


Abstract
Generated by 3′ end cleavage and polyadenylation at alternative polyadenylation (poly(A)) sites, alternative terminal exons account for much of the variation between human transcript isoforms. More than a dozen protocols have been developed so far for capturing and sequencing RNA 3′ ends from a variety of cell types and species. In previous studies, we have used these data to uncover novel regulatory signals and cell type-specific isoforms. Here we present an update of the PolyASite (https://polyasite.unibas.ch) resource of poly(A) sites, constructed from publicly available human, mouse and worm 3′ end sequencing datasets by enforcing uniform quality measures, including the flagging of putative internal priming sites. Through integrated processing of all data, we identified and clustered sites that are closely spaced and share polyadenylation signals, as these are likely the result of stochastic variations in processing. For each cluster, we identified the representative - most frequently processed - site and estimated the relative use in the transcriptome across all samples. We have established a modern web portal for efficient finding, exploration and export of data. Database generation is fully automated, greatly facilitating incorporation of new datasets and the updating of underlying genome resources.
INTRODUCTION
The cleavage of 3′ ends and the addition of a polyadenosine tail are necessary for the maturation of most eukaryotic messenger RNAs (mRNAs) (1). In mammals, such as mouse and human, most genes have multiple sites where cleavage and polyadenylation can occur (2), enabling the production of distinct transcript isoforms in various cell types. Along with alternative first exons resulting from the choice of promoters, alternative terminal exons contribute most to the variation between human transcript isoforms (3). Interest in the regulation of polyadenylation (poly(A)) site choice was boosted by an observation made a decade ago, namely that 3′ end processing at coding region-proximal poly(A) sites leads to the expression of transcripts with short 3′ untranslated regions (3′ UTRs) in proliferating cells; in contrast, the use of distal poly(A) sites in resting cells leads to transcripts carrying long 3′ UTRs (4). Induction of pluripotency in somatic cells is also associated with a systematic shortening of 3′ UTRs (5) and, conversely, 3′ UTRs become longer during cell differentiation (6). As numerous RNA-binding proteins regulate gene expression by binding to 3′ UTRs (7), transcript isoforms that differ only in the length of their 3′ UTRs can nevertheless vary widely with regard to properties such as their stability, subcellular localization and others, in spite of encoding the same protein. To chart the 3′ UTR landscape in individual cell types and to explore its response to perturbations, many laboratories have developed methods that take advantage of the long poly(A) tails to capture and sequence RNA 3′ ends. This has led to >400 samples and over 1.6 billion sequenced reads being available in public databases. These data have not been integrated in a common resource, but rather a few databases have been developed to hold poly(A) sites obtained with individual experimental methods (Table 1; reviewed in (8)).
Table 1.
Comparison of databases of poly(A) sites
| PolyASite 2.0 | PolyA_DB 3 | APASdb | APADB | PolyASite | |
|---|---|---|---|---|---|
| Content | |||||
| Organisms | H. sapiens | H. sapiens | H. sapiens | H. sapiens | H. sapiens |
| M. musculus | M. musculus | M. musculus | M. musculus | M. musculus | |
| C. elegans | G. gallus | D. rerio | G. gallus | ||
| R. norvegicus | B. belcheri | ||||
| Genome version | hg38 | hg19 | hg19 | hg19 | hg19 |
| mm10 | mm9 | mm9 | mm10 | mm10 | |
| ce11 | galgal4 | Zv_9 | galgal4 | ||
| Rnor_5.0 | v7h2 | ||||
| Number of samples | 221 | 107 | 33 | 8 | 78 |
| 178 | 246 | 8 | 3 | 110 | |
| 22 | 11 | 8 | 1 | ||
| 9 | 2 | ||||
| Number of protocols | 12 | 1 | 1 | 1 | 9 |
| Number of poly(A) clusters | 569 005 | 290 168 | NA | NA | 392 912 |
| 301 001 | 384 337 | 183 225 | |||
| 20 931 | 61 905 | ||||
| 65 909 | |||||
| Functionality | |||||
| Gene search | yes | yes | yes | yes | no |
| Genomic region search | yes | no | no | no | no |
| Genome browser | UCSC | UCSC | GBrowse | JBrowse | - |
| Atlas (sample integration) | yes | yes | no | no | yes |
| Individual samples | yes | no | yes | yes | yes |
| Web | |||||
| Url | https://polyasite.unibas.ch/ | http://exon.umdnj.edu/polya_db/v3/ | http://genome.bucm.edu.cn/utr/ | http://tools.genxpro.net/apadb/ | http://polyasite-v1.scicore.unibas.ch |
| https | yes | no | no | no | no |
| Responsive design | yes | no | no | no | no |
