


Abstract
Chloroplast transcription requires numerous quality control steps to generate the complex but selective mixture of accumulating RNAs. To gain insight into how this RNA diversity is achieved and regulated, we systematically mapped transcript ends by developing a protocol called Terminome-seq. Using Arabidopsis thaliana as a model, we catalogued >215 primary 5′ ends corresponding to transcription start sites (TSS), as well as 1628 processed 5′ ends and 1299 3′ ends. While most termini were found in intergenic regions, numerous abundant termini were also found within coding regions and introns, including several major TSS at unexpected locations. A consistent feature was the clustering of both 5′ and 3′ ends, contrasting with the prevailing description of discrete 5′ termini, suggesting an imprecision of the transcription and/or RNA processing machinery. Numerous termini correlated with the extremities of small RNA footprints or predicted stem-loop structures, in agreement with the model of passive RNA protection. Terminome-seq was also implemented for pnp1–1, a mutant lacking the processing enzyme polynucleotide phosphorylase. Nearly 2000 termini were altered in pnp1–1, revealing a dominant role in shaping the transcriptome. In summary, Terminome-seq permits precise delineation of the roles and regulation of the many factors involved in organellar transcriptome quality control.
INTRODUCTION
The sophisticated interplay of factors regulating chloroplast gene expression results from over a billion year symbiosis between the plastid and nucleus. Transcription of the entire plastome (1,2) combined with inefficient termination (3,4) leads to a complex primary transcriptome that undergoes numerous maturation steps. These may include 5′ and 3′ end processing, intergenic cleavage of polycistronic transcripts, intron removal through RNA splicing and RNA editing to convert specific cytosines into uracils (5–7). Among the protein factors involved in maturation, a variety of endo- and exoribonucleases (RNases) are responsible for processing and cleavage (8), while RNA-binding proteins (RBPs) counteract their activities to stabilize some transcript ends (9) or participate in splicing or editing protein complexes.
Legacy chloroplast RNA maturation analyses have used tedious gene by gene molecular techniques, restricting most detailed RNA analyses to several mono- or polycistronic transcripts including psbA, rbcL, atpI-atpH-atpF-atpA and psbB-psbT-psbN-psbH-petB-petD (10), along with the ribosomal RNA operon, which together may not be fully representative of plastid RNA maturation pathways. Gene-by-gene analyses of processing factors may also have limited value. For example, the RNase CSP41a was shown to possess strong endoribonuclease activity in vitro (11), however in vivo mutant analysis suggests regulatory roles that may have little to do with RNase activity per se (12–15). Additionally, while mutants for the endoribonuclease RNase E, its specificity partner RHON1 and the 5′→3′ exoribonuclease and endoribonuclease RNase J accumulate novel and apparently unprocessed transcripts, pleiotropic effects are prone to masking their precise sites of cleavage or interaction (16–18).
To overcome some of these limitations, we and others have increasingly employed RNA-seq-based approaches that yield genome-wide cataloging and mechanistic insights into chloroplast transcription, editing, splicing and translation (19–28). Our own results, for example, revealed a large number of non-coding RNAs, which additional research suggests may include a class that exerts its functions through sense-antisense RNA pairing (19,29,30). It is difficult to fully understand transcript function, however, without knowledge of the 5′ and 3′ termini which together help define promoter sequences, regulatory UTRs and the potential for sense-antisense pairing. On a genome-wide scale, we refer to these 5′ and 3′ ends as the (RNA) terminome, and the associated technique as Terminome-seq.
Efforts to define the chloroplast RNA terminome have been limited to date, with the most comprehensive study focused on 5′ ends in barley, a model that has been effectively used to dissect the respective roles of nucleus- and chloroplast-encoded RNA polymerases (31,32). On a genome-wide level, barley chloroplasts were found to possess larger than expected numbers of both primary and processed 5′ termini, consistent with a highly complex transcriptional landscape (20). We chose Arabidopsis for our analysis, because of its broad use to dissect post-transcriptional RNA events in the chloroplast, including the analysis of RNase mutants, pentatricopeptide repeat (PPR) and other helical repeat proteins, and RNA editing and splicing factors. We found that the Arabidopsis chloroplast terminome is complex, and in some cases surprising. For example, both known and new transcription start sites (TSS) were identified, sometimes internal or antisense to known transcripts and a general imprecision of both processed 5′ and 3′ ends was observed. To highlight the comparative potential of Terminome-seq, we examined the pnp1–1 mutant which lacks the major 3′ processing enzyme polynucleotide phosphorylase (33,34), revealing a largely reshaped terminome. Overall, our results showcase Terminome-seq as a valuable addition to the organelle gene expression analysis toolkit.
MATERIALS AND METHODS
Plant material
Arabidopsis thaliana Col-0 and pnp1–1 seeds were germinated on MS medium with 16 h of light per day at 23°C. Three-week old leaf material was flash-frozen in liquid nitrogen, and total RNA was isolated using TRI Reagent according to the manufacturer's instructions (www.sigmaaldrich.com).
Terminome library synthesis and analysis
All libraries were produced from 1 μg of DNase I-treated RNA (www.neb.com), and for TAP-treated samples, tobacco acid phosphorylase (TAP; www.epibio.com) was used according to the manufacturer's instructions with heat inactivation at the end of the incubation period. Library synthesis was carried out using the Illumina TruSeq Small RNA library preparation kit (www.illumina.com) intended to capture the RNA population containing a 5′ phosphate and 3′ hydroxyl group. Minor modifications were made to the protocol depending on whether a native 5′ or 3′ end was the target (Supplementary Figure S1). Libraries intended for native 3′ end capture, followed the protocol with initial 3′ adapter ligation using T4 RNA ligase 2, a deletion mutant that can only ligate a 3′ hydroxyl group to a 5′ adenylated RNA, consistent with the 3′ RNA adapter chemistry. After ligation, the RNA was fragmented using a Covaris sonicator (www.covaris.com), with a target size of 200 nt, followed by ethanol precipitation for concentration and 5′ adapter ligation with T4 RNA ligase. Libraries intended for native 5′ end capture required further adjustments. The order of adapter ligation was reversed: 5′ adapter ligation (with T4 RNA ligase)—sonication—ethanol precipitation—3′ adapter ligation (with T4 RNA ligase 2). In this case, excess 5′ adapter remaining following the sonication and ethanol precipitation could ligate to added 3′ adapter, but not to any new 5′ ends created through sonication as the new 5′ ends would not be adenylated. This resulted in unwanted adapter dimers that were preferentially amplified during library amplification (PCR1) due to their small size (∼133 bp). Therefore, size selection was performed on the products from PCR1, retaining only products over 200 bp using Pippin Prep (www.sagescience.com). A second polymerase chain reaction amplification (PCR2) was executed on these products. Quality control was performed after Pippin size selection and before library submission for sequencing using an Agilent BioAnalyzer (www.agilent.com). Details about the procedure and the Pippin Prep are available at https://github.com/BenoitCastandet/Terminome_Seq. The final cDNA libraries were purified using magnetic AMPure beads (www.beckman.com) following the manufacturer's protocol. Multiple steps in the above protocol, including fragmentation, ethanol precipitation, Pippin size selection (5′ libraries only) and AMPure purification of cDNA libraries, resulted in a bias toward the retention, and therefore sequencing, of fragments >67 nt. As a consequence, ends of small RNAs (smRNAs) and tRNAs would be expected to be underrepresented in the results. Additionally, a minor bias was introduced because the RT primer is fully complementary to the 3′ adapter, ending in a sequence complementary to TGG. Illegitimate priming by the adapter resulted in an estimated 52 additional 3′ ends terminating in TGG which are included in our data as they cannot be distinguished from legitimate 3′ termini ending in T(U)GG. While this bias was inevitable, the overall interpretation of the results was not affected.
Libraries were pooled and sequenced on a NextSeq500 Sequencer (www.illumina.com) using the v3 kit, with paired-end reads generating 40 bp long R1 reads and 35 bp long R2 reads for all libraries. R1 reads are only of use for libraries generated to obtain 5′ related data, while R2 reads contain data related to 3′ ends and therefore are only relevant for libraries generated to obtain 3′ data. Raw sequences have been deposited on the SRA database with the number PRJNA533962 and can be accessed here https://www.ncbi.nlm.nih.gov/sra/PRJNA533962. The detailed pipeline used to analyze the relevant reads is available at https://github.com/BenoitCastandet/Terminome_Seq. Briefly, the quality of relevant reads was checked using fastq-mcf (https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md) followed by alignment to the chloroplast reference genome, Arabidopsis TAIR10 version modified to add the first exon of the chloroplast gene ycf3, using tophat2 (https://ccb.jhu.edu/software/tophat/index.shtml). Two customized scripts allowed us to extract the positions of the 5′ and 3′ termini and the results were normalized according to the numbers of reads aligned to the chloroplast genome. Normalized data are available in Supplementary Table S1.
5′ RACE
Total RNA was isolated from mature leaf tissue using TRI Reagent® and treated with DNase I (Ambion; http://www.thermofisher.com). 5′ Rapid Amplification of cDNA Ends (RACE) reactions were completed as described (35) with some modifications. For analysis of primary transcripts, 4 μg of RNA was treated with TAP (0.5U/4 μg RNA; Epicenter, http://www.epibio.com) for 1 h at 37°C with RNaseOUT (40U; Invitrogen, http://www.thermofisher.com), followed by phenol/chloroform extraction and ethanol precipitation. The 5′ RACE adapter (Supplementary Table S2) was then ligated to the 5′ ends of the + or −TAP treated RNA with T4 RNA ligase (Ambion). For this, 10 μM of 5′ RACE adapter and 4 μg of ±TAP-treated RNA were incubated for 5 min at 65°C. Samples were chilled on ice and then 5U of T4 RNA ligase, 1× ligase buffer, 1 mM adenosine triphosphate (ATP) and 40U of RNaseOUT were added and the reactions were incubated for 1 h at 37°C. Ligated RNA was phenol/chloroform purified and precipitated with ethanol, and cDNA was generated using SuperScript III (Invitrogen) with random hexamers according to the manufacturer's protocol. Transcripts were amplified by PCR using the 3′ gene-specific primers indicated in the figure legends and the 5′ RACE primer (Supplementary Table S2). Amplicons were visualized after separation through a 1% agarose gel and gel purified. Purified bands were used for a second, nested PCR reaction with the indicated 3′ RACE primer and the RACE 5′ nested primer to ensure amplified sequences were specific to the intended target and/or increase amplicon intensity as the bands were directly sequenced, or cloned and sequenced. The nested PCR reactions were visualized in 1% agarose gels, and specific bands were gel purified, cloned and sequenced.
