


Abstract
The integrity of our DNA is challenged daily by a variety of chemicals that cause DNA base alkylation. DNA alkylation repair is an essential cellular defence mechanism to prevent the cytotoxicity or mutagenesis from DNA alkylating chemicals. Human oxidative demethylase ALKBH3 is a central component of alkylation repair, especially from single-stranded DNA. However, the molecular mechanism of ALKBH3-mediated damage recognition and repair is less understood. We report that ALKBH3 has a direct protein-protein interaction with human RAD51 paralogue RAD51C. We also provide evidence that RAD51C–ALKBH3 interaction stimulates ALKBH3-mediated repair of methyl-adduct located within 3′-tailed DNA, which serves as a substrate for the RAD51 recombinase. We further show that the lack of RAD51C–ALKBH3 interaction affects ALKBH3 function in vitro and in vivo. Our data provide a molecular mechanism underlying upstream events of alkyl adduct recognition and repair by ALKBH3.
INTRODUCTION
DNA alkylating agents are present in the environmental contaminants, e.g. haloalkane (1,2), industrial chemicals, e.g. ethylene oxide (3), or they may be generated intracellularly during normal metabolic reaction (4), e.g. S-adenosylmethionine. In double-stranded DNA, simple methylating agents such as methyl methanesulphonate (MMS) methylates guanine to N7-methylguanine (7meG) and adenine to N3-methlyladenine (3meA) (5). 3meA is removed by the base excision repair (BER) pathway DNA glycosylases to yield abasic site (6). Cleavage of abasic sites by apurinic/apyrimidinic endonuclease (APE1) creates single-strand break (SSB), followed by DNA synthesis and DNA ligation (7,8). MMS exposure in single-stranded DNA (ssDNA) results in N1-methyladenine (1meA) and N3-methylcytosine (3meC) (9). These adducts are repaired directly by Fe(II) and 2-oxoglutarate(2OG)-dependent dioxygenase known as AlkB in Escherichia coli (10) AlkB family of proteins are ubiquitous, and human cells have at least eight AlkB family members (ALKBH1-8) (11,12). However, only two AlkB homologs, ALKBH2 and ALKBH3, are oxidative DNA demethylase (13). ALKBH2 associates with proliferating cell nuclear antigen (PCNA) (14,15) and shows a preference for double-stranded DNA (16,17). ALKBH3 requires unwinding activity of a DNA helicase known as activating signal cointegrator complex-3 (ASCC3) to demethylate the duplex DNA (16,18,19). ALKBH3 has an important role in prostate and lung cancer, where it is overexpressed, and knockdown of ALKBH3 causes reduced cell proliferation and apoptosis (18,20). Biochemical evidence suggests that ALKBH3 prefers for ssDNA as substrate (21). Although the importance of protecting ssDNA from alkylation damage is obvious, whether ALKBH3 is required for this remains unknown. Nevertheless, locating such recombination-associated ssDNA region could be a challenging task for ALKBH3.
MMS-induced DNA alkylation can also result in DNA strand breaks due to collapsed replication fork at the alkyl adduct or when two closely-opposed abasic sites are processed into SSB (22). One of the pathways of double-strand break (DSB) repair is HR. In HR, DSBs are resected by nucleolytic cleavage, generating 3′ ssDNA tail onto which the RAD51 recombinase forms filament. This RAD51 nucleoprotein structure invades homologous DNA, which is used as a template for repair DNA synthesis (23). Consistently, the mutation of HR genes display MMS sensitivity. In E. coli, RecA recombinase mediates the HR process (24). HR in human cells relies on recombinase RAD51 and modulated by BRCA2 and RAD51 paralogues, including RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 (25–27). However, all the function of the RAD51 paralogues are still not completely known (28).
In this study, we aimed to improve our understanding of DNA alkylation repair by ALKBH3. Earlier we demonstrated a stable association between E. coli DNA alkylation repair protein AlkB and recombinational repair protein RecA. We also found that RecA stimulates the DNA alkylation repair by recruiting AlkB to the alkylated ssDNA (29). In light of our previous study, it seemed intuitive that a similar mechanism might be present in human cells. We hypothesised that, by analogy to RecA, human recombinase RAD51, or its paralogues, might interact with human homologues of AlkB. To test this idea, we examined the interaction between human RAD51 and RAD51 paralogues and oxidative demethylases ALKBH2 and ALKBH3. We demonstrate that ALKBH3 has a direct protein–protein interaction with RAD51C. Our results suggest that RAD51C stimulates ALKBH3-mediated demethylation of alkyl-adduct in 3′-tailed DNA. We also provide evidence to suggest that RAD51C–ALKBH3 interaction promotes ALKBH3 function in vivo.
MATERIALS AND METHODS
Yeast two-hybrid analysis
The full length human RAD51 and its paralogs namely RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 were PCR amplified from Human cDNA library (Clontech). The Yeast two-hybrid plasmid constructs namely pACT2-RAD51, pACT2-RAD51B, pACT2-RAD51C, pACT2-RAD51D, pACT2-XRCC2 and pACT2-XRCC3 (activation domain) plasmid was co-transformed with pGBKT7-ALKBH2 or pGBKT7-ALKBH3 (binding domain) plasmid into yeast strain pJ69-4A. The deletion constructs of RAD51C, namely, pACT2-RAD51C Δ1–18, pACT2-RAD51C Δ19–32, pACT2-RAD51C Δ33–41, pACT2-RAD51C Δ42–52, pACT2-RAD51C Δ53–66, pACT2-RAD51C NTD, pACT2-RAD51C ΔNTD, pACT2-RAD51C 33–41, pACT2-RAD51C 42–52 and pACT2-RAD51C 33–52 was co-transformed with pGBKT7-ALKBH3 to generate strain J69AR1 (pGBKT7-ALKBH3+ pACT2-RAD51C Δ1–18), J69AR2 (pGBKT7-ALKBH3+ pACT2-RAD51C Δ19–32), J69AR3 (pGBKT7-ALKBH3 + pACT2-RAD51C Δ33–41), J69AR4 (pGBKT7-ALKBH3 + pACT2-RAD51C Δ42–52), J69AR5 (pGBKT7-ALKBH3 + pACT2-RAD51C Δ53–66), J69AR6 (pGBKT7-ALKBH3 + pACT2-RAD51C NTD), J69AR7 (pGBKT7-ALKBH3 + pACT2-RAD51C ΔNTD), J69AR8 (pGBKT7-ALKBH3 + pACT2-RAD51C 33–41), J69AR9 (pGBKT7-ALKBH3 + pACT2-RAD51C 42–52) and J69AR10 (pGBKT7-ALKBH3 + pACT2-RAD51C 33–52). Similarly, deletion constructs of ALKBH3 namely; pGBKT7-ALKBH3 1–70, pGBKT7-ALKBH3 71–115, pGBKT7-ALKBH3 116–174 and pGBKT7-ALKBH3 175–286 was co-transformed to generate strain J69AR11 (pGBKT7-ALKBH3 1–70 + pACT2-RAD51C), J69AR12 (pGBKT7-ALKBH3 71–115 + pACT2-RAD51C), J69AR13 (pGBKT7-ALKBH3 116–174 + pACT2-RAD51C), J69AR14 (pGBKT7-ALKBH3 175–286 + pACT2-RAD51C). The transformants were plated onto synthetic defined (SD) -Leu -Trp dropout plates and incubated at 30°C for 2–3 days. Expression of the HIS and ADE2 reporter genes were analysed by spotting the transformants on three dropout (SD -leucine, -tryptophan and -histidine) or 4 dropout (SD -leucine, -tryptophan, -histidine and -adenine) plates. Also, the expression of the lacZ reporter was evaluated qualitatively by the X-gal filter assay as described previously (29).
