

ABSTRACT
The liver is the most frequent site of metastatic spread in malignancies that arise from the digestive system, including pancreatic ductal adenocarcinoma (PDAC). Metastasis to the liver is a major cause of morbidity and mortality in cancer patients, yet mechanisms that govern this process remain poorly understood. Until recently, liver tropism of metastasis was believed to be driven by mechanical factors that direct the passive flow of circulating cancer cells to the liver. However, emerging evidence now shows that liver metastasis is a dynamic process that is, at least in part, dependent on the formation of a “pro-metastatic niche”. Key features of this niche are myeloid cells and fibrosis that support cancer cell colonization and growth. Inflammatory responses that are mounted early during primary tumor development are critical for the recruitment of myeloid cells and the deposition of extracellular matrix (ECM) proteins within the liver. Intriguingly, the inflammatory processes that direct the formation of a pro-metastatic niche share remarkable resemblance to mechanisms of liver injury and regeneration, suggesting that cancer co-opts physiological liver functions to support metastasis. Therefore, therapeutic strategies that target key elements of liver inflammation that form the basis of a pro-metastatic niche may lead to effective treatments for metastatic cancer.
KEYWORDS: Liver, metastasis, cancer, hepatocytes, inflammation, pro-metastatic niche
Metastasis is the most common cause of morbidity and mortality in cancer patients. This is especially evident in gastrointestinal (GI) malignancies, which most commonly spread to the liver. Apart from focal or isolated disease that can be surgically resected, metastatic disease typically portends a grim prognosis. In PDAC, for example, combination chemotherapies have improved the median overall survival by up to 5 months [1,2], but the 5-year overall survival rate remains at 3% [3]. Furthermore, immune checkpoint blockade [4–6], cancer vaccines [7,8], and chimeric antigen receptor (CAR)-modified T cell therapies [9,10] have not provided a major clinical benefit to patients with metastatic PDAC. High mortality in PDAC is, at least in part, attributable to the propensity of cancer cells to spread to the liver early in the disease, even when the primary tumor is small [11,12]. In addition to GI malignancies, cancers that arise from many non-GI organs, including the breast, ovary, lung, and skin, frequently metastasize to the liver. The presence of liver metastases is associated with worse outcomes in patients [13] and reduced response to immunotherapies [14]. Thus, metastatic disease, particularly liver metastasis, poses a significant barrier to effective cancer therapies.
Mechanisms that determine the spread of cancer cells to the liver are now starting to be elucidated. In PDAC, metastatic lesions in the liver are believed to emerge from primary tumor cells that acquire distinct genetic mutations [15]. While these lesions typically harbor identical driver gene mutations [16], liver lesions show a heterogeneous response to cancer treatments [17]. Even across different patients, PDAC has limited heterogeneity in mutations, but patients display varying patterns of metastases and disease behavior [18]. Together, these results suggest that the metastatic behavior of cancer cells is likely determined by both tumor-intrinsic as well as tumor-extrinsic factors. Therefore, understanding mechanisms that direct the spread of cancer cells to the liver and reversal of this process may lead to effective therapies for metastatic cancers. In this Review, we discuss key determinants of liver metastasis, with a focus on cancer cell-extrinsic mechanisms that form the basis of liver tropism of metastasis.
Mechanical determinants of liver metastasis
Metastatic spread of cancer cells is a multi-step process. The traditional model of cancer metastasis delineates a linear process in which primary cancer cells grow and invades their surrounding vasculature. Single cancer cells or aggregates then detach from the primary tumor, enter the circulation, and eventually become embedded in the capillary beds of a distant organ, including the liver. Here, cancer cells undergo extravasation, invade into the distant organ parenchyma, and proliferate. For metastases to form, all steps must be fulfilled, and each step may be rate-limiting [19]. Unique architectural features of the liver have long been believed to enable pooling and seeding of circulating tumor cells. For example, in contrast to many organs in the body, the liver has a dual blood supply. The portal vein, which drains most digestive organs, including the pancreas and colon, provides 60–70% of hepatic blood flow, while the hepatic artery supplies the remaining blood flow. Within the liver, these vessels further ramify through 17 to 20 orders of branches, and this extensive vascular network is believed to function as a “mechanical trap” that captures circulating tumor cells within the liver [20] (Figure 1).
Figure 1.
