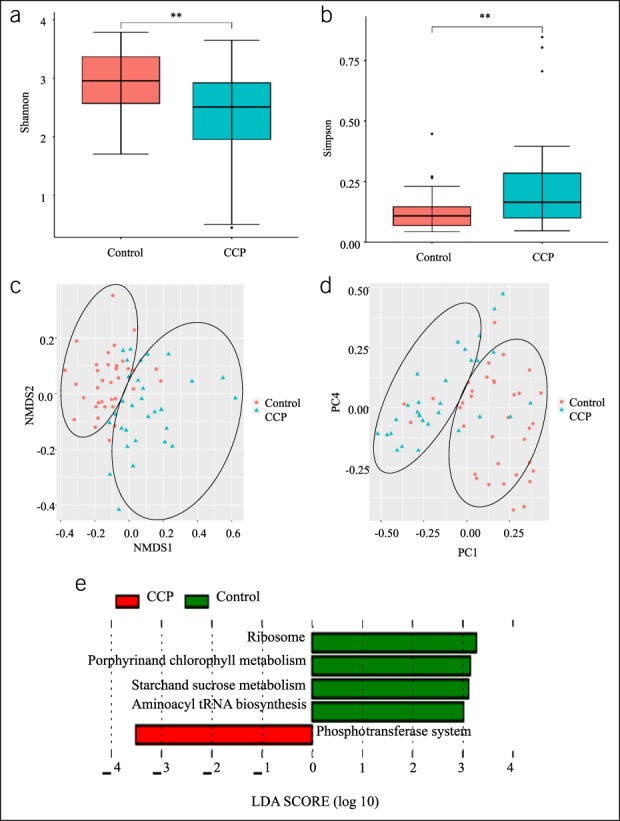
Alterations of the gut microbiota diversity in CCP. (a and b), Alpha diversity in patients and controls was calculated using the Shannon index and the Simpson index, and a two-tailed Wilcoxon signed-rank test was used for comparisons. **P < 0.01. (c and d), Beta diversity in patients and controls was calculated using PCoA and NMDS. (e), KEGG orthology (KO), showing differences in the functional pathways of gut microbiota in patients and controls. LEFSe was used for comparison. The predetermined threshold on the logarithmic linear discriminant analysis (LDA) score for discriminative features was set at >3.0; the predetermined α-value for the factorial Kruskal-Wallis test was set at >0.01. CCP, children with chronic pancreatitis; NMDS, nonmetric multidimensional scaling; PCoA, principal coordinate analysis.
Identification of disordered genera
We identified 28 genera with different abundances in the CCP and HC groups. Relative to the HCs, CP patients had 19 genera with decreased abundance and 9 genera with increased abundance (see Table 2, Supplementary Digital Content 1, http://links.lww.com/CTG/A229). Faecalibacterium, Subdoligranulum, Phascolarctobacterium, Bifidobacterium, Collinsella, and Eubacterium were the 6 genera with the greatest decreases in absolute abundance in CCP patients (Figure 2a).
Figure 2.

Alterations in the abundances of gut microbial genera in CCP. (a), Six genera whose abundances had the greatest reductions in CCP patients relative to HCs, with comparisons by a two-tailed Wilcoxon signed-rank test (sum of the mean <2.3, difference of the mean >2.5, see Table 2, Supplementary Digital Content 1, http://links.lww.com/CTG/A229). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (b), The AUROC curve analysis of the performance of 6 genera in the diagnosis of CCP. Faecalibacterium: AUC = 0.80 (0.68–0.80, P < 0.001), Subdoligranulum: AUC = 0.65 (0.52–0.65, P < 0.05), Phascolarctobacterium: AUC = 0.71 (0.59–0.71, P < 0.01), Bifidobacterium: AUC = 0.65 (0.51–0.65, P = 0.07), Collinsella: AUC = 0.67 (0.54–0.67, P < 0.67) Eubacterium: AUC = 0.70 (0.56–0.70, P < 0.01). The use of each of the 6 individual arcsine square root transformed abundance values, together with the coefficients from multivariate logistic regression: AUC = 0.92 (0.84–0.92, P < 0.001). AUC, area under curve; CCP, children with chronic pancreatitis; HC, healthy control.