Expression and purification of proteins
For tag-less recombinant expression, ALKBH3 and RAD51C coding DNA was amplified by PCR from human cDNA (Clontech) and cloned in NcoI–XhoI and NheI–XhoI sites of pTYB3 respectively. pTYB3-RAD51c Δ42–52 construct was generated by PCR mediated internal deletion method. The active site mutant pTYB3-ALKBH3 (H191A D193A H257A) construct was generated by site-directed mutagenesis using appropriate primers. In the case of His-tag fusion proteins used in the pull-down experiments, ALKBH3 and RAD51C were cloned into pET-28a (Novagen) using BamHI and XhoI restriction sites. GST fusion proteins were generated by cloning PCR amplified DNA was in BamHI and XhoI sites of vector pGEX-6P1. Plasmids were transformed into the E. coli strain BL21-CodonPlus(DE3)-RIL (Agilent), and proteins were overexpressed by induction with 1 mM IPTG at 16°C for 18 h. For tag-less protein production, ALKBH3, mutant ALKBH3, RAD51C and RAD51C Δ42–52 were expressed as a C-terminal chitin-fusion protein and purification were performed using chitin agarose, as recommended by the manufacturer (NEB). All the His-tagged proteins were purified using Ni-NTA agarose (Qiagen) and dialysed against 20 mM Tris–HCl pH 8.0, 500 mM NaCl and 5% glycerol. Proteins were analysed by SDS-PAGE and concentrations were estimated by Bradford assay (Bio-Rad).
GST pull-down experiments
For GST pull-down experiments, full-length RAD51C and truncated mutant RAD51C Δ42–52 were expressed as His-tag protein and ALKBH3 was expressed as GST-fusion protein. 150μg of GST-ALKBH3 bound to 50 ul glutathione sepharose beads (Thermo Scientific) was incubated with ∼100 μg of His-RAD51C or His-RAD51C Δ42–52 in 500 μl binding buffer containing 25 mM Tris pH 7.5, 1 mM CaCl2, 500 mM NaCl at room temperature for 2 h. To eliminate any residual DNA in the GST pull-down system, GST-ALKBH3 bound beads incubated with HIS-RAD51C in the presence of 10 U Micrococcal nuclease (Mnase). In case of competition experiment with the peptide, 50, 250 and 500 μM of RAD51C 42–52 or RAD51C 33–41 peptide was incubated with His-RAD51C and glutathione sephrose beads bound ALKBH3 for 2 h. Protein complexes were then pulled down and analyzed on SDS-PAGE followed by western blot analysis using an anti-6xHis antibody (1:1000; Thermo) as described previously (29,30).
Isothermal titration calorimetry (ITC) measurements
Calorimetric binding measurements were conducted on a MicroCal iTC200 (Malvern Instruments Ltd, UK) at 25°C. Synthetic peptides p33–41 (QTAEELLEV) and p42-52 (KPSELSKEVGI) were obtained (GM Research Foundation Pvt. Ltd). All the ITC experiments were performed with 12 consecutive injections (3 μl) of RAD51C peptides (1 mM) into the sample cell containing 180 ul of ALKBH3 (15 μM) with a spacing of 150s between the successive injections. The rotation speed of the syringe was maintained at 450 rpm. The integration of the heat pulses obtained from each titration data was analysed by the MicroCal Origin programme (version 5.0) to determine the dissociation constant.
CD spectroscopy
The circular dichroism (CD) experiments were carried out using a JASCO J-1500 instrument. RAD51C 33–41 and RAD51C 42–52 were taken in a 1 mm path length quartz cell and spectra were recorded at room temperature in buffer containing 20 mM Tris–HCl, pH 8.0 and 100 mM NaCl.
Demethylation assay by direct detection of formaldehyde
ALKBH3-mediated demethylation activity was measured by the repair of 3meC present in two different types of chemically synthesized oligonucleotide substrates: (a) ssDNA substrate, 40-mer single 3meC containing (5′ TTTTTTTTTTTTTTTTTTT3meCTTTTTTTTTTTTTTTTTTTT 3′) and (b) 3′ overhang (tailed) substrate, 41-mer oligo 5′ TCCTTTTGATAAGAGGTCAATTTTTTTTTT-3meC-TTTTTTTTTT 3′ was annealed with the complementary 20mer oligo 5′ TTGACCTCTTATCAAAAGGA 3′ generating a 20 nucleotide single-stranded overhang with a single 3meC positioned in the centre, as described before (31,32). DNA oligonuleotides were dissolved in annealing buffer (10 mM Tris pH 8 and 1 mM EDTA) by brief heating at 95°C for 5 min and then followed by slow cooling to 37°C for 1 h to form this 3′ overhang structure. ALKBH3-mediated repair was monitored by incubating the substrate (4μM) in reaction buffer-A (20 mM Tris–HCl pH 8.0, 0.2 mM 2-oxoglutarate, 20 μM Fe(NH4)2(SO4)2, 2 mM l-ascorbate, 1 mM MgCl2, 0.2 mM ATP and 1 mM DTT) for 1 h at 37°C. Demethylation repair reaction product was mixed with 40 μl of 5 M ammonium acetate and 10 μl of 0.5 M acetoacetanilide. The fluorescent dihydropyridine derivative (Amax 365 nm, Emax 465 nm) was measured using a 96-well microplate and Synergy (Biotek Instrument) multi-mode reader and the released formaldehyde was directly quantified as described previously (29,33).
Demethylation assay by FDH-coupled indirect detection of formaldehyde
Formaldehyde dehydrogenase (FDH)-coupled DNA repair assay was performed to monitor the formaldehyde production as described before (33,34). Briefly, ALKBH3 and 3meC containing oligonucleotide ssDNA(4μM) or 3′ overhang substrate (4 μM) was incubated with reaction buffer-A containing 1 mM NAD+ and 0.01 U FDH. NADH generated has a characteristic absorption peak at 340 nm which was monitored using a 96-well plate Synergy (Biotek Instrument) multimode reader.
Demethylation assay by dot-blot analysis
ALKBH3 (0.4 μM) and 3meC containing oligonucleotide ssDNA (8 μM) or 3′ overhang substrate (8μM) was incubated with reaction buffer-A in the presence and absence of RAD51C (8 μM). Modified oligonucleotides were treated with 2 N NaOH at 4°C for 30 min to denature DNA and transferred to Hybond-N+ nylon membrane using a dot-blot apparatus. DNA was crosslinked by incubating at 80°C for 30 min and probed with the anti-3meC antibody (61180, Active Motif).