| Latest release | September 2019 | August 2018 | September 2014 | June 2014 | October 2015 |
The utility of poly(A) site databases in unraveling the regulation of 3′ end processing has been underscored in many recent studies. For example, they have enabled the discovery of novel polyadenylation signals (9,10) and of novel types of isoforms, such as those generated by ‘intronic’ polyadenylation in immune cells (11,12). Application of machine learning techniques to large data on condition-dependent poly(A) site usage has further lead to the discovery of RNA-binding protein modulators of poly(A) site usage (13). Given the prevalence of polyadenylation site changes in cancers (reviewed in (8)), it is likely that the interest in the regulation and consequences of alternative polyadenylation will continue to grow, further catalyzed by the availability of individual human genomes and of standardized methods to sequence 3′ ends up to the resolution of single cells.
To catalyze further discoveries in this field, here we present an extensive update of PolyASite (https://polyasite.unibas.ch), a database of poly(A) sites in the human, mouse and worm genomes that has been constructed by integrating currently available 3′ end sequencing data. These data cover many cell types and conditions and have been generated with many distinct protocols. In constructing PolyASite, we have used an updated set of poly(A) signals (9), as well as uniform criteria for distinguishing well-supported poly(A) sites from background. Internal priming, caused by the hybridization of the poly(T) primer to oligo(A) stretches that are internal to transcripts rather than part of the poly(A) tail, is a common artifact that leads to spurious poly(A) sites. Here, we applied consistent criteria for flagging reads and poly(A) sites that could be the result of internal priming across samples. We have also clustered sites that are closely spaced and share regulatory signals, as these are likely the result of some degree of imprecision in 3′ end processing (14). We provide overall and per-sample quantification of poly(A) site usage at the level of the clusters.
Since the previous release of PolyASite (9) we have more than doubled the number of contributing samples, included three new 3′ end sequencing protocols, included Caenorhabditis elegans data, and updated genome and annotation versions. Also in contrast to the initial version, PolyASite 2.0 is generated from the primary sequencing data in a fully automated manner, through containerized workflows. This greatly facilitates the maintenance of the resource and the update of the underlying genome information (genome versions and gene annotation releases). It will also allow efficient incorporation of new datasets, including for organisms that are not yet represented in the resource. The redesigned modern and responsive user interface enables users to quickly and conveniently find, explore, and export information, whether they be the annotated poly(A) sites for a gene of interest, the entire set of sites for systematic analyses, or information about 3′ end sequencing protocols and corresponding samples. Table 1 shows a summary of the comparison of PolyASite versions.
MATERIALS AND METHODS
3′ end sequencing libraries originating from different tissues, organisms and protocols are processed in three broad steps, (i) a protocol-specific pre-processing of reads, (ii) a uniform analysis of putative poly(A) sites in each sample and (iii) a final analysis of the pooled data. The generation of the database is fully automated through a containerized pipeline that allows fast, portable and reproducible updates (see Figure 1). The most important aspects of the processing pipeline are described in detail in the following sections.
Figure 1.

Schematic representation of the data processing underlying PolyASite. An integrated pipeline is used to process all input samples, regardless of 3′ end sequencing technique used for their generation, cell or tissue as well as species from which they originated. Data processing is done in three main parts. The first part of the pipeline consists of protocol specific modules, which cater to the different processing requirements of different 3′ end sequencing techniques in terms of adapter- and poly(A) tail removal, reverse complementation, etc. The second part consists of quality control, alignment and filtering of reads likely originating from internal priming sites, using uniform criteria for all samples, to ensure comparability. Finally, in the third part of the pipeline samples are pooled by species and the poly(A) site clusters are computed, annotated and quantified. The resulting species atlases are accessible through the PolyASite 2.0 web resource (https://polyasite.unibas.ch).