RESULTS
Terminome sequencing strategy and overview
Identification of RNA termini has traditionally been performed by sequencing individual clones from RACE products, primer extension, or nuclease protection assays. A recent improvement was the substitution of high-throughput sequencing for product-by-product analysis (36–40). Because of their focus on nucleus-encoded RNAs, these protocols rely on the presence of 3′ poly(A) tails, rendering them unsuitable for studying organellar RNAs where such tails mark transcripts for degradation (27,41,42). Our experimental design included the sequencing of three distinct libraries devoted to the identification of both primary and processed 5′ ends, or 3′ ends. The protocol (Supplementary Figure S1) was modified from the Illumina TruSeq Small RNA kit, which relies on sequential ligation of adaptors prior to cDNA synthesis and amplification. For Terminome-seq, the initial ligation of 5′ or 3′ adapter captures the native 5′ and 3′ termini, respectively, then the RNA is fragmented followed by a second adapter ligation step before amplification. To differentiate and identify primary transcripts that correspond to TSS, RNA was pretreated with TAP prior to 5′ adaptor ligation. TAP converts triphosphorylated 5′ ends, uniquely found in primary transcripts, to monophosphates, making them amenable to adapter ligation. Thus, libraries created with or without TAP pretreatment (+TAP and −TAP) can be compared to identify TSS. Duplicate libraries were constructed from Arabidopsis thaliana Col-0 biological replicates and the results show high correlation (Supplementary Figure S2). Native transcript ends were defined as the first nucleotide of each read, following suitable manipulation of the primary sequencing. The full coverage for Terminome-seq at single nucleotide resolution for 5′ (±TAP) and 3′ ends generated for this manuscript can be accessed in Supplementary Table S1. It is important to note that transcripts with post-transcriptional modifications, such as tRNAs with a post-transcriptionally added CCA tail, or RNA degradation intermediates bearing polynucleotide tails, would not fully align to the genome and are therefore excluded from our curated dataset.
A genome-wide view of end distribution is depicted in Figure 1. Clusters of 5′ and 3′ ends, covering a remarkable 13 and 16% of the genome, respectively, are clearly discernable, but are largely absent from areas antisense to known transcripts. In contrast, the full chloroplast genome is covered by reads from total RNA-seq experiments (2,19,20,23). However, when considering only positions representing at least 0.001% of total reads (10 reads per million, or RPM), <0.7% of the genome (1628 processed 5′ ends and 1299 3′ ends, respectively) was represented (Figure 2A). Thus, the vast majority of termini are stochastically found at low levels, probably representing RNA degradation products or processing intermediates.
Figure 1.

Plastome-scale view of Terminome-seq results End coverages are the average of two Col-0 biological replicates and given in RPM. 5′ ends obtained with or without TAP treatment are red and blue, respectively, and 3′ ends are displayed in green. Gene models are indicated between the tracks corresponding to the plus and minus strands of the plastome. One copy of the large inverted repeat is omitted for clarity. Selected TSS described in more detail in the main text are marked by arrows and labeled, as are the psbB and rRNA operons. Tick marks are every 1000 nt.
Figure 2.

Coverage and distribution of transcript termini (A) Comparison of genome coverage between RNA-seq and Terminome-seq. While the plastome is almost fully covered by at least one read in RNA-seq, only 12.7 and 15.8% is covered by 5′ and 3′ ends, respectively. Data for RNA-seq correspond to the average of two previously published WT replicates (19). Termini coverage at >10 RPM is marked and discussed in the text. (B) Terminome-seq read distribution in the WT. The results are the average of two biological replicates. Reads antisense to exons (as-exon) refers to reads mapping to the antisense strand of a known coding region.
To compare the abundance of termini across regions of the chloroplast genome, we informatically divided them into six types: rRNAs, tRNAs, exons (corresponding to protein-coding regions), introns, intergenic regions (anything else) and positions on the antisense strand of exons. When categorized this way, Figure 2B shows that ∼85% of the 5′ and 3′ ends were found in the rRNAs. The high degree of rRNA transcript ends was expected due to the elevated expression level of the rRNA operon and lack of rRNA depletion during the library preparation. Addition of an rRNA depletion step could help to map low abundance transcript ends that may have been overlooked in this study. Ends corresponding to intergenic regions were overrepresented compared to exons (Figure 2B). Indeed, even though intergenic regions cover around 25% of the genome (43), they contained 10.2 and 8.8% of the −TAP 5′ and 3′ ends, respectively, whereas only 1.7 and 1.5% of the ends mapped within exons. TAP treatment, which allowed TSS to be represented, significantly altered these proportions, with the proportion of 5′ ends mapping to rRNAs decreasing to 65%, mainly in favor of an increase in reads mapping to introns (12.4%) and antisense to exons (8.3%; Figure 2B). Remarkably, an overabundance of TSS is found in the first clpP intron (TSS internal-clpP-intron 1) and antisense to the ndhF transcript (as-ndhF; highlighted in Figure 1).
The chloroplast genome contains at least 215 TSS
Because the 5′ ends of most primary transcripts are marked by a 5′ triphosphate, they should be highly over-represented in +TAP-treated versus −TAP libraries. We therefore defined a TSS as any 5′ end that was at least 10 times more abundant in +TAP libraries, a calculation that generated 352 positions (Figure 3A). Because of the generally precise mechanism of transcription initiation, a true TSS should be a single predominant nucleotide rather than a cluster. We therefore filtered out putative TSS where the first nucleotide (the TSS itself) represented <50% of the coverage over a 5-nt stretch where it was the first nucleotide, as was done for barley (20). This reduced the number of TSS being considered to 215, with the others potentially representing stochastic initiation (Supplementary Table S3). Among the 215 defined TSS, 81% of the initiating nucleotides were purines (119 A’s and 55 G’s; Figure 3A). This trend is consistent with barley, where purines defined >80% of TSS (20). Our data identified 45 previously described Arabidopsis TSS, 16 of which are in orthologous positions in barley (20), meeting our expectations considering that more closely related species like Arabidopsis thaliana and Nicotiana tabacum differ in their promoter usage for some genes (44).
Figure 3.

TSS analysis (A) The abundance of 5′ ends at each position for both genome strands was compared between +TAP and −TAP libraries. The dashed line separates the 352 ends that have a +TAP/−TAP ratio >10 from those with a lower ratio. Putative TSS were filtered to remove any ends that did not reach 50% of the coverage of the most represented read within a 5 nt stretch. The pie chart graphs the initiating nucleotide of the remaining 215 TSS. (B) The novel TSS detected within clpP intron 1 were mapped using 5′ RACE. 5′ RACE was completed with (red) and without (blue) prior TAP treatment, and sequenced clones are represented by colored arrowheads above/below the nucleotide sequence. The stained gel of the corresponding PCR reactions is shown at right. The gene model between exons 1 and 2 is shown with Terminome-seq results. +TAP 5′ ends are in red, −TAP ends in blue and 3′ ends are in green. The X-axis is genomic position and the Y-axis is RPM coverage. Black arrowheads P1 and P2 represent the 3′ primers used for 5′ RACE.
Most plastid genes belong to polycistronic units and have classically been described as being co-transcribed. A few genes, such as psbA, rbcL and ndhF, are considered to be monocistronic, which is also the tendency for tRNAs outside the rRNA operon (45,46). TSS could be identified at the 5′ ends of all but one of the 20 main transcriptional units, with petL-petG-psaJ-rpl33-rps18 being the exception. The processed petL 5′ end is easily identifiable, however (see below). This suggests rapid maturation of the primary transcript, a phenomenon that is more prevalent in Chlamydomonas reinhardtii chloroplasts, where a recent transcriptome analysis revealed only 23 TSS, albeit using an entirely different method (47). Terminome-seq also confirmed our earlier discovery of TSS upstream of trnC, trnF and trnN (22). All transcriptional units also contained internal TSS, similar to barley (20), which could reflect mechanisms to allow for differential expression of genes within a larger cluster as is particularly well-documented for the psbD-C segment of the barley psbK gene cluster (48).
TAP treatment revealed three genomic areas with unexpectedly massive numbers of initiation events. As mentioned above, these lie in the first clpP intron and on the sense and antisense strands of ndhF (Figure 1 and Supplementary Table S3). TSS antisense to ndhF are distributed over nearly 300 nt and some of them were also characterized in barley and tobacco (20,49). The TSS internal to clpP intron 1 and on the sense strand of ndhF are distributed over ∼160 and 600 nt, respectively, and to our knowledge have not been previously described. As a validation step, we confirmed that at least some of these ends could be amplified by 5′ RACE following TAP treatment (Figure 3B and Supplementary Figure S3A).
Although we attempted to obtain an exhaustive catalog of TSS, the data filters we used to reduce the false discovery rate eliminated a few known initiation sites (Supplementary Figure S3B). For instance, the PpsbN -32 promoter (50) and PatpE -430 (51,52) are absent from our list because they have a +TAP:−TAP ratio of <10 (9.9 for PatpE and 2.5 for PpsbN). On the other hand, our data shed light on the uncertainty of whether PpsbD -186 (position 32525) is a genuine promoter or processed end (53–55), with our results being strongly diagnostic of a post-transcriptional processing site. The opposite is true for the ndhA -66 (position 122076) 5′ end that has been described as being created post-transcriptionally in maize through the action of PPR53 (56), but is a TSS based on our data.
The case of the psbB operon
The psbB operon is particularly well studied, containing five genes on the plus strand (psbB-psbT-psbH-petB-petD, the last two containing introns) and one on the minus strand (psbN). Altogether, over 20 accumulating transcripts generated from this operon have been characterized (5,10,57), making it an attractive testbed for the ability of Terminome-seq to reflect such a complex landscape accurately.