Expression constructs
The epitope-tagged RAD51C expression plasmids were constructed by cutting the PCR-amplified wild-type RAD51C cDNA with NheI and NotI, and then cloned in-frame and downstream of the sequence encoding the FLAG and HA epitope in pIRES-puro (Clontech). RAD51C Δ42–52 deletion mutation was generated by inverse-PCR using specific primers. To construct epitope-tagged ALKBH3 expression plasmids, the PCR-amplified wild-type ALKBH3 cDNA was cut with NotI and EcoRI, and then cloned in-frame and downstream of the sequence encoding the MYC epitope in pIRES-puro (Clontech). RAD51C and ALKBH3 shRNA constructs were generated using reported sequences (shRAD51C#1 5′ CACCTTCTGTTCAGCACTA 3′, shRAD51C#2 5′ CTCCTAGAGGTGAAACCCT 3, shALKBH3 #1 5′-GAGAGGATATAACTTATCA-3′, and shALKBH3 #2 5′-GTGTTCAAAGAACCTCAGC-3′) and cloned into the pSilencer 2.1-U6 puro shRNA vector. For overexpression analysis in knockdown cells, RNAi-resistant Flag-HA-RAD51C and Flag-HA-RAD51C Δ42–52 constructs were generated by site-directed mutagenesis as described before (35).
Cell culture and transfection
PC-3 cells were maintained in Ham's F-12 GlutaMAX and 293T, NCI-H23 cells were maintained in DMEM (Invitrogen) with 10% FBS at 37°C in a humidified air containing 5% CO2. All plasmid transfections for stable and transient expression were performed using a Bio-Rad gene pulsar X cell (260 V and 960 μF). Short hairpin RNA(shRNA)-mediated down-regulation was achieved by cloning the sequence targeting RAD51C or ALKBH3 into the pSilencer 2.1-U6 puro shRNA expression vector. PC3 and NCI-H23 cells were transfected by electroporation and followed by selection with 1 μg/ml puromycin for 30 days. Control transfection was performed using the pSilencer 2.1-U6 puro vector expressing shRNA targeting Luciferase (5′-CGCTGAGTACTTCGAAATG-3′).
Immunoprecipitation
The co-immunoprecipitation (IP) of endogenous proteins from PC-3 whole cell extract was carried out using RAD51C antibody (Novus Biologicals, 2H11/6) or control IgG. The immunocomplex was bound to protein A/G agarose beads (20421, Thermo Scientific Pierce) to detect the bound ALKBH3 using western blotting. The co-IP experiments with overexpressed proteins were carried out by Flag-RAD51C complex pull down. Whole-cell extract was prepared by resuspension of the cells in hypotonic buffer (20 mM HEPES pH 7.4, 10 mM KCl and 1 mM MgCl2) containing protease inhibitors and passing the extract through a 25-gauge needle several times to induce osmotic lysis. Next, KCl was adjusted to 200 mM and Triton X-100 was added to final concentration of 1% and incubated on ice for 30 min. After removing the insoluble materials by centrifugation, the extract was then incubated with Anti-Flag M2-agarose resin (Sigma) for 4 h at 4°C and centrifuged. The supernatant was harvested and considered as the unbound fraction. The beads were washed extensively with PBS and bound material was eluted using SDS loading buffer.
Cell survival assays
Cells were seeded onto 96-well plates at a density of 5000 cells per well in triplicate. Cells were treated continuously with MMS (50–400 μM) for 48 h. Later, media was removed, and 100 μl of 0.5 mg/ml MTT reagent (Cell Growth Determination Kit, CGD1, Sigma-Aldrich) was added to each well. The cultures were incubated for 4 h at 37°C in the dark. The formazan crystals generated were solubilized using 100 ul of solubilization buffer provided in the kit. Absorbance at 570 nm was measured using Synergy (Biotek Instrument) multi-mode reader.
Calcein-AM staining
After the indicated treatments with MMS as described above, cancer cells were incubated for 30 min at 37°C with 2 μM Calcein AM (C1359, Sigma-Aldrich) diluted in PBS. Fluorescence emitted by stained cells was then observed using an inverted fluorescence microscope (EVOS FL Auto, ThermoFisher Scientific).
Immunofluorescence microscopy
Cells were grown on chamber slides with 400 μM MMS for 48 h. Cells were then washed with PBS and fixed using 4% formaldehyde for 15 min at RT. After fixation, cells were treated with 1.5 M HCl for 30 min, followed by neutralization with sodium borate (pH 8.5). Cells were permeabilized with 0.1% Triton X-100 and blocked with 1% BSA for 1 h. After blocking, cells were incubated with primary antibodies against 3meC (rabbit, Abcam, ab231795) for overnight at 4°C. Next, samples were incubated with secondary antibody Alexa-Fluor 594 goat anti-rabbit (ab150080, Abcam) for 1 h. Nuclear DNA was stained using DAPI (sc-3598, Genetix). Cells were then washed with PBS, mounted with 4 μl ProlongDiamond antifade mountant (P36970, Thermo), and sealed with nail polish. Cells were washed between all steps. Microscopy was performed with a fluorescence microscope (EVOS FL Auto, ThermoFisher Scientific) under 20× and 40× magnification. The images were processed using ImageJ. For overexpression analysis in RAD51C-knockdown cells, RNAi-resistant Flag-HA-RAD51C and Flag-HA-RAD51C Δ42–52 constructs were electroporated in PC3 and NCI-H23 cells. After 72 h, cells were probed with anti-HA (12cE5) for the detection of Flag-HA-RAD51C or Flag-HA-RAD51C Δ42–52; 3meC antibody was used for the detection of alkylation damage.
RESULTS
RAD51C interacts with ALKBH3
Previously we showed that E. coli recombinase RecA interacts with DNA alkylation repair protein AlkB (29). Because E. coli AlkB is a functional homologue of human ALKBH2 and ALKBH3 and all the RAD51 family members are homologous to the E. coli RecA, we wanted to know if there are any interactions between ALKBH2 or ALKBH3 and RAD51 family proteins. First, we used yeast two-hybrid assays to test interactions between these proteins. We cloned the RAD51 and RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3) fused to GAL4 activation domain and ALKBH2 and ALKBH3 fused to GAL4 DNA binding domain. In this screening, we could not detect any interaction of ALKBH2 with RAD51 or RAD51 paralogues (Figure 1A). However, we observed robust interaction of ALKBH3 with RAD51C, but not with RAD51 and other RAD51 paralogues (Figure 1B). Human RAD51C consists of a 66 amino acid flexible N-terminal domain (NTD) and a core catalytic domain containing the ATP-binding motif. To map the interacting region, we divided the protein into two segments; NTD consisting of 1–66 amino acid residues and ΔNTD consisting of remaining amino acid residues (67–376) (Figure 2A). In the initial experiments, we observed a robust interaction between ALKBH3 and RAD51C-NTD and truncation of the N-terminal 66 amino acid residues in RAD51C (ΔNTD) completely abrogated the interaction (Figure 2B). This could imply that ALKBH3 interacts with the N-terminal domain of RAD51C and bulk of RAD51C protein is dispensable for this interaction. Five more RAD51C deletion mutants within NTD (residues 1–66) were generated lacking amino acid residues 1–18, 19–32, 33–41, 42–52 and 53–66, respectively, to narrow down the RAD51C-interacting domain further. These deletion mutants were tested for their abilities to interact with ALKBH3 by the yeast two-hybrid analysis. Notably, the interactions of RAD51CΔ33–41 was partially affected, while the interactions of RAD51CΔ42–52 was completely abolished (Figure 2B). We also observed that binding of RAD51CΔ1–18, RAD51CΔ19–32, RAD51CΔ53–66 proteins to ALKBH3 remained unaffected (Figure 2B). This result indicates that ALKBH3 interacting region could be confined within 33–52 amino acids of RAD51C. To confirm the involvement of amino acid residues 33–52 of RAD51C, three RAD51C fragments were constructed across NTD: RAD51C33–41, RAD51C42–52 and RAD51C33–52 represented amino acid residues 33–41, 42–52 and 33–52, respectively. As expected, all of these RAD51C constructs with amino acid residues between 33 and 51 showed binding to ALKBH3 (Figure 2C). Lack of growth in adenine dropout plates could probably indicate weaker interaction (36).