Protocol-specific pre-processing of reads
Publicly available 3′ end sequencing samples for human, mouse and worm were identified by querying the sequence read archive (SRA) database of NCBI (15) with the expression ‘polyadenylation BUTNOT (‘RNA-Seq’ OR ‘miRNA-Seq’ OR ‘ChIP-Seq’ [STRATEGY])’. To ensure comparability of the estimates of poly(A) site usage, we considered only samples that were generated by 3′ end sequencing from total RNA. They have been obtained with a variety of protocols: DRS (16), (13 datasets), PAS-Seq (17), (2 datasets), 3P-seq (18), (27 datasets), SAPAS (19), (34 datasets), A-seq (20), (14 datasets), PolyA-seq (21), (23 datasets), 2P-seq (22), (12 datasets), 3′-seq (23), (59 datasets), 3′READS (24), (148 datasets), PAPERCLIP (10), (50 datasets), PAT-seq (25), (12 datasets), and the commercially available QuantSeq (Lexogen, Austria), (27 datasets). After programmatic download of unprocessed fastq files, reads were processed to remove adaptors, poly(A) tracts and bar codes according to the descriptions of sample preparation steps in the respective studies. Details on protocols and protocol-specific processing are provided in a GitHub repository containing the pipeline used for sample processing (https://github.com/zavolanlab/polyAsite_workflow), as well as the sections on protocols (https://polyasite.unibas.ch/protocols) and samples (https://polyasite.unibas.ch/samples) on PolyASite.
Uniform analysis of putative poly(A) sites in individual samples
Following protocol-specific pre-processing of reads, each sample was subjected to the following analysis. ‘Clean’ reads that were longer than 15 nucleotides and did not contain more than two uncalled nucleotides (‘N’) or >80% adenines were mapped contiguously to both the genome (GRCh38, GRCm38 and WBcel235 assembly versions for human, mouse and worm, respectively) and corresponding transcriptomes (Ensembl release 96) with the Segemehl (26) program. Best mappings with up to 10% error were kept and used to recompute non-redundant genome coordinates of reads, recovering genome coordinates from alignments across splice boundaries of transcripts. Reads that mapped to unique genomic locations were processed further to obtain putative 3′ ends with their respective count from each sample. To exclude reads that possibly originated from internal priming, a read was discarded if more than 6 consecutive adenines, or more than seven adenines were present in the 10 genomically-encoded nucleotides downstream of the mapped 3′ end of the read. A final uniform criterion was imposed on the inclusion of putative poly(A) sites from each library into the atlas. Namely, because 80–90% of poly(A) sites are expected to contain a polyadenylation signal (PAS) at ∼21 nucleotides upstream of the cleavage site (9,27) and spurious sites should have low counts, we sorted the unique 3′ ends in each library in decreasing order of the number of supporting reads and grouped high abundance sites with all lower abundance sites located within ±25 nucleotides. We kept only as many of the most abundantly supported site groups as necessary to ensure that 90% of them had a poly(A) signal in the region of −60 to +10 nucleotides from a putative poly(A) site in the group. The rest of the reads, corresponding to lowest abundance sites, were discarded. Statistics regarding the proportion of each library that was retained at every step of processing and finally contributed to the atlas are shown in Supplementary Figure S1.
Clustering of closely spaced sites
As we and others found a certain degree of micro-heterogeneity in poly(A) sites (9,14), we grouped together closely spaced sites into clusters to infer distinct poly(A) sites. Specifically, we collected all the unique 3′ end processing sites from all libraries and tabulated their total counts in each library. We next sorted the sites in the order of overall read support and, in cases of equal counts, by the number of protocols supporting the sites. We then traversed this list from the most to the least supported site, associating sites that were within a window of −12 to +12 nucleotides of a site with stronger support (20). The most supported site in the cluster was chosen as the ‘representative’ processing site of the cluster. We then annotated each cluster with the PAS that were present within a window of −60 to +10 nucleotides relative to the poly(A) site. We identified a subset of sites where 3′ end processing seemed to have occurred within the PAS itself and flagged these as ‘putative internal priming (IP) sites’. This flag was removed when we could associate the site with a site downstream that was not flagged as IP site and that shared a PAS with the putative IP site. Finally, poly(A) site clusters with annotated PAS were merged if the total width of the cluster did not surpass 25 nucleotides, the range of micro-heterogeneity that we considered above. Clusters without annotated PAS, for which evidence of distinct regulation was thus not available, were merged more permissively, namely if they were within 25 nucleotides of each other.