The AUROC analysis showed that when each genus was considered individually, the diagnostic performance of Faecalibacterium (AUC = 0.80, 95% confidence interval [CI]: 0.68–0.80, P < 0.001), Phascolarctobacterium (AUC = 0.71, 95% CI: 0.59–0.71, P < 0.01), and Eubacterium (AUC = 0.70, 95% CI: 0.56–0.70, P < 0.01) were “fair”; and the diagnostic performance of Subdoligranulum (AUC = 0.65, 95% CI: 0.52–0.65, P < 0.01), Bifidobacterium (AUC = 0.65, 95% CI: 0.51–0.65, P = 0.07), and Collinsella (AUC = 0.67, 95% CI: 0.54–0.67, P < 0.05) were “poor.” The use of each individual arcsine square root transformed abundance, together with the coefficients from multivariate logistic regression, led to “excellent” diagnostic performance (AUC = 0.92, 95% CI: 0.84–0.92, P < 0.001) (Figure 2b).
Relationship of host functional mutations with gut microbiota
Relative to the HCs, the abundance of Butyricicoccus was significantly lower in group 1, but not significantly different in group 2 and group 3 (Figure 3a). The abundance of Ruminococcaceae was significantly greater in group 2, but not significantly different in group 1 and group 3 (Figure 3b). The abundance of Veillonella was greater and the abundance of Phascolarctobacterium was lower in group 3, but not significantly different in group 1 and group 2 (Figure 3c,d).
Figure 3.
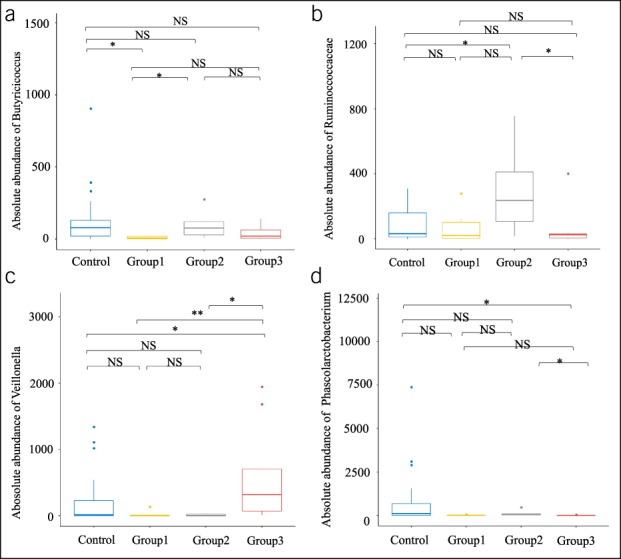
Relationship of gut microbial genera with functional mutations in hosts who have CCP. (a), Butyricicoccus was significantly less abundant in group 1 (CFTR mutations and mutations in CASR, CTSB, SPINK1, and/or PRSS1). (b), Ruminococcaceae was significantly more abundant in group 2 (mutations only in CASR, CTSB, SPINK1, and/or PRSS1). (c and d). Veillonella was significantly more abundant and Phascolarctobacterium was significantly less abundant in group 3 (no mutations of the tested genes). Box plots were drawn using R software, and a two-tailed Wilcoxon singed-rank test was used for comparisons. *P < 0.05, **P < 0.01. CCP, children with chronic pancreatitis.
DISCUSSION
This is the first study of the gut microbiota of CCP patients who have mutations in different specific functional genes. There were several major findings. First, CCP was associated with a significantly reduced gut microbial alpha diversity, and the subsequent KEGG analysis indicated that the disordered gut microbiota probably contributed to the pathogenesis of CCP. Second, our comparison of the gut microbiota of CCP patients and HCs using the AUROC curve analysis indicated the presence of 4 new genera (Subdoligranulum, Phascolarctobacterium, Collinsella, and Eubacterium) and 2 known genera (Faecalibacterium and Bifidobacterium) (10,21). The use of all 6 genera together led to excellent diagnostic performance (AUC = 0.92, 95% CI: 0.84–0.92, P < 0.001). Thus, determination of the abundances of these 6 genera might be useful as a diagnostic marker of CCP, and these genera may also be considered as therapeutic targets for treatment of CCP. Third, Butyricicoccus, Ruminococcaceae, Veillonella, and Phascolarctobacterium are associated with different functional gene mutations in the host that are linked to CCP, thus confirming that specific functional gene mutations in the host might affect the composition of gut microbiota. This further supports the hypothesis that the presence of disordered gut microbiota is related to the pathogenesis of CCP. Further in-depth studies of gut microbiota are thus necessary to investigate the pathogenic mechanisms of CCP and to develop new treatments for CCP.