Figure 1.

ALKBH3 associates with RAD51C. Yeast two-hybrid analysis depicting the interaction of RAD51 and RAD51 paralogues with ALKBH2 (A) and ALKBH3 (B). Yeast two-hybrid reporter strains were co-transformed with two plasmids, one encoding Gal4 activation domain (pACT2), or fused to RAD51, RAD51B, RAD51C, RAD51D, XRCC2 or XRCC3; and the other encoding Gal4 binding domain (pGBKT7), or fused to full-length (A) ALKBH2 and (B) ALKBH3. Yeast strains were grown on SD plates without tryptophan and leucine (for the selection of bait and prey plasmids) but with histidine (top row). Interaction of the two fusion proteins, indicated by growth on SD plates without histidine and adenine. Interaction was further confirmed by the activation of expression of the lacZ reporter gene which is detected by the β-galactosidase activities using X-Gal filter assay (bottom row). For lacZ assay, two separate pieces of filter papers were used and put together for the figure.
Figure 2.

Mapping the regions important for the RAD51C–ALKBH3 interaction. (A) Yeast two-hybrid constructs used in the interaction analysis. RAD51C was divided into its major subdomains, N-terminal domain (NTD) and C-terminal domain (CTD). Five more internal deletion mutations were created within NTD. Fragments containing RAD51C amino acids 33–52, 33–41 and 41–51 were also generated. (B) Examination of the RAD51C NTD or different internal deletion constructs and ALKBH3 interaction. (C) Confirmation of interaction domain located with 33–52 amino acids of RAD51C (see more in Supplementary Figure S1).
To gain a better understanding of the molecular details of the interaction, we performed molecular docking experiments using the human ALKBH3 (PDB entry 2IUW) and RAD51C, which was modelled using Sulfolobus solfataricus Rad-A (PDB: 2ZUB) as the template. To dock these two proteins, we used the HADDOCK 2.2 web interface and included the relevant inter-protein constraints. A model of ALKBH3-RAD51C is illustrated in Supplementary Figure S1A (Supporting Information). Our model suggested that ALKBH3 forms hydrogen bond and ionic interactions predominantly with the amino acids 42–52 located in the N-terminal domain of RAD51C. This result is consistent with the previous result (Figure 2C), where deletion of amino acids 42–52 had the profound effect on the interaction. The binding region of ALKBH3 was also investigated by yeast two-hybrid analysis. For this, we divided the 286 amino acid ALKBH3 into four portions, amino acid residues 1–70, 71–115, 116–174 and 175–286, respectively (Supplementary Figure S1B, Supporting Information). Interestingly, ALKBH3 fragments corresponding to amino acid residues 1–174 failed to interact with RAD51C (Supplementary Figure S1C, Supporting Information). Only the C-terminal segment of ALKBH3 (amino acids 175–286) interacted with RAD51C (Supplementary Figure S1C and D, Supporting Information). We could not narrow down the interacting region of ALKBH3 further, as truncations within the amino acid residues 174–286 could adversely affect the structural integrity of jelly-roll core domain involved in iron and 2OG binding (21). We wanted to know whether RAD51C remained functional after deletion of amino acid residues 42–52. By using yeast two hybrid assay we examined whether RAD51CΔ42–52 is able to interact with other RAD51 paralogues. We observed that both RAD51C and RAD51CΔ42–52 interacted strongly with RAD51B and XRCC3 and retained weak interaction with RAD51 and RAD51D as reported earlier (37–40) (Supplementary Figure S2, Supporting Information). This result confirmed that interaction of RAD51C with other RAD51 paralogue is not affected by the deletion of amino acid residues 42–52 of RAD51C.
To further confirm whether human RAD51C physically interacts with ALKBH3, we used the GST pull-down assay. To test the specificity of the interaction, we used synthetic peptides corresponding to amino acid residues 33–41 (QTAEELLEV) and 42–52 (KPSELSKEVGI) of RAD51C. The secondary structures analysis of the peptides by circular dichroism (CD) in the far-UV range showed that the peptides were folded (Figure 3A). Deconvolution of this spectrum indicated that the p33–41 consists of 83.59% α-helix and 16.41% random coil and p42–52 consists of 84.27% α-helix and 15.73% random coil. This is consistent with the predicted nature of the p33–41 and p42–52 (Figure 3A, top). For GST pull-down assay, bead-bound GST-ALKBH3 fusion protein was incubated with purified HIS-tagged RAD51C protein. To eliminate residual DNA in the GST pull-down system, proteins were incubated in the presence of micrococcal nuclease (MNase). We observed that the HIS-RAD51C could be pulled down by GST-ALKBH3 protein (Figure 3B, lane 1). When the increasing concentration of synthetic p42–52 peptide was added, diminishing binding of ALKBH3 was observed confirming that this peptide competitively inhibited ALKBH3 interaction (Figure 3B, lanes 2–5). Notably, no inhibition of ALKBH3 binding was observed when the p33–41 was used (Figure 3C). To confirm whether the residues 42–52 of RAD51C is indeed required for ALKBH3 interaction, we prepared another HIS-RAD51C construct lacking amino acid residues 42–52 of RAD51C (HIS-RAD51CΔ42–52) and examined its ability to interact with ALKBH3 in a GST pull-down assay. As shown in Figure 3D (lanes 2 and 3), HIS-RAD51CΔ42–52 could not be pulled down by GST-ALKBH3, whereas, HIS-RAD51C could bind to GST-ALKBH3 under similar condition (Figure 3D, lane 6). Thus, in vitro pull-down assays with recombinant proteins recapitulated the results from yeast two-hybrid studies and show that the ALKBH3 associates with RAD51C and amino acid residues 42–52 of RAD51C are critical for this interaction.
Figure 3.