Figure 4A shows the positions of the eight TSS (numbered in red), along with at least 12 processed 5′ ends (numbered in blue) and 17 3′ ends (numbered in green), which are annotated in Table 1. A consistent feature of 5′ ends is their clustered organization around a dominant peak, whereas the 3′ termini are more discrete. Heterodisperse 5′ ends are reminiscent of degradation intermediates, perhaps created by an enzyme such as RNase J that would progressively stall as it encountered secondary structures or bound proteins. 3′ ends that were more discrete could be identified in both coding and intergenic regions, probably representing a mix of degradation intermediates and mature 3′ ends. Among the processed 5′ ends and 3′ termini found by Terminome-seq, several had been previously described. These include the 5′ ends psbB -51 (57), psbH -44 (58,59) and -67 (60) and petB -47 (61,62), as well as the 3′ termini psbT +60 (30), +223 (60), psbH +109 (61,62), petB +67 (63), petD +94 (64) and psbN +39 (30). Thus, there was excellent overlap between our data and previously published work.
Figure 4.
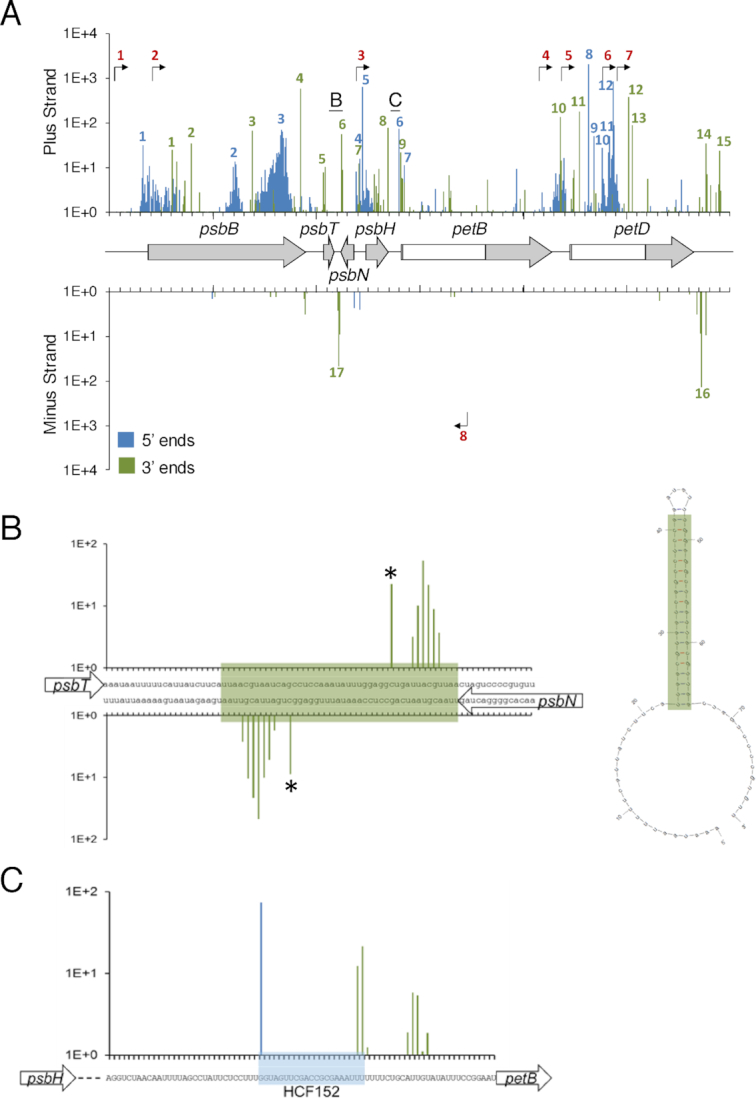
Transcript termini of the psbB operon highlighting the role of a secondary structure and RNA binding protein in defining ends (A) Terminome-seq coverage of the psbB operon with the corresponding gene models, with exons in gray and introns in white. −TAP 5′ ends are in blue and 3′ ends are in green; bent arrows represent TSS inferred from +TAP data. Underlined letters mark the locations that are expanded in panels B and C; numbered peaks and promoters refer to features listed in Table 1. (B) 3′ end coverage for a stem-loop structure between psbT and psbN. The stem is highlighted in green in the nucleotide sequence and the Mfold (119) predicted secondary structure is at right. Asterisks highlight the previously described ends (30). (C) The gene model, nucleotide sequence and end coverage for the HCF152 binding site. Reads accumulate at both the 5′ and 3′ ends of the binding site on the plus strand, indicative of a protected RNA fragment. The color code is the same as in panel A.
Table 1.
Description of transcript ends originating from the psbB operon
| End number | Genome position | Notes |
|---|---|---|
| TSS, +TAP 5′ ends | ||
| 1 | 72 200 | PpsbB -171, described in (57). Seen in barley |
| 2 | 72 409 | Internal to psbB. Distal promoter for psbT? |
| 3 | 74 393 | PpsbH -92, described in (60) |
| 4 | 76 153 | Internal to petB exon 2. Distal promoter for petD? |
| 5 | 76 375–76 376 | Upstream petD |
| 6 | 76 391 | Internal to petD intron |
| 7 | 76 780 | Internal to petD intron |
| 8 | 75 482 | Antisense to petB intron. Distal promoter for psbN ? |
| Processed 5′ ends, −TAP 5′ends | ||
| 1 | 72 320 | psbB -51 mature end, described in (57) |
| 2 | 73 211 | Internal to psbB. Highest peak from a region with multiple 5′ends. Degradation intermediate? |
| 3 | 73 658 | Internal to psbB. Highest peak from a region with multiple 5′ends. Degradation intermediate? |
| 4 | 74 418 | psbT-psbH intergenic region, psbH -67 mature end, described as a precise endoribonuclease cleavage in (60). See also 3′ end #7 |
| 5 | 74 441 | psbT-psbH intergenic region, psbH -44 mature end. Main psbH 5′ end, processing depends on HCF107, described in (58,59) |
| 6 | 74 794 | psbH-petB intergenic region, petB -47 mature end. Processing depends on HCF152, described in (61,62). See 3′ end #9 |
| 7 | 74 847 | first nucleotide of petB intron. Sign of a hydrolytic splicing? |
| 8 | 76 627 | internal to petD intron. Degradation intermediates? |
| 9 | 76 679 | internal to petD intron. Degradation intermediates? |
| 10 | 76 760 | internal to petD intron. Degradation intermediates? |
| 11 | 76 830 | internal to petD intron. Degradation intermediates? |
| 12 | 76 863 | internal to petD intron. Highest peak from a region with multiple 5′ ends. Degradation intermediates? |
| 3′ ends | ||
| 1 | 72 601 | Internal to psbB. Degradation intermediate? |
| 2 | 72 786 | Internal to psbB. Degradation intermediate? |
| 3 | 73 371 | Internal to psbB. Degradation intermediate? Downstream of numerous 5′ends, see 5′end #2, maybe the signature of an endo-ribonuclease cleavage? |
| 4 | 73 838 | Internal to psbB. Degradation intermediate? Downstream of numerous 5′ends, see 5′end #3, maybe the signature of an endo-ribonuclease cleavage? |
| 5 | 74 082 | First nucleotide of psbT, might be the 3′ end of a cDNA identified in (30) |
| 6 | 74 242 | 3′ end of psbT, psbT +60, defined by a stem loop that also defines the 3′ end of the antisense psbN transcript, see 3′ end #17. Described in (30) |
| 7 | 74 405 | psbT-psbH intergenic region, 3′ end of psbT, psbT +223, described as a precise endo-ribonuclease cleavage in (60). See also 5′ end #4 |
| 8 | 74 687 | Internal to psbH, 20 nt upstream of the stop codon. Degradation intermediate? |
| 9 | 74 814 | psbH-petB intergenic region, psbH +109 mature end. Processing depends on HCF152, described in (61,62). See 5′ end #6 |
| 10 | 76 358 | petB-petD intergenic region, petB +67 mature end. Processing depends on CRP1, described in (63) |
| 11 | 76 543 | internal to petD intron. Degradation intermediates? |
| 12 | 77 014 | internal to petD intron. Degradation intermediates? 5 nt downstream of a smRNA footprint. |
| 13 | 77 047 | internal to petD intron. Degradation intermediates? |
| 14 | 77 765 | 3′ end of petD, petD +94, defined by a stem loop that also defines the 3′end of the antisense rpoA transcript, see 3′ end #16. Processing requires mTERF6, described in (64) |
| 15 | 77 892 | 3′ end of petD, petD +221. |
| 16 | 77 716 | 3′ end of rpoA, rpoA +185, defined by a stem loop that also defines the 3′end of the antisense petD transcript, see 3′ end #14. Processing requires mTERF6, described by (64) |
| 17 | 74 211 | 3′ end of psbN, psbN +39, defined by a stem loop that also defines the 3′ end of the antisense psbT transcript, see 3′ end #6. Described in (30) |
We conducted a similar analysis for the atpI-atpH-atpF-atpA and ndhH-ndhA-ndhI-ndhG-ndhE-psaC-ndhD gene clusters (Supplementary Figure S4 and Table S4). Five TSS were identified in the atpI cluster, including what might be specific promoters for atpH and atpA, and 15 TSS in the ndhH cluster, which were located predominantly toward the distal end. Most of these 3′ and 5′ termini were previously identified in a thorough investigation of the atpI cluster processing (52). The existence of an accumulating monocistronic atpI transcript in Arabidopsis has been debated (52,65) and our data suggest that the main atpI 3′ end is 584 nt downstream of the stop codon, inside atpH. This 3′ end is more abundant than the one at position +493, whose processing depends on PPR10 (52,66). Twenty seven termini in addition to TSS were found for the ndhH cluster. No detailed information was previously published on the transcript population, however it is quite complex based on gel blot analysis (67), in keeping with our results.
