

Abstract
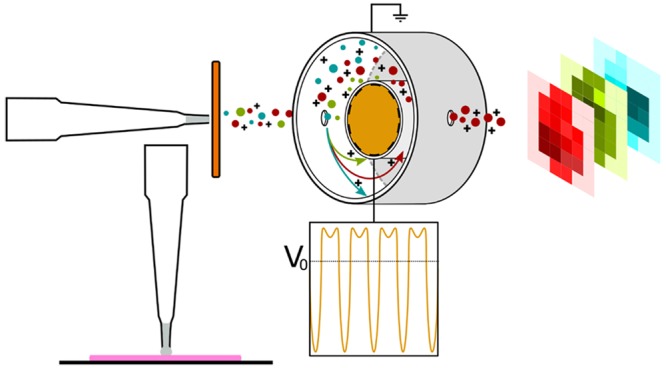
The benefits of high field asymmetric waveform ion mobility spectrometry (FAIMS) for mass spectrometry imaging of intact proteins in thin tissue sections have been demonstrated previously. In those works, a planar FAIMS device coupled with a Thermo Elite mass spectrometer was employed. Here, we have evaluated a newly introduced cylindrical FAIMS device (the FAIMS Pro) coupled with a Thermo Fusion Lumos mass spectrometer for liquid extraction surface analysis mass spectrometry imaging of intact proteins in thin tissue sections from rat testes, kidney, and brain. The method makes use of multiple FAIMS compensation values at each location (pixel) of the imaging array. A total of 975 nonredundant protein species were detected in the testes imaging dataset, 981 in the kidney dataset, and 249 in the brain dataset. These numbers represent a 7-fold (brain) and over 10-fold (testes, kidney) improvement on the numbers of proteins previously detected in LESA FAIMS imaging, and a 10-fold to over 20-fold improvement on the numbers detected without FAIMS on this higher performance mass spectrometer, approaching the same order of magnitude as those obtained in top-down proteomics of cell lines. Nevertheless, high throughput identification within the LESA FAIMS imaging workflow remains a challenge.
Liquid extraction surface analysis (LESA)1 is an ambient mass spectrometry technique which is particularly well suited to the analysis of intact proteins from a range of biological substrates including thin tissue sections. The benefits of interrogation of intact proteins rather than their proteolytic peptides include retention of all information relating to primary structure, including single nucleotide polymorphisms, and presence and connectivity of post-translational modifications. A challenge for LESA of thin tissue sections is the inherent complexity of the extracted sample. That challenge can be addressed through integration of ion mobility separation, such as high field asymmetric waveform ion mobility spectrometry2,3 (FAIMS; also known as differential mobility spectrometry (DMS)), in the workflow. FAIMS separates gas-phase ions at atmospheric pressure by exploiting differences in their mobilities in high and low electric fields. Ions are passed between parallel electrodes, to which an asymmetric waveform is applied, by a carrier gas. As a result of their differential mobilities, the ions will deviate from their original trajectory. This deviation can be corrected by superposition of a dc compensation voltage (CV). It is possible to selectively transmit ions of particular differential mobility by tuning the CV.
Mass spectrometry imaging (MSI)4 provides information on the spatial distribution of analytes across substrates such as a thin tissue sections, allowing insights into analyte colocalization and the molecular basis of tissue features. We have previously demonstrated LESA FAIMS mass spectrometry imaging of intact proteins in sections of mouse brain and mouse liver.5 At each tissue location in the imaging array, the CV was kept constant. In that work, ∼30 proteins were detected in each tissue type. We subsequently demonstrated LESA FAIMS MSI of thin tissue sections from rat kidney and testes, in which multiple CV settings were employed for analysis at each tissue location.6 That approach enabled detection of ∼60 intact proteins from the kidney samples and ∼75 for the testes samples. Both of these previous studies made use of a planar miniaturized ultrahigh field FAIMS device and a Thermo Elite orbitrap mass spectrometer. Recently, a cylindrical FAIMS device (FAIMS Pro) has been introduced which offers improved ion mobility resolution and high transmission efficiency.7,8 FAIMS Pro has been shown to be advantageous for bottom-up proteomic analyses, when coupled with liquid chromatography tandem mass spectrometry (LC-MS/MS), by improving signal-to-noise and extending proteome coverage.9,10 Here, we have performed LESA FAIMS MSI of intact proteins in thin tissue sections of rat testes, kidney, and brain by use of the FAIMS Pro coupled with a Thermo Fusion Lumos mass spectrometer and demonstrate significant improvements in the numbers of proteins detected. The results are applicable not only for mass spectrometry imaging of intact proteins but also more generally for top-down proteomics experiments11,12 in which complex mixtures of intact proteins isolated from cell lines or homogenized tissue are analyzed by liquid chromatography tandem mass spectrometry.