Annotation and quantification of poly(A) sites
As each 3′ end read originated in one transcript, we calculated the ‘abundance’ of each poly(A) site in each sample as  (transcripts per million, TPM), where
(transcripts per million, TPM), where  is the number of reads from sample
is the number of reads from sample 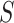 ending at site
ending at site 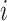 and
and 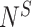 is the total number of reads from sample
is the total number of reads from sample 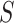 that contributed to the atlas. The abundance of a cluster was calculated as the sum of abundances of all sites in the cluster. We also estimated the relative usage of a cluster within the corresponding gene by dividing its abundance to the total abundance of all clusters located within the gene.
that contributed to the atlas. The abundance of a cluster was calculated as the sum of abundances of all sites in the cluster. We also estimated the relative usage of a cluster within the corresponding gene by dividing its abundance to the total abundance of all clusters located within the gene.
The genome coordinate of the cluster representative was used to annotate the clusters. First, transcript features that were annotated at that location in the genome (based on Ensembl release 96) were identified, and then one of these features was associated with the poly(A) site according to the following priority scheme: TE, terminal exon; EX, exonic; IN, intronic; DS, 1,000 nt downstream of an annotated terminal exon; AE, anti-sense to an exon; AI, anti-sense to an intron; AU, 1000 nt upstream in antisense direction of a transcription start site; IG, intergenic. Beyond the more expected cases (TE, IG), these categories facilitate extraction of poly(A) sites that may correspond to less abundant but previously observed classes of transcripts such as those ending prematurely (IN), longer versions of currently annotated isoforms (DS), antisense transcripts (AE, AI), including those that result from bidirectional promoters (AU). Supplementary Table S1 shows a summary of the atlas including the relative frequency of various types of poly(A) sites.
RESULTS
Website roadmap
PolyASite 2.0 (https://polyasite.unibas.ch) is accessible through an encrypted connection and features a user-friendly and responsive interface that allows it to be explored from desktop, tablet and phone alike. The main functionalities that are provided are database search, links to the UCSC genome browser (28) as well as to Ensembl (29), and bulk downloads of the atlases. Further information about the primary data that was used to construct the atlas, as well as tools that exploit the datasets are also provided. In the following section we briefly discuss the main functions implemented in the web site:
The ‘Search’ functionality allows users to retrieve information (such as poly(A) signals, annotation and average usage) about poly(A) sites located within specified genomic regions or associated with a gene (specified either by an HGNC/MGNC symbol or by an Ensembl/Wormbase identifier). For each poly(A) site cluster, the number of distinct protocols and the fraction of samples in which sites contributing to the cluster were detected is given as an indicator of confidence. The dynamic search results can be further expanded to show the quantification of cluster usage across samples, visualized in the UCSC genome browser, or downloaded for further analyses. Links to the Ensembl database are also provided to facilitate the exploration of the sites. For an example of a search query, the resulting table of poly(A) site clusters and the corresponding data tracks in the UCSC genome browser, see Figure 2.
Figure 2.

Use case: Search for poly(A) sites in a genomic region. Shown is an example search from PolyASite 2.0 and its results, including a screenshot from the UCSC genome browser. After a genomic region was entered into the search field (1) and the organism of interest was selected (2), the search was triggered (3). The figure shows the first rows of the retrieved table of poly(A) site clusters, which has been customized by toggling specific columns via the ‘Column visibility’ button and by sorting the results according to the ‘Usage’ and ‘Protocols’ columns (by clicking/tapping the respective column headers). The arrow on the black background to the left of a row leads to an expanded table, which contains information on the expression/usage of the respective cluster in individual samples (not shown). Selecting ‘Explore region in UCSC browser’ (4) yielded the genome view depicted on top, with distinct browser tracks for atlas sites on the plus and minus strands. The location of the most abundant cluster in the results table and in the browser view is indicated by the curved arrow. Descriptions of the visible columns are indicated at the bottom.