The roles of RNA secondary structures and RNA-binding proteins in shaping the terminome
Chloroplast RNA termini are known to be stabilized by stem-loop structures, as well as by sequence-specific and general RBPs (6,7,68). Both of these mechanisms act on transcripts from the psbB operon, for which termini were described above. While the strategy used here to make the terminome libraries would be biased against smRNAs, the processing sites from longer, precursor RNAs could be detected. The psbT-psbN intergenic region, for example, forms a stem-loop that defines the 3′ ends of transcripts encoded on opposite strands (30). Terminome-seq confirmed the previously identified 3′ ends inside the stem and additionally showed staggered 3′ ends closer to the base of the stem, an expected phenomenon given the tendency of exoribonucleases to stall at such positions (Figure 4B). Another stem-loop, originally described as a ‘twin terminator’ in spinach (69), defines the 3′ ends of the petD and rpoA transcripts encoded on opposite strands, and a similar structure exists for petA and psbJ (Supplementary Figure S5). The co-existence of two sets of 3′ ends associated with the psbT - psbN stem-loop is also true for the transcripts of rbcL (ends in positions 56 485 and 56 488; see below) and psbA (ends in positions 293 and 285; Supplementary Figure S5), probably reflecting breathing in AU-rich regions of the secondary structures.
The psbH-petB intergenic region is known to be bound by HCF152 (61,62), a PPR protein that defines the psbH 3′ and petB 5′ ends (70). In agreement with these results, Terminome-seq data analysis identified a single major 5′ end for petB correlating with the 5′ end of a smRNA established as the footprint protected by HCF152 binding (9,66,71). The Terminome-seq psbH 3′ end also correlates with HCF152 binding, with an additional cluster of 3′ ends found ∼10 nt downstream, possibly reflecting the different stalling characteristics of the exoribonucleases PNPase and RNase II (Figure 4C).
The correlation between RBPs, their footprints found in smRNAs and Terminome-seq ends can be extended beyond HCF152 to at least 13 RBPs that have been described as involved in RNA maturation (Supplementary Figure S6). These include PPR10, which protects the two adjacent transcripts atpI and atpH and assists in the processing of both their 5′ and 3′ ends (66,70,72); and HCF107 and CRP1 which, like HCF152, target the psbB operon, in particular the psbH -44 5′ end and the petB +67 3′ end. We can further generalize this phenomenon, since, on a plastome-wide level, termini are enriched in areas containing smRNAs, which are in many cases likely to be marks of RBPs that still await identification (Supplementary Table S5).
PNPase deficiency has broad impacts on both 5′ and 3′ termini
The roles of PNPase in chloroplast transcript 3′ maturation, intron degradation, and tRNA processing, as well as an ancillary role in phosphorus metabolism, have been well documented (22,33,34,73–75). Although RNA-seq shows that most chloroplast RNAs contain 3′ extensions in the pnp1–1 null mutant (22), the individual termini were not systematically compared between WT and mutant. Such a comparison could reveal more precisely the types of sequences and structures that impede PNPase activity, and highlight its overall impact on the plastid terminome. We proceeded to analyze duplicate pnp1–1 Terminome-seq libraries, and decided against performing TAP treatment to capture primary 5′ ends because PNPase is not known or expected to affect transcription initiation.
A plastome-wide view of pnp1–1 termini is presented in Figure 5A. While the WT and pnp1–1 patterns show substantial overlap, there are numerous areas where reads are specific to pnp1–1. This is quantified as genome coverage of reads (Figure 5B) where, regardless of the RPM threshold applied, coverage is higher in pnp1–1 than in WT. In aggregate, 5′ end coverage increases from 12.7 to 26.1%, and 3′ coverage from 26.1 to 39.5%. When a threshold of >10 RPM is applied to remove low abundance termini, coverage is ∼1% in the mutant, with 2420 5′ termini and 2744 3′ termini exceeding the threshold. This, total of 5164 >10 RPM termini in pnp1–1 represents an increase of 76% over the combined 2927 termini found in the WT. The locations of termini are also dramatically altered between genotypes (compare Figures 2B and 5C). Another noticeable difference is the decline in the proportion of termini found in rRNA, which can be rationalized as a relative increase in non-rRNA termini in the mutant. In the −TAP 5′ termini population, the mutant differs by having an increase in intronic ends and a decrease in tRNA ends, the latter of which holds true for 3′ termini as well.
Figure 5.

Distribution, coverage and location of transcript termini in pnp1–1 (A) Plastome-scale view of end coverages from the average of two Col-0 and pnp1–1 biological replicates in RPM. 5′ ends obtained without TAP treatment are blue (WT) and pink (pnp1–1), and 3′ ends are displayed in green (WT) and orange (pnp1–1). Gene models are indicated between the tracks corresponding to the plus and minus strands of the plastome. One copy of the large inverted repeat is omitted for clarity. Tick marks are every 1000 nt. (B) Comparison of Terminome-seq coverage for WT and pnp1–1. While 12.7 and 15.8% of the WT plastome is represented by 5′ and 3′ ends, respectively, these numbers increase to 26.1 and 39.5%, respectively, in pnp1–1. Termini coverage at >10 RPM (0.94 and 1.07% for 5′ and 3′ ends, respectively, in pnp1–1) is marked and discussed in the text. (C) Terminome-seq read distribution in pnp1–1. The results are the average of two biological replicates. as-exon refers to reads mapping to the antisense strand of known coding regions.
A more pronounced effect on 3′ versus 5′ ends in pnp1–1 can be gleaned from Figure 6A, which shows that 349 5′ termini and 1348 3′ termini are at least 10 times more abundant in the mutant compared to the WT, whereas only 96 and 150, respectively, decrease at least 10-fold in the mutant. The tendency of termini to accumulate in the mutant is in keeping with the degradative function of PNPase, with the expected bias toward 3′ termini. The strong effect of PNPase on the 5′ terminome was unexpected and somewhat counterintuitive, given that it is a 3′→5′ exonuclease. We have previously shown, however, that PNPase degrades tRNA 5′ leader sequences following their liberation by RNase P cleavage (22), and Terminome-seq-based evidence of this phenomenon is shown in Supplementary Figure S7. We speculate that other 5′ termini that hyperaccumulate in pnp1–1 represent the upstream termini of other endonucleolytic cleavage products that are usually removed by the 3′→5′ activity of PNPase.
Figure 6.

Terminome-seq coverage in pnp1–1 (A) The RPM abundance of −TAP 5′ ends and 3′ ends was compared between WT and pnp1–1 at a genomic level. The dashed lines separate ends that are at least 10-fold more abundant in a given genotype. For example, 349 5′ ends are more abundant in the PNPase mutant. (B) Terminome-seq coverage upstream of the rbcL gene. Color coding of ends is provided in an inset. Genome position 54 958 is the rbcL coding region 5′ end according to the TAIR10 annotation. The rbcL processed 5′ end (position 54 889) correlates with the 5′ end of the smRNA footprint of MRL1 (highlighted in blue). (C) Terminome-seq coverage downstream of the rbcL gene, with labeling as in Panel B. Genome position 56 397 is the 3′ end of the coding region. The stem-loop downstream of the gene (positions 56 437–56 488) is highlighted in green and matches a smRNA (66). Other genome positions discussed in the text are also labeled.
To illustrate the utility of Terminome-seq for characterizing a ribonuclease mutant at the individual gene level, results are shown for the monocistronic rbcL transcript (Figure 6B and C), which in plants accumulates as two species with primary and processed 5′ ends. The processed 5′ end is protected by the PPR protein MRL1, which prevents degradation by RNase J (76,77). MRL1 leaves a smRNA footprint (Figure 6B, blue shading), whose 5′ end is represented as similarly abundant termini in WT and pnp1–1 at genome position 54 889. There is, however, a cluster of 3′ ends located ∼40 nt downstream of the MRL1 binding site (with a peak at position 54 956) that is present only in the mutant, even though there is a less abundant 3′ end slightly downstream (position 54 989) in both genetic backgrounds. This indicates that PNPase likely degrades rbcL mRNA until it is stalled by MRL1 and/or nearby RNA secondary structures, as such termini are absent in the WT. The 3′ end at position 54 901 may be an RNase II stall site, given the known cooperation of RNase II and PNPase in 3′→5′ RNA decay (78). A comparison of WT and pnp1–1 ends in the vicinity of all described RBP sites is provided in Supplementary Figure S6.
At its 3′ end, the rbcL transcript is defined by a highly conserved stem-loop (79,80) represented as a smRNA (66,71). RNA-seq showed higher coverage in pnp1–1 compared to the WT beginning about 10 nt downstream of this structure (22). Using Terminome-seq results, we identified three different 3′ ends in WT plants, two of them directly at the 3′ base of the stem (positions 56 485 and 56 488) and one 36 nt downstream (position 56 524; Figure 6C). The 3′ end at position 56 485 could also be identified in pnp1–1, although it was less abundant than in WT, suggesting that its production is not fully dependent on PNPase. On the contrary, ends at positions 56 488 and 56 524 are certainly produced through the action of PNPase because they are missing in the mutant. The most abundant 3′ ends in the mutant cluster around position 56 608, 120 nt downstream of the stem-loop (Figure 6C), accounting for the 3′ extension previously noted in RNA gel blots, which represents the stall point of RNase II (78). Terminome-seq evidence for several other putative 3′ extensions is presented in Supplementary Figure S8.
At last, we present an overview of rRNA operon ends for the WT and pnp1–1 (Supplementary Figure S9). This analysis revealed termini corresponding to known processing sites (6) for all four rRNAs, and also showed clear peaks in pnp1–1 for the well-known 23S rRNA extension in this mutant (73). Of note, the first hidden break of the 23S rRNA was not clearly delineated compared to the second 23S rRNA hidden break, in keeping with previously mapped ends in this region (19). Surprisingly, in the 5′ part of 16S rRNA there were numerous, staggered 5′ termini indicative of a processing or degradation event, albeit at 1–2 logs lower abundance than the mature 5′ and 3′ ends. RNA processing at this position, however, has not been previously described and further analysis of this region is needed to evaluate the origins of these termini.
DISCUSSION
The chloroplast TSS landscape
In this work, we systematically sequenced Arabidopsis thaliana chloroplast RNA 3′ termini as well as primary and processed 5′ ends, using TAP treatment to discriminate between them. Such strategies are under continual improvement, as transcriptome analyses generally endeavor to be as comprehensive and quantitative as possible. In our case, transcript chemistry, size and secondary structure will all introduce biases to the dataset ultimately used to draw the main conclusions. In addition, factors such as ligation bias—the preferential ligation of adapters to certain sequences (81,82), undoubtedly impacted the selection and ratios of the transcript ends we have reported. Therefore, our conclusions—particularly quantitative ones – should be evaluated with those caveats in mind.