Methods
Samples
Rat tissue was the kind gift of Dr. Richard Goodwin (AstraZeneca). Brain, testes, and kidney tissues were obtained from control (vehicle-dosed) male Hans Wistar rats. Animals were euthanized by cardiac puncture under isofluorane anesthetic 2 h post dose. All tissue dissection was performed by trained AstraZeneca staff (project license 40/3484, procedure number 10). Tissues were snap frozen in dry ice chilled isopentane and stored at −80 °C. Tissues were subsequently cryosectioned at a thickness of 10 μm using a CM1810 Cryostat (Leica Microsystems, Wetzlar, Germany) and thaw mounted onto glass slides.
Acetonitrile, ethanol, water (all Optima LC/MS grade), and formic acid were purchased from Fisher Scientific (Waltham, MA).
LESA
Rat brain sections were prewashed in 70% ethanol for 10 s (to remove abundant lipid species) before air drying and loading onto a universal LESA adapter plate. Other tissue sections (which do not contain similarly high levels of lipids) were not prewashed. Samples were placed into the TriVersa Nanomate chip-based electrospray device (Advion, Ithaca, NY). The extraction/ionization solvent comprised 40:60 acetonitrile:water with 1% formic acid. Contact-LESA13 was performed as follows: 4 μL of solvent was aspirated, and 2 μL was dispensed onto the sample for 10 s before 2.5 μL was reaspirated. Samples were mixed twice and introduced into the mass spectrometer via the TriVersa NanoMate with gas pressure of 0.3 psi and tip voltage of 1.70 kV. All MSI experiments were acquired at 1 mm × 1 mm spacing.
FAIMS Mass Spectrometry Imaging
The Triversa Nanomate was coupled to a Thermo Fisher FAIMS Pro device (Thermo Fisher Scientific, San Jose, CA) which was coupled to a Thermo Fisher Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The FAIMS dispersion voltage (DV) was −5 kV. For each tissue type, optimization experiments were performed in which the compensation voltage (CV) was stepped in 10 V increments from −120 to +50 V. For each imaging experiment, multistep compensation workflows (optimized for tissue type) were applied. At each location in the testes imaging data set, data were acquired for 30 s with the FAIMS voltages off, followed by 30 s at each of CV = −70, −60, −50, −40, −30, and −20 V, and a final 30 s with the FAIMS voltages off, for a total acquisition time of 4 min. For the kidney data set, data were acquired for 30 s with the FAIMS voltages off, followed by 30 s at each of CV = −80, −70, −60, −50, −40, and −30 V, and a final 30 s with the FAIMS voltages off, for a total acquisition time of 4 min/location. For the brain data set, data were acquired for 30 s with the FAIMS voltages off, followed by 30 s at each of CV = −90, −80, −70, and −60, and a final 30 s with the FAIMS voltages off, for a total acquisition time of 3 min/location. Mass spectra were recorded in full scan mode at a resolution of 120 000 at m/z 200 in the m/z range of 350–2000. To avoid imaging bias introduced by use of automated gain control (AGC), an AGC target of 5 × 106 charges and a maximum injection time of 100 ms was applied. (The combination of extremely high AGC target and moderate injection time ensures that the AGC target is never reached, and each scan comprises identical accumulation times (i.e., 100 ms)). Each scan was comprised of five coadded microscans. For top-down MS/MS experiments, electron transfer high-energy collision-induced dissociation (EThcD) was performed. For precursor m/z 661, the ETD reaction time was 20 ms, and HCD was performed at 15% normalized collision energy (NCE). The MS/MS spectrum comprises 80 scans. For precursors m/z 706 and 558, the ETD reaction time was 10 ms, and HCD was performed at 30% NCE. The MS/MS spectra comprise 20 and 12 scans, respectively. For precursor m/z 1036, the ETD reaction time was 20 ms, and CID was performed at 30% NCE. The MS/MS spectrum comprises 15 scans.
