


Abstract
Significant improvements have been made in the efficiency and accuracy of RNA 3D structure prediction methods during the succeeding challenges of RNA-Puzzles, a community-wide effort on the assessment of blind prediction of RNA tertiary structures. The RNA-Puzzles contest has shown, among others, that the development and validation of computational methods for RNA fold prediction strongly depend on the benchmark datasets and the structure comparison algorithms. Yet, there has been no systematic benchmark set or decoy structures available for the 3D structure prediction of RNA, hindering the standardization of comparative tests in the modeling of RNA structure. Furthermore, there has not been a unified set of tools that allows deep and complete RNA structure analysis, and at the same time, that is easy to use. Here, we present RNA-Puzzles toolkit, a computational resource including (i) decoy sets generated by different RNA 3D structure prediction methods (raw, for-evaluation and standardized datasets), (ii) 3D structure normalization, analysis, manipulation, visualization tools (RNA_format, RNA_normalizer, rna-tools) and (iii) 3D structure comparison metric tools (RNAQUA, MCQ4Structures). This resource provides a full list of computational tools as well as a standard RNA 3D structure prediction assessment protocol for the community.
INTRODUCTION
RNA 3D structure prediction, which dates back to the late 1960s (1), is nowadays being widely studied with the help of computer science. An increasing number of programs with different prediction approaches are being designed and continuously improved (2,3). Like in protein 3D structure prediction, it is important to benchmark the prediction programs to assess the capabilities of the prediction and the bottleneck in the field. CASP (Critical Assessment of Protein Structure Prediction) (4) is the largest worldwide event of protein structure prediction. And RNA-Puzzles (5–7) is a CASP-like assessment of RNA 3D structure prediction, which is supported by dozens of research groups around the world.
RNA has its own structural and evolutionary features. Most importantly, the RNA secondary structure, determined by the set of cis-Watson-Crick base pairs, can be generally determined using sequence comparisons (8,9). However, the formation of a 3D structure requires, in addition, non-Watson-Crick base pairs (10), structural modules (11), and sometimes pseudoknots (12). Thus, the secondary structure description of RNA structure is insufficient. Precise sequence and covariation analysis (13), and/or chemical/enzymatic probing (14,15) are therefore necessary to predict relevant 3D structures. In RNA-Puzzles, we highlight the fact that 3D structure models can severely deviate from the reference structures even if the model retains perfect secondary structure (100% correct in terms of cis-Watson-Crick base pairing) (6) (see Supplementary Figure S1). In this context, RNA 3D structure prediction needs independent benchmarking systems that include both datasets and assessment metrics.
With the progress in protein structure prediction, many benchmark datasets and assessment metrics have been curated and developed (16). One available dataset for RNA structure benchmarking is the non-redundant dataset maintained by Leontis and Zirbel (17). Alternatively, the Rfam database, which links RNA sequence families with crystallographic structures when available, can also be used in prediction benchmarking (18). However, only 99 Rfam families have their 3D structures available. Such benchmarks are not blind and are biased towards RNAs with many homologous sequences. This is not always the case in prediction: some rare RNA structures do not necessarily have homologous sequence available, e.g. Varkud satellite ribozyme (19), in which case sequence alignment-dependent prediction methods may not be helpful. The RNA-Puzzles benchmark sets have been successfully used in developing RNA quality assessment methods (20) to identify the models similar to experimental structures without reference. Potentially, they will also serve as decoy sets for proposing structure-based force field or scoring functions, RNA design and other utilities.
Reliable evaluation of dozens of RNA 3D models cannot be performed manually and is usually preceded by normalization to comply with a common 3D structure representation. Since the start of RNA-Puzzles, a good number of RNA structure manipulation tools and structure comparison metrics, some of which are being used by the RNA-Puzzles community, have been conceived and designed. They are helpful in various ways, including structure analysis, comparison, and function inference. Here, we gather and summarize a computational resource ‘RNA-Puzzles toolkit’ that includes a set of datasets and various computational tools accumulated in the practice of RNA-Puzzles, which cover important aspects to understand RNA structure. RNA-Puzzles toolkit includes tools for structure formatting, analysis, manipulation, visualization, mutagenesis study and structure comparison. This computational resource will benefit biologists working with RNA structure and RNA structure prediction. All the datasets and codes are available as open-source on GitHub (https://github.com/RNA-Puzzles).
MATERIALS AND METHODS
Datasets
We provide three datasets derived from RNA-Puzzles: (i) raw_dataset - a dataset of raw submissions, which were generated by various prediction methods, (ii) for-evaluation_dataset - dataset used for official evaluation of the prediction methods in RNA-Puzzles, which does not change the coordinates of the predicted structures or add missing atoms, and (iii) standardized_dataset -a standardized dataset optimized with rna-tools, which not only unified the residue and atom names but also completed the missing atoms in incomplete RNA structures to standardize all the structures to the same format. All the datasets follow the same rules to name the structural files, which is a combination of the RNA-Puzzles identifier, prediction group name, and the structure model number, e.g. 19_RNAComposer_3.pdb means the third model predicted by RNAComposer (21) for Puzzle 19 in RNA-Puzzles. The reference structures were obtained from the crystallographers, renamed according to the puzzle name and marked as ‘solution’, e.g. 19_solution_0 means the first reference model of Puzzle 19. If one sequence has multiple solved structures or multiple chains in the asymmetric biological unit, all of them are used as reference structures. And the one with the lowest root mean square deviation (RMSD) to a given model is used as the reference structure to report the scores for that model.
RNA_format, RNA_normalizer and RNA_assessment
RNA_format, RNA_normalizer and RNA_assessment constitute a set of computational tools for the data formatting, processing and evaluation in RNA-Puzzles. They are implemented as Python packages making use of the BioPython (22) structure I/O library. The algorithms to compute RMSD, P-value (23), Deformation Profile, and Interaction Network Fidelity (24) are implemented in the Python package RNA_assessment, which makes use of BioPython, MC-Annotate (25) and NumPy (26). Deformation Profile was also implemented as an independent Python package.
rna-tools
rna-tools is a core library written in Python and a set of command-line programs execute various functions to process structural files in the PDB format but also to process RNA sequences, folding simulations, sequence alignments. Some tools in rna-tools are dependent on other programs or libraries such as ModeRNA (27), ClaRNA (28), BioPython (22).
RNAQUA
RNAQUA (RNA QUality Assessment tool) is a RESTful web service client developed in Java using Jersey (https://jersey.github.io/). It provides services for RNA 3D structure normalization and comparison, including the metrics of RMSD, P-value (23), Deformation Profile, Interaction Network Fidelity (24) and clash score (29). It uses selected functions from RNAlyzer (30) and RNAssess (31), both of which are in the RNApolis platform (32).