The strategy as implemented led to the description of 215 TSS meeting defined expression thresholds, which are widely distributed in the plastome (Figure 1) and is consistent in number with the 176 TSS mapped in mature barley leaves (20). These TSS are created by two RNA polymerase types, a bacterial-like, plastid-encoded RNA polymerase (PEP) and two phage-like, nucleus-encoded, RNA polymerases (NEP). NEP and PEP operate simultaneously, however our samples were taken from tissue populated by mature chloroplasts, where PEP activity predominates (83). Since NEP transcription primarily occurs during early differentiation, the TSS we describe likely underestimate the number of promoters utilized over the course of chloroplast differentiation. In barley, the use of the albostrians mutant with sectors lacking PEP allowed the discovery of 254 additional NEP-dependent TSS, giving a total of 398 unique TSS when overlap between PEP and NEP TSS was considered. At the same time, NEP is known to become promiscuous when PEP has been eliminated genetically (1,84). The proportion of NEP promoters active under these circumstances that are also utilized in WT plants remains to be determined.
If we assume a total of ∼400 TSS in Arabidopsis when taking PEP and ‘developmentally hidden’ NEP promoters into account, this would average to a TSS every ∼600 nt when both strands, but only one of the large inverted repeats, are used as a basis. This frequency is not surprising, and such phenomenon occurs in bacteria as well (85,86). Given the AT-rich content of the plastid genome, functional PEP promoter −10 elements are likely to be present by chance, along with the short and highly variable elements that seem to constitute NEP promoters. The average 600 nt spacing we calculate would be reduced if we changed the 10-fold ratio threshold used to define TAP versus non-TAP-dependent transcripts. Some known TSS such as PatpE -430, PpsbN -32 and PpsbD -948 (87), have a ±TAP ratio <10 and were therefore not formally considered to be TSS (Supplementary Figure S3B). Perhaps the biggest surprise from this Terminome-seq dataset is the evidence for massive transcription initiation activity in the 3′ part of the ndhF gene, antisense to ndhF and within the first intron of clpP. The former two areas represent a staggering ∼20% of the reads derived from primary transcripts, and could have functional implications in the chloroplast.
Do some TSS mark transcripts with novel coding potential?
Chloroplasts are known to contain plastid non-coding RNAs (pncRNAs), with >100 in Arabidopsis (88), whose functions remain mostly unclear. Although many pncRNAs appear to be generated by read-through from adjacent genes, some are known to be primary transcripts (20,23), and the high number of TSS identified here potentially increases the count of pncRNA promoters. For example, the internal ndhF TSS (Supplementary Figure S3A) probably initiates a 300–600 nt RNA that ends at the ndhF termini 110 331 and 109 924, the latter position being protected by an RBP or stem-loop since a smRNA from this position was identified (66). This pncRNA overlaps ycf1 on the antisense strand and was previously thought to originate from ndhF readthrough (23). Initiation antisense to ndhF was also described in tobacco (89) and more recently in barley (20), and is likely responsible for additional Arabidopsis pncRNAs (nc89 and nc90) which were validated by RT-PCR and gel blots (23). The tobacco ndhF antisense TSS was proposed to be a proximal TSS for the downstream rpl32 gene (89), which might also be the case in Arabidopsis.
Many pncRNAs are antisense to coding sequences, and for a few there is evidence they exert a regulatory function at the RNA level (19,29,60). On the other hand, many pncRNAs contain small open reading frames (ORFs) and could therefore encode unknown chloroplast proteins. Such ORFs were discarded from the chloroplast genome sequences first obtained from tobacco and Marchantia (90,91), specifically ORFs shorter than 70 nt unless the products were known. In contrast, the functions of the longer, conserved, hypothetical coding frames (ycfs) were still discussed. Whether pncRNA-encoded ORFs are represented in the proteome remains to be determined, however data from bacterial and animal systems suggests that at least some antisense or intergenic RNAs harbor hidden genetic functions (92–94). In this case, the retention of TSS would be expected.
Among the better-studied YCFs, our data shows that ycf15 (also annotated as ORF77) contains its own TSS (position 93 369, Supplementary Table S3) and shares an abundant 3′ end with the upstream ycf2 transcript at position 93 750, probably defined by an RBP (Supplementary Table S5). ycf15 is a short ORF downstream of ycf2 whose functionality as a protein-coding gene has been debated (95–98); however the presence of discrete 5′ and 3′ ends would be consistent with functionality.
Terminome-seq corroborates the production of smRNA footprints from post-transcriptional processing
Despite their high number, TSS only account for a small fraction of 5′ end diversity. Instead, most 5′ and 3′ ends are the result of a maturation and winnowing process where RBPs and secondary structures selectively protect RNAs from degradation by RNases with low sequence specificity (6,8). Many of these RBPs are members of plant-specific or plant-amplified helical repeat protein families (99), most prominently the PPR family (100). The correlation between smRNA footprints and transcript termini is a key argument which posits that target sequences and secondary structures largely exist to define the ends of functional transcripts (66,71,101). It follows that the presence of termini correlating with an smRNA represents an effective way to distinguish true footprints from other smRNAs that are more scattered or whose termini are much more ragged.
A good example of using Terminome-seq to distinguish smRNA footprints is the psbB transcriptional unit (Figure 4), which gives rise to eight accumulating smRNAs (66). These include footprints for three RBPs: HCF107, HCF152 and CRP1; two derived from stem-loops downstream of psbT/psbN and petD, and three smRNAs complementary to the petD intron. All but the three intron antisense smRNAs, which may be too unstable to be found in the longer transcripts we sequenced, correlate with termini (Table 1). Another example is the recently described PPR10-mediated protection of the maize psaI 3′ end (72), which correlates with a smRNA from an analogous position in Arabidopsis (genomic position 59 475, see Supplementary Figure S6 and Table S5). Across the transcriptome, we were able to identify (using the 10 RPM threshold) 45 5′ ends and 44 3′ ends which correlate with the termini of 81 smRNAs (8 footprints correlate with both 5′ and 3′ ends, 37 with 5′ ends only and 36 with 3′ ends only), accounting for 33.5% of the 242 smRNAs previously described (66,71). Because of the way in which our libraries were made, these ends belong to the longer transcripts from which the smRNAs were ultimately derived. The ability to look for bona fide footprints, along with the elucidation of the PPR code explaining their binding specificity (102–105), will allow the prediction and discovery of new PPR proteins involved in chloroplast RNA stability. Not all of the hundreds of chloroplast PPRs generate accumulating footprints, as demonstrated by PGR3, which is required for rpl14 3′ end formation (72).
According to the footprint model, a single RBP can protect the 5′ and 3′ ends of overlapping transcripts. The original example of this phenomenon is maize PPR10, which in binding the atpI-atpH intergenic region protects the atpI 3′ and atpH 5′ ends (70,106). Although Terminome-seq could identify the expected atpH 5′ end, only a minor 3′ end mapped to the PPR10-dependent atpI 3′ end (Supplementary Figure S6). The major Terminome-seq 3′ end is located further downstream, inside the atpH coding region and does not correlate with a smRNA. Systematic mapping of the atp operon termini already revealed that transcripts with overlapping ends are a minority (52) and co-immunoprecipitation showed that PPR10 is preferentially associated with processed atpH mRNA rather than processed atpI mRNA (66). At a genome-wide level, only ∼10 footprints (including PPR38 and HCF152) can be linked to simultaneous stabilization of 5′ and 3′ ends, suggesting that this is the exception rather than the rule. Secondary structures, on the other hand, can stabilize the 3′ ends of two adjacent transcripts, for example in the cases of psbT-psbN, petD-rpoA and petA-psbJ (Figure 4B and Supplementary Figure S5). Currently, the degradation pathway of RBP footprints is unknown. Evidence derived from Terminome-seq is consistent with PNPase participating in this mechanism, as there are more termini correlating with smRNAs in the PNPase mutant than in WT (42.5% versus 33.5% of all termini; Supplementary Table S5), however a direct analysis of smRNAs in both genotypes would be required to substantiate this possibility.
Terminome-seq of the PNPase mutant points to its potential use in reverse genetics
mRNA processing up to the border of the region protected by RBPs or secondary structures is principally performed by three enzymes, RNase J for 5′ ends and PNPase and RNase II for 3′ ends (76,78,106). As expected, Terminome-seq of the PNPase mutant showed a more pronounced effect on 3′ versus 5′ termini, although many termini of both types were affected (Figures 5 and Figure 6A). We could confirm that PNPase is required to remove tRNA precursor 5′ extensions subsequent to RNase P processing (Supplementary Figures S7,(22)). Similarly, we confirmed that the 3′ extensions observed in the PNPase mutant (22,34,73) correlate with 3′ ends distal to secondary structures or RBP binding sites (Supplementary Figures S5, 6 and 8), two elements known to stall PNPase (106,107).
Using pnp1–1 as a proof of concept opens the door to understanding the roles of other chloroplast gene regulators using Terminome-seq. Concerning 5′ termini, this might include the roles of the sigma factors that specify PEP initiation sites (108) and other PEP-associated proteins (PAPs; 109), a closer view at RNase J specificity, or a better understanding of some of the numerous factors that have been implicated in rRNA maturation (6). Terminome-seq would be an excellent choice to gain additional insight into poorly-characterized RNases whose targets have only begun to be described (110–112).