RAD51C binds to ALKBH3 in vitro and in vivo. (A) Circular dichroism (CD) spectroscopy of synthetic peptides corresponding to the 11 amino acids RAD51C interaction region identified in the yeast two-hybrid assays (p42–52) and the adjacent nine amino acids (p33–41). A far UV spectrum was recorded over the range 190–260 nm. (B) GST pull-down experiments, where RAD51C pull-down with the ALKBH3 under increasing concentration of competing peptides p42–52. (C) RAD51C pull-down with the ALKBH3 under increasing concentration of peptide p33–41 (negative control). (D) GST pull-down experiments, where RAD51C and RAD51CΔ42–52 were pull-down with the ALKBH3. (E) The binding affinity of p42–52 peptide with ALKBH3 protein was examined by isothermal titration calorimetry assay. The p42–52 peptide corresponding to the binding residues titrating into the ALKBH3. The binding constant (KD) value was determined by fitting of the titration curve to a 1-site binding mode. (F) The control peptide p33–41 corresponding to region adjacent to the binding residues titrating into the ALKBH3. Affinities for each binding are indicated in data plot.
To obtain more quantitative values for ALKBH3-binding affinity, binding of p33–41 and p42–52 peptides to ALKBH3 was analysed by isothermal titration calorimetry (ITC). Each peptide was titrated into a 15 μM solution of ALKBH3. The binding isotherm (Figure 3E) revealed the exothermic binding characteristic of p42–52 with a calculated KD of 4 μM and stoichiometry of one protein monomers bound per peptide. In contrast, peptide p33–41 failed to bind ALKBH3 under these conditions (Figure 3F).
To verify whether ALKBH3 physically interacts with RAD51C in vivo, we performed a co-immunoprecipitation (co-IP) assay with whole-cell extracts in human cells. First, we examined the interaction of endogenous proteins in prostate cancer cell line PC-3. We observed that ALKBH3 could indeed be immunoprecipitated with RAD51C (Figure 4A). To further verify the specificity of this interaction, HEK-293T cells transiently co-expressing Flag-HA-RAD51C or Flag-HA-RAD51CΔ42–52 and Myc-ALKBH3 was used for co-IP experiment. Consistently, ALKBH3 was immunoprecipitated with RAD51C but not with RAD51CΔ42–52 (Figure 4B). We also performed co-IP in a lung cancer cell line NCI-H23 and prostate cancer cell line PC-3 as these cell lines were reported to have confirmed ALKBH3 expression (18). As expected, we found that RAD51C immunoprecipitated endogenous ALKBH3 in both the cell lines. Conversely, a significantly reduced amount of endogenous ALKBH3 was immunoprecipitated with RAD51CΔ42–52 (Figure 4C and D). Together, these co-IP results confirm our in vitro ALKBH3-RAD51C interaction results.
Figure 4.
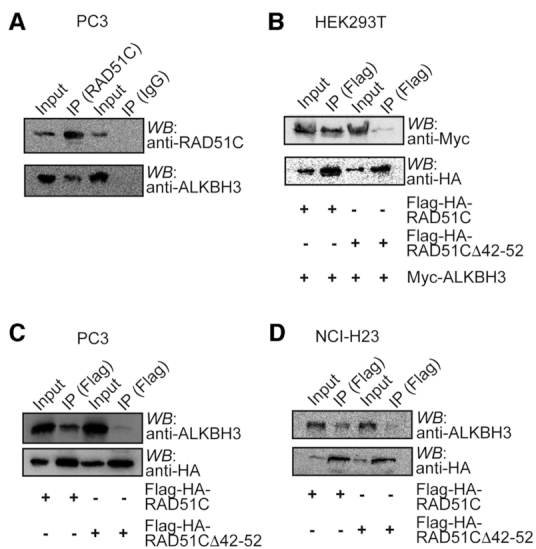
Co-immunoprecipitation of ALKBH3 and RAD51C proteins. (A) RAD51C was immunoprecipitated (IP) from PC3 cell extract using anti-RAD51C antibody and the presence of ALKBH3 was detected by western blot using anti-ALKBH3 antibody. (B) Myc-ALKBH3 and Flag-HA-RAD51C or Flag-HA-RAD51C Δ42–52 were overexpressed in HEK293T cells as indicated and the lysates were subjected to immunoprecipitation using anti-Flag beads. IP and whole-cell extracts (input) were western blotted with the indicated antibodies. (C) Flag-HA-RAD51C or Flag-HA-RAD51C Δ42–52 were overexpressed in NCI-H23 cells as indicated and the lysates were subjected to immunoprecipitation using anti-Flag beads. IP and whole-cell extracts (input) were western blotted with the indicated antibodies. (D) Flag-HA-RAD51C or Flag-HA-RAD51C Δ42–52 were overexpressed in PC3 cells and whole-cell extract (input) was immunoprecipitated with anti-Flag beads and western blotted as shown with the indicated antibodies.
RAD51C enhances ALKBH3 activity in a substrate-specific manner
We reported earlier that E. coli recombinase RecA interacts with AlkB and stimulate AlkB activity (29). Here, we investigated whether the physical interaction between ALKBH3 and RAD51C might mediate a similar change in ALKBH3 activity. We used purified tag-less human RAD51C, as well as ALKBH3 expressed in E. coli and purification was carried out using chitin agarose as described before (34). Loading of Rad51 requires a duplex DNA with 3′-overhang (31) and RAD51 paralogues, including RAD51C, also has specificity to 3′-tailed duplex (41). Therefore, we used a 3′-tailed DNA containing a 3meC adduct in the 3′-ssDNA overhang to determine the effect of RAD51C on ALKBH3 activity. As negative control, we used ssDNA containing a single 3meC adduct. To monitor demethylation activity we used three previously reported methods (Figure 5A), (i) direct detection of formaldehyde by acetoacetanilide (AAA), which is converted to a fluorescent product in the presence of ammonia and formaldehyde (42) (ii) indirect detection of formaldehyde by FDH to convert formaldehyde to formic acid using the coenzyme nicotinamide adenine dinucleotide (NAD+) as a co-substrate. NAD+ is reduced by FDH to NADH, which has an detectable absorption peak at 340 nm (43), and (iii) detection of 3meC by dot blot analysis with a 3meC-specific antibody (18,44).
Figure 5.

RAD51C stimulates ALKBH3 mediated repair in a substrate-dependent manner. (A) Schematic diagram depicting three different techniques used to monitor ALKBH3 activity (i) detection of formaldehyde by acetoacetanilide (AAA) and ammonia (NH3); formaldehyde forms a fluorescent compound with peak emission at 465 nm (ii) indirect detection of formaldehyde by formaldehyde dehydrogenase (FDH)-coupled reduction of its NAD+ cofactor to NADH. The reaction can be monitored continuously for the production of NADH with peak absorption at 340 nm. (iii) dot blot analysis using the 3meC specific antibody. (B) Fluorescence emission spectra of formaldehyde released during demethylation of ALKBH3 alone (0.2 μM) or in the presence of RAD51C (2.4–4.0 μM) with 3′-tailed DNA as substrate (4 μM). (C) FDH-coupled demethylation assays of ALKBH3 (0.2 μM) with increasing concentration of RAD51C (2.4–4.0 μM). The repair reaction was coupled to the FDH reaction by adding 0.1 mM NAD+ and 0.01 units of FDH. (D) Detection of formaldehyde released during the repair of 3meC 40-mer ssDNA substrate (4 μM) with either ALKBH3 (0.2 μM) alone or in the presence of RAD51C (4 μM) by acetoacetanilide method. (E) Demethylation assay of ALKBH3 (0.2 μM) alone or in the presence of Rad51C (4 μM) with 3meC 40-mer ssDNA substrate (4 μM) monitored using FDH-coupled assay. (F) Representative dot blot using 3meC specific antibody to detect repair of 3meC in 40mer ssDNA DNA (8 μM) or 3′-tailed DNA (8 μM) by ALKBH3 (0.4 μM) alone or in the presence of RAD51C (8 μM). Methylene blue staining was used to assess the equal loading of the DNA (**P <0.01 assessed by Student's t-test; error bars are represented as mean ± SE for duplicate experiments from two independent assays).