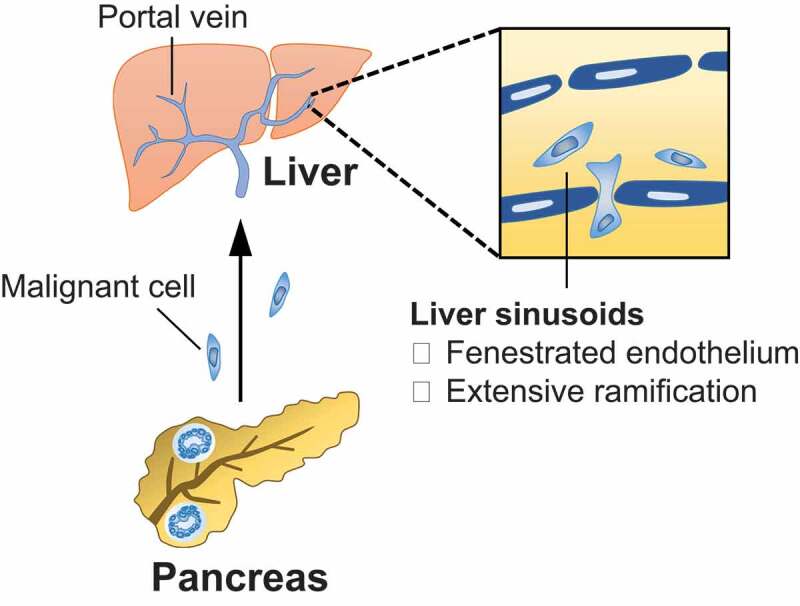
Mechanical determinants of liver metastasis. Primary cancer cells that are released into the circulation drain to the liver via the portal vein. The extensive vascular network and fenestrated endothelial layer of the liver sinusoids act as a mechanical trap that capture circulating tumor cells.
Blood supplies to the liver are especially pertinent to the spread of GI malignancies, since the portal vein serves as a direct conduit between many digestive organs and the liver. In patients with PDAC, for example, circulating tumor cells are detected in all portal vein blood samples, whereas less than 25% of peripheral blood samples contain tumor cells [21]. Similarly, patients with colorectal carcinoma (CRC) display a higher number of circulating cancer cells in the mesenteric vein, which drains to the portal vein, compared to the peripheral vein [22]. These findings support the notion that the liver may capture cancer cells that are released into the circulation by the primary tumor. The fenestrated endothelial layer of the liver sinusoids is also believed to facilitate the invasion of cancer cells into the parenchyma. Compared to other organs in the body, the duration of tumor cell extravasation is typically shorter in the liver due to unique structural and migratory properties of liver endothelial cells. In a mouse model of metastasis, liver endothelial cells were found to migrate and directly engage tumor cells that are dislodged within the liver sinsoids [23]. Mediated by cytoplasmic projections on liver endothelial cells, this interaction facilitates extravasation of tumor cells into the liver parenchyma. Taken together, liver tropism of metastasis is, at least in part, determined by distinct anatomic structures of the liver.
Immunological determinants of liver metastasis
While metastatic spread of cancer may depend on structural features of the liver, metastatic tropism is a complex process that cannot be described using mechanical rationale alone. The liver is a frequent site of cancer spread in many malignancies, including those that arise from organs without a direct vascular connection to the liver. For example, liver metastases are detected in 14% and 9% of patients with breast and ovarian cancers, respectively [13]. This observation was first made by Stephen Paget in 1889, when he described that breast cancer and malignancies of the female reproductive tract metastasize to the liver at a much higher frequency than to the spleen, though both organs are vascular in nature [24]. Based on this observation, Paget posited that metastasis requires a proper “soil” that nurtures the growth of “seeds” (i.e. tumor cells). Another finding supportive of the “seed and soil” hypothesis is that metastasis is a remarkably inefficient process. Previous studies showed that primary tumors may shed more than a million cells per gram of tissue, but less than 0.1% of these cells form metastatic lesions [25–27]. Even though disseminated tumor cells may become dislodged in distant organs, their presence is insufficient to predict the subsequent development of metastases [28]. Moreover, these studies demonstrated that metastases preferentially occur in the liver, suggesting the liver provides a fertile environment (i.e. pro-metastatic niche) for circulating tumor cells to seed and grow.
The molecular and cellular basis of a pro-metastatic niche in the liver is an active area of investigation. The formation of a pro-metastatic niche is initiated by molecules that are released by the primary tumor, including exosomes [29,30], tissue inhibitor of metalloproteinases-1 (TIMP1) [31,32], and interleukin 6 (IL-6) [33]. Exosomes are small membrane vesicles (30–150 nm in size) that contain biomolecules derived from cancer cells, including proteins and nucleic acids [34]. In contrast, TIMP1 and IL-6 are proteins secreted by malignant cells and stromal cells that reside within the primary tumor [31–33]. Even though these molecules are biologically distinct, they all converge on inflammatory responses that induce myeloid cell accumulation and fibrosis within the liver, and these changes in concert create a pro-metastatic niche (Figure 2). Liver-resident cells, including Kupffer cells, hepatic stellate cells (HSCs), and hepatocytes, are critical determinants of this process. Below, we discuss in detail mechanisms by which tumor-derived exosomes, TIMP1, and IL-6 initiate the establishment of a pro-metastatic niche in the liver.