Transcription termination analysis with Terminome-seq
Transcription termination in plastids has recently been reviewed (113). The endosymbiont hypothesis would predict that chloroplast termination would resemble that of bacteria, which use both Rho-dependent and Rho-independent mechanisms. Rho-independent termination occurs in AT-rich sequences downstream of GC-rich stem-loops, however in vitro and in vivo assays found that PEP terminates inefficiently at chloroplast stem-loops, although it does recognize certain bacterial terminators. This is in keeping with the longstanding hypothesis that chloroplast stem-loops are involved in transcript stability rather than transcription termination (3), a finding well illustrated by the correlation between 3′ ends and secondary structures (Figure 4B and Supplementary Figure S5). On the other hand, Rho-dependent termination, which works through destabilization of the elongating polymerase (114), would have to rely on an alternative cofactor since Rho is not found in organelles. Such a factor was recently identified by its partial similarity to Rho, RHON1, which assists in transcription termination downstream of rbcL (115), binding the transcript between two termini identified here (Figure 6C). The 3′ end in the PNPase mutant is further downstream, suggesting a model similar to Escherichia coli where Rho factors initiate termination and exoribonucleases like PNPase produce the precise, mature 3′ ends directly downstream of stem-loops or RBP sites (116). Similarly, MTERF6, a member of a gene family related to a human mitochondrial transcription termination factor, acts in the trnI and petD-rpoA regions (64,113,117). As additional factors involved in termination are discovered, Terminome-seq offers a tool to decipher their sites of action.
CONCLUSION
Several high-throughput RNA-seq-based strategies have recently been used to gain an unprecedented genome-level understanding of chloroplast RNA biology. It is now possible to study RNA abundance, splicing and editing (26–28), to monitor translation rates with ribosome profiling (24,118) and to infer protein binding sites from smRNA sequencing (66,71,101). Terminome-seq now creates access to a single-nucleotide resolution map of the full set of chloroplast RNA termini. We anticipate that this strategy, combined with the other plastome-wide approaches, will be instrumental in deciphering mechanisms such as transcription (from initiation to termination), as well as the roles of RNases and RBPs in shaping the chloroplast transcriptome.
DATA AVAILABILITY
Raw sequences have been deposited on the SRA database with the number PRJNA533962 and can be accessed here https://www.ncbi.nlm.nih.gov/sra/PRJNA533962.
Supplementary Material
Notes
Present address: Arnaud Germain, Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, of the U.S. Department of Energy [DE-FG02–10ER20015, in part]; LabEx Saclay Plant Sciences-SPS [ANR-10-LABX-0040-SPS to B.C.]. Funding for open access charge: Internal Funds (to D.B.S).
Conflict of interest statement. None declared.
REFERENCES
- 1. Legen J., Kemp S., Krause K., Profanter B., Herrmann R.G., Maier R.M.. Comparative analysis of plastid transcription profiles of entire plastid chromosomes from tobacco attributed to wild-type and PEP-deficient transcription machineries. Plant J. 2002; 31:171–188. [DOI] [PubMed] [Google Scholar]
- 2. Sanitá Lima M., Smith D.R.. Pervasive, genome-wide transcription in the organelle genomes of diverse plastid-bearing protists. G3. 2017; 7:3789–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stern D.B., Gruissem W.. Control of plastid gene expression: 3′ inverted repeats act as mRNA processing and stabilizing elements, but do not terminate transcription. Cell. 1987; 51:1145–1157. [DOI] [PubMed] [Google Scholar]
- 4. Mullet J.E., Klein R.R.. Transcription and RNA stability are important determinants of higher plant chloroplast RNA levels. EMBO J. 1987; 6:1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barkan A. Expression of plastid genes: organelle-specific elaborations on a prokaryotic scaffold. Plant Physiol. 2011; 155:1520–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Germain A., Hotto A.M., Barkan A., Stern D.B.. RNA processing and decay in plastids. Wiley Interdiscip. Rev. RNA. 2013; 4:295–316. [DOI] [PubMed] [Google Scholar]
- 7. Stern D.B., Goldschmidt-Clermont M., Hanson M.R.. Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 2010; 61:125–155. [DOI] [PubMed] [Google Scholar]
- 8. Stoppel R., Meurer J.. The cutting crew - ribonucleases are key players in the control of plastid gene expression. J. Exp. Bot. 2012; 63:1663–1673. [DOI] [PubMed] [Google Scholar]
- 9. Manavski N., Schmid L.-M., Meurer J.. RNA-stabilization factors in chloroplasts of vascular plants. Essays Biochem. 2018; 62:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stoppel R., Meurer J.. Complex RNA metabolism in the chloroplast: an update on the psbB operon. Planta. 2013; 237:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang J., Schuster G., Stern D.B.. CSP41, a sequence-specific chloroplast mRNA binding protein, is an endoribonuclease. Plant Cell. 1996; 8:1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beligni M.V., Mayfield S.P.. Arabidopsis thaliana mutants reveal a role for CSP41a and CSP41b, two ribosome-associated endonucleases, in chloroplast ribosomal RNA metabolism. Plant Mol. Biol. 2008; 67:389–401. [DOI] [PubMed] [Google Scholar]
- 13. Bollenbach T.J., Sharwood R.E., Gutierrez R., Lerbs-Mache S., Stern D.B.. The RNA-binding proteins CSP41a and CSP41b may regulate transcription and translation of chloroplast-encoded RNAs in Arabidopsis. Plant Mol. Biol. 2009; 69:541–552. [DOI] [PubMed] [Google Scholar]
- 14. Bollenbach T.J., Tatman D.A., Stern D.B.. CSP41a, a multifunctional RNA-binding protein, initiates mRNA turnover in tobacco chloroplasts. Plant J. 2003; 36:842–852. [DOI] [PubMed] [Google Scholar]
- 15. Qi Y., Armbruster U., Schmitz-Linneweber C., Delannoy E., de Longevialle A.F., Rühle T., Small I., Jahns P., Leister D.. Arabidopsis CSP41 proteins form multimeric complexes that bind and stabilize distinct plastid transcripts. J. Exp. Bot. 2012; 63:1251–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stoppel R., Manavski N., Schein A., Schuster G., Teubner M., Schmitz-Linneweber C., Meurer J.. RHON1 is a novel ribonucleic acid-binding protein that supports RNase E function in the Arabidopsis chloroplast. Nucleic Acids Res. 2012; 40:8593–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walter M., Piepenburg K., Schöttler M.A., Petersen K., Kahlau S., Tiller N., Drechsel O., Weingartner M., Kudla J., Bock R.. Knockout of the plastid RNase E leads to defective RNA processing and chloroplast ribosome deficiency. Plant J. 2010; 64:851–863. [DOI] [PubMed] [Google Scholar]
- 18. Sharwood R.E., Halpert M., Luro S., Schuster G., Stern D.B.. Chloroplast RNase J compensates for inefficient transcription termination by removal of antisense RNA. RNA. 2011; 17:2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hotto A.M., Castandet B., Gilet L., Higdon A., Condon C., Stern D.B.. Arabidopsis chloroplast Mini-Ribonuclease III participates in rRNA maturation and intron recycling. Plant Cell. 2015; 27:724–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhelyazkova P., Sharma C.M., Forstner K.U., Liere K., Vogel J., Börner T.. The primary transcriptome of barley chloroplasts: numerous noncoding RNAs and the dominating role of the plastid-encoded RNA polymerase. Plant Cell. 2012; 24:123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruwe H., Castandet B., Schmitz-Linneweber C., Stern D.B.. Arabidopsis chloroplast quantitative editotype. FEBS Lett. 2013; 587:1429–1433. [DOI] [PubMed] [Google Scholar]
- 22. Castandet B., Hotto A.M., Fei Z., Stern D.B.. Strand-specific RNA sequencing uncovers chloroplast ribonuclease functions. FEBS Lett. 2013; 587:3096–3101. [DOI] [PubMed] [Google Scholar]
- 23. Hotto A.M., Schmitz R.J., Fei Z., Ecker J.R., Stern D.B.. Unexpected diversity of chloroplast noncoding RNAs as revealed by deep sequencing of the Arabidopsis transcriptome. G3. 2011; 1:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chotewutmontri P., Barkan A.. Dynamics of chloroplast translation during chloroplast differentiation in maize. PLoS Genet. 2016; 12:e1006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guillaumot D., Lopez-Obando M., Baudry K., Avon A., Rigaill G., Falcon de Longevialle A., Broche B., Takenaka M., Berthomé R., De Jaeger G. et al.. Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:8877–8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michel E.J.S., Hotto A.M., Strickler S.R., Stern D.B., Castandet B.. A guide to the chloroplast transcriptome analysis using RNA-seq. Methods Mol Biol. 2018; 1829:295–313. [DOI] [PubMed] [Google Scholar]
- 27. Castandet B., Hotto A.M., Strickler S.R., Stern D.B.. ChloroSeq, an optimized chloroplast rna-seq bioinformatic pipeline, reveals remodeling of the organellar transcriptome under heat stress. G3. 2016; 6:2817–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malbert B., Rigaill G., Brunaud V., Lurin C., Delannoy E.. Bioinformatic analysis of chloroplast gene expression and RNA posttranscriptional maturations using RNA sequencing. Methods Mol. Biol. 2018; 1829:279–294. [DOI] [PubMed] [Google Scholar]
- 29. Hotto A.M., Huston Z.E., Stern D.B.. Overexpression of a natural chloroplast-encoded antisense RNA in tobacco destabilizes 5S rRNA and retards plant growth. BMC Plant Biol. 2010; 10:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zghidi-Abouzid O., Merendino L., Buhr F., Malik Ghulam M., Lerbs-Mache S.. Characterization of plastid psbT sense and antisense RNAs. Nucleic Acids Res. 2011; 39:5379–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hübschmann T., Börner T.. Characterisation of transcript initiation sites in ribosome-deficient barley plastids. Plant Mol. Biol. 1998; 36:493–496. [DOI] [PubMed] [Google Scholar]