Data Analysis
Data were analyzed by use of Xcalibur software and BioPharma Finder 3.1 (both Thermo Fisher Scientific). All mass spectra were deconvoluted using the Xtract algorithm in BioPharma Finder in the batch processing mode. Source spectra were defined by the “Average Over Selected Retention Time” method in which the RT range correlated with the data acquisition time at each CV. That is, for each pixel, multiple deconvolutions were performed, each of which corresponded to a particular CV. The parameters for the search were a minimum signal-to-noise ratio of 3 (“FAIMS on”) or 4 (“FAIMS off”) and a fit factor of 80%. All other settings were left as default. Output masses were reported as the neutral species M. The resulting output files were collated in MATLAB (version 2013a, The MathWorks Inc., Natick, MA) such that a list of all unique detected masses and their frequency of occurrence was produced (see File S1, File S2, and File S3 of the Supporting Information). Masses detected across multiple CV steps and/or locations and within a tolerance of 0.5 Da were considered as a single (mean) mass. The noise level was determined using the median of the frequency, and masses which had a signal-to-noise ratio ≤3 were discarded.
For image generation, single location .raw data files were converted to .mzML using MS convert and then converted to the imzML format and loaded into MATLAB using imzMLConverter14 and SpectralAnalysis software.15 t-SNE plots were generated using Python 3.7 and the SciKitLearn library.16 Prior to embedding, spectra were linearly interpolated and median normalized. t-SNE parameters were as follows: perplexity = 35, initialization = random, learning rate = 500, early exaggeration = 10. The distance metric was Euclidean. UpSet plots were created using the UpSet R package.17
Top-down protein identification was performed with ProSightPC software, version 3.0 (Thermo Fisher Scientific, Bremen, Germany). MS/MS spectra were deconvoluted by the THRASH algorithm at a signal-to-noise ratio of 3 and matched against the Rattus norvegicus database available via UniProt. Putative protein assignments were confirmed by manual analysis, in which experimental fragment m/z values were compared with theoretical m/z values calculated by Protein Prospector (http://prospector.ucsf.edu/) and sequence coverages calculated.
Results and Discussion
Thin tissue sections of rat testes, kidney, and brain were subjected to LESA FAIMS MSI. The workflow is shown in Figure S1 of the Supporting Information. For each dataset, the mass spectra obtained at each CV value at each location (pixel) were deconvoluted using the Xtract algorithm in the BioPharma Finder software resulting in multiple lists of intact protein masses associated with each CV/location. To determine the total number of intact protein masses detected across the entire image, or within a single pixel, masses detected within multiple CV steps and/or locations that fell within a tolerance of 0.5 Da were considered as a single (mean) mass. (Although the Biopharma Finder software does offer the facility for Multiconsensus reporting, i.e., merging the deconvolution results from multiple raw files, which would in principle remove the requirement for this step, it is limited to a maximum of 10 data files. This limit is incompatible with a FAIMS MSI data set. For example, the kidney image data set comprised 117 pixels, with six CV steps, i.e., a total of 702 data files). Concatenated mass lists (either entire image or single pixel) were filtered such that all masses that were detected with a frequency S/N of ≤3 were discarded. Noise levels differed between tissue types and for the FAIMS data were determined to be 4 (testes), 8 (kidney), and 1 (brain). For “FAIMS voltages off” data, the noise levels were 1 (testes), 2 (kidney), and 2 (brain). It is important to note that the Xtract algorithm does not take into account the presence of adducts; therefore, the numbers of proteins reported herein include both proteoforms and any adducts thereof (e.g., sodium or potassium adducts) that may be present. A potential solution would be software that enables removal of adducts by consideration of exact mass shifts, an approach that is commonly applied in metabolomics studies; however, this approach would require retention of accurate masses in the deconvolution step. We have used a broad tolerance (±0.5 Da) to prevent overestimation of protein numbers; however, further development of the deconvolution software itself (i.e., raising the limit of data files allowable for multiconsensus reporting) might enable this approach to be implemented.