Figure 2.
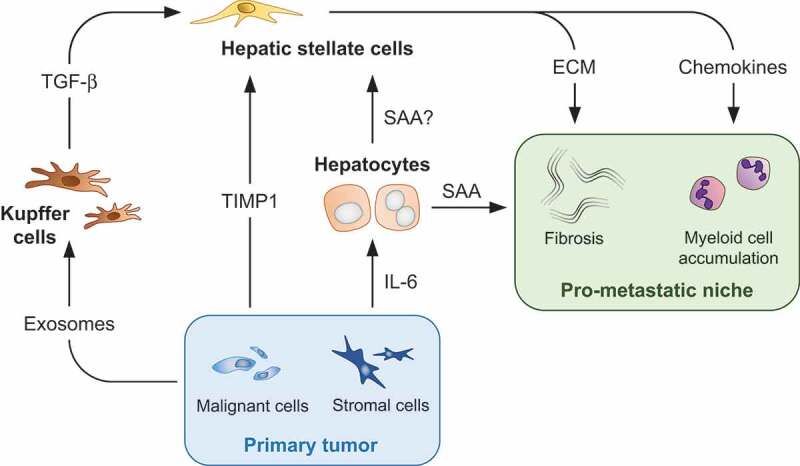
Immunological determinants of liver metastasis. Malignant cells and stromal cells that reside within the primary tumor release factors that engage liver-resident cells, including Kupffer cells, hepatocytes, and hepatic stellate cells. Inflammatory responses mounted by these cells induce myeloid cell accumulation and fibrosis, which form the basis of a pro-metastatic niche in the liver. ECM, extracellular matrix; IL-6, interleukin 6; SAA, serum amyloid A; TGF-β, transforming growth factor β; TIMP1, tissue inhibitor of metalloproteinases-1.
A role for exosomes in directing liver tropism of metastasis was first described by David Lyden and colleagues [29]. Their study showed that macrophage migration inhibitory factor (MIF)-positive exosomes are released into the circulation by primary cancer cells. Exosomes are subsequently phagocytosed by Kupffer cells, which are liver-resident macrophages whose primary function is to bind and internalize pathogens and associated molecules [35]. Kupffer cells in turn produce transforming growth factor β (TGF-β) that induces HSCs to deposit fibronectin within the liver. Much akin to a role for fibronectin in recruiting myeloid cells into the lung [36], fibronectin in the liver enhances the recruitment of F4/80+ myeloid cells that promote cancer cell seeding and growth. Disruption of this signaling cascade via depletion of MIF from exosomes or blockade of TGF-β receptor inhibits liver metastasis. In a follow up study, exosomes that direct the spread of cancer cells to the liver were also shown to express the integrin αvβ5, which is distinct from exosomes that direct metastasis to other organs, including the brain and lung [30]. Once phagocytosed by Kupffer cells, αvβ5-expressing exosomes induce the expression of pro-inflammatory S100 proteins, which recruit myeloid cells into the liver. Collectively, these studies identified myeloid cell accumulation and fibrosis as key elements of a pro-metastatic niche.
TIMP1 is another factor that has been proposed by Achim Krüger and colleagues to initiate the formation of a pro-metastatic niche in the liver [31,32]. Associated with poor prognosis in patients with metastatic cancer, TIMP1 is secreted into the circulation by tumor cells starting in early stages of primary tumorigenesis. Once in the liver, TIMP1 binds its receptor CD63 on HSCs, and this engagement induces the activation of HSCs via phosphatidylinositol 3-kinase (PI3K). Activated HSCs express α-smooth muscle actin (α-SMA) and desmin, in essence becoming myofibroblasts that induce fibrogenesis in the liver [37]. Activated HSCs also secrete chemokine (C-X-C motif) ligand 12 (CXCL12), which in turn, recruits Ly6G+ myeloid cells into the liver in a chemokine (C-X-C motif) receptor 4 (CXCR4)-dependent manner. Autocrine production of TIMP1 by activated HSCs further reinforces the formation of a pro-metastatic niche in the liver by propagating a positive feedback loop. Studies also showed that genetic ablation of Timp1 or its receptor Cd63 prevents activation of HSCs and subsequent myeloid cell accumulation [31,32]. Without these cellular changes, metastatic seeding of pancreatic cancer cells within the liver is inhibited. These results, along with previous studies on exosomes, identify liver inflammation as a key driver of metastasis.