MCQ4Structures
MCQ4Structures is a set of computational tools for RNA 3D structure comparison in the torsion angle space. It includes algorithms to compute Mean of Circular Quantities (MCQ) (33) and Longest Continuous Segments in Torsion Angle space (LCS-TA) (34) that compare structures, compute structure similarity, cluster and visualize the results, identify similar structural fragments, and rank the structural models. The package is implemented in Java, while functional modules of structure I/O and geometric statistics, on which both MCQ and LCS-TA depend, are implemented as separate packages of BioCommons (https://github.com/tzok/BioCommons) and Circular (https://github.com/tzok/Circular).
RESULTS
The overview of the resource
Our computational resource includes (i) the benchmark datasets from RNA-Puzzles, (ii) structure analysis, manipulation, visualization, clustering and normalization tools, (iii) and 3D structure comparison metrics (Figure 1). Considering an RNA structure comparison workflow given both a list of predicted structures and several reference structures, it is first necessary to standardize the predicted and reference structures to the same length and the same format. Structural features, such as clash score, which is based on the structure model, can be calculated and compared with the scores derived from the reference structures. Furthermore, our resource provides a set of tools for RNA structure manipulation and visualization, which can greatly facilitate manual inspection of the structures. Finally, our structure comparison metrics demonstrate the similarity/dissimilarity between the prediction and the reference structures in various aspects. The tools can be accessed via command-line, Jupyter Notebook, Docker image or web service. The user-friendly interfaces enable different usage scenarios throughout the community. Supplementary Table S1 gives a list of the datasets and computational tools in this resource, which are described in detail in the next sections.
Figure 1.
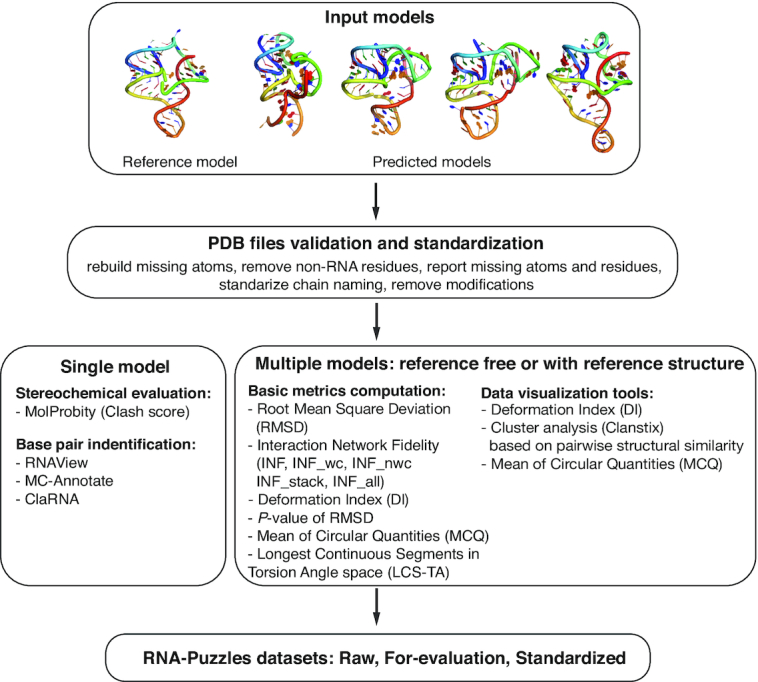
Scheme of the RNA-Puzzles toolkit. The toolkit is composed of three parts: tools for validation and standardization of PDB structure files, tools for analyzing the models, and the dataset of standardized submissions to the RNA-Puzzle. The user can start an analysis with single or multiple models. The first step is to standardize the formatting of analyzed structural models. Then, the user can run an analysis for a single model, such as Clash Score evaluation or base pair identification using various methods; or, for multiple models, various comparison methods are implemented. The tools can be accessed via command-line or Jupyter. The toolkit can be also executed as a Docker image that can be easily used.
Benchmark datasets of RNA 3D structure
In a structure prediction scenario, a good predictor should be robust in predicting structures of different types accounting for the characteristics of each prediction target. Therefore, a good benchmark must cover diverse structures (Figure 2A). The datasets from RNA-Puzzles, as listed in Supplementary Table S2, cover crucial aspects for the selection of puzzles, such as symmetry (35), ion binding (36), ligand binding (37,38), protein binding (39), the conformational change (40), and structural modules (7). Our datasets include 972 decoy RNA structures for 20 RNAs. They can be used as: (i) a standard dataset to compare with existing prediction methods, e.g. (41); (ii) a decoy dataset to develop effective structure scoring function, e.g. (20). The theoretical models were generated by the best existing RNA 3D structure prediction programs (21,42–46). The similarities of these theoretical models to crystal structures range from low quality to the near-native (cf. Figure 2 and Supplementary Table S2), which provides a wide range of decoy structures that exist during structure modeling. The presented benchmark dataset can benefit the development of energy function or scoring function to discriminate the near-native structures from those far away decoys. This is an important step to identify high-quality prediction when the reference structure is unknown. In RNA-Puzzles, each group (or each prediction method) provides five candidate models (in the first 17 challenges, up to 10 models were allowed) and ranks these models according to its own prediction reliability index. However, some of the near-native structures are not ranked as the top models. The detection of such instances would improve prediction accuracy. In RNA-Puzzles, the scores for ‘quality prediction’ were obtained in Puzzles 4, 7, 8, 12, 13 and 14. The structure data from this resource is a good starting point for developing and benchmarking model ranking methods (20). According to the RMSD distribution (Figure 2C), longer structures are more difficult to predict unless homologous templates are available. Although this is consistent with the previous report (47), RNA-Puzzles includes the best RNA structure prediction approaches and demonstrates better performance in de novo prediction. Further, the Interaction Network Fidelity distribution highlights the insufficient prediction of non-Watson–Crick interactions. Other available datasets of the same kind are: (i) RASP (48) dataset, which includes 85 RNAs with 500 decoys for each structure and (ii) the KB (49) dataset, which includes 23 950 decoys for 20 RNAs. However, the decoy structures in these datasets were generated using only a couple of prediction methods, while our dataset covers a much wider variability in RNA structure prediction.
Figure 2.

The structure diversity and comparison of the dataset. (A) The dataset is composed of 18 Puzzles of different types of RNA. (B) Most of them are one-chain or two-chain structures, except Puzzle 2 is of eight chains. (C) Correlation plot between the lengths of RNAs and the RMSD distributions, shown as violin plots, indicates that shorter RNA structures tend to be easier to predict. The RMSD, deformation index and clash score are shown in (D–F). The distributions of Interaction Network Fidelities are shown in (G), including stacking interactions (INF stacking), Watson-Crick interactions (canonical) (INF wc), non-Watson-Crick interactions (non-canonical) (INF nwc) and all interactions (INF all). MCQ assesses structure similarity based on torsion angles (H).