- 32. Hess W.R., Prombona A., Fieder B., Subramanian A.R., Börner T.. Chloroplast rps15 and the rpoB/C1/C2 gene cluster are strongly transcribed in ribosome-deficient plastids: evidence for a functioning non-chloroplast-encoded RNA polymerase. EMBO J. 1993; 12:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marchive C., Yehudai-Resheff S., Germain A., Fei Z., Jiang X., Judkins J., Wu H., Fernie A.R., Fait A., Stern D.B.. Abnormal physiological and molecular mutant phenotypes link chloroplast polynucleotide phosphorylase to the phosphorus deprivation response in Arabidopsis. Plant Physiol. 2009; 151:905–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Germain A., Herlich S., Larom S., Kim S.H., Schuster G., Stern D.B.. Mutational analysis of Arabidopsis chloroplast polynucleotide phosphorylase reveals roles for both RNase PH core domains in polyadenylation, RNA 3′-end maturation and intron degradation. Plant J. 2011; 67:381–394. [DOI] [PubMed] [Google Scholar]
- 35. Steglich C., Futschik M.E., Lindell D., Voss B., Chisholm S.W., Hess W.R.. The challenge of regulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet. 2008; 4:e1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Olivarius S., Plessy C., Carninci P.. High-throughput verification of transcriptional starting sites by Deep-RACE. Biotechniques. 2009; 46:130–132. [DOI] [PubMed] [Google Scholar]
- 37. Denise H., Moschos S.A., Sidders B., Burden F., Perkins H., Carter N., Stroud T., Kennedy M., Fancy S.-A., Lapthorn C. et al.. Deep Sequencing Insights in therapeutic shRNA processing and siRNA target cleavage precision. Mol. Ther. Nucleic Acids. 2014; 3:e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoff A.M., Johannessen B., Alagaratnam S., Zhao S., Nome T., Løvf M., Bakken A.C., Hektoen M., Sveen A., Lothe R.A. et al.. Novel RNA variants in colorectal cancers. Oncotarget. 2015; 6:36587–36602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lagarde J., Uszczynska-Ratajczak B., Santoyo-Lopez J., Gonzalez J.M., Tapanari E., Mudge J.M., Steward C.A., Wilming L., Tanzer A., Howald C. et al.. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat. Commun. 2016; 7:12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park D., Morris A.R., Battenhouse A., Iyer V.R.. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 2014; 42:3736–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rorbach J., Bobrowicz A., Pearce S., Minczuk M.. Rorbach J, Bobrowicz JA. Polyadenylation in bacteria and organelles. Polyadenylation: Methods and Protocols. 2014; Totowa, NJ: Humana Press; 211–227. [DOI] [PubMed] [Google Scholar]
- 42. Slomovic S., Schuster G.. Exonucleases and endonucleases involved in polyadenylation-assisted RNA decay. Wiley Interdiscip. Rev. RNA. 2011; 2:106–123. [DOI] [PubMed] [Google Scholar]
- 43. Sato S., Nakamura Y., Kaneko T., Asamizu E., Tabata S.. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 1999; 6:283–290. [DOI] [PubMed] [Google Scholar]
- 44. Swiatecka-Hagenbruch M., Liere K., Börner T.. High diversity of plastidial promoters in Arabidopsis thaliana. Mol. Genet. Genomics. 2007; 277:725–734. [DOI] [PubMed] [Google Scholar]
- 45. Sugita M., Sugiura M.. Regulation of gene expression in chloroplasts of higher plants. Plant Mol. Biol. 1996; 32:315–326. [DOI] [PubMed] [Google Scholar]
- 46. Shahar N., Weiner I., Stotsky L., Tuller T., Yacoby I.. Prediction and large-scale analysis of primary operons in plastids reveals unique genetic features in the evolution of chloroplasts. Nucleic Acids Res. 2019; 47:3344–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cavaiuolo M., Kuras R., Wollman F., Choquet Y., Vallon O.. Small RNA profiling in Chlamydomonas: insights into chloroplast RNA metabolism. Nucleic Acids Res. 2017; 45:10783–10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christopher D.A., Kim M., Mullet J.E.. A novel light-regulated promoter is conserved in cereal and dicot chloroplasts. Plant Cell. 1992; 4:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vera A., Hirose T., Sugiura M.. A ribosomal protein gene (rpl32) from tobacco chloroplast DNA is transcribed from alternative promoters: Similarities in promoter region organization in plastid housekeeping genes. Mol. Gen. Genet. 1996; 251:518–525. [DOI] [PubMed] [Google Scholar]
- 50. Zghidi W., Merendino L., Cottet A., Mache R., Lerbs-Mache S.. Nucleus-encoded plastid sigma factor SIG3 transcribes specifically the psbN gene in plastids. Nucleic Acids Res. 2007; 35:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kapoor S., Wakasugi T., Deno H., Sugiura M.. An atpE-specific promoter within the coding region of the atpB gene in tobacco chloroplast DNA. Curr. Genet. 1994; 26:263–268. [DOI] [PubMed] [Google Scholar]
- 52. Ghulam M.M., Courtois F., Lerbs-Mache S., Merendino L., Merendino L.. Complex processing patterns of mRNAs of the large ATP synthase operon in arabidopsis chloroplasts. PLoS One. 2013; 8:e78265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hanaoka M., Kanamaru K., Takahashi H., Tanaka K.. Molecular genetic analysis of chloroplast gene promoters dependent on SIG2, a nucleus-encoded sigma factor for the plastid-encoded RNA polymerase, in Arabidopsis thaliana. Nucleic Acids Res. 2003; 31:7090–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoffer P.H., Christopher D.A.. Structure and blue-light-responsive transcription of a chloroplast psbD promoter from Arabidopsis thaliana. Plant Physiol. 1997; 115:213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nagashima A., Hanaoka M., Shikanai T., Fujiwara M., Kanamaru K., Takahashi H., Tanaka K.. The multiple-stress responsive plastid sigma factor, SIG5, directs activation of the psbD blue light-responsive promoter (BLRP) in Arabidopsis thaliana. Plant Cell Physiol. 2004; 45:357–368. [DOI] [PubMed] [Google Scholar]
- 56. Zoschke R., Watkins K.P., Miranda R.G., Barkan A.. The PPR-SMR protein PPR53 enhances the stability and translation of specific chloroplast RNAs in maize. Plant J. 2016; 85:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Westhoff P., Herrmann R.G.. Complex RNA maturation in chloroplasts: the psbB operon from spinach. Eur. J. Biochem. 1988; 171:551–564. [DOI] [PubMed] [Google Scholar]
- 58. Felder S., Meierhoff K., Sane A.P., Meurer J., Driemel C., Plucken H., Klaff P., Stein B., Bechtold N., Westhoff P.. The nucleus-encoded HCF107 gene of Arabidopsis provides a link between intercistronic RNA processing and the accumulation of translation-competent psbH transcripts in chloroplasts. Plant Cell. 2001; 13:2127–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hammani K., Cook W.B., Barkan A.. RNA binding and RNA remodeling activities of the half-a-tetratricopeptide (HAT) protein HCF107 underlie its effects on gene expression. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:5651–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chevalier F., Ghulam M.M., Rondet D., Pfannschmidt T., Merendino L., Lerbs-Mache S.. Characterization of the psbH precursor RNAs reveals a precise endoribonuclease cleavage site in the psbT/psbH intergenic region that is dependent on psbN gene expression. Plant Mol. Biol. 2015; 88:357–367. [DOI] [PubMed] [Google Scholar]
- 61. Nakamura T., Meierhoff K., Westhoff P., Schuster G.. RNA-binding properties of HCF152, an Arabidopsis PPR protein involved in the processing of chloroplast RNA. Eur. J. Biochem. 2003; 270:4070–4081. [DOI] [PubMed] [Google Scholar]
- 62. Meierhoff K., Felder S., Nakamura T., Bechtold N., Schuster G.. HCF152, an arabidopsis RNA Binding pentatricopeptide repeat protein involved in the processing of chloroplast psbB-psbT-psbH-petB-petD RNAs. Plant Cell. 2003; 15:1480–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ferrari R., Tadini L., Moratti F., Lehniger M.-K., Costa A., Rossi F., Colombo M., Masiero S., Schmitz-Linneweber C., Pesaresi P.. CRP1 Protein: (dis)similarities between Arabidopsis thaliana and Zea mays. Front. Plant Sci. 2017; 8:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Y., Cui Y.L., Zhang X.L., Yu Q.B., Wang X., Yuan X.B., Qin X.M., He X.F., Huang C., Yang Z.N.. A nuclear-encoded protein, mTERF6, mediates transcription termination of rpoA polycistron for plastid-encoded RNA polymerase-dependent chloroplast gene expression and chloroplast development. Sci. Rep. 2018; 8:11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Malik Ghulam M., Zghidi-Abouzid O., Lambert E., Lerbs-Mache S., Merendino L.. Transcriptional organization of the large and the small ATP synthase operons, atpI/H/F/A and atpB/E, in Arabidopsis thaliana chloroplasts. Plant Mol. Biol. 2012; 79:259–272. [DOI] [PubMed] [Google Scholar]
- 66. Ruwe H., Wang G., Gusewski S., Schmitz-Linneweber C.. Systematic analysis of plant mitochondrial and chloroplast small RNAs suggests organelle-specific mRNA stabilization mechanisms. Nucleic Acids Res. 2016; 44:7406–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meurer J., Berger A., Westhoff P.. A nuclear mutant of Arabidopsis with impaired stability on distinct transcripts of the plastid psbB, psbD/C, ndhH, and ndhC operons. Plant Cell. 1996; 8:1193–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shi X., Hanson M.R., Bentolila S.. Functional diversity of Arabidopsis organelle-localized RNA-recognition motif-containing proteins. Wiley Interdiscip. Rev. RNA. 2017; 8:e1420. [DOI] [PubMed] [Google Scholar]
- 69. Sijben-Müller G., Hallick R.B., Alt J., Westhoff P., Herrmann R.G.. Spinach plastid genes coding for initiation factor IF-1, ribosomal protein S11 and RNA polymerase alpha-subunit. Nucleic Acids Res. 1986; 14:1029–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pfalz J., Bayraktar O.A., Prikryl J., Barkan A.. Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J. 2009; 28:2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ruwe H., Schmitz-Linneweber C.. Short non-coding RNA fragments accumulating in chloroplasts: footprints of RNA binding proteins. Nucleic Acids Res. 2012; 40:3106–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rojas M., Ruwe H., Miranda R.G., Zoschke R., Hase N., Schmitz-Linneweber C., Barkan A.. Unexpected functional versatility of the pentatricopeptide repeat proteins PGR3, PPR5 and PPR10. Nucleic Acids Res. 2018; 46:10448–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Walter M., Kilian J., Kudla J.. PNPase activity determines the efficiency of mRNA 3′-end processing, the degradation of tRNA and the extent of polyadenylation in chloroplasts. EMBO J. 2002; 21:6905–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yehudai-Resheff S., Zimmer S.L., Komine Y., Stern D.B.. Integration of chloroplast nucleic acid metabolism into the phosphate deprivation response in Chlamydomonas reinhardtii. Plant Cell. 2007; 19:1023–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zimmer S.L., Schein A., Zipor G., Stern D.B., Schuster G.. Polyadenylation in Arabidopsis and Chlamydomonas organelles: the input of nucleotidyltransferases, poly(A) polymerases and polynucleotide phosphorylase. Plant J. 2009; 59:88–99. [DOI] [PubMed] [Google Scholar]