In addition to Kupffer cells and HSCs, hepatocytes play a major role in directing liver tropism of metastasis. Previously, claudin-2 that is expressed on the surface of hepatocytes was shown to enhance liver metastasis by facilitating adhesion between circulating tumor cells and hepatocytes [38]. Our recent work also highlights the importance of hepatocytes in inducing the formation of a pro-metastatic niche in the liver [33]. Early during pancreatic cancer development in mice, stromal cells that reside within the primary tumor release IL-6 into the circulation. IL-6 drains to the liver through the portal vein and subsequently activates signal transducer and activator of transcription 3 (STAT3) signaling in hepatocytes. In turn, hepatocytes produce acute phase reactants serum amyloid A1 and A2 (referred to collectively as SAA). Overexpression of SAA by hepatocytes also occurs in patients with PDAC, and many patients with locally advanced and metastatic disease have elevated levels of circulating SAA compared to healthy individuals. Our results are consistent with a previous study demonstrating an association between increased circulating levels of acute-phase reactants and pancreatic cancer development [39]. We also found that high levels of circulating SAA correlate with worse outcomes in patients with metastatic PDAC. Intriguingly, overexpression of SAA by hepatocytes occurs in patients with metastatic CRC, suggesting that SAA may regulate liver metastasis across various malignancies.
Our study further showed that SAA is critical for the deposition of fibronectin and collagen within the liver. SAA also induces the expression of myeloid chemoattractants, including S100 proteins and chemokine (C-C motif) ligand 6 (CCL6). Through these functions, SAA engenders the accumulation of F4/80+ and Ly6G+ myeloid cells and liver fibrosis, which in concert establish a pro-metastatic niche. Genetic ablation or blockade of any component of IL-6 – STAT3 – SAA signaling effectively prevents this process and inhibits liver metastasis. In addition, even though SAA released by hepatocytes enters the systemic circulation, the SAA-mediated formation of a pro-metastatic niche is specific to the liver, and genetic ablation of Saa has no bearing on lung metastasis. Based on this result, cellular targets of SAA are most likely liver-resident cells that have the capacity to either directly or indirectly recruit myeloid cells and induce fibrotic changes in the liver. Identification of such liver-resident cells is an area of active investigation in our laboratory. One promising target is HSCs, which produce chemokines and promote fibrosis in the liver upon stimulation by SAA [40]. In summary, SAA released by hepatocytes in response to IL-6 derived from stromal cells within the primary tumor orchestrates liver metastasis.
Even after a pro-metastatic niche in the liver is fully formed and metastasis has already occurred, myeloid cells and the fibrotic microenvironment of the liver continue to have a major role in supporting liver metastasis. Consistent with studies showing that tumor cells release chemotactic factors to recruit myeloid cells [41,42], metastatic tumor cells attract pro-tumorigenic myeloid cells to the liver. A recent study showed that myeloid cells associated with these tumor cells secrete granulin, which activates HSCs to differentiate into myofibroblasts [43]. Myofibroblasts then release periostin, an ECM component that sustains a fibrotic microenvironment needed for the proliferation of metastatic tumor cells. In the same study, inhibition of granulin halted the growth of metastatic lesions and, by doing so, prevented metastasis to the liver. Myeloid cell accumulation and fibrotic changes continue as metastatic lesions grow, and these lesions ultimately mirror the microenvironment of the primary tumor [44]. Thus, myeloid cells and fibrosis are critical determinants of liver metastasis and are essential to all stages of this process.
Myeloid cell recruitment and ECM alterations are key features of pro-metastatic niches in other organs as well, such as the lung [34,45]. Much akin to the formation of a pro-metastatic niche in the liver, molecules released from the primary tumor initiate cellular and stromal alterations in the lung. For instance, the hypoxic microenvironment of the primary tumor drives tumor cells to secrete lysyl oxidase (LOX), which enhances the capacity of tumor cells to migrate and invade into distant organs [46]. LOX that is released into the circulation also crosslinks collagen fibers and creates within the lung a fibrotic microenvironment that induces the recruitment of CD11b+ myeloid cells [47]. These cells then further modify ECM components in the lung to facilitate cancer cell seeding and growth. Interestingly, LOX-mediated fibrosis also enhances metastatic colonization of the liver in a model of breast cancer [48], suggesting that LOX may be fundamental to pro-metastatic niche formation not only in the lung, but also the liver.