Standardizing the structure format considering all types of variations is the first step of a fair structure comparison. Different prediction methods result in a wide range of variations in the format of the predicted structures, ranging from nomenclature (chain names, residue names, atom names and their ordering) to structural variations (i.e. the structure at the 5′ and 3′ ends). For example, some prediction methods may use the molecular dynamics force field to minimize the energy of the predicted structure at their final steps, thus the output format depends on the force field used. Besides, the predicted structures need to be normalized according to the reference structure allowing unsolved fragments.
The RNA-Puzzles dataset can be used as (i) a standard dataset to benchmark with existing prediction methods; (ii) a decoy dataset to develop and test effective structure scoring function. To fulfill these two tasks, we provide standardized_dataset including structural data standardized and missing atoms completed using rna-tools. rna-tools was used to (i) add the missing atoms, especially at the 5′ and 3′ ends; (ii) mutate variant nucleotides in the predictions to make them consistent with the sequence of the reference structure. All the steps of processing and the detailed analysis of the differences between predicted models and the references, such as gaps, mismatches, etc. are described in the README files provided with the structures. The standardized_dataset is under active maintenance. The advanced users can also use rna-tools to process their own datasets.
RNA 3D structure formatting, manipulation, analysis and visualization tools
RNA_normalizer and rna-tools are two RNA oriented structure format tools providing semi-automatic RNA structure processing workflows.
RNA_normalizer
RNA_normalizer is an RNA structure formatting tool used in RNA-Puzzles evaluation workflow. It can: (i) normalize the residue names and atom names; (ii) order residues and atoms; (iii) extract pre-defined regions of an RNA structure. RNA_normalizer uses mapping dictionaries to normalize the non-canonical residue and atom names to the standard nomenclature. The idea of RNA_normalizer is to keep the maximum number of fragments that can be compared while keeping the prediction structures untouched. In a couple of cases, the sequence used in prediction slightly differ from the sequence of the crystal structure: e.g., single nucleotides variants or chain break because of the unsolved dynamic region in the reference structure. RNA_normalizer focuses on the consensus structure regions between the crystal sequence and the sequence in prediction. However, the skipped nucleotide makes the structure incomplete. Considering the need of complete structures for scoring function testing or molecular dynamics simulation, we provide rna-tools to add the missing atoms in the structures. After normalizing the structure formats, we suggest to use ‘RNA_format’ or ‘diffpdb’ from rna-tools (Figure 3E) to check the consistency between the results and the standard format.
Figure 3.

rna-tools is a set of tools dedicated to RNA structural file manipulation and analysis. (A) Mutate functionality allows for exchanging bases, in this case, C:G pair was replaced with A:U pair from two chains. (B) Contact classification for selected residues can be performed directly in PyMOL. In this case, trans Sugar-Hoogsteen interaction was detected for closing residues of a tetraloop. (C) One of the tools implemented in rna-tools, clanstix. Clanstix can be used for visualizing RNA 3D structures based on pairwise structural similarity (as RMSD) with CLANS. The tool can be used for interactive clustering analysis when various RMSD thresholds can be tested. Here, the clustering of submission for RNA-Puzzle 8 was visualized. Dokholyan submitted four models very different from each other. Models of Chen and of Adamiak were similar respectively and made separate clusters. Models of Ding were similar to each other, and additionally, clustered with models of Das and Bujnicki. When the reference structure was released, it could be added to the visualization. Interestingly, the reference model clustered with two structures of Bujnicki and Das. (D) The functions of the package can be accessed from command-line, from Python scripts, and from Jupyter Notebooks, giving multiple ways to access the functionality. (E) diffpdb checks the consistency between annotations of two structural files. The tool ignores 3D coordinates of atoms and compares only text-content of two files in the PDB to identify the difference in the annotation of atoms, missing atoms (missing the O2′ atom) and missing fragment (shown on the left side with the gray-red bar). (F) Multiple wrappers are implemented allowing for secondary structure prediction performed directly in Jupyter Notebooks, with methods such as RNAsubopt, IPknot, Centroidfold and Contextfold. (G) For RNA alignments it is possible to select only a subset of columns and work on them as a new alignment (in this case on the 1st to the 9th column). Sequences from RNA alignments and their secondary structures can be visualized with VARNA including gaps (H) and without gaps (I). The algorithm checks if residues are ‘paired’ with a gap position (‘−’) (position in red circle) for proper extraction of secondary structure. In this case, after wrong gap removal (J), G (in blue circle) is incorrectly paired with C (in green circle) and all other pairings are shifted by one.
rna-tools
rna-tools includes a set of tools dedicated to (i) RNA structural handling and manipulating, i.e. rebuilding missing atoms, (ii) structure clustering, (iii) standardization of RNA structures, (iv) visualization of secondary RNA structures, i.e. drawing RNA arc diagrams of secondary structure, (v) visualization of RNA sequence alignments, and more.
The core library shared with the tools
The core part of the rna-tools package is the ‘rna_pdb_toolsx.py’ program that was used to prepare the standardized_dataset. The program facilitates many tedious operations on structural files. For example, one tool is the ‘get-rnapuzzle-ready’, which is used to get a standardized naming of atoms, residues, chains to be compatible with the format required by RNA-Puzzles. All structures from the standardized_dataset are compatible with this format, which makes it easy to compare them and use for further analysis. Another example of structure manipulation is introducing mutations. The rna-tools package uses ModeRNA (25) to introduce single or double mutations in structures. But it overcomes ModeRNA’s limitation in processing only one chain at the time (Figure 3A). Multiple mutations in multiple chains can be introduced.
Furthermore, rna-tools includes tools operating on various levels of RNA data: sequences, secondary structures, alignments, and 3D structures. rna-tools includes a collection of almost one hundred functionalities that facilitate common operations in RNA structural bioinformatics. It can be easily imported into 3rd party programs or pipelines. The full list of functionalities can be found in Supplementary Table S3.
RNA sequence tools
The first group of tools deals with RNA sequences. The tools help to perform searches using both Blast (50) on the PDB database and Infernal (51) on the Rfam database (52). Furthermore, multiple wrapper tools of RNA secondary structure prediction are implemented (Figure 3f), including RNAsubopt, RNAeval, RNAfold from ViennaRNA (45), CentroidFold (46), ContextFold (47), MC-Fold (53) and IPknot (54). All tools are compatible with Jupyter Notebook.