To determine the effect of RAD51C on ALKBH3 activity, we incubated fixed amount of ALKBH3 (0.2 μM) to increasing amounts of RAD51C before the addition of 3′-tailed DNA containing a single 3meC (4 μM). As shown in Figure 5B, the ALKBH3 activity was enhanced significantly by RAD51C protein in a dose-dependent manner. The presence of RAD51C (2.4 μM) increased the activity of ALKBH3 by a factor of 3, compared to the ALKBH3 alone (Figure 5B). Similar dose-dependent effect of RAD51C on ALKBH3 activity was also observed with FDH-coupled reaction which indirectly detects the formaldehyde produced (Figure 5C). To further confirm if RAD51C could indeed stimulate DNA dealkylation by ALKBH3, we performed dot blot analysis of 3meC using an anti-3-meC-specific antibody (18,44). The representative results in Figure 5F show that when RAD51C was present with ALKBH3, the reduction in the levels of 3meC in 3′-tailed DNA was higher compared to the ALKBH3 alone. Interestingly, when we used 3meC-containing ssDNA as the substrate, we failed to observe any effect of RAD51C on demethylation activity (Figure 5D–F). Thus, these results suggest an active role for RAD51C in promoting ALKBH3-mediated DNA repair activity. RAD51C contain ATP binding motifs and hydrolyse ATP. In these experiments, ATP and magnesium ion was included in the reaction buffer used, but the exclusion of ATP or its substitution with the nonhydrolyzable ATP-γ-S did not affect the results (Supplementary Figure S3A). Similarly, catalytically inactive RAD51C K131A mutant was also equally active in promoting ALKBH3 activity (Supplementary Figure S3A). This result is not surprising because while ATP binding or hydrolysis activity of RAD51C is required for modulating HR (45), they are dispensable for DNA binding (46) and apparent strand-exchange activity of RAD51C (41). To rule out any nonspecific effect of RAD51C, we also carried out the repair reaction with RAD51, in the presence and absence of RAD51C. As expected, RAD51 could not stimulate the ALKBH3 reaction (Supplementary Figure S3B). This result suggests that the role of RAD51C in DNA alkylation repair is distinct from its role in recombinational repair. While RAD51C has a DNA-dependent ATPase activity relevant to HR, its ATP-independent DNA melting and strand-exchange activity could be relevant to stimulating the ALKBH3 function.
RAD51C stimulates ALKBH3 through protein–protein interaction
In our analysis, RAD51C lacking the amino acid residues 42–52 did not interact with ALKBH3. To examine the functional significance of this interaction, we expressed tag-less RAD51CΔ42–52 in E. coli and purified it to homogeneity. We then assessed the ability of RAD51CΔ42–52 to stimulate ALKBH3 activity using 3′-overhang substrate (4 μM). By analyzing the amount of formaldehyde produced, we observed that repair of 3meC is much lower with the RAD51CΔ42–52 (4 μM) compared with that obtained with wild-type RAD51C (4 μM) (Figure 6A). As expected, the presence of only RAD51C did not result in any formaldehyde release (Figure 6A). Similar results were also observed with FDH-coupled reaction, which indirectly detects the formaldehyde produced (Figure 6B). Dot blot analysis of 3′overhang DNA also confirmed that the presence of RAD51CΔ42–52 with ALKBH3 could not result in any significant reduction in the levels of 3meC compared to ALKBH3 alone.
Figure 6.

RAD51C–ALKBH3 interaction is critical for stimulation of AlkBH3 activity. (A) Fluorescence emission spectra of formaldehyde released during demethylation of AlkBH3 (0.2 μM) with RAD51C (4 μM) or RAD51CΔ42–52 (4 μM) monitored by Acetoacetanilide method. (B) Comparison of DNA repair activity of AlkBH3 (0.2 μM) with RAD51C (4 μM) or RAD51CΔ42–52 (4 μM) by FDH-coupled assay. Demethylation assay of ALKBH3 (0.2 μM) with competitor peptide p42–52 (50 μM) in the presence or absence of RAD51C (4 μM), monitored by (C) Acetoacetanilide method and (D) FDH-coupled assay. (E) Dot blot using 3meC specific antibody to detect repair of 3meC in 3′-tailed DNA (8 μM) by ALkBH3 (0.4 μM) in the presence of RAD51C (8 μM) or RAD51CΔ42–52 (8 μM). Methylene blue staining was used to validate the equal loading of the DNA. (F) Michaelis–Menten analysis of steady-state kinetics for ALKBH3-mediated repair of 3′-tailed DNA (0.6–9.6 μM) with RAD51C (4 μM) or RAD51CΔ42–52 (4 μM). Curves represent nonlinear regressions to the Michaelis–Menten equation obtained using GraphPad Prism 7.0 software (**P <0.01 assessed by Student's t-test; error bars are represented as mean ±SE for duplicate experiments from two independent assays).
To further support the role of RAD51C amino acid residues 42–52 in promoting ALKBH3 activity, peptide competition assays were employed. We observed that peptide p42–52 and RAD51C competes for binding with ALKBH3 (Figure 3B). We wanted to examine the effect of excess p42–52 on RAD51C-mediated stimulation of ALKBH3 activity. As shown in Figure 6C, the presence of p42–52 (50 μM) along with RAD51C (2.4 μM) and ALKBH3 caused significant reduction in the level of formaldehyde release compared to RAD51C with ALKBH3. However, the presence of p42–52 alone did not stimulate ALKBH3 activity and formaldehyde release (Figure 6C). Similar inhibitory effect of p42–52 peptide on RAD51C-mediated stimulation of ALKBH3 activity was also observed with the FDH-coupled indirect formaldehyde detection (Figure 6D). Dot blot analysis also confirmed that the presence of p42–52 abated RAD51C-mediated stimulation of ALKBH3 activity (Figure 6E). Together, these competition experiments indicate that p42–52 peptide appears to bind ALKBH3 and compete with RAD51C protein, confirming an active role for amino acid residues 42–52 of RAD51C in promoting ALKBH3-mediated DNA repair activity. Absence of any stimulation with the interacting peptide indicates that the peptide p42–52 lacks allosteric effect.