Among the genera of gut microbiota that had significantly decreased abundance in CCP, Faecalibacterium (10) and Bifidobacterium (21) were also less abundant in adults with CP. Faecalibacterium prausnitzii can produce butyrate (22) and salicylic acid (23), participate in the inflammatory process by blocking NF-kB activation and IL-8 production (24), and negatively correlate with the plasma endotoxin and glycemic status (10). A reduced level of Faecalibacterium is also associated with Crohn's disease (24), colitis (25), and colorectal cancer (26). A decreased abundance of Faecalibacterium might promote CP by weakening the inflammatory process. Bifidobacterium is a well-known probiotic that can improve the nutritional status and enhance the immune capacity of patients with severe acute pancreatitis (27). Therefore, these 2 groups of gut microbiota should be considered as novel therapeutic targets in future applications that seek to prevent or treat CCP.
No previous studies of CP have examined 4 other genera of gut microbiota that we examined, Subdoligranulum, Phascolarctobacterium, Eubacterium, and Collinsella. However, as potentially beneficial bacteria, previous research reported that a reduction of Subdoligranulum was related to glycemic control, inflammatory processes (28), and neoplasms of the stomach, colon, and rectum (29). In addition, the abundance of Subdoligranulum increases after the administration of vitamin D (30) and is associated with a healthy eating index (31). These results suggest that CP patients might benefit from vitamin D supplements and healthy eating. Phascolarctobacterium is a genus of proinflammatory bacteria that is associated with the onset and development of inflammation in metabolic diseases and mental illnesses (32,33). In addition, patients with nephrolithiasis have a greater abundance of Phascolarctobacterium than HCs, and the abundance of these bacteria correlates with the concentration of trace elements in blood, including potassium, sodium, calcium, and chlorinum (34). Eubacterium, a potential probiotic, is a butyrate-producing bacterium whose level is decreased in Crohn's disease (35) but increased in primary biliary cholangitis (36). The abundance of Collinsella is closely related to high levels of alpha aminoadipate and asparagine and the production of the proinflammatory cytokine IL-17A, and animal studies indicated it has a role in altering gut permeability and disease severity in arthritis (37). On the other hand, an increased abundance of Collinsella is associated with symptomatic atherosclerosis (38).
To our knowledge, no previous studies have examined the relationship of gut microbiota with functional gene mutations in CP patients. Thus, one of our important findings is that 4 genera of gut microbiota appear to be related to different functional mutations in their hosts. We found that the abundance of Butyricicoccus was significantly decreased patients with mutations in CFTR, which manifests as abnormal secretions of the pancreatic duct. Butyricicoccus is a butyrate-producing clostridial cluster IV genus (39). Previous research showed that its abundance was reduced in the stools of patients with ulcerative colitis. In addition, Butyricicoccus-conditioned medium can prevent the cytokine-induced increase of epithelial permeability in vitro, and a specific strain of Butyricicoccus pullicaecorum can reduce intestinal inflammation in a rat model of colitis (40). Butyrate and B. pullicaecorum-conditioned medium can reduce the CLDN1 expression while preserving the epithelial integrity (41). These results suggest that Butyricicoccus may affect gene expression in its host, thereby altering physiological function and inducing disease, thus indirectly confirming our hypothesis that the presence of disordered gut microbiota is related to the pathogenesis of CCP.
The abundance of Ruminococcaceae was significantly increased CP patients who had mutations in CASR, CTSB, SPINK1, and/or PRSS1, each of which is associated with abnormal activation of trypsinogen. A reduced abundance of these bacteria is associated with increased innate immune responses and the levels of proinflammatory cytokines (IL-6 and TNFα) (42). A reduced abundance of Ruminococcaceae appears to be implicated in food sensitization and to precede the development of atopic eczema (43). Han et al. showed that a decreased abundance of Ruminococcaceae had a positive correlation with the Treg/Th17 ratio, thus suggesting that intestinal microbiota might induce acquired graft-vs-host disease by altering the Treg/Th17 balance (44). Several other studies demonstrated that host heredity affects the abundance of gut Ruminococcaceae (7,8).