RNA secondary structure tools
The second group of tools aims to facilitate operations on RNA secondary structure that can be executed from Jupyter Notebooks (Figure 3F). The functionalities include visualization of a sequence and a structure with VARNA (55), evaluation of free energy, parsing secondary structure into a list of pairs, and various tools for secondary structure format conversions, etc.
RNA alignment tools
The third group includes tools that process RNA sequence alignments. The analysis of RNA sequence alignment is a crucial part of the structure prediction process used in RNA-Puzzle. To process and analyze RNA sequence alignments, rna-tools includes a collection of tools to load alignments, subset columns (Figure 3G) or sequences (rows), save a subset to a new file, plot an RNA arc diagrams (Figure 3D) (56), obtain a secondary structure in the dot-bracket notation, and visualize the data using VARNA of each of sequences in the alignment. Sequences and their secondary structures can be visualized with gaps (Figure 3H) and without gaps (Figure 3I). The algorithm checks if residues are ‘paired’ with a gap position (‘–’) to avoid the common problem with other tools with the wrong secondary structure after gap removal (Figure 3J).
RNA 3D structure tools
The last group of tools operates on RNA 3D structure. This group includes (i) tools for the analysis of 3D models (such as contact classifications) and (ii) tools for RNA 3D structure prediction, including the whole pipeline of structure prediction. First, to perform contact classifications, we provide two wrappers, that are ClaRNA (28) and 3DNA/DSSR (57). Using the wrappers together with the PyMOL4RNA tool in rna-tools, it is possible to perform contact classifications in PyMOL for a selected set of residues (Figure 3B). Second, the package contains scripts to help the RNA 3D structure prediction processes, both for SimRNA (42) (including SimRNAweb (58)), and Rosetta (59). Tools for SimRNA and Rosetta help to prepare input files, run modeling, cluster results, and extract models from trajectory files. Moreover, the program for SimRNAweb allows the users to download SimRNAweb prediction models and trajectory files. For processing trajectories of SimRNA, a Python interface is provided to parse trajectories into atoms, residues, simulation frames to prepare for further analysis. At the final step of a structure modeling process, a user can run the RNA refinement procedure implemented in a wrapper of QRNAS (60).
Auxiliary tools
In the package, there is a set of auxiliary tools of novel functions. One of them is diffpdb. It is a simple tool to compare two files of PDB format to identify the difference in the annotation of atoms, missing atoms, missing fragments (Figure 3e). Another standalone tool implemented in rna-tools is Clanstix. Clanstix can be used to interactively visualize the clustering results from CLANS (49). CLANS uses the Fruchterman–Reingold graph layout algorithm to visualize pairwise sequence similarities in either two-dimensional or three-dimensional space. The program was initially designed to calculate pairwise attraction values to compare protein sequences. However, it is possible to load a matrix of precomputed attraction values and thereby display any type of data based on pairwise interactions. Therefore, the Clanstix program from the rna-tools package can convert the all-vs-all distance (e.g., Root Mean Square Deviation) matrix into an input file for CLANS. An example of Clanstix is shown in Figure 3C, which is the result for RNA-Puzzle Puzzle 8. Models with a pairwise distance of RMSD lower than 8 Å are connected. The reference structure was added to this clustering. Interestingly, the reference structure was mapped to the small cluster with two models from Das's group and two models from Bujnicki's group. The visualization can provide useful insights into a set of analyzed models or models obtained from a simulation trajectory. Another example of the usage of Clanstix can be found in the publication of EvoClustRNA (61), which shows how 3D models of various homologous sequences are clustered with respect to each other and the reference models.
The documentation with step-by-step tutorials
The description in this publication only briefly reports functionalities implemented in rna-tools. To facilitate the finding of the right tool, the package is well documented in both online documentation and tutorials that will walk the users through various use cases. The step-by-step tutorial that explains how to prepare files for the submission to RNA-Puzzles is also included.
Extensibility by design
The rna-tools package was developed with the goal in mind of providing a framework for various tools specifically to support extensibility. A new script can be easily drafted just by copying-pasting to a new folder in ‘rna_tools/tools/<new tool>’. Many core functionalities are coded in the ‘rna_tools_lib.py’ file that is shared between scripts; hence, the functions can be imported to new scripts. This design speeds up the development of new programs since many of them need some low-level common functionalities, e.g., Python engine for parsing selection of residues, atoms, parsing/converting various types of data.
Example of a complete analysis of the blind prediction of the RNA-Puzzle Puzzle 19
The functionality implemented in rna-tools can be accessed via command-line, imported in Python scripts or in Jupyter Notebooks (Figure 3D). One such notebook is released together with rna-tools and illustrates the steps performed by the Bujnicki group to collect information about the RNA-Puzzles Puzzle 19, the Twister Sister ribozyme (62) (https://github.com/mmagnus/rna-tools/blob/master/rp19.ipynb). The analysis started with the secondary structure prediction using multiple wrappers implemented in rna-tools followed by the Rfam search for an RNA family that the sequence belongs to. At the time of this analysis, no RNA family for the sequence of the puzzle was presented in the Rfam database. A useful piece of information was provided by a successful hit in the PDB database, to the structure in the PDB database, Xrn1-resistant RNA from the 3′ untranslated region of a flavivirus (PDB: 4PQV) (63). This structure was considered as a homolog of the Puzzle and was used for comparative modeling.
Metrics in RNA 3D structure comparison
Root mean square deviation (RMSD)
Root Mean Square Deviation (RMSD) is a widely used metric for 3D structure comparison. The RMSD calculation aligns all the atoms that are found both in the predicted structure and the reference structure. A superimposition is performed based on these aligned atoms, and the result is calculated as the Root Mean Square Deviation based on the Euclidean distances of the aligned atoms.
Although RMSD is a well-established metric in structure comparison, it generalizes the errors over the whole structure. Thus, the final result can be misleading. When a linker region takes a different path or a hairpin loop has a different angle with respect to the core region, the overall RMSD may be large even if the core region is properly folded. In addition, RNA structure has more degrees of freedom in the backbone than proteins do and the accuracy of the base-pair interactions requires inspection. To overcome the limitations of the RMSD metric, the concepts of Interaction Network Fidelity (INF) and Deformation Profile (DP) were introduced (24). These metrics, RMSD, INF, DP and P-value (23) are included in the packages of RNA_assessment and RNAQUA.
