

Abstract
Photoactivation in the Photoactive Yellow Protein, a bacterial blue-light photoreceptor, proceeds via photoisomerization of the double C=C bond in the covalently attached chromophore. Quantum chemistry calculations, however, have suggested that in addition to double-bond photoisomerization, the isolated chromophore and many of its analogues can isomerize around a single C–C bond as well. Whereas double-bond photoisomerization has been observed with X-ray crystallography, experimental evidence of single-bond photoisomerization is currently lacking. Therefore, we have synthesized a chromophore analogue, in which the formal double bond is covalently locked in a cyclopentenone ring, and carried out transient absorption spectroscopy experiments in combination with nonadiabatic molecular dynamics simulations to reveal that the locked chromophore isomerizes around the single bond upon photoactivation. Our work thus provides experimental evidence of single-bond photoisomerization in a photoactive yellow protein chromophore analogue and suggests that photoisomerization is not restricted to the double bonds in conjugated systems. This insight may be useful for designing light-driven molecular switches or motors.
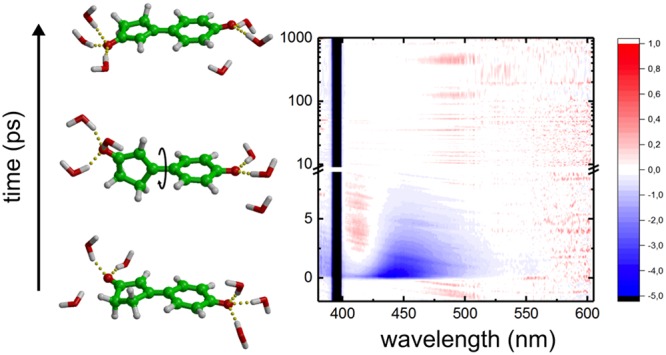
Photoactive yellow protein (PYP), a 125-residue water-soluble protein, is believed to act as the primary photoreceptor in the negative phototaxis response of the Halorhodospira halophila bacterium to blue light.1 The photocycle of PYP is initiated by an ultrafast photoisomerization of the conjugated p-coumaric acid chromophore that is covalently bound to the protein via a thioester linkage. Time-resolved crystallography2,3 and molecular dynamics simulations4 suggest that the photoisomerization occurs around the double bond (Figure 1).
Figure 1.
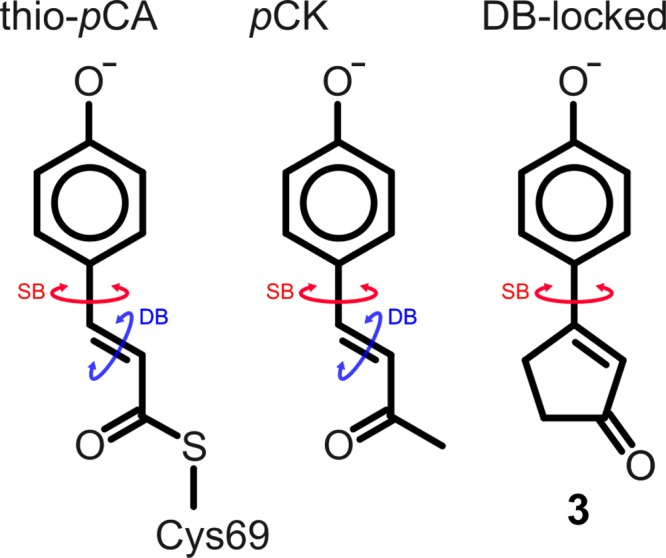
In PYP, the para-coumaric acid (pCA) chromophore is deprotonated and covalently attached to the protein via a thioester bond with a cysteine side chain (thio-pCA, left structure). The pCK analogue (middle) has a methyl group instead of a thioester but exhibits very similar optical properties compared with thio-pCA.5 In this work, the double-bond (DB)-locked analogue (3, right) was studied. The cyclic structure prevents isomerization around the double bond, and only single-bond (SB) isomerization is possible.