To analyse the mechanism by which RAD51C stimulates oxidative demethylation by ALKBH3, we determined how RAD51C affects the steady-state kinetic parameter KM, for 3′-tailed DNA substrates. For this, the initial velocity of the reaction was measured using tag-less ALKBH3 (0.2 μM), RAD51C (4 μM) protein and varying concentration of duplex 3′-tailed DNA with single 3meC was used (0.6–9.6 μM). As a negative control, RAD51CΔ42–52 was used along with ALKBH3. For determining KM, the initial rates obtained from FDH-coupled reaction at different substrate DNA concentrations were plotted as a function of DNA concentration, and the data were fit to the Michaelis–Menten equation (Figure 6E). KM for ALKBH3 (1.4 μM) with 3′-tailed DNA was in close agreement with the previously published values for ALKBH3 protein on the ssDNA substrate (1.9 μM) (47). It was observed that the presence of RAD51C did not alter the KM significantly (1.7 μM). However, the RAD51C increased the Vmax for ALKBH3 reaction, highlighting the strong stimulatory effect of RAD51C. We then estimated the kcat/KM ratio as an approximate measure of the catalytic efficiency for ALKBH3 either on its own or together with RAD51C. kcat/KM for ALKBH3 (1.01 min−1 μM−1) with 3′-tailed DNA was similar to previously published values for ALKBH3 protein on the ssDNA substrate (0.87 min−1 μM−1) (47). We found that RAD51C stimulated the catalytic activity of ALKBH3, as kcat/KM was determined to be 2.6 min−1 μM−1 (Figure 6F). As expected, RAD51CΔ42–52 failed to significantly increase the catalytic efficiency of ALKBH3 reaction, with kcat/KM being 1.42 min−1 μM−1. Mechanistically, this result suggests that the preferential binding of RAD51C to 3′-tailed DNA likely to results in a more efficient turnover of ALKBH3 enzyme without significantly affecting the binding of 3meC.
Stimulation of ALKBH3 by RAD51C is recruitment-dependent
Our results with yeast two-hybrid interaction show that the ALKBH3 substrate-binding site is distant from the RAD51C interaction site. Nevertheless, the effects of RAD51C binding could, in principle, be transmitted through structural elements of ALKBH3 leading to conformational change altering both the catalytic and substrate-binding of ALKBH3. To explore whether RAD51C binding has any effect on enzyme catalysis, we examined the effect of ferrozine and succinate on ALKBH3 activity. ALKBH3 activity is inhibited by iron chelator ferrozine which sequesters cofactor (Fe2+) and succinate, which compete with co-substrate (2-oxoglutarate). ALKBH3 activity is also known to be inhibited by mutation of the conserved catalytic site residues, including two histidine residues and an aspartate residue (H191, D193 and H257). As shown in Figure 7A, the presence of RAD51C could not alleviate the inhibitory effect of ferrozine and succinate. We also generated mutant AlKBH3 construct (H191A, D193A and H257A) and examined the effect of RAD51C. We observed that the demethylation activity of the mutant ALKBH3 in the presence of RAD51C was indistinguishable from the mutant ALKBH3 alone (Figure 7B). These results and the inability of RAD51C to enhance the reactivity of ALKBH3 with canonical ssDNA substrate, indicate that RAD51C interaction does not change the general mechanism of ALKBH3 reaction.
Figure 7.

RAD51C recruits ALKBH3 to 3′ tailed DNA substrate. (A) Fluorescence emission spectra of formaldehyde released during demethylation of ALKBH3 (0.2μM) with RAD51C (4.0μM) in the presence of iron chelator ferrozine (1 mM) or competitive inhibitor succinate (5 mM). (B) Comparison of DNA repair activity of ALKBH3 (0.2 μM) or catalytically inactive mutant ALKBH3 (0.2 μM) with Rad51C (4.0 μM). (C) Demethylation assay of ALKBH3 with competitor undamaged DNA monitored by acetoacetanilide method. Comparison of repair activity of ALKBH3 (0.2 μM) in the presence or absence of RAD51C (4 μM) with 3meC containing 3′-tailed DNA (4 μM) and undamaged 3′-tailed DNA (20 μM). (D) Demethylation assay of ALKBH3 with competitor undamaged 40-mer ssDNA (20 μM) and 3meC 40-mer ssDNA substrate (4 μM) in presence or absence of RAD51C. (E) Comparison of effect of RAD51C and RAD51CΔ288–305 on demethylation activity of ALKBH3. (F) Demethylation of ALKBH3 with increasing concentration of competitor RAD51CΔ288–305 and RAD51C.
As an alternative mechanism by which RAD51C might increase the demethylation reaction of ALKBH3 could involve the recruitment of ALKBH3 to 3′-overhang substrate via association with RAD51C. To determine whether the stimulatory effect of RAD51C is recruitment-dependent, we compared the reaction rates in the presence and absence of excess undamaged with 3′-tailed DNA (Figure 7C). As judged by formaldehyde release, stimulation of ALKBH3 activity by RAD51C was significantly diminished in the presence of an excess amount of undamaged 3′-tailed DNA (Figure 7C). However, the excess amount of 3′-overhang DNA in the absence of RAD51C had no effect on ALKBH3. Interestingly, when 3meC containing ssDNA was used as a substrate, excess undamaged DNA had no significant effect on the presence or absence of RAD51C (Figure 7D). These results indicate that the undamaged 3′-tailed DNA acted as decoy substrate for RAD51C. When the majority of RAD51C was titrated by excess amount of undamaged 3′-tailed DNA, it resulted in the diminishing effect of RAD51C. This finding is consistent with the recruitment-dependent rate acceleration mechanism. To further validate this model, we generated a mutant RAD51C lacking DNA binding region. Structural comparison with human RAD51 revealed that RAD51C DNA binding region is located within amino acid residues 288–305 (Supplementary Figure S4A and B, Supporting Information). We generated recombinant RAD51CΔ288–305 (Supplementary Figure S4C, Supporting Information) and circular dichroism (CD) spectra of RAD51CΔ288–305 mutants show similar profiles as the RAD51C wild-type form, probably indicating the formation of similar structure (Supplementary Figure S4D, Supporting Information). While wild-type RAD51C could bind 3′-tailed DNA (Supplementary Figure S4E, Supporting Information), deletion of DNA binding region severely affected DNA binding ability of RAD51CΔ288–305 mutant (Supplementary Figure S4F, Supporting Information). When RAD51CΔ288–305 mutants were added to ALKBH3, very little stimulation of ALKBH3 activity was observed (Figure 7E, Supporting Information). When increasing concentration of RAD51CΔ288–305 mutant protein was added in the presence of RAD51C, stimulatory effect of RAD51C was gradually diminished (Figure 7F, Supporting Information). Thus, DNA binding domain mutant RAD51CΔ288–305 recapitulated the phenotype observed with RAD51CΔ42–52 mutant. Together, these results indicate that RAD51C binding appears to retain ALKBH3 in the proximity of resected ssDNA and this local enrichment might accelerate the repair rate.