Interaction Network Fidelity (INF)
The whole RNA structure can be considered as a large interaction network composed of Watson-Crick interactions, non-Watson–Crick interactions and base stackings. The correct prediction of all these interactions determines the success of the prediction. The interactions of an RNA structure can be extracted by programs such as MC-Annotate (25) and 3DNA (64). The Interaction Network Fidelity (INF) is defined as the Matthews correlation coefficient (MCC) between the interactions of the reference structure and that of the predicted structure. A higher INF score indicates higher consistency between the prediction and the reference structure in terms of interactions. The Interaction Network Fidelity can also assess a specific type of interaction. Thus, INF_wc, INF_nwc, INF_stack and INF_all, which define the Interaction Network Fidelity of Watson–Crick interactions, non-Watson–Crick interactions, stackings, and overall interactions, are used in the evaluation of RNA-Puzzles. Further, to account for the relationship between RMSD and INF, Deformation Index (DI) is defined as the ratio between RMSD and INF.
Deformation profile (DP)
To complement single value evaluation metrics, Deformation Profile is a 2D distance matrix representing the average distance between a prediction and the reference structure (Figure 4). The deformation profile matrix calculation includes two steps: (i) computing 1-nt superimposition of predicted model over reference structure for each aligned nucleotide; (ii) computing the average distance between each base in the reference structure and the corresponding base in a predicted structure for each superimposition. The Deformation Profile displays the regions that depart most from the rest of the structure.
Figure 4.

Deformation Profile comparison between predicted structure and reference structure. (A) Deformation Profile heatmap aligned with average B factor histogram, showing the Puzzle 8 (6) solution structure (PDB ID: 4L81) compared to the model 3 predicted by Das lab. (B) Electron density map of the high B factor region, G36–A39, shown in the red circle region in (A). This region is highly mobile, while A37 and A39 do not have a full density in the 2fo – fc electron density map to support the coordinates proposed by the crystal structure.
The deformation profile is effective in detecting the ‘poorly predicted’ regions. All comparisons between the model and the reference structure determined experimentally (e.g., by X-ray crystallography) rely on an assumption that the reference structure is 100% accurate, which may not always be true. Figure 4 shows that a poorly predicted region in the deformation profile (in red) corresponds to a region with a high B factor and insufficient electron density. One cannot exclude an error in the native structure during the modeling and fitting of the native structure.
P-value
P-value represents the confidence that a prediction is significantly different from a randomly generated RNA 3D structure (23). It was designed as a quality measure for RNA 3D structure prediction resulting from empirical relations for RMSD distribution as a function of RNA length. Therefore, it is independent of the molecule size. P-value is capable to differentiate de novo algorithms predicting all interactions from those who require to input base-pairing information. Normally, P-value lower than 0.01 indicates a successful prediction.
Clash score
Clash score (29) reports serious steric clashes identified in the RNA 3D structure. The score is computed as the number of disallowed (<0.4 Å) overlaps of atom pairs per thousand atoms. All-atom contacts are computed by PROBE (65) that uses van der Waals atom radii and identifies probes intersecting any not-covalently-bonded atom. In general, the existence of interatomic clashes indicates that a local conformation is not stereochemically accurate and should be refined. A high clash score indicates more severe steric clashes. However, clashes can exist also in high-resolution structures. Moreover, even if the global 3D fold of a modeled structure is close to the native one, the clash score value can be quite high when base-base interactions are not accurately reconstructed. Clash score is computed by MolProbity (29) incorporated into RNAQUA.
Mean of circular quantities (MCQ)
In the practice of RNA structure modeling, several approaches try to represent the RNA structure with simplified models, such as a network model (66), and reconstruct the RNA 3D structure with standard bond lengths and bond angles. Assuming the standard bond lengths and bond angles are constant values, it is important to understand the accuracy of the torsion angles, which are the only degrees of freedom in the modeling in this context. Therefore, the Mean of Circular Quantities (MCQ) is a metric to compare RNA 3D structures in the torsion angle space. A nucleotide can be described by six torsion angles from the backbone, while the δ dihedral is constrained by the sugar ring (Figure 5A). The residue-wise comparison in the torsion angle space highlights the dissimilarity in local structure. We divide the torsion angle difference into four bins: <15°, 15–30°, 30–60° and >60°. MCQ value <15° means the best similarity, while >60° implies severe structural change. Dissimilar regions can be highlighted on the secondary structure plot by coloring the four bins in gradient color (Figure 5B). MCQ can measure the similarity between whole structures or selected fragments. It also allows multiple models comparison with the reference structure (Figure 5C).
Figure 5.

MCQ and LCS-TA assess structure similarity in torsion angle space. MCQ and LCS-T compare structures based on (A) torsion angles defined for RNA structure. (B) MCQ supports assessing torsion angle-based similarity on a residue level and allows to visualize the results on the secondary structure diagram (here P3 stem characteristic to SAM-I/IV structures predicted in model 4 by Bujnicki lab (top) and Dokholyan lab (bottom) has been compared to the target fragment in Puzzle 8 (6)). (C) Heatmap shows the results of MCQ for the same P3 stem with PK-2 residues in bold, computed for all models submitted in Puzzle 8 and sorted by rank in reference to the target. (D) Clustering (colors) and visualization of models by different groups (markers) in Puzzle 8 upon MCQ distance matrix. LCS-TA finds structure fragments with torsion angle similarity threshold. (E) The resulting backbone fragment for LCS-TA in sequence-dependent mode with a threshold equal to 15° for model 4 (blue) by Bujnicki lab (left) and Dokholyan lab (right) aligned with the target (green) in Puzzle 8 and (F) positions of two LCS-TA-identified fragments marked with ‘1’ inserted in the appropriate places of the sequence, while unaligned regions are marked as ‘−’.
When the reference structure is unknown, clustering the structures to identify consensus structural cores may give biological insights to the folding and function of the RNA structure. MCQ enables structure clustering in the torsion angle space. Pairwise MCQ comparison scores are used as similarity distance and structures can be clustered using the resulted distance matrix (Figure 5D).
Longest continuous segments in torsion angle space (LCS-TA)
In the comparison of two RNA 3D structures, LCS-TA (34) identifies the longest continuous segments that display local similarity in the torsion angle space (Figure 5F). Two segments from different structures are considered similar if their angular distance (MCQ) does not exceed a predefined MCQ threshold which ranges between 10° and 20°. LCS-TA performs an iterative search using a slide-window approach until the longest continuous segment is found.
The structure comparison performed by LCS-TA can be either independent or dependent on the sequence. Sequence-dependent comparison assumes the same sequence in both the prediction and the reference structure and it finds similar segments with the same sequence. Sequence-independent comparison attempts to perform a structural alignment to identify the longest continuous segments which are similar in torsion angle space ignoring the sequence. In this mode, LCS-TA finds similar fragments with different sequences. When more than one segment is found to be similar in the sequence-independent comparison, all possible segments are listed. LCS-TA is also capable of performing a global comparison: with a fixed MCQ threshold, the prediction model with a longer identified segment has a higher similarity to the reference structure (Figure 5E).