Chromophore analogues also undergo ultrafast deactivation in solution, but for many analogues, no photoproduct is detected.5−10 This paradox was addressed with nonadiabatic molecular dynamics simulations of the pCK analogue in water.11,12 These simulations suggested that photoisomerization can also occur around the single bond adjacent to the phenolate ring (SB, Figure 1) without forming a photoproduct. Despite support from high-level ab initio quantum chemistry calculations,13−17 direct experimental evidence of single-bond isomerization is, however, still lacking. Experimentally demonstrating the existence of single-bond photoisomerization in the chromophore of PYP is important not only to validate theoretical methods but also to better understand the basic principles and versatility of photoisomerization in general.
Previously, experiments on locked analogues of important biological chromophores, in which isomerization around a selected bond is prevented by means of a covalent bridge, have provided valuable insights into the isomerization process in rhodopsins,18 phytochromes,19 and PYP.20,21 It was found that when PYP is reconstituted with chromophore analogues, with either the single bond adjacent to the phenolate ring (SB, Figure 1)21 or both single and double bonds locked,20 the protein is still photoactive, albeit with a lower quantum yield. In contrast, gas-phase photoelectron spectroscopy experiments on a pCA analogue in which the double bond is locked suggest that photoisomerization is suppressed in favor of electron photodetachment.22
To experimentally test whether photoisomerization around the single bond provides an alternative decay channel for the photoexcited chromophore, as suggested by computations,11−16 we have synthesized a chromophore analog in which the double bond is locked within a rigid cyclopentenone ring (Figure 1, DB-locked, compound 3). Details of the synthesis and structural characterization of 3 can be found in the Supporting Information (SI). Although this chromophore lacks the thioester moiety that is present in the protein, previous experiments and computations on thio-pCA and pCK have shown that the photochemistries of these chromophores are very similar.5,13 Therefore, results obtained for the double-bond-locked (DB-locked) chromophore should be transferable to the native chromophore of PYP.
In Figure 2, we compare the steady-state absorption and emission spectra of the DB-locked chromophore (3) and of pCK in aqueous solution at pH >10. Because the pKa values of pCK and the DB-locked chromophore are around 8 and 9, respectively (SI), both chromophores are fully deprotonated in our measurements. The similarity between the absorption spectra suggests that the covalent bridge that locks the double bond has no significant effect on the electronic structure of the chromophore in the ground (S0) or excited (S1) state. However, comparing the fluorescence spectra reveals that the DB-locked chromophore has a smaller Stokes shift than pCK. We attribute this difference to the more rigid structure of the DB-locked chromophore (SI).
Figure 2.

Left panels: Normalized absorption (black) and fluorescence (red) spectra of the DB-locked chromophore (a) and pCK (d) in water at pH >10. Middle panels: 2D plots showing transient absorption as a function of time after excitation with a 120 fs laser pulse at 395 nm of the DB-locked chromophore (b) and pCK (e). Part of the spectral region is masked by scattering from the pump pulse. Right panels: Transient absorption spectra for selected delay times after 120 fs excitation at 395 nm of the DB-locked chromophore (c) and pCK (f).