Primary tumor cells may also release into the circulation exosomal RNAs, which engage Toll-like receptor (TLR) 3 in lung epithelial cells [49]. As a result, lung epithelial cells express chemokines that promote myeloid cell accumulation within the lung. Activation of TLR3 in lung epithelial cells is also associated with fibronectin deposition, and, together with myeloid cell accumulation, these changes establish a pro-metastatic niche in the lung. Versicans are yet another tumor-derived molecule that has been implicated in driving the metastatic spread of cancer cells to the lung [50]. Upon engaging TLR2 on myeloid cells, versicans induce myeloid cells to produce tumor-necrosis factor α (TNF-α) that creates a pro-inflammatory milieu hospitable for metastatic growth. Future studies should further explore roles for these tumor-derived molecules in regulating the spread of cancer cells to the liver and crosstalks that may exist between processes that establish a pro-metastatic niche in various distant organs.
Common themes in liver regeneration and metastasis
Solid malignancies, including PDAC and CRC, are described to be in a perpetual state of “wound healing” because of pervasive immune cell infiltration and fibrosis that are typically associated with tissue repair [51]. Central to this principle is the notion that physiological processes that are beneficial to our health may in another context be co-opted for tumorigenesis. We find that this principle applies not only to primary tumor development but also to processes that direct liver metastasis. This is particularly relevant to IL-6 – STAT3 signaling in hepatocytes. While this signaling is critical for the formation of a pro-metastatic niche in the liver, IL-6 – STAT3 signaling is also important for coordinating liver repair and regeneration after injury [52]. In mouse models of liver injury, including partial hepatectomy [53], sclerosing cholangitis [54,55], and steatohepatitis [56], IL-6 – STAT3 signaling protects hepatocytes from apoptosis and is required for their proliferation. Genetic ablation of either Il-6 or Stat3 predisposes the liver to tissue necrosis and metabolic derangements that eventually lead to liver failure.
Comparable to myeloid cell accumulation that occurs within the liver early during primary tumor development, liver injury engenders robust recruitment of myeloid cells into the liver, especially in response to trauma or infection [57–59]. Liver injury induces hepatocytes to produce acute phase reactants, which facilitate elimination of pathogens and tissue repair to restore homeostasis. SAA is a major acute phase reactant whose circulating levels may increase by more than a 1,000-fold in response to inflammatory stimuli [60]. Evolutionarily conserved in mammals [61,62] and other vertebrate species [63], SAA is believed to be an archetypal acute phase reactant. In addition to serving as an opsonin for bacteria [64], SAA binds a range of structurally distinct receptors that are expressed on the surface of myeloid cells and fibroblasts, including TLR2, TLR4, and formyl peptide receptor 2 (FPR2). Through these interactions, SAA induces the expression of pro-inflammatory cytokines and migration of myeloid cells [65]. Within the liver, myeloid cells attenuate inflammatory responses mediated by other innate immune cells and T cells to minimize liver damage while supporting tissue repair [57–59].
Another molecule that serves a key determinant of both liver metastasis and regeneration is CXCL12 [66]. Following liver injury, HSCs and liver sinusoidal endothelial cells release CXCL12, which in turn mobilizes bone marrow-derived mesenchymal stem cells [67]. Upon engraftment into the liver, these cells transdifferentiate into hepatocyte-like cells to facilitate liver regeneration. A balanced interplay between CXCL12 and its receptors CXCR4 and CXCR7 is believed to promote liver regeneration while minimizing liver injury [68]. In addition to promoting liver regeneration, CXCL12 supports the progression of hepatocellular carcinoma (HCC). CXCL12 is expressed early during the invasion of HCC and remains upregulated throughout the invasion process [69]. Produced primarily by liver cells that are located adjacent to HCC [70], CXCL12 recruits pro-tumorigenic Gr-1+ myeloid cells and activates HSCs to induce liver fibrosis [71]. Taken together, molecules that mediate the formation of a pro-metastatic niche in the liver have major roles in liver metastasis as well as liver regeneration.
ECM alterations that follow liver injury also resemble the deposition of ECM proteins that occurs during the establishment of a pro-metastatic niche in the liver. Recovery of liver injury requires coordinated ECM remodeling to ensure proper restoration of liver tissue. In response to injury, the liver shows increased deposition of ECM proteins, including fibronectin and collagen, and alterations in non-structural proteins [72,73]. Together, these changes increase liver stiffness, which is believed to promote the migration and proliferation of bone marrow-derived cells necessary for liver regeneration [74,75]. In addition, fibronectin that is deposited within the liver in response to injury improves the survival of hepatocytes [76] and promotes liver sinusoid repair by enhancing the adhesion of liver sinusoidal endothelial cells to injured tissues [77]. Fibronectin also prevents the liver from becoming excessively fibrotic by regulating the availability of TGF-β to HSCs, thereby ensuring optimal levels of fibrosis necessary for liver repair [78]. Hence, even though the formation of a pro-metastatic niche and liver regeneration are biologically distinct, parallels can be draw in that both processes depend on myeloid cell accumulation and ECM remodeling. Interestingly, these changes are also observed in the liver of female mice after weaning, providing potential rationale for the higher frequency of liver metastases in women with postpartum breast cancer [79]. Collectively, these parallels suggest that cancer usurps physiological liver functions to promote the spread of tumor cells to the liver.