The total number of proteins detected across the entire testes imaging data set (FAIMS on) was 975, with a mean of 517 proteins detected at each CV. The number of proteins detected across the imaging data set when the FAIMS voltages were off was 48. (It is worth noting that in a previous study of LESA planar FAIMS MS of fresh frozen rat testes the number of proteins detected when the FAIMS voltages were off was five.6 The improvement observed here when the FAIMS voltages were off is due to the improved performance of the mass spectrometer). The molecular weight range was from 1.8 to 21 kDa (File S1, Supporting Information). The distribution of proteins across CV values is shown in Figure 1a. The overlap in protein masses at the various CV values is shown in the UpSet plot in Figure S2 of the Supporting Information.17 The greatest overlap was 72 proteins, corresponding to 7.4% of the total proteins, detected at CV = −20, −30, −40, and −50 V. The majority of the proteins (54%) were observed at ≤3 CV values. The complementarity of the various CV steps and therefore the benefits of FAIMS integration can be further visualized by t-distributed stochastic neighbor embedding (t-SNE) (Figure 1b). Each point corresponds to a pixel at a particular CV value, or in the absence of FAIMS. There is clear separation between the CV values. The various CV values are also generally clearly separated from the “FAIMS voltages off” data, with some similarity between “FAIMS voltages off” and CV = −30 V and CV = −40 V. Figure 1c shows an overlay of the mass spectra obtained at the different CV values for a representative pixel (pixel 59). For that pixel, the total number of proteins detected was 382 (FAIMS voltages on) and 5 (FAIMS voltages off).
Figure 1.

LESA FAIMS MSI of thin tissue sections from testes, kidney, and brain. (A), (D), (G) Distribution of protein masses across CV values. (B), (E), (H) t-SNE plot of LESA FAIMS MSI data. Each point corresponds to a pixel at a particular CV value. (C), (F), (I) Overlay of the mass spectra obtained at the different CV values for pixel 59 (testes), pixel 70 (kidney), and pixel 35 (brain).
Similar analyses were performed for the kidney and brain imaging data sets (Figure 1d–i). For kidney, a total of 981 proteins were detected across the entire imaging data set with the “FAIMS on” compared with 44 with the “FAIMS off”. The molecular weight range was from 1.7 to 18 kDa (File S2, Supporting Information). The UpSet plot (Figure S3, Supporting Information) reveals that the majority of the proteins (90%) were detected at three or fewer CV values, and the greatest overlap was 203 proteins (21% of total proteins detected) observed at CVs = −30, −40, and −50 V. t-SNE reveals clear separation between the CV values, and the greatest similarity between the “FAIMS off” and CV= −40 V. For brain, a total of 249 proteins were detected across the entire imaging data set with FAIMS voltages on, and 21 proteins were detected with FAIMS voltages off. The molecular weight range was from 2.2 to 19 kDa (File S3, Supporting Information). The UpSet plot (Figure S4, Supporting Information) reveals that 65% of proteins were detected at individual CV values. The greatest set overlap (18%) was observed for CVs = −60 and −70 V. t-SNE reveals clear separation between the various CV values and the “FAIMS voltages off” data. Within CV values, CVs = −60 and −70 V are clearly separated, and CVs = −80 and −90 V show the greatest similarity.
A well-established advantage of mass spectrometry imaging is its broad specificity: There is no requirement for a priori knowledge of the analyte to be imaged, and ion images may be generated for any ion detected. Clearly, the greater the sensitivity of the imaging technique is, the greater the possibilities are for mining the imaging data set (i.e., the greater the number of analytes that can be imaged). To illustrate, Figure 2 shows a mass spectrum obtained at CV = −60 V from a single location within the testes data set, together with example ion images. As mentioned above, the total analysis time per pixel was 4 min for the testes and kidney samples and 3 min for the brain sample. The total image acquisition times were 624 min (testes), 468 min (kidney), and 252 min (brain). Previous LESA FAIMS MSI5,6 using the planar FAIMS device had single pixel analyses times of 1–2 min but were limited to one or two CV steps. Although the image acquisition time is longer in the current work, that feature is offset by the improved numbers of proteins detected.