The abundance of Veillonella was significantly increased and that of Phascolarctobacterium was significantly decreased in CCP patients who had no mutations in any of the tested genes. Veillonella is a genus of anaerobic Gram-negative cocci and considered as part of the normal flora of the animal and human mouth, gastrointestinal tract, and genitourinary tract (45). Matera et al. demonstrated that Veillonella parvula lipopolysaccharides (LPS) are recognized by toll-like receptors (TLR4) and that activation of MAP kinases plays an important role in V. parvula LPS-stimulated signaling of human peripheral blood mononuclear cells (46). Disorders of these cells are associated with many inflammatory diseases, such as chronic anaerobic pneumonitis (47), acute pyelonephritis, and secondary bacteraemia during pregnancy (48).
Our results showed that 8 groups of gut microbiota are potential probiotics because of their anti-inflammatory potential. Disruption of these microbes is associated with many different inflammatory diseases. The first human intervention trial that used a butyrate-producing Clostridium cluster IV isolate demonstrated that B. pullicaecorum 25-3T administration was safe and well tolerated by healthy participants (49). This safety study paved the way for the further development of this strain as a next-generation probiotic. We found no association between these genera and the CP phenotype in the GMrepo database, which continues to annotate the human gut macrogenome (50). Thus, our findings are a useful complement to the GMrepo database.
There were some limitations in our study. We examined only a small number of patients, and we therefore need to verify the relationships of the different functional mutations of hosts with different genera of gut microbes by examining more patients. The sequencing and comparison of fecal samples from CCP patients who improve or do not improve during treatment and follow-up would also be useful. Deep sequencing, including metagenomic sequencing, is also necessary. The anti-inflammatory and therapeutic effects of probiotics still need to be verified in vivo and in vitro.
Our major findings are that the presence of disordered gut microbiota occurs in CCP and that specific functional gene mutations in the host appear to influence the gut microbiota composition. These findings may be helpful for further investigations into the pathogenic mechanisms of CP and provide several promising targets for the diagnosis and treatment of CP.
CONFLICTS OF INTEREST
Guarantor of the article: Chundi Xv, MD, Wei Wang, MD, and Bai-Yong Shen, MD.
Specific author contributions: Study concept and design: W.W., Sequencing and Analysis of Specimens: W.W. and Y.X., Specimen and data collection: Y.X., X.W., Y.Z., T.W., and W.W., Study supervision: B.-Y.S., C.X. and W.W.
Financial support: This study was supported by grants from the Shanghai Charity Cancer Research Center (2017).
Potential competing interests: None to report.
Availability of data and materials: The data sets analyzed are available from the corresponding author on reasonable request.
Study Highlights.
WHAT IS KNOWN
✓ CP is a serious condition, particularly in children because it reduces dietary intake and delays growth and development. However, the pathogenesis of CP is still unclear. Interactions of host genetics with gut microbiota underlie many diseases, but little is known about these interactions in the pathogenesis of CP, especially in CCP, in whom external pathogenic factors relative are less important.
WHAT IS NEW HERE
✓ This is the first study of gut microbiota in CCP to analyze the impact of different functional gene mutations in the host. CCP is characterized by gut microbiota with remarkably reduced alpha diversity compared to age-matched HCs. Receiver operating characteristic analysis indicated that 6 genera, including 4 newly described genera, were significantly less abundant in CCP. Patients with CCP who had different functional gene mutations had different gut microbiota.
TRANSLATIONAL IMPACT
✓ These findings may help to identify new biomarkers or therapeutic targets for CP.