Therapeutic strategies and future directions
In describing liver tropism of metastasis in 1889, Paget stated that “he who turns over the records of cases of cancer is only a ploughman, but his observation of the properties of the soil may also be helpful [24].” Recent studies are beginning to provide insight on mechanisms underlying the formation of a fertile “soil” that supports cancer cell colonization and growth in the liver. Based on these studies, hepatocytes, HSCs, and Kupffer cells have emerged as key liver-resident cells that orchestrate myeloid cell accumulation and fibrosis. Therefore, therapeutic strategies that target specific molecular and cellular components of the liver pro-metastatic niche may prevent liver metastasis and, by doing so, significantly improve patient outcomes. For instance, hepatocyte-mediated formation of a pro-metastatic niche in the liver presents multiple opportunities for therapeutic intervention [80]. Given that IL-6 initiates pro-metastatic niche formation, antibodies that target IL-6 or IL-6 receptor (IL-6R) offer an effective means to prevent liver metastasis. In addition, molecules that target components of IL-6 – STAT3 signaling, including Janus kinase 1/2 (JAK1/2) and STAT3 inhibitors, provide an approach to inhibit metastasis with high specificities. Other molecules that drive the formation of a pro-metastatic niche in the liver, including TGF-β, may also be targeted using small molecules [29].
Apart from antibody- and small molecule-based therapies, nanoparticles that enable liver-specific expression of antibody-like proteins (so-called “traps”) that bind specific molecules offer an alternative means to inhibit the formation of a pro-metastatic niche. In a recent study, CXCL12 traps were utilized to prevent the recruitment of myeloid cells and seeding of CXCR4+ cancer cells within the liver [81]. Similar strategies may be applied to neutralize key chemoattractants that have been implicated in pro-metastatic niche formation, including SAA and S100 proteins. Therapeutic agents that can reverse liver fibrosis may also be used in conjunction with modalities that inhibit myeloid cell recruitment to further prevent liver metastasis. CD40 agonists [82,83] as well as focal adhesion kinase (FAK) inhibitors [84,85] are promising therapeutic agents that may be used to reverse ECM deposition within the liver. In general, strategies that target the pro-metastatic niche in the liver may be combined with conventional chemo- and radiation therapies. However, one must exercise caution because some cancer treatments, including anti-vascular endothelial growth factor (VEGF) therapy [86], are known to induce liver fibrosis and may counteract therapies designed to prevent the formation of a pro-metastatic niche in the liver. Medications that are prescribed for common chronic conditions, such as diabetes, may also accelerate tumor metastasis [87]. Therefore, additional studies are needed to determine optimal cancer treatment regimens that can effectively tackle the primary cancer as well as metastatic disease in the liver.
Future studies should also focus on understanding the impact of liver pro-metastatic niche on adaptive immune responses against the primary tumor and metastatic lesions. The fact that patients with liver metastases respond less to immunotherapies [14] suggests that the formation of a pro-metastatic niche in the liver may suppress local as well as systemic T cells responses against tumor cells. Supportive of this idea is the capacity of SAA to regulate T cell migration [88], which may impact T cell infiltration into tumor tissue and subsequent interactions between T cells and tumor cells. Myeloid cells are also an important determinant of cancer dormancy [89], and therapeutic strategies that target myeloid cells in the liver may alter recognition and elimination of tumor cells by T cells. Together, additional studies on mechanisms that direct the formation of a pro-metastatic niche and its impact on anti-tumor immune responses may lead to effective therapies for cancer.
Funding Statement
This work is supported by National Institutes of Health grants [R01 CA197916 (G.L.B.), R01 CA245323 (G.L.B.), T32 HL007439 (J.W.L.), F30 CA196106 (J.W.L.)]; The Pancreatic Cancer Action Network-AACR Career Development Award [15-20-25-BEAT] supported by an anonymous foundation (G.L.B.); and the Stand Up to Cancer (SU2C) Innovative Research Grant [SU2C-AACR-IRG 13-17 (G.L.B)].
Disclosure statement
G.L.B. is a consultant/advisory board member for Seattle Genetics, Aduro Biotech, AstraZeneca, Bristol-Myers Squibb, Genmab, Merck, and BiolineRx; reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, Verastem, Halozyme, Biothera, Newlink, Novartis, and Janssen. G.L.B. is an inventor of intellectual property and recipient of royalties related to CAR T cells that are licensed by the University of Pennsylvania to Novartis. No additional potential conflicts of interest were disclosed by J.W.L.