Figure 2.

LESA FAIMS MSI of section of testes. Mass spectrum obtained at CV = −60 V for pixel 58. Inset: example ion images and photograph of tissue section after LESA sampling.
The protein numbers reported here are a significant improvement (over 10-fold for testes and kidney and 7-fold for brain) on those previously reported for intact protein FAIMS MSI5,6,18 and approach the same order of magnitude as those obtained in top-down proteomics of protein extracted from cell lines.19,20 In those studies, the extracted proteins were separated by gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) prior to liquid chromatography MS using a 90 min gradient. Here, the MS analysis time at each pixel was 3 or 4 min. It is also useful to consider the numbers of proteins detected with those obtained in bottom-up proteomics studies of homogenized tissue. For example, we detected 981 proteins in kidney, which compares well with the 991 quantifiable proteins detected in a recent proteomic analysis of rat kidney;21 however, the number of testes proteins detected here is approximately 10% of those recently reported in a bottom-up proteomics study of homogenized testis tissue.22
Although the numbers of proteins detected here may be comparable to top-down or bottom-up proteomics studies, it is important to note that in the proteomics studies the proteins are identified. Protein identification is a challenge for intact protein MSI. Typically, LESA MSI of a thin tissue section is followed by LESA sampling of an adjacent tissue section and top-down tandem mass spectrometry. We applied that approach to several of the protein ions detected here, focusing specifically on ions that have not been detected in previous LESA or LESA FAIMS analyses of thin tissue sections. For example, 14+ ions with m/z 661 (MWmeas 9242 Da) detected at CV = −50 V in kidney were selected for electron transfer higher energy collision dissociation (EThcD) (Figure S5, Supporting Information). The protein was identified as high mobility group nucleosomal binding domain 2 (HMGN2) with a sequence coverage of 25%. A protein with this molecular weight has been observed previously by LESA FAIMS MSI; however, it was not identified.6 Three ions were selected for EThcD from the testis tissue (Figure S6, Supporting Information) and identified as β-thymosin 10 (with N-terminus acetylation; sequence coverage 63%), a 24 amino acid peptide fragment arising from the N-terminal region of serum albumin (sequence coverage 91%), and a phosphatidyl-ethanolamine binding protein (with N-terminus acetylation; sequence coverage 8%). This low-throughput approach is clearly not compatible with the hundreds of proteins detected and is a limitation that will need to be addressed. One approach perhaps is to perform unsupervised multivariate analysis followed by targeted identification of the differentiating proteins. Alternatively, a data-independent approach, whereby all protein ions detected are fragmented simultaneously, could be developed.
Conclusion
Integration of the cylindrical FAIMS Pro device in the LESA mass spectrometry imaging workflow results in significant improvements in the number of proteins detected (over 10-fold for testes and kidney and 7-fold for brain) when compared with previous LESA FAIMS imaging in which a planar FAIMS device was employed. For testes and kidney, the improvement in protein numbers detected between “FAIMS voltages on” and “FAIMS voltages off” was over 20-fold, with an over 10-fold improvement observed for brain. The benefits of FAIMS and complementarity of results obtained at various CV values was visualized through use of UpSet plots and t-SNE. Currently, the LESA FAIMS MSI workflow does not lend itself to high throughput identification of proteins; nevertheless, we employed LESA EThcD MS/MS on adjacent tissue sections to assign three previously unidentified proteins.
Acknowledgments
The authors thank Dr. Daniel Eikel (Advion) for technical assistance. H.J.C. is an EPSRC Established Career Fellow (EP/L023490/1 and EP/S002979/1). R.L.G. is funded by EPSRC (EP/L023490/1). J.W.H. is funded by the EPSRC Physical Sciences for Health Doctoral Training Centre (EP/L016346/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05124.