Time-resolved transient absorption experiments were performed for both the DB-locked chromophore and pCK with a ∼120 fs pump pulse centered at 395 nm. The chirp-corrected spectra are shown in Figure 2b,c and Figure 2e,f. The transient absorption spectrum of pCK has been previously reported by Changenet-Barret and coworkers.10 Our results are in good agreement with theirs, despite a lower laser power in our experiments (SI). We chose our excitation energies to be low enough to prevent the formation of free electrons due to a two-photon photoionization process that can occur at higher laser intensities.10
The spectra presented in Figure 2 show similar features for both chromophores. The most prominent feature is the negative signal at 420–480 nm for the DB-locked chromophore and at 440–520 nm for pCK. On the basis of the steady-state fluorescence spectra, we attribute this signal to stimulated emission. The negative bands around 400 nm are assigned to the ground-state bleach (GSB) signal, centered at ∼380 nm. However, in our measurements, the GSB was largely masked by the scattering from the pump pulse. We also observe a distinct positive feature at 410 nm for the DB-locked chromophore and at 430 nm for pCK, which grows initially but then decays, leaving no long-lived absorption signal. This transient signal is more prominent in pCK than in the DB-locked chromophore. We attribute this band to the absorption of the vibrationally hot ground state that is formed immediately after excited-state decay via a conical intersection, as in previous work.10
A full global analysis of the transient absorption data is prevented by overlapping spectral features and their dynamic shifts, which would lead to wavelength-dependent decay kinetics. We therefore did not perform such analysis. Instead, we can immediately conclude from the raw data presented in Figure 2 that the excited-state decay is ultrafast in both chromophores: All transient signals disappear within ∼10 ps, leaving no long-lived signals associated with photoproducts. The ultrafast relaxation of the DB-locked analogue is consistent with the single-bond photoisomerization pathway predicted for pCK.11,12 Because we also do not observe any spectral features associated with hydrated electrons, we can furthermore rule out that in water, electron photodetachment occurs, in contrast with the situation in the gas phase.22−24 This difference was already rationalized by Gromov and coworkers, who showed that hydrogen bonding between the chromophore and water molecules brings the energy of the S1 state significantly below that of the ionized state.13 We therefore conclude, in line with results from computations,11−16 that the chromophore decays via a rotation around the single bond adjacent to the phenolate ring, followed by internal conversion through a conical intersection.
To confirm that the ultrafast decay observed in our experiments is indeed due to single-bond photoisomerization, we also performed 100 nonadiabatic molecular dynamics simulations of the deprotonated DB-locked chromophore in water. (See the SI for details.) In all but one of the simulations, the chromophore rapidly twists by 90° around the single bond after photoexcitation. Figure 3 shows the time evolution of the single-bond torsion angle in one of the QM/MM trajectories. As in pCK,12 shortly after reaching the 90° single-bond twisted conformation in the excited state, the system decays to the ground state via a conical intersection. The existence of this conical intersection in the presence of water was confirmed at a higher level of theory (SI). After the transition, which was modeled as a diabatic surface hop,25 the chromophore either relaxes back to the initial conformation or continues the single-bond isomerization process (as in Figure 3). However, because the phenolate ring is symmetric, there is no distinction between this photoproduct and the initial conformation, and thus the isomerization quantum yield is zero. In one trajectory, a higher energy conical intersection is accessed at which the phenolate ring is puckered (SI). However, because this decay pathway was observed in only 1% of the trajectories, we consider it a minor decay channel and hence insignificant. Although the number of trajectories is too small to be statistically relevant, an exponential fit to the decay times yields an excited-state lifetime of 1.3 ps (SI), in qualitative agreement with our experiments (Figure 2c).
Figure 3.
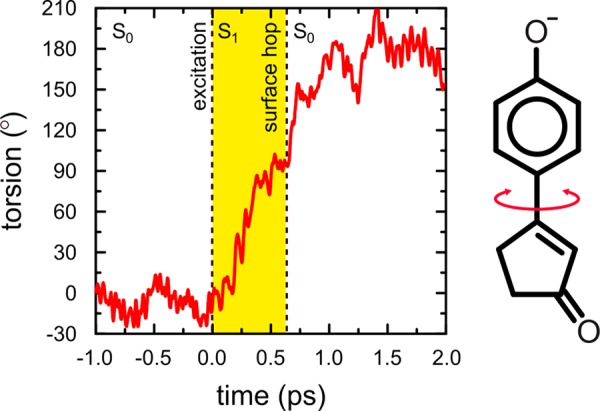
Time evolution of the single bond, adjacent to the phenolate ring in excited-state QM/MM MD simulations of the DB-locked chromophore in water. At 0.0 ps, the system is excited. The period in the excited state (S1) is highlighted in yellow, and the surface hop back to the ground state (S0) is indicated by the vertical dashed line at 0.646 ps. An animation of this trajectory is available as Supporting Information.