References
- [1].Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. [DOI] [PubMed] [Google Scholar]
- [2].Hoff VD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- [4].Royal RE, Levy C, Turner K. et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aglietta M, Barone C, Sawyer MB, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014;25:1750–1755. [DOI] [PubMed] [Google Scholar]
- [7].Le DT, Brockstedt DG, Nir-Paz R, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin. Cancer Res. 2012;18:858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. Journal of Clinical Oncology. 2015;33:1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beatty GL, Haas AR, Maus MV. et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T Cells induce antitumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beatty GL, O’Hara MH, Lacey SF, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155:29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. [DOI] [PubMed] [Google Scholar]
- [12].Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009;133:413–422. [DOI] [PubMed] [Google Scholar]
- [13].Schroeder A, Heller DA, Winslow MM, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2011;12:39–50. [DOI] [PubMed] [Google Scholar]
- [14].Tumeh PC, Hellmann MD, Hamid O, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. 2017;5:417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017;49:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013;19:6286–6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sahin IH, Elias H, Chou JF, et al. Pancreatic adenocarcinoma: insights into patterns of recurrence and disease behavior. BMC Cancer. 2018;18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. [DOI] [PubMed] [Google Scholar]
- [20].Weiss L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin Exp Metastasis. 1992;10:191–199. [DOI] [PubMed] [Google Scholar]
- [21].Catenacci DVT, Chapman CG, Xu P, et al. Acquisition of portal venous circulating tumor cells from patients with pancreaticobiliary cancers by endoscopic ultrasound. Gastroenterology. 2015;149:1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Denève E, Riethdorf S, Ramos J, et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin. Chem. 2013;59:1384–1392. [DOI] [PubMed] [Google Scholar]
- [23].Paku S, Döme B, Tóth R, et al. Organ-specificity of the extravasation process: an ultrastructural study. Clin Exp Metastasis. 2000;18:481–492. [DOI] [PubMed] [Google Scholar]
- [24].Paget S. Distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–573. [PubMed] [Google Scholar]
- [25].Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2ʹ-deoxyuridine. J Natl Cancer Inst. 1970;45:773–782. [PubMed] [Google Scholar]
- [26].Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512–516. [PubMed] [Google Scholar]
- [27].Luzzi KJ, MacDonald IC, Schmidt EE, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998;153:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015;34:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Costa-Silva B, Aiello NM, Ocean AJ, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hoshino A, Costa-Silva B, Shen T-L, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Seubert B, Grünwald B, Kobuch J, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology. 2015;61:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grünwald B, Harant V, Schaten S, et al. Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via CD63 signaling to create a premetastatic niche in the liver. Gastroenterology. 2016;151:1011–1024.e7. [DOI] [PubMed] [Google Scholar]
- [33].Lee JW, Stone ML, Porrett PM, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature. 2019;567:249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Becker A, Thakur BK, Weiss JM, et al. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30:836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006. [DOI] [PubMed] [Google Scholar]
- [36].Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tabariès S, Dupuy F, Dong Z, et al. Claudin-2 promotes breast cancer liver metastasis by facilitating tumor cell interactions with hepatocytes. Mol. Cell. Biol. 2012;32:2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Brand RE, Nolen BM, Zeh HJ, et al. Serum biomarker panels for the detection of pancreatic cancer. Clin. Cancer Res. 2011;17:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Siegmund SV, Schlosser M, Schildberg FA, et al. Serum amyloid A induces inflammation, proliferation and cell death in activated hepatic stellate cells. PLoS ONE. 2016;11:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Qian B-Z, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nielsen SR, Quaranta V, Linford A. et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aiello NM, Bajor DL, Norgard RJ. et al. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat Commun. 2016;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gao Y, Bado I, Wang H, et al. Metastasis organotropism: redefining the congenial soil. Dev Cell. 2019;49:375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. [DOI] [PubMed] [Google Scholar]
- [47].Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cox TR, Bird D, Baker A-M, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73:1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu Y, Gu Y, Han Y, et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell. 2016;30:243–256. [DOI] [PubMed] [Google Scholar]
- [50].Kim S, Takahashi H, Lin -W-W, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Flier JS, Underhill LH, Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. [DOI] [PubMed] [Google Scholar]
- [52].Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64:1403–1415. [DOI] [PubMed] [Google Scholar]
- [53].Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. [DOI] [PubMed] [Google Scholar]