Figure S1: Schematic showing the workflow for tissue analysis via liquid extraction surface analysis coupled to cylindrical FAIMS mass spectrometry imaging. Figure S2: UpSet plot showing set intersections of the filtered Xtract results for intact proteins across the testes image and all compensation field values. Figure S3: UpSet plot showing set intersections of the filtered Xtract results for intact proteins across the kidney image and all compensation field values. Figure S4: UpSet plot showing set intersections of the filtered Xtract results for intact proteins across the brain image and all compensation field values. Figure S5: LESA FAIMS EThCD MS/MS of ions with m/z 661 (14+, 9242.00 Da) detected in kidney (CV = −50 V). Figure S6: LESA FAIMS EThCD MS/MS of ions detected in testes: (A) m/z 706 (7+, 4933.53 Da) (CV = −70 V), (B) m/z 558 (5+, 2788.48 Da) (CV = −70 V), (C) m/z 1036 (20+, 20698.49 Da) (CV = −30 V). Table S1: Fragment ion assignments for high mobility group nucleasomal binding domain 2 (precursor m/z 661; 14+). Table S2: Fragment ion assignments for β-thymosin 10 (precursor m/z 706; 7+). Table S3: Fragment ion assignments for N-terminal region peptide of serum albumin (precursor m/z 558; 5+). Table S4: Fragment ion assignments for phosphatidyl-ethanolamine binding protein (precursor m/z 1036; 20+). (PDF)
File S1: Protein masses detected in testes imaging data set. (XLSX)
File S2: Protein masses detected in kidney imaging data set. (XLSX)
File S3: Protein masses detected in brain imaging data set. (XLSX)
Author Present Address
# School of Pharmacy, University of Nottingham, Nottingham NG7 2RD, United Kingdom
Author Present Address
∇ The Barnett Institute, Northeastern University, Huntington Avenue, Boston, Massachusetts 02115, United States
Author Contributions
○ R.L.G. and J.W.H. contributed equally.
Supplementary data supporting this research is openly available from the University of Birmingham data archive at https://doi.org/10.25500/edata.bham.00000423.
The authors declare the following competing financial interest(s): S.E.A. was an employee of Thermo Fisher Scientific. M.W.B. is an employee of Thermo Fisher Scientific.
Supplementary Material
References
- Kertesz V.; Van Berkel G. J. Fully automated liquid extraction-based surface sampling and ionization using a chip-based robotic nanoelectrospray platform. J. Mass Spectrom. 2010, 45 (3), 252–260. 10.1002/jms.1709. [DOI] [PubMed] [Google Scholar]
- Purves R. W.; Guevremont R.; Day S.; Pipich C. W.; Matyjaszczyk M. Mass spectrometric chracterization of a high-field asymmetric waveform ion mobility spectrometer. Rev. Sci. Instrum. 1998, 69 (12), 4094–4105. 10.1063/1.1149255. [DOI] [Google Scholar]
- Guevremont R. High-field asymmetric waveform ion mobility spectrometry: A new tool for mass spectrometry. J. Chromatogr. A 2004, 1058, 3–19. 10.1016/S0021-9673(04)01478-5. [DOI] [PubMed] [Google Scholar]
- Buchberger A. R.; DeLaney K.; Johnson J.; Li L. Mass spectrometry imaging: A review of emerging advancements and future insights. Anal. Chem. 2018, 90, 240–265. 10.1021/acs.analchem.7b04733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R. L.; Creese A. J.; Race A. M.; Bunch J.; Cooper H. J. LESA FAIMS mass spectrometry for the spatial profiling of proteins from tissue. Anal. Chem. 2016, 88, 6758–6766. 10.1021/acs.analchem.6b01060. [DOI] [PubMed] [Google Scholar]
- Griffiths R. L.; Simmonds A. L.; Swales J. G.; Goodwin R. J. A.; Cooper H. J. LESA MS imaging of heat-preserved and frozen tissue: Benefits of multistep static FAIMS. Anal. Chem. 2018, 90, 13306. 10.1021/acs.analchem.8b02739. [DOI] [PubMed] [Google Scholar]
- Prasad S.; Belford M. W.; Dunyach J.-J.; Purves R. W. On an aerodynamic mechanism to enhance ion transmission and sensitivity of FAIMS for nano-electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25, 2143–2153. 10.1007/s13361-014-0995-8. [DOI] [PubMed] [Google Scholar]