DISCUSSION
The ability to predict RNA 3D structure attracts lots of attention because it opens great opportunities for new developments in biotechnology and basic science. The establishment of RNA-Puzzles boosted the improvement in RNA 3D structure prediction methods, as reported. Furthermore, through active and dynamic collaborations among research groups in RNA-Puzzles (5–7), new ideas were generated, validated and valuable tools were developed and implemented in the past eight years. These tools cover various functions that may be useful for RNA structure formatting, analysis, manipulation, visualization and comparison, which can be used in new exploratory studies.
Although biophysical rules are being learned from the experimentally determined RNA structures, the prediction of RNA structure is a data-driven problem. Unbiased assessment of a prediction is the key to understand its performance and usability. It is beneficial to have a standard dataset, which can be used to benchmark the performance of a new method against all other prediction approaches. The RNA-Puzzles toolkit directly provides such a benchmark and has been used to demonstrate the accuracy of a novel prediction (46). Although it is possible to run RNA structure prediction programs on other public datasets, such as Rfam and non-redundant dataset (17), RNA-Puzzles prediction stands for the best state-of-the-art blind prediction performance and includes structural diversity. In addition, selecting the top-quality model from a set of models generated by different prediction methods is another important step for an accurate prediction. Our benchmark set has also proved its usability in developing such a scoring model (20). Our datasets can be used as a standard test allowing for methods development and comparison.
Moreover, we provide a unified kit of tools used already by our groups in previous research projects. RNA_format, RNA_normalizer and RNA_assessment were used before to support all calculations in the RNA-Puzzle experiment. The rna-tools package was used in various scientific projects, to calculate stability of various U6 RNAs of the spliceosome (67), to process input files for SimRNAweb (RNA 3D structure prediction method) (58) and NPDock (RNA/DNA-protein docking method) (68), and to analyze data for RNArchitecture database (a classification system of RNA families with a focus on structural information) (69) and EvoClustRNA (RNA 3D structure prediction using multiple sequence alignment information) (61). MCQ-based methods were used i.a. to evaluate models in the second (7) and third (6) round of RNA-Puzzles, to identify structural patterns in plant pre-miRNAs (70), to build a database of conformers within the RNAfitme system (71,72). For the first time, we describe these tools and show how they can be integrated into one robust pipeline giving the users a way to provide a broad perspective on an RNA structure.
The installation of computational tools is non-trivial and can sometimes cost much time even for computational experts. A user-friendly implementation will greatly help the use of a computational tool. Considering that users may have diverse preferences, our resource tools provide both command-line executives and Jupyter Notebook (73) based tutorials, while all the tools are documented. Furthermore, we installed all the tools on a Docker image that can be easily downloaded and launched by the user, in particular, a biologist without programming skills. The Docker image saves the complicated actions required for installing all the tools. Finally, we release all of our datasets and computational tools at GitHub, which can be continuously updated if any bugs are detected. The ‘fork’ function of Github also facilitates novel computational methods or datasets being developed based on our resource, i.e. RNA-ligand interaction prediction.
The Jupyter Notebook (74) workflow in the resource provides a standard example for RNA structure prediction evaluation. Jupyter Notebook is an open-source web application that allows users to create and share documents that contain live code, equations, visualizations, and explanatory text. The tools implemented in the toolkit can be imported to such notebooks to create reproducible analyses that can be uploaded online and shared with the RNA structural bioinformatics community. One example of such analysis was described in the Result section for rna-tools. This approach of describing RNA bioinformatic analyses should help scientists to share their pipelines, e.g., protocols used for modeling in RNA-Puzzles, that can be later reproduced and/or improved by others. And since the Jupyter Notebook has support for over 40 programming languages, including those popular in Data Science such as Python, R, Julia and Scala, this is a great approach to incorporate the toolkit into pipelines written in other languages. In this way, all the RNA structure analysis work can be efficiently shared and reproduced. In addition, RNAQUA provides all the RNA structure comparison tools as a web service, which can alleviate the burden of software installation for non-computationally oriented users.
RNA structure comparison metrics have been developed since a decade ago (24). The availability of these metrics as computational tools is limited and not systematic, which highlights the importance of our toolkit. We also share every detail in a standard workflow accepted by the RNA-Puzzles community, i.e., when multiple structures have been solved for the same sequence, it is fair to consider all of them as native structures and use the nearest one to the prediction as the reference. Secondary structure analysis and visualization are useful aspects in understanding RNA 3D structure: rna-tools implements the easy transformation from 3D structure visualization in PyMOL(75) to 2D structure contacts annotation, thus enabling the intuitive comprehension from the biophysics aspects.
Our resource brings various tools and datasets into one unified resource that can be easily downloaded and used by biologists interested in RNA 3D structure prediction and analysis. We think that the toolkit with its open code should be considered as a library of functions and tools rather than a complete package with a fixed set of functionalities. The toolkit is a framework of various functions. The users are invited to extend it with their scripts on the top of the existing tools. In this way, it is possible to adapt our tools for future cases. For example, to have a particular wrapper or variant of tools that can be used for a very specific application saving time and brainpower of the user to write the code from scratch. We believe that the RNA-Puzzle Toolkit will prompt new advances in the applications of the RNA 3D structure prediction and in method development.
DATA AVAILABILITY
All the datasets, computational tools, and related documentation are available as open-source at https://github.com/RNA-Puzzles.
Supplementary Material
ACKNOWLEDGEMENTS
Authors contributions: M.M. developed rna-tools, combined the workflow, created the standardized_dataset and wrote parts of the manuscript. M.A. developed and tested RNAQUA, and wrote parts of the manuscript, P.L. supervised the development of RNAQUA, T.Z. implemented MCQ algorithm, J.W. and T.Z. implemented LCS-TA. Y.C. provided technical support to web development. J.M.B. supervised the development of rna-tools. M.S. developed a concept for MCQ and LCS-TA algorithms, supervised their implementation and wrote parts of the manuscript. E.W. supervised the entire project and drafted the manuscript. Z.M. conceived the project, cleaned up the datasets, implemented the RNA structure format tool, format check tool and evaluation metrics (INF, DP), designed the website and wrote parts of the manuscript. All authors contributed to manuscript preparation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Polish National Science Centre [NCN, 2015/17/N/NZ2/03360 to M.M., 2016/23/B/ST6/03931 to M.S., 2016/23/N/ST6/03779 to T.Ż., 2017/26/A/NZ1/01083 to J.M.B.]; Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund [MAB/20172 carried out within the International Research Agendas Program]; Institute of Computing Science, Poznan University of Technology [09/91/SBAD/0681 to M.A.]; Single Cell Gene Expression Atlas grant from the Wellcome Trust [108437/Z/15/Z]. Funding for open access charge: Shanghai Fourth People's Hospital.
Conflict of interest statement. None declared.