- [54].Mair M, Zollner G, Schneller D, et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology. 2010;138:2499–2508. [DOI] [PubMed] [Google Scholar]
- [55].Plum W, Tschaharganeh DF, Kroy DC, et al. Lack of glycoprotein 130/signal transducer and activator of transcription 3-mediated signaling in hepatocytes enhances chronic liver injury and fibrosis progression in a model of sclerosing cholangitis. Am. J. Pathol. 2010;176:2236–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kroy DC, Beraza N, Tschaharganeh DF, et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology. 2009;51:463–473. [DOI] [PubMed] [Google Scholar]
- [57].Klein C, Wüstefeld T, Assmus U, et al. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang H, Park O, Lafdil F, et al. Interplay of hepatic and myeloid signal transducer and activator of transcription 3 in facilitating liver regeneration via tempering innate immunity. Hepatology. 2009;51:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sander LE, Sackett SD, Dierssen U, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010;207:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gabay C, Kushner I, Epstein FH. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. [DOI] [PubMed] [Google Scholar]
- [61].Uhlar CM, Burgess CJ, Sharp PM, et al. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics. 1994;19:228–235. [DOI] [PubMed] [Google Scholar]
- [62].Uhlar CM, BLACK IL, SHIELDS DC, et al. Wallaby serum amyloid A protein: cDNA cloning, sequence and evolutionary analysis. Scand. J. Immunol. 1996;43:271–276. [DOI] [PubMed] [Google Scholar]
- [63].Jensen LE, Hiney MP, Shields DC, et al. Acute phase proteins in salmonids: evolutionary analyses and acute phase response. J Immunol. 1997;158:384–392. [PubMed] [Google Scholar]
- [64].Shah C, Hari-Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood. 2006;108:1751–1757. [DOI] [PubMed] [Google Scholar]
- [65].Eklund KK, Niemi K, Kovanen PT. Immune functions of serum amyloid A. Crit Rev Immunol. 2012;32:335–348. [DOI] [PubMed] [Google Scholar]
- [66].Liepelt A, Tacke F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J Physiol Gastrointest Liver Physiol. 2016;311:G203–9. [DOI] [PubMed] [Google Scholar]
- [67].DeLeve LD, Wang X, Wang L. VEGF-sdf1 recruitment of CXCR7+ bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2016;310:G739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ding B-S, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chen R-X, Song H-Y, Dong YY, et al. Dynamic expression patterns of differential proteins during early invasion of hepatocellular carcinoma. PLoS ONE. 2014;9:e88543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Neve Polimeno M, Ierano C, D’Alterio C, et al. CXCR4 expression affects overall survival of HCC patients whereas CXCR7 expression does not. Cell Mol Immunol. 2014;12:474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen Y, Huang Y, Reiberger T, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ogata H, Chinen T, Yoshida T, et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-β1 production. Oncogene. 2006;25:2520–2530. [DOI] [PubMed] [Google Scholar]
- [73].Klaas M, Kangur T, Viil J, et al. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci Rep. 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tse JR, Engler AJ, Leipzig ND. Stiffness gradients mimicking in vivo tissue variation regulate mesenchymal stem cell fate. PLoS ONE. 2011;6:e15978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vincent LG, Choi YS, Alonso-Latorre B, et al. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol J. 2013;8:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Moriya K, Sakai K, Yan MH, et al. Fibronectin is essential for survival but is dispensable for proliferation of hepatocytes in acute liver injury in mice. Hepatology. 2012;56:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sackey-Aboagye B, Olsen AL, Mukherjee SM, et al. Fibronectin extra domain A promotes liver sinusoid repair following hepatectomy. PLoS ONE. 2016;11:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kawelke N, Vasel M, Sens C, et al. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-β. PLoS ONE. 2011;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Goddard ET, Hill RC, Nemkov T. et al. The rodent liver undergoes weaning-induced involution and supports breast cancer metastasis. Cancer Discov. 2017;7:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lee JW, Beatty GL. Hepatocytes prepare the soil for liver metastasis. Mol Cell Oncol. 2019;6:e1632686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Goodwin TJ, Zhou Y, Musetti SN, et al. Local and transient gene expression primes the liver to resist cancer metastasis. Sci Transl Med. 2016;8:364ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Long KB, Gladney WL, Tooker GM, et al. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6:400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhao X-K, Yu L, Cheng ML. et al. Focal adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci Rep. 2017;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jiang H, Hegde S, Knolhoff BL. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rahbari NN, Kedrin D, Incio J, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci. Transl. Med. 2016;8:360ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang H, Liu X, Long M, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med. 2016;8:334ra51. [DOI] [PubMed] [Google Scholar]
- [88].Xu L, Badolato R, Murphy WJ, et al. A novel biologic function of serum amyloid A. Induction of T lymphocyte migration and adhesion. J Immunol. 1995;155:1184–1190. [PubMed] [Google Scholar]
- [89].Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]