- Purves R. W.; Prasad S.; Belford M.; Vandenberg A.; Dunyach J.-J. Optimization of a new aerodynamic cylindrical FAIMS device for small molecule analysis. J. Am. Soc. Mass Spectrom. 2017, 28, 525–538. 10.1007/s13361-016-1587-6. [DOI] [PubMed] [Google Scholar]
- Hebert A. S.; Prasad S.; belford M. W.; Bailey D. J.; McAlister G. C.; Abbatiello S. E.; Huguet R.; Wouters E. R.; Dunyach J.-J.; Brademan D. R.; Westphall M. S.; Coon J. J. Comprehensive single-shot proteomics with FAIMS on a hybrid orbitrap mass spectrometer. Anal. Chem. 2018, 90, 9529–9537. 10.1021/acs.analchem.8b02233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfammatter S.; Bonneil E.; McManus F. P.; Prasad S.; Bailey D. J.; Belford M.; Dunyach J.-J.; Thibault P. A novel differential ion mobility device expands the depth of proteome coverage and the sensitivity of multiplex proteomic measurements. Mol. Cell. Proteomics 2018, 17, 2051–2067. 10.1074/mcp.TIR118.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Brown K. A.; Lin Z.; Ge Y. Top-down proteomics: Ready for prime time?. Anal. Chem. 2018, 90, 110–127. 10.1021/acs.analchem.7b04747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby T. K.; Fornelli L.; Kelleher N. L. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall E. C.; Bunch J.; Cooper H. J. Direct analysis of intact proteins from Escherichia coli colonies by liquid extraction surface analysis mass spectrometry. Anal. Chem. 2014, 86 (21), 10504–10510. 10.1021/ac503349d. [DOI] [PubMed] [Google Scholar]
- Race A. M.; Styles I. B.; Bunch J. Inclusive sharing of mass spectrometry imaging data requires a converter for all. J. Proteomics 2012, 75 (16), 5111–5112. 10.1016/j.jprot.2012.05.035. [DOI] [PubMed] [Google Scholar]
- Race A. M.; Palmer A. D.; Dexter A. J.; Steven R. T.; Styles I. B.; Bunch J. SpectralAnalysis: software for the masses. Anal. Chem. 2016, 88, 9451. 10.1021/acs.analchem.6b01643. [DOI] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay E. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 2011, 12, 2825–2830. [Google Scholar]
- Lex A.; Gehlenborg N.; Strobelt H.; Vuillemot R.; Pfister H. UpSet: Visualization of Intersecting Sets. IEEE Trans Vis Comput. Graph 2014, 20 (12), 1983–92. 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza K. Y.; Feider C. L.; Klein D. R.; Rosenberg J. A.; Brodbelt J. S.; Eberlin L. S. Desorption electrospray ionization mass spectrometry imaging of proteins directly from biological tissue sections. Anal. Chem. 2018, 90 (13), 7785–7789. 10.1021/acs.analchem.8b00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin K. R.; Fornelli L.; Fellers R. T.; Doubleday P. F.; Narita M.; Kelleher N. L. Quantitation and identification of thousands of human proteoforms below 30 kDa. J. Proteome Res. 2016, 15, 976–982. 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornelli L.; Durbin K. R.; Fellers R. T.; Early B. P.; Greer J. B.; LeDuc R. D.; Compton P. D.; Kelleher N. L. Advancing top-down analysis ofthe human proteome using a benchtop quadrupole-orbitrap mass spectrometer. J. Proteome Res. 2017, 16, 609–618. 10.1021/acs.jproteome.6b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkawi A. K.; Masood A.; Shinwari Z.; Jacob M.; Benabdelkamel H.; Matic G.; Almuhanna F.; Dasouki M.; Alaiya A. A.; Rahman A. M. A. Proteomic analysis of morphologically changed tissues after prolonged dexamthasone treatment. Int. J. Mol. Sci. 2019, 20, 3122. 10.3390/ijms20133122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Xia Y. Q.; Wang G.; Zhou T.; Guo Y.; Zhang C.; An X.; Sun Y.; Guo X.; Zhou Z.; Sha J. In-depth proteomics analysis of whole testis tissue from the adult rhesus macaque. Proteomics 2014, 14, 1393–1402. 10.1002/pmic.201300149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.