Supplementary Material
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A229
REFERENCES
- 1.Whitcomb DC, Frulloni L, Garg PK, et al. Chronic pancreatitis: An international draft consensus proposal for a new mechanistic definition. Pancreatology 2016;16(2):218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho J, Walia M, Scragg R, et al. Frequency and risk factors for mental disorders following pancreatitis: A nationwide cohort study. Curr Med Res Opin 2019;35(7):1157–64. [DOI] [PubMed] [Google Scholar]
- 3.Abu-El-Haija M, Uc A, Werlin SL, et al. Nutritional considerations in pediatric pancreatitis: A position paper from the NASPGHAN pancreas committee and ESPGHAN cystic fibrosis/pancreas working group. J Pediatr Gastroenterol Nutr 2018;67(1):131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lévy P, Domínguez-Muñoz E, Imrie C, et al. Epidemiology of chronic pancreatitis: Burden of the disease and consequences. United European Gastroenterol J 2014;2(5):345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013;144(6):1252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang LW, Li ZS, Li SD, et al. Prevalence and clinical features of chronic pancreatitis in China: A retrospective multicenter analysis over 10 years. Pancreas 2009;38(3):248–54. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Y, Yuan W, Yu B, et al. Targeted gene next-generation sequencing in Chinese children with chronic pancreatitis and acute recurrent pancreatitis. J Pediatr 2017;191:158–63.e3. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Sun XT, Weng XL, et al. Comprehensive screening for PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations in Chinese paediatric patients with idiopathic chronic pancreatitis: A cohort study. BMJ Open 2013;3(9):e003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blekhman R, Goodrich JK, Huang K, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol 2015;16:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jandhyala SM, Madhulika A, Deepika G, et al. Altered intestinal microbiota in patients with chronic pancreatitis: Implications in diabetes and metabolic abnormalities. Sci Rep 2017;7:43640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorovits ES, Tokareva EV, Khlynova OV, et al. Complex evaluation of intestine microbiocenosis condition in patients with chronic pancreatitis [in Russian]. Zh Mikrobiol Epidemiol Immunobiol 2013;4:73–6. [PubMed] [Google Scholar]
- 12.Wüst J, Smid I, Salfinger M. Experience of gas-liquid chromatography in clinical microbiology. Ann Biol Clin (Paris) 1990;48(6):416–9. [PubMed] [Google Scholar]
- 13.Farrell JJ, Zhang L, Zhou H, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012;61(4):582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savitskaia KI, Mel'nikova EF, Vorob'ev AA, et al. Evaluation of the colonic content microenvironment in patients with chronic pancreatitis [in Russian]. Vestn Ross Akad Med Nauk 2002(4):20–3. [PubMed] [Google Scholar]
- 15.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490(7418):55–60. [DOI] [PubMed] [Google Scholar]
- 16.Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44(D1):D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegyi P, Rakonczay Z. The role of pancreatic ducts in the pathogenesis of acute pancreatitis. Pancreatology 2015;15(4 Suppl):S13–7. [DOI] [PubMed] [Google Scholar]
- 18.Ishiguro H, Steward MC, Naruse S, et al. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol 2009;133(3):315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sofia VM, Surace C, Terlizzi V, et al. Trans-heterozygosity for mutations enhances the risk of recurrent/chronic pancreatitis in patients with cystic fibrosis. Mol Med 2018;24(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kummen M, Holm K, Anmarkrud JA, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017;66(4):611–9. [DOI] [PubMed] [Google Scholar]
- 21.Memba R, Duggan SN, Ni Chonchubhair HM, et al. The potential role of gut microbiota in pancreatic disease: A systematic review. Pancreatology 2017;17(6):867–74. [DOI] [PubMed] [Google Scholar]
- 22.Miquel S, Martín R, Bridonneau C, et al. Ecology and metabolism of the beneficial intestinal commensal bacterium Faecalibacterium prausnitzii. Gut Microbes 2014;5(2):146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miquel S, Leclerc M, Martin R, et al. Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. MBio 2015;6(2):e00300–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 2008;105(43):16731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 2009;15(8):1183–9. [DOI] [PubMed] [Google Scholar]
- 26.Konstantinov SR, Kuipers EJ, Peppelenbosch MP. Functional genomic analyses of the gut microbiota for CRC screening. Nat Rev Gastroenterol Hepatol 2013;10(12):741–5. [DOI] [PubMed] [Google Scholar]