The ‘Atlas’ provides summaries of the data for the three species, including the number of samples and reads that contributed to the atlas, and the number of poly(A) clusters and their annotation. We considered the following categories of sites, depending on their location within or outside of genes: sites located in annotated terminal exons, in all other exons, in introns, downstream of genes, in intergenic regions, or antisense to exons, introns or upstream genomic regions. The most common annotations are terminal exons, introns and intergenic (Supplementary Table S1). Bed-formatted files of the poly(A) site clusters can be downloaded for bulk analyses, and TPMs of clusters can be visualized in the UCSC genome browser across the entire genome.
The ‘Samples’ section of the web site enables the user to easily retrieve data on a per-sample basis, either as a bed-formatted file of poly(A) clusters with their sample-specific abundance, or as a links to the UCSC genome browser. Furthermore, information on the sample is given, such as the cell type from which the sample was prepared, whether any treatments were applied, or the fraction of sequenced reads that contributed to the atlas. Links to the original publications and to the GEO or SRA records for the samples are also provided.
Substantial documentation about the protocols that were used for sample preparation is provided in the ‘Protocol’ section of the web site, while the ‘Tools’ section describes tools that we have developed to further unravel the regulation of poly(A) site choice, taking advantage of the PolyASite atlas and associated quantification of poly(A) site usage in various conditions (11,13).
DISCUSSION
PolyASite 2.0 is a searchable, easy-to-use resource of 3′ end processing sites in the human, mouse and worm genomes that aims to be as comprehensive as possible, by integrating publicly available datasets, irrespective of the protocols that were used to obtain them. With this approach, artifacts due to technical biases in individual methods should be minimized. The fact that the nucleotide composition around sites that are supported by multiple samples and protocols more clearly conforms to expectations (20,30) compared to sites supported by a single protocol (Supplementary Figures S2–S4) indicates that our approach of data integration is valid and underscores the importance of such a resource. The automated workflow for atlas generation, which includes uniform criteria for data quality, flagging of potential artifacts and clustering of sites, greatly facilitates the maintenance and development of the resource, as well as the update of the underlying genome resources (genome versions and gene annotation releases). In the immediate future, our main focus will be the incorporation of new datasets (31), especially given that commercial solutions for sequencing RNA 3′ ends have become available, up to the resolution of single cells. In this context, it will be essential to incorporate cell type/tissue ontologies to be able to comparatively analyze large datasets. Additional model organisms like Danio rerio and Drosophila melanogaster will also be included in future releases, to make the PolyASite resource usable for a broader audience. As a substantial proportion of the poly(A) sites that have been reproducibly detected in multiple experiments are not yet associated with terminal exons, identifying the transcripts in which these sites reside will be an important goal in the future, to further expand our understanding of gene expression, on a cell type-specific basis. Finally, the server can be enhanced with additional functionality, for example for comparing the usage of poly(A) sites between conditions or for identifying regulatory signals in the vicinity of specific subsets of sites.
DATA AVAILABILITY
PolyASite 2.0 is accessible at https://polyasite.unibas.ch. The pipeline used to process the data for the atlas is available on GitHub: https://github.com/zavolanlab/polyAsite_workflow. The code for the website itself is available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the many members of the Zavolan lab that have provided input that led to the development of PolyASite over the years, in particular Andreas R. Gruber, Manuel Belmadani, Maria Katsantoni, Foivos Gypas, Paula Iborra de Toledo, Georges Martin and Walter Keller.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swiss National Science Foundation [31003A_170216 to M.Z., 51NF40_141735 in part to M.Z.]. Funding for open access charge: Swiss National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Edmonds M., Vaughan M.H. Jr, Nakazato H.. Polyadenylic acid sequences in the heterogeneous nuclear RNA and rapidly-labeled polyribosomal RNA of HeLa cells: possible evidence for a precursor relationship. Proc. Natl. Acad. Sci. U.S.A. 1971; 68:1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tian B., Hu J., Zhang H., Lutz C.S.. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005; 33:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reyes A., Huber W.. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018; 46:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B.. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer MicroRNA target sites. Science. 2008; 320:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ji Z., Tian B.. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS One. 2009; 4:e8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ji Z., Lee J.Y., Pan Z., Jiang B., Tian B.. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kishore S., Luber S., Zavolan M.. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief. Funct. Genomics. 2010; 9:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gruber A.J., Zavolan M.. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019; 20:599–614. [DOI] [PubMed] [Google Scholar]