In summary, the results of our experiments and simulations strongly suggest that single-bond photoisomerization is possible in the chromophore of Photoactive Yellow Protein. Although the single-bond isomerization pathway was predicted more than a decade ago,4,11,13,15,25 it is now also supported by the ultrashort excited-state lifetime of the DB-locked chromophore in our experiments. With the lock on the double bond, the only remaining candidate for the ultrafast decay process is single-bond isomerization. The lack of photoproduct for this deactivation channel is further supported by the total recovery of the original spectrum within picoseconds (Figure 2c). Whereas it remains to be seen whether the single-bond isomerization channel is also active inside PYP, our results confirm that in addition to double-bond isomerization, single-bond isomerization can also be considered when designing light-driven molecular switches or motors.26
Acknowledgments
We thank the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533), the CSC-IT center in Espoo, Finland, as well as PRACE for awarding us access to resource Cartesius based in The Netherlands at SURFsara and to resource Curie based in France at GENCI. We thank Antti Lahdenperä for his early contributions to the synthesis of 3. This work has been done as part of the BioExcel CoE (www.bioexcel.eu), a project funded by the European Union contracts H2020-INFRAEDI-02-2018-823830 and H2020-EINFRA-2015-1-675728. In addition, we received funding from the Academy of Finland (grants 297874, 290677, and 304455).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c00060.
Complete description of the synthesis of the DB-locked chromophore (3), structural characterizations of intermediates and 3, NMR spectra of intermediates and 3, complete description of the transient absorption experiments, pH-dependent absorption spectra of pCK and 3, complete description of the molecular dynamics simulations, and results of 100 trajectories (PDF)
Animation of photoisomerization trajectory (MOV)
Author Contributions
† S.M. and D.M. contributed equally to this work. G.G., M.P., and P.M.P. designed and supervised the project. H.L.L., R.R.P., and P.M.P. performed the chromophore synthesis and analysis. S.M. performed the spectroscopy measurements. S.M. and P.M. analyzed the spectroscopy data. D.M. performed and analyzed the QM/MM simulations. G.G. wrote the manuscript with contributions from all other authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Hellingwerf K. J.; Hendriks J.; Gensch T. Photoactive Yellow Protein, A New Type of Photoreceptor Protein: Will This ”Yellow Lab” Bring Us Where We Want to Go?. J. Phys. Chem. A 2003, 107, 1082–1094. 10.1021/jp027005y. [DOI] [Google Scholar]
- Schotte F.; Cho H. S.; Kaila V. R. I.; Kamikubo H.; Dashdorj N.; Henry E. R.; Graber T. J.; Henning R.; Wulff M.; Hummer G.; Kataoka M.; Anfinrud P. A. Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 19256–19261. 10.1073/pnas.1210938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande K.; Hutchison C. D. M.; Groenhof G.; Aquila A.; Robinson J. S.; Tenboer J.; Basu S.; Boutet S.; DePonte D. P.; Liang M.; White T. A.; Zatsepin N. A.; Yefanov O.; Morozov D.; Oberthuer D.; Gati C.; Subramanian G.; James D.; Zhao Y.; Koralek J.; Brayshaw J.; Kupitz C.; Conrad C.; Roy-Chowdhury S.; Coe J. D.; Metz M.; Xavier P. L.; Grant T. D.; Koglin J. E.; Ketawala G.; Fromme R.; Šrajer V.; Henning R.; Spence J. C. H.; Ourmazd A.; Schwander P.; Weierstall U.; Frank M.; Fromme P.; Barty A.; Chapman H. N.; Moffat K.; van Thor J. J.; Schmidt M. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science 2016, 352, 725–729. 10.1126/science.aad5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenhof G.; Bouxin-Cademartory M.; Hess B.; de Visser S. P.; Berendsen H. J. C.; Olivucci M.; Mark A. E.; Robb M. A. Photoactivation of the Photoactive Yellow Protein: Why Photon Absorption Triggers a Trans-to-Cis Isomerization of the Chromophore in the Protein. J. Am. Chem. Soc. 2004, 126, 4228–4233. 10.1021/ja039557f. [DOI] [PubMed] [Google Scholar]
- Espagne A.; Paik D. H.; Changenet-Barret P.; Plaza P.; Martin M. M.; Zewail A. H. Ultrafast light-induced response of photoactive yellow protein chromophore analogues. Photochem. Photobiol. Sci. 2007, 6, 780–787. 10.1039/b700927e. [DOI] [PubMed] [Google Scholar]
- Larsen D. S.; Vengris M.; van Stokkum I. H. M.; van der Horst M. A.; Cordfunke R. A.; Hellingwerf K. J.; van Grondelle R. Initial photo-induced dynamics of the photoactive yellow protein chromophore in solution. Chem. Phys. Lett. 2003, 369, 563–569. 10.1016/S0009-2614(03)00003-4. [DOI] [Google Scholar]
- Larsen D. S.; Vengris M.; van Stokkum I. H. M.; van der Horst M. A.; de Weerd F. L.; Hellingwerf K. J.; van Grondelle R. Photoisomerization and photoionization of the photoactive yellow protein chromophore in solution. Biophys. J. 2004, 86, 2538–2550. 10.1016/S0006-3495(04)74309-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengris M.; van der Horst M. A.; Zgrablic G.; van Stokkum I. H. M.; Haacke S.; Chergui M.; Hellingwerf K. J.; van Grondelle R.; Larsen D. S. Contrasting the excited-state dynamics of the photoactive yellow protein chromophore: Protein versus solvent environments. Biophys. J. 2004, 87, 1848. 10.1529/biophysj.104.043224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengris M.; Larsen D. S.; van der Horst M. A.; Larsen O. F. A.; Hellingwerf K. J.; van Grondelle R. Ultrafast dynamics of isolated model photoactive yellow protein chromophores: ”Chemical perturbation theory” in the laboratory. J. Phys. Chem. B 2005, 109, 4197–4208. 10.1021/jp045763d. [DOI] [PubMed] [Google Scholar]
- Changenet-Barret P.; Lacombat F.; Plaza P. Reaction-coordinate tracking in the excited-state deactivation of the photoactive yellow protein chromophore in solution. J. Photochem. Photobiol., A 2012, 234, 171–180. 10.1016/j.jphotochem.2012.03.011. [DOI] [Google Scholar]
- Virshup A. M.; Punwong C.; Pogorelov T. V.; Lindquist B. A.; Ko C.; Martínez T. J. Photodynamics in Complex Environments: Ab Initio Multiple Spawning Quantum Mechanical/Molecular Mechanical Dynamics. J. Phys. Chem. B 2009, 113, 3280–3291. 10.1021/jp8073464. [DOI] [PubMed] [Google Scholar]
- Boggio-Pasqua M.; Robb M. A.; Groenhof G. Hydrogen Bonding Controls Excited-State Decay of the Photoactive Yellow Protein Chromophore. J. Am. Chem. Soc. 2009, 131, 13580–13581. 10.1021/ja904932x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromov E. V.; Burghardt I.; Hynes J. T.; Köppel H.; Cederbaum L. S. Electronic structure of the photoactive yellow protein chromophore: Ab initio study of the low-lying excited singlet states. J. Photochem. Photobiol., A 2007, 190, 241–257. 10.1016/j.jphotochem.2007.04.033. [DOI] [Google Scholar]
- Gromov E. V.; Burghardt I.; Köppel H.; Cederbaum L. S. Photoinduced Isomerization of the Photoactive Yellow Protein (PYP) Chromophore: Interplay of Two Torsions, a HOOP Mode and Hydrogen Bonding. J. Phys. Chem. A 2011, 115, 9237–9248. 10.1021/jp2011843. [DOI] [PubMed] [Google Scholar]