- 27.Jin Y, Xu H, Chen Y, et al. Therapeutic effect of Bifidobacterium combined with early enteral nutrition in the treatment of severe acute pancreatitis: A pilot study. Eur Rev Med Pharmacol Sci 2018;22(12):4018–24. [DOI] [PubMed] [Google Scholar]
- 28.Chumpitazi BP, Hollister EB, Oezguen N, et al. Gut microbiota influences low fermentable substrate diet efficacy in children with irritable bowel syndrome. Gut Microbes 2014;5(2):165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Youssef O, Lahti L, Kokkola A, et al. Stool microbiota composition differs in patients with stomach, colon, and rectal neoplasms. Dig Dis Sci 2018;63(11):2950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schäffler H, Herlemann DP, Klinitzke P, et al. Vitamin D administration leads to a shift of the intestinal bacterial composition in Crohn's disease patients, but not in HCs. J Dig Dis 2018;19(4):225–34. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Ajami NJ, El-Serag HB, et al. Dietary quality and the colonic mucosa-associated gut microbiome in humans. Am J Clin Nutr 2019;110(3):701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cattaneo A, Cattane N, Galluzzi S, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 2017;49:60–8. [DOI] [PubMed] [Google Scholar]
- 33.Lim MY, You HJ, Yoon HS, et al. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 2017;66(6):1031–8. [DOI] [PubMed] [Google Scholar]
- 34.Tang R, Jiang Y, Tan A, et al. 16S rRNA gene sequencing reveals altered composition of gut microbiota in individuals with kidney stones. Urolithiasis 2018;46(6):503–14. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi K, Nishida A, Fujimoto T, et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn's disease. Digestion 2016;93(1):59–65. [DOI] [PubMed] [Google Scholar]
- 36.Abe K, Takahashi A, Fujita M, et al. Dysbiosis of oral microbiota and its association with salivary immunological biomarkers in autoimmune liver disease. PLoS One 2018;13(7):e0198757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Wright K, Davis JM, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med 2016;8(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Zhang J, Wu C, et al. Unique features of ethnic Mongolian gut microbiome revealed by metagenomic analysis. Sci Rep 2016;6:34826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eeckhaut V, Van Immerseel F, Teirlynck E, et al. Butyricicoccus pullicaecorum gen. nov., sp. nov.,an anaerobic, butyrate-producing bacteriumisolated from the caecal content of a broiler chicken. Int J Syst Evol Microbiol 2008;58(Pt 12):2799–802. [DOI] [PubMed] [Google Scholar]
- 40.Eeckhaut V, Machiels K, Perrier C, et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013;62(12):1745–52. [DOI] [PubMed] [Google Scholar]
- 41.Devriese S, Eeckhaut V, Geirnaert A, et al. Reduced mucosa-associated Butyricicoccus activity in patients with ulcerative colitis correlates with aberrant claudin-1 expression. J Crohns Colitis 2017;11(2):229–36. [DOI] [PubMed] [Google Scholar]
- 42.West CE, Rydén P, Lundin D, et al. Gut microbiome and innate immune response patterns in IgE-associated eczema. Clin Exp Allergy 2015;45(9):1419–29. [DOI] [PubMed] [Google Scholar]
- 43.Simonyte Sjödin K, Vidman L, Rydén P, et al. Emerging evidence of the role of gut microbiota in the development of allergic diseases. Curr Opin Allergy Clin Immunol 2016;16(4):390–5. [DOI] [PubMed] [Google Scholar]
- 44.Han L, Jin H, Zhou L, et al. Intestinal microbiota at engraftment influence acute graft-versus-host disease via the Treg/Th17 balance in allo-HSCT recipients. Front Immunol 2018;9:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Periasamy S, Kolenbrander PE. Aggregatibacter actinomycetemcomitans builds mutualistic biofilm communities with fusobacterium nucleatum and Veillonella species in saliva. Infect Immun 2009;77(9):3542–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matera G, Muto V, Vinci M, et al. Receptor recognition of and immune intracellular pathways for Veillonella parvula lipopolysaccharide. Clin Vaccin Immunol 2009;16(12):1804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah A, Panjabi C, Nair V, et al. Veillonella as a cause of chronic anaerobic pneumonitis. Int J Infect Dis 2008;12(6):e115–7. [DOI] [PubMed] [Google Scholar]
- 48.Yagihashi Y, Arakaki Y. Acute pyelonephritis and secondary bacteraemia caused by Veillonella during pregnancy. BMJ Case Rep 2012;2012:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boesmans L, Valles-Colomer M, Wang J, et al. Butyrate producers as potential next-generation probiotics: Safety assessment of the administration of Butyricicoccus pullicaecorum to healthy volunteers. mSystems 2018;3(6):e00094–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu S, Sun C, Li Y, et al. GMrepo: A database of curated and consistently annotated human gut metagenomes. Nucleic Acids Res 2020;48(D1):D545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.