- 9. Gruber A.J., Schmidt R., Gruber A.R., Martin G., Ghosh S., Belmadani M., Keller W., Zavolan M.. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016; 26:1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hwang H.-W., Park C.Y., Goodarzi H., Fak J.J., Mele A., Moore M.J., Saito Y., Darnell R.B.. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 2016; 15:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gruber A.J., Gypas F., Riba A., Schmidt R., Zavolan M.. Terminal exon characterization with TECtool reveals an abundance of cell-specific isoforms. Nat. Methods. 2018; 15:832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh I., Lee S.-H., Sperling A.S., Samur M.K., Tai Y.-T., Fulciniti M., Munshi N.C., Mayr C., Leslie C.S.. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 2018; 9:1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gruber A.J., Schmidt R., Ghosh S., Martin G., Gruber A.R., van Nimwegen E., Zavolan M.. Discovery of physiological and cancer-related regulators of 3′ UTR processing with KAPAC. Genome Biol. 2018; 19:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hon C.-C., Weber C., Sismeiro O., Proux C., Koutero M., Deloger M., Das S., Agrahari M., Dillies M.-A., Jagla B. et al.. Quantification of stochastic noise of splicing and polyadenylation in Entamoeba histolytica. Nucleic Acids Res. 2013; 41:1936–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leinonen R., Sugawara H., Shumway M. International Nucleotide Sequence Database Collaboration . The sequence read archive. Nucleic Acids Res. 2011; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ozsolak F., Kapranov P., Foissac S., Kim S.W., Fishilevich E., Monaghan A.P., John B., Milos P.M.. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010; 143:1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shepard P.J., Choi E.-A., Lu J., Flanagan L.A., Hertel K.J., Shi Y.. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011; 17:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jan C.H., Friedman R.C., Ruby J.G., Bartel D.P.. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature. 2011; 469:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu Y., Sun Y., Li Y., Li J., Rao X., Chen C., Xu A.. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011; 21:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin G., Gruber A.R., Keller W., Zavolan M.. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012; 1:753–763. [DOI] [PubMed] [Google Scholar]
- 21. Derti A., Garrett-Engele P., Macisaac K.D., Stevens R.C., Sriram S., Chen R., Rohl C.A., Johnson J.M., Babak T.. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012; 22:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spies N., Burge C.B., Bartel D.P.. 3′ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res. 2013; 23:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lianoglou S., Garg V., Yang J.L., Leslie C.S., Mayr C.. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013; 27:2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Park J.Y., Yehia G., Tian B.. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods. 2013; 10:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elewa A., Shirayama M., Kaymak E., Harrison P.F., Powell D.R., Du Z., Chute C.D., Woolf H., Yi D., Ishidate T. et al.. POS-1 Promotes Endo-mesoderm Development by Inhibiting the Cytoplasmic Polyadenylation of neg-1 mRNA. Dev. Cell. 2015; 34:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoffmann S., Otto C., Doose G., Tanzer A., Langenberger D., Christ S., Kunz M., Holdt L.M., Teupser D., Hackermüller J. et al.. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014; 15:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beaudoing E., Freier S., Wyatt J.R., Claverie J.M., Gautheret D.. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000; 10:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haeussler M., Zweig A.S., Tyner C., Speir M.L., Rosenbloom K.R., Raney B.J., Lee C.M., Lee B.T., Hinrichs A.S., Gonzalez J.N. et al.. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019; 47:D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S. et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Retelska D., Iseli C., Bucher P., Jongeneel C.V., Naef F.. Similarities and differences of polyadenylation signals in human and fly. BMC Genomics. 2006; 7:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogorodnikov A., Levin M., Tattikota S., Tokalov S., Hoque M., Scherzinger D., Marini F., Poetsch A., Binder H., Macher-Göppinger S. et al.. Transcriptome 3′end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018; 9:5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PolyASite 2.0 is accessible at https://polyasite.unibas.ch. The pipeline used to process the data for the atlas is available on GitHub: https://github.com/zavolanlab/polyAsite_workflow. The code for the website itself is available upon request.