- Ko C.; Virshup A. M.; Martínez T. J. Electrostatic control of photoisomerization in the photoactive yellow protein chromophore: Ab initio multiple spawning dynamics. Chem. Phys. Lett. 2008, 460, 272–277. 10.1016/j.cplett.2008.05.029. [DOI] [Google Scholar]
- Gromov E. V.; Burghardt I.; Köppel H.; Cederbaum L. S. Native hydrogen bonding network of the photoactive yellow protein (PYP) chromophore: Impact on the electronic structure and photoinduced isomerization. J. Photochem. Photobiol., A 2012, 234, 123–134. 10.1016/j.jphotochem.2012.01.007. [DOI] [Google Scholar]
- Knoch F.; Morozov D.; Boggio-Pasqua M.; Groenhof G. Steering the excited state dynamics of a photoactive yellow protein chromophore analogue with external electric fields. Comput. Theor. Chem. 2014, 1040–1041, 120–125. 10.1016/j.comptc.2014.04.005. [DOI] [Google Scholar]
- Kandori H.; Sasabe H.; Nakanishi K.; Yoshizawa T.; Mizukami T.; Shichida Y. Real-time Detection of 60-fs Isomerization in a Rhodopsin Analog Containing Eight-Membered-Ring Retinal. J. Am. Chem. Soc. 1996, 118, 1002–1005. 10.1021/ja951665h. [DOI] [Google Scholar]
- Inomata K.; Hammam M. A. S.; Kinoshita H.; Murata Y.; Khawn H.; Noack S.; Michael N.; Lamparter T. Sterically Locked Synthetic Bilin Derivatives and Phytochrome Agp1 from Agrobacterium tumefaciens Form Photoinsensitive Pr- and Pfr-like Adducts. J. Biol. Chem. 2005, 280, 24491–24497. 10.1074/jbc.M504710200. [DOI] [PubMed] [Google Scholar]
- Cordfunke R.; Kort R.; Pierik A.; Gobets B.; Koomen G. J.; Verhoeven J. W.; Hellingwerf K. J. trans/cis (Z/E) photoisomerization of the chromophore of photoactive yellow protein is not a prerequisite for the initiation of the photocycle of this photoreceptor protein. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7396–7401. 10.1073/pnas.95.13.7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl A. D.; Hospes M.; Singhal K.; van Stokkum I.; van Grondelle R.; Groot M. L.; Hellingwerf K. J. On the Involvement of Single-Bond Rotation in the Primary Photochemistry of Photoactive Yellow Protein. Biophys. J. 2011, 101, 1184–1192. 10.1016/j.bpj.2011.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley A.; Patel A. M.; Parkes M. A.; Anderson J. C.; Fielding H. H. Role of Photoisomerization on the Photodetachment of the Photoactive Yellow Protein Chromophore. J. Phys. Chem. A 2018, 122, 8222–8228. 10.1021/acs.jpca.8b07770. [DOI] [PubMed] [Google Scholar]
- Lee I.-R.; Lee W.; Zewail A. H. Primary steps of the photoactive yellow protein: Isolated chromophore dynamics and protein directed function. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 258–262. 10.1073/pnas.0510015103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull J. N.; Anstöter C. S.; Verlet J. R. R. Ultrafast valence to non-valence excited state dynamics in a common anionic chromophore. Nat. Commun. 2019, 10, 5820. 10.1038/s41467-019-13819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggio-Pasqua M.; Burmeister C. F.; Robb M. A.; Groenhof G. Photochemical reactions in biological systems: probing the effect of the environment by means of hybrid quantum chemistry/molecular mechanics simulations. Phys. Chem. Chem. Phys. 2012, 14, 7912–7928. 10.1039/c2cp23628a. [DOI] [PubMed] [Google Scholar]
- Gerwien A.; Schildhauer M.; Thumser S.; Mayer P.; Dube H. Direct evidence for hula twist and single-bond rotation photoproducts. Nat. Commun. 2018, 9, 2510. 10.1038/s41467-018-04928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.