REFERENCES
- 1. Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature. 1969; 224:759–763. [DOI] [PubMed] [Google Scholar]
- 2. Miao Z., Westhof E.. RNA structure: advances and assessment of 3D structure prediction. Annu. Rev. Biophys. 2017; 46:483–503. [DOI] [PubMed] [Google Scholar]
- 3. Dawson W.K., Bujnicki J.M.. Computational modeling of RNA 3D structures and interactions. Curr. Opin. Struct. Biol. 2016; 37:22–28. [DOI] [PubMed] [Google Scholar]
- 4. Moult J., Fidelis K., Kryshtafovych A., Schwede T., Tramontano A.. Critical assessment of methods of protein structure prediction (CASP)-Round XII. Proteins. 2018; 86:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cruz J.A., Blanchet M.-F., Boniecki M., Bujnicki J.M., Chen S.-J., Cao S., Das R., Ding F., Dokholyan N.V., Flores S.C. et al.. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. RNA. 2012; 18:610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miao Z., Adamiak R.W., Antczak M., Batey R.T., Becka A.J., Biesiada M., Boniecki M.J., Bujnicki J.M., Chen S.-J., Cheng C.Y. et al.. RNA-Puzzles Round III: 3D RNA structure prediction of five riboswitches and one ribozyme. RNA. 2017; 23:655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miao Z., Adamiak R.W., Blanchet M.-F., Boniecki M., Bujnicki J.M., Chen S.-J., Cheng C., Chojnowski G., Chou F.-C., Cordero P. et al.. RNA-Puzzles Round II: assessment of RNA structure prediction programs applied to three large RNA structures. RNA. 2015; 21:1066–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noller H.F., Woese C.R.. Secondary structure of 16S ribosomal RNA. Science. 1981; 212:403–411. [DOI] [PubMed] [Google Scholar]
- 9. Haas E., Morse D., Brown J., Schmidt F., Pace N.. Long-range structure in ribonuclease P RNA. Science. 1991; 254:853–856. [DOI] [PubMed] [Google Scholar]
- 10. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruz J.A., Westhof E.. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat. Methods. 2011; 8:513–521. [DOI] [PubMed] [Google Scholar]
- 12. Kucharík M., Hofacker I.L., Stadler P.F., Qin J.. Pseudoknots in RNA folding landscapes. Bioinformatics. 2016; 32:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michel F., Westhof E.. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J. Mol. Biol. 1990; 216:585–610. [DOI] [PubMed] [Google Scholar]
- 14. Brunel C., Romby P., Westhof E., Ehresmann C., Ehresmann B.. Three-dimensional model of Escherichia coli ribosomal 5 S RNA as deduced from structure probing in solution and computer modeling. J. Mol. Biol. 1991; 221:293–308. [DOI] [PubMed] [Google Scholar]
- 15. Westhof E., Romby P., Romaniuk P.J., Ebel J.P., Ehresmann C., Ehresmann B.. Computer modeling from solution data of spinach chloroplast and of Xenopus laevis somatic and oocyte 5 S rRNAs. J. Mol. Biol. 1989; 207:417–431. [DOI] [PubMed] [Google Scholar]
- 16. Rychlewski L., Fischer D.. LiveBench-8: the large-scale, continuous assessment of automated protein structure prediction. Protein Sci. 2005; 14:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leontis N.B., Zirbel C.L.. Nonredundant 3D structure datasets for RNA knowledge extraction and benchmarking. Nucleic Acids Mol. Biol. 2012; 27:281–298. [Google Scholar]
- 18. Weinreb C., Riesselman A.J., Ingraham J.B., Gross T., Sander C., Marks D.S.. 3D RNA and functional interactions from evolutionary couplings. Cell. 2016; 165:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suslov N.B., DasGupta S., Huang H., Fuller J.R., Lilley D.M.J., Rice P.A., Piccirilli J.A.. Crystal structure of the Varkud satellite ribozyme. Nat. Chem. Biol. 2015; 11:840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J., Zhu W., Wang J., Li W., Gong S., Zhang J., Wang W.. RNA3DCNN: Local and global quality assessments of RNA 3D structures using 3D deep convolutional neural networks. PLoS Comput. Biol. 2018; 14:e1006514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antczak M., Popenda M., Zok T., Sarzynska J., Ratajczak T., Tomczyk K., Adamiak R.W., Szachniuk M.. New functionality of RNAComposer: an application to shape the axis of miR160 precursor structure. Acta Biochim. Pol. 2016; 63:737–744. [DOI] [PubMed] [Google Scholar]
- 22. Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al.. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hajdin C.E., Ding F., Dokholyan N.V., Weeks K.M.. On the significance of an RNA tertiary structure prediction. RNA. 2010; 16:1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parisien M., Cruz J.A., Westhof E., Major F.. New metrics for comparing and assessing discrepancies between RNA 3D structures and models. RNA. 2009; 15:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gendron P., Lemieux S., Major F.. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001; 308:919–936. [DOI] [PubMed] [Google Scholar]
- 26. Oliphant T.E. A Guide to NumPy. USA: Trelgol Publishing. 2006; https://www.scipy.org/citing.html. [Google Scholar]
- 27. Rother M., Rother K., Puton T., Bujnicki J.M.. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 2011; 39:4007–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waleń T., Chojnowski G., Gierski P., Bujnicki J.M.. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res. 2014; 42:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davis I.W., Leaver-Fay A., Chen V.B., Block J.N., Kapral G.J., Wang X., Murray L.W., Arendall W.B. 3rd, Snoeyink J., Richardson J.S. et al.. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007; 35:W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lukasiak P., Antczak M., Ratajczak T., Bujnicki J.M., Szachniuk M., Adamiak R.W., Popenda M., Blazewicz J.. RNAlyzer–novel approach for quality analysis of RNA structural models. Nucleic Acids Res. 2013; 41:5978–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lukasiak P., Antczak M., Ratajczak T., Szachniuk M., Popenda M., Adamiak R.W., Blazewicz J.. RNAssess—a web server for quality assessment of RNA 3D structures. Nucleic Acids Res. 2015; 43:W502–W506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szachniuk M. RNApolis: computational platform for RNA structure analysis. Found. Comput. Decision Sci. 2019; 44:241–257. [Google Scholar]
- 33. Zok T., Popenda M., Szachniuk M.. MCQ4Structures to compute similarity of molecule structures. Central Eur. J. Oper. Res. 2014; 22:457–473. [Google Scholar]
- 34. Wiedemann J., Zok T., Milostan M., Szachniuk M.. LCS-TA to identify similar fragments in RNA 3D structures. BMC Bioinformatics. 2017; 18:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dibrov S.M., McLean J., Parsons J., Hermann T.. Self-assembling RNA square. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:6405–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ren A., Vušurović N., Gebetsberger J., Gao P., Juen M., Kreutz C., Micura R., Patel D.J.. Pistol ribozyme adopts a pseudoknot fold facilitating site-specific in-line cleavage. Nat. Chem. Biol. 2016; 12:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baird N.J., Zhang J., Hamma T., Ferre-D’Amare A.R.. YbxF and YlxQ are bacterial homologs of L7Ae and bind K-turns but not K-loops. RNA. 2012; 18:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peselis A., Serganov A.. Structural insights into ligand binding and gene expression control by an adenosylcobalamin riboswitch. Nat. Struct. Mol. Biol. 2012; 19:1182–1184. [DOI] [PubMed] [Google Scholar]