- 76. Luro S., Germain A., Sharwood R.E., Stern D.B.. RNase J participates in a pentatricopeptide repeat protein-mediated 5′ end maturation of chloroplast mRNAs. Nucleic Acids Res. 2013; 41:9141–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Johnson X., Wostrikoff K., Finazzi G., Kuras R., Schwarz C., Bujaldon S., Nickelsen J., Stern D.B., Wollman F.-A., Vallon O.. MRL1, a conserved pentatricopeptide repeat protein, is required for stabilization of rbcL mRNA in Chlamydomonas and Arabidopsis. Plant Cell. 2010; 22:234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Germain A., Kim S.H., Gutierrez R., Stern D.B.. Ribonuclease II preserves chloroplast RNA homeostasis by increasing mRNA decay rates, and cooperates with polynucleotide phosphorylase in 3′ end maturation. Plant J. 2012; 72:960–971. [DOI] [PubMed] [Google Scholar]
- 79. Zurawski G., Perrot B., Bottomley W., Whitfeld P.R.. The structure of the gene for the large subunit of ribulose 1,5-bisphosphate carboxylase from spinach chloroplast DNA. Nucleic Acids Res. 1981; 9:3251–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zurawski G., Clegg M.T.. Evolution of higher-plant chloroplast DNA-encoded genes: implications for structure-function and phylogenetic studies. Ann. Rev. Plant Physiol. 1987; 38:391–418. [Google Scholar]
- 81. Lama L., Cobo J., Buenaventura D., Ryan K.. Small RNA-seq: The RNA 5′-end adapter ligation problem and how to circumvent it. J. Biol. Methods. 2019; 6:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu H., Yao J., Wu D.C., Lambowitz A.M.. Improved TGIRT-seq methods for comprehensive transcriptome profiling with decreased adapter dimer formation and bias correction. Sci. Rep. 2019; 9:7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liebers M., Grübler B., Chevalier F., Lerbs-Mache S., Merendino L., Blanvillain R., Pfannschmidt T.. Regulatory shifts in plastid transcription play a key role in morphological conversions of plastids during plant development. Front. Plant Sci. 2017; 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Silhavy D., Maliga P.. Mapping of promoters for the nucleus-encoded plastid RNA polymerase (NEP) in the iojap maize mutant. Curr. Genet. 1998; 33:340–344. [DOI] [PubMed] [Google Scholar]
- 85. Lloréns-Rico V., Cano J., Kamminga T., Gil R., Latorre A., Chen W.-H., Bork P., Glass J.I., Serrano L., Lluch-Senar M.. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016; 2:e1501363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Georg J., Hess W.R.. Widespread antisense transcription in prokaryotes. Microbiol. Spectr. 2018; 6:doi:10.1128/microbiolspec.RWR-0029-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shimmura S., Nozoe M., Kitora S., Kin S., Matsutani S., Ishizaki Y., Nakahira Y., Shiina T.. Comparative analysis of chloroplast psbD promoters in terrestrial plants. Front. Plant Sci. 2017; 8:1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hotto A.M., Germain A., Stern D.B.. Plastid non-coding RNAs: emerging candidates for gene regulation. Trends Plant Sci. 2012; 17:737–744. [DOI] [PubMed] [Google Scholar]
- 89. Vera A., Matsubayashi T., Sugiura M.. Active transcription from a promoter positioned within the coding region of a divergently oriented gene: the tobacco chloroplast rpl32 gene. Mol. Gen. Genet. 1992; 233:151–156. [DOI] [PubMed] [Google Scholar]
- 90. Ohyama K., Fukuzawa H., Kohchi T., Shirai H., Sano T., Sano S., Umesono K., Shiki Y., Takeuchi M., Chang Z. et al.. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature. 1986; 322:572–574. [Google Scholar]
- 91. Shinozaki K., Ohme M., Tanaka M., Wakasugi T., Hayashida N., Matsubayashi T., Zaita N., Chunwongse J., Obokata J., Yamaguchi-Shinozaki K. et al.. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986; 5:2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bazzini A.A., Johnstone T.G., Christiano R., Mackowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C. et al.. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Washietl S., Findeiss S., Müller S.A., Kalkhof S., von Bergen M., Hofacker I.L., Stadler P.F., Goldman N.. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011; 17:578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Slavoff S.A., Mitchell A.J., Schwaid A.G., Cabili M.N., Ma J., Levin J.Z., Karger A.D., Budnik B.A., Rinn J.L., Saghatelian A.. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2013; 9:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wicke S., Schneeweiss G.M., dePamphilis C.W., Müller K.F., Quandt D.. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 2011; 76:273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Shi C., Liu Y., Huang H., Xia E.-H., Zhang H.-B., Gao L.-Z.. Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. PLoS One. 2013; 8:e59620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schmitz-Linneweber C., Maier R.M., Alcaraz J.P., Cottet A., Herrmann R.G., Mache R.. The plastid chromosome of spinach (Spinacia oleracea): complete nucleotide sequence and gene organization. Plant Mol. Biol. 2001; 45:307–315. [DOI] [PubMed] [Google Scholar]
- 98. Raubeson L.A., Peery R., Chumley T.W., Dziubek C., Fourcade H.M., Boore J.L., Jansen R.K.. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007; 8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hammani K., Bonnard G., Bouchoucha A., Gobert A., Pinker F., Salinas T., Giegé P.. Helical repeats modular proteins are major players for organelle gene expression. Biochimie. 2014; 100:141–150. [DOI] [PubMed] [Google Scholar]
- 100. Barkan A., Small I.. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014; 65:415–442. [DOI] [PubMed] [Google Scholar]
- 101. Zhelyazkova P., Hammani K., Rojas M., Voelker R., Vargas-Suarez M., Borner T., Barkan A.. Protein-mediated protection as the predominant mechanism for defining processed mRNA termini in land plant chloroplasts. Nucleic Acids Res. 2012; 40:3092–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yagi Y., Hayashi S., Kobayashi K., Hirayama T., Nakamura T.. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One. 2013; 8:e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Takenaka M., Zehrmann A., Brennicke A., Graichen K.. Improved Computational target site prediction for pentatricopeptide repeat RNA editing factors. PLoS One. 2013; 8:e65343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yan J., Yao Y., Hong S., Yang Y., Shen C., Zhang Q., Zhang D., Zou T., Yin P.. Delineation of pentatricopeptide repeat codes for target RNA prediction. Nucleic Acids Res. 2019; 47:3728–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Barkan A., Rojas M., Fujii S., Yap A., Chong Y.S., Bond C.S., Small I.. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012; 8:e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Prikryl J., Rojas M., Schuster G., Barkan A.. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yehudai-Resheff S., Portnoy V., Yogev S., Adir N., Schuster G.. Domain analysis of the chloroplast polynucleotide phosphorylase reveals discrete functions in RNA degradation, polyadenylation, and sequence homology with exosome proteins. Plant Cell. 2003; 15:2003–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chi W., He B., Mao J., Jiang J., Zhang L.. Plastid sigma factors: their individual functions and regulation in transcription. Biochim. Biophys. Acta Bioenerg. 2015; 1847:770–778. [DOI] [PubMed] [Google Scholar]
- 109. Pfannschmidt T., Blanvillain R., Merendino L., Courtois F., Chevalier F., Liebers M., Grubler B., Hommel E., Lerbs-Mache S.. Plastid RNA polymerases: orchestration of enzymes with different evolutionary origins controls chloroplast biogenesis during the plant life cycle. J. Exp. Bot. 2015; 66:6957–6973. [DOI] [PubMed] [Google Scholar]
- 110. Zhou W., Lu Q., Li Q., Wang L., Ding S., Zhang A., Wen X., Zhang L., Lu C.. PPR-SMR protein SOT1 has RNA endonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E1554–E1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yang Z., Hou Q., Cheng L., Xu W., Hong Y., Li S., Sun Q.. RNase H1 cooperates with DNA Gyrases to restrict R-Loops and maintain genome integrity in arabidopsis chloroplasts. Plant Cell. 2017; 29:2478–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Condon C., Piton J., Braun F.. Distribution of the ribosome associated endonuclease Rae1 and the potential role of conserved amino acids in codon recognition. RNA Biol. 2018; 15:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ji D., Manavski N., Meurer J., Zhang L., Chi W.. Regulated chloroplast transcription termination. Biochim. Biophys. Acta Bioenerg. 2019; 1860:69–77. [DOI] [PubMed] [Google Scholar]
- 114. Porrua O., Boudvillain M., Libri D.. Transcription termination: variations on common themes. Trends Genet. 2016; 32:508–522. [DOI] [PubMed] [Google Scholar]
- 115. Chi W., He B., Manavski N., Mao J., Ji D., Lu C., Rochaix J.D., Meurer J., Zhang L.. RHON1 mediates a Rho-Like activity for transcription termination in plastids of arabidopsis thaliana. Plant Cell. 2014; 26:4918–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dar D., Sorek R.. High-resolution RNA 3′-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res. 2018; 46:6797–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Romani I., Manavski N., Morosetti A., Tadini L., Maier S., Kühn K., Ruwe H., Schmitz-Linneweber C., Wanner G., Leister D. et al.. A member of the arabidopsis mitochondrial transcription termination factor family is required for maturation of chloroplast transfer RNA Ile (GAU). Plant Physiol. 2015; 169:627–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zoschke R., Watkins K.P., Barkan A.. A rapid ribosome profiling method elucidates chloroplast ribosome behavior in vivo. Plant Cell. 2013; 25:2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequences have been deposited on the SRA database with the number PRJNA533962 and can be accessed here https://www.ncbi.nlm.nih.gov/sra/PRJNA533962.