- 39. Zhang J., Ferré-D’Amaré A.R.. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013; 500:363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ren A., Xue Y., Peselis A., Serganov A., Al-Hashimi H.M., Patel D.J.. Structural and dynamic basis for low-affinity, high-selectivity binding of L-glutamine by the glutamine riboswitch. Cell Rep. 2015; 13:1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watkins A.M., Das R.. FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds. 2019; 10 September 2019, preprint: not peer reviewed 10.1101/764449. [DOI] [PMC free article] [PubMed]
- 42. Boniecki M.J., Lach G., Dawson W.K., Tomala K., Lukasz P., Soltysinski T., Rother K.M., Bujnicki J.M.. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016; 44:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng C.Y., Chou F.-C., Das R.. Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol. 2015; 553:35–64. [DOI] [PubMed] [Google Scholar]
- 44. Sharma S., Ding F., Dokholyan N.V.. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008; 24:1951–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao C., Xu X., Chen S.-J.. Predicting RNA Structure with Vfold. Methods Mol. Biol. 2017; 1654:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watkins A.M., Geniesse C., Kladwang W., Zakrevsky P., Jaeger L., Das R.. Blind prediction of noncanonical RNA structure at atomic accuracy. Sci Adv. 2018; 4:eaar5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kerpedjiev P., Siederdissen Höner Zu, Hofacker I.L.. Predicting RNA 3D structure using a coarse-grain helix-centered model. RNA. 2015; 21:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Capriotti E., Norambuena T., Marti-Renom M.A., Melo F.. All-atom knowledge-based potential for RNA structure prediction and assessment. Bioinformatics. 2011; 27:1086–1093. [DOI] [PubMed] [Google Scholar]
- 49. Bernauer J., Huang X., Sim A.Y.L., Levitt M.. Fully differentiable coarse-grained and all-atom knowledge-based potentials for RNA structure evaluation. RNA. 2011; 17:1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 51. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kalvari I., Argasinska J., Quinones-Olvera N., Nawrocki E.P., Rivas E., Eddy S.R., Bateman A., Finn R.D., Petrov A.I.. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018; 46:D335–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parisien M., Major F.. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008; 452:51–55. [DOI] [PubMed] [Google Scholar]
- 54. Sato K., Kato Y., Hamada M., Akutsu T., Asai K.. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics. 2011; 27:i85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lai D., Proctor J.R., Zhu J.Y.A., Meyer I.M.. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012; 40:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hanson R.M., Lu X.-J.. DSSR-enhanced visualization of nucleic acid structures in Jmol. Nucleic Acids Res. 2017; 45:W528–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Magnus M., Boniecki M.J., Dawson W., Bujnicki J.M.. SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 2016; 44:W315–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Das R., Baker D.. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:14664–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stasiewicz J., Mukherjee S., Nithin C., Bujnicki J.M.. QRNAS: software tool for refinement of nucleic acid structures. BMC Struct. Biol. 2019; 19:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Magnus M., Kappel K., Das R., Bujnicki J.M.. RNA 3D structure prediction guided by independent folding of homologous sequences. BMC Bioinformatics. 2019; 20:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu Y., Wilson T.J., Lilley D.M.J.. The structure of a nucleolytic ribozyme that employs a catalytic metal ion. Nat. Chem. Biol. 2017; 13:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chapman E.G., Costantino D.A., Rabe J.L., Moon S.L., Wilusz J., Nix J.C., Kieft J.S.. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science. 2014; 344:307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lu X.-J., Olson W.K.. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003; 31:5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Word J.M., Lovell S.C., LaBean T.H., Taylor H.C., Zalis M.E., Presley B.K., Richardson J.S., Richardson D.C.. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 1999; 285:1711–1733. [DOI] [PubMed] [Google Scholar]
- 66. Kim N., Petingi L., Schlick T.. Network theory tools for RNA Modeling. WSEAS Trans. Math. 2013; 9:941–955. [PMC free article] [PubMed] [Google Scholar]
- 67. Eysmont K., Matylla-Kulińska K., Jaskulska A., Magnus M., Konarska M.M.. Rearrangements within the U6 snRNA core during the transition between the two catalytic steps of splicing. Mol. Cell. 2019; 75:538–548. [DOI] [PubMed] [Google Scholar]
- 68. Tuszynska I., Magnus M., Jonak K.. NPDock: a web server for protein–nucleic acid docking. Nucleic Acids Res. 2015; 43:W425–W430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Boccaletto P., Magnus M., Almeida C., Zyla A., Astha A., Pluta R., Baginski B., Jankowska E., Dunin-Horkawicz S., Wirecki T.K. et al.. RNArchitecture: a database and a classification system of RNA families, with a focus on structural information. Nucleic Acids Res. 2018; 46:D202–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miskiewicz J., Tomczyk K., Mickiewicz A., Sarzynska J., Szachniuk M.. Bioinformatics study of structural patterns in plant MicroRNA precursors. Biomed. Res. Int. 2017; 2017:6783010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zok T., Antczak M., Riedel M., Nebel D., Villmann T., Lukasiak P., Blazewicz J., Szachniuk M.. Building the library of RNA 3D nucleotide conformations using the clustering approach. Int. J. Appl. Math. Comput. Sci. 2015; 25:689–700. [Google Scholar]
- 72. Antczak M., Zok T., Osowiecki M., Popenda M., Adamiak R.W., Szachniuk M.. RNAfitme: a webserver for modeling nucleobase and nucleoside residue conformation in fixed-backbone RNA structures. BMC Bioinformatics. 2018; 19:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yakimchik A.I. Jupyter Notebook: a system for interactive scientific computing. Geofizicheskiy Zhurnal. 2019; 41:121. [Google Scholar]
- 74. Basu A. Reproducible research with jupyter notebooks. Authorea. doi:10.22541/au.151460905.57485984. [Google Scholar]
- 75. Rigsby R.E., Parker A.B.. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016; 44:433–437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the datasets, computational tools, and related documentation are available as open-source at https://github.com/RNA-Puzzles.