The role of RAD51C in DNA alkylation repair
The biochemical analysis described in this work identifies RAD51C interaction and stimulation of ALKBH3 activity as for the repair of the alkyl-adduct on 3′-tailed DNA. We tested the in vivo functional correlation of these observations by determining the level of 3meC adduct in genomic DNA and DNA strand break following treatment with MMS. For this, we selected prostate cancer cell line PC3 and non-small cell lung adenocarcinoma cell line NCI-H23. Previous studies showed ALKBH3 is relatively overexpressed in these cell lines (18). In line with that, we investigated the impact of RAD51C on the accumulation of 3meC by stably knockdown RAD51C in PC3 and NCI-H23 cells. For this, we utilized small hairpin RNAs (shRNAs) to target RAD51C (35,48) or ALKBH3 (49). The efficiency of stable RAD51C and ALKBH3 knockdown was confirmed at the protein level by western blotting (Figure 8A). Having obtained stable knockdown PC3 and NCI-H23 cell lines, we examined whether the depletion of RAD51C and ALKBH3 generated an expected phenotype. First, we tested if there was any diminished cell survival in the presence of MMS. As demonstrated by the MTT assay, the proliferation of PC3 and NCI-H23 cells were significantly affected by MMS when RAD51C and ALKBH3 were knocked down (Figure 8B and C). The combined knockdown caused a severe cell survival defect, indicating that the lack of both RAD51C and ALKBH3 inhibit cell proliferation in a synergistic way. Cell viability was also analysed using cell membrane permeant dye Calcein-AM. Stable knockdown of RAD51C, ALKBH3, or both were found to render PC3 or NCI-H23 cells MMS sensitive (Figure 8D–G). This MMS sensitive phenotype is similar to MMS sensitivity of mouse embryo fibroblasts (MEFs) (50,51), Chinese hamster ovary (CHO) and chicken (DT40) (52) cell lines lacking RAD51C and cancer cells lacking ALKBH3 (18). Together, these results confirm the phenotypes expected from ALKBH3 and RAD51C knockdown.
Figure 8.

Stable knockdown of ALKBH3 and RAD51C results in severe MMS-induced cytotoxicity in PC3 and NCI-H23 cells. (A) HEK293T was transiently transfected with shRNA for 72 h. Knockdown of ALKBH3 and RAD51C was confirmed by western blotting with indicated antibodies. Effect of stable knockdown of ALKBH3 and RAD51C on (B) PC-3 and (C) NCI-H23. Cells with indicated shRNAs were exposed to various concentrations of MMS (0, 50, 100, 200, 300 and 400 μM) for 48 h; then cell viability was evaluated by MTT assay. Error bar represent the means ± SD of percentage cell viability from four representative experiments. (D) Analysis of MMS sensitivity using Calcein-AM assay. Representative images of intracellular Calcein AM esterase activity of PC-3 and (F) NCI-H23 cells. Cells were treated with MMS(400uM) for 48h and fluorescent images were captured using an inverted microscope. Bar chart represents the percentage (%) of MMS-induced cytotoxicity of (E) PC-3 and (G) NCI-H23 cells for the indicated shRNAs.
Having confirmed the stable knockdown of ALKBH3 and RAD51C, we wanted to determine whether the absence of RAD51C could affect ALKBH3 activity in vivo by quantifying global 3meC levels using the 3meC-specific antibody. We used an imaging-based assay for the quantification of 3meC on ssDNA (53). We observed no 3meC signal in stable ALKBH3 or RAD51C knockdown PC3 cell line (Figure 9A). When PC3 cells expressing control shRNA were cultured in the presence of MMS, no visible signal for 3meC were detected in the nucleus, indicating a robust repair by ALKBH3 (Figure 9B). However, in ALKBH3-depleted PC3 cell line, a number of cells showed 3meC accumulation in the nucleus after adding MMS to the culture medium (Figure 9A and B). Interestingly, knockdown of RAD51C in PC3 cells also had resulted in increased global 3meC level in these cell nuclei, albeit the effect was ∼50% less than ALKBH3 knockdown. This result suggests that RAD51C is important for 3meC repair in these specific cancer cell lines, although ALKBH3 is the critical component. Combined knockdown of RAD51C and ALKBH3, led to 3meC accumulation in the nucleus in a manner epistatic with ALKBH3 knockdown. The absence of any additive effect of double knockdown on 3meC accumulation further indicates a supportive role for RAD51C in oxidative DNA demethylation and rule out the involvement of HR in removal of 3meC. Similar results were also obtained with ALKBH3 or RAD51C knockdown NCI-H23 cell line (Figure 9C and D). Together, these observations indicate that ALKBH3-mediated repair is indirectly affected in the absence of RAD51C. These observations further raise the question of whether ALKBH3 by itself is competent to repair adducts on ssDNA.
Figure 9.
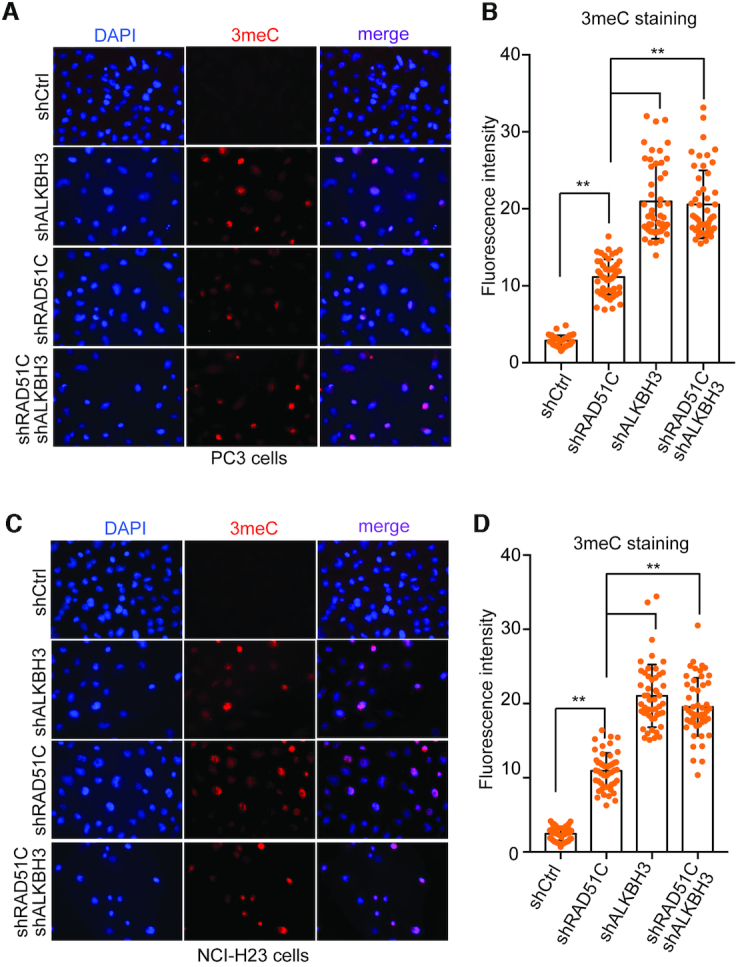
Knockdown of RAD51C exacerbate accumulation of 3-methylcytosine damage following MMS treatment. Immunofluorescent staining for 3-methylcytosine in PC3 cells (A) and NCI-H23 cells (C) with stable RAD51C or ALKBH3 or combined knockdown of both RAD51C and ALKBH3. Cells were treated with MMS (400 μM) for 48 h and analyzed using fluorescent microscopy. Data are shown as representative results of three independent experiments with 3-methylcytosine (red), and the nuclei are visualized by DAPI staining (blue). The histogram represents quantification of 3-methylcytosine signal in PC3 cells (B) and NCI-H23 cells (D) (**P <0.01 assessed by Student's t-test; error bar are represented as mean ± SE from three independent assays).
To test whether RAD51C–ALKBH3 interaction is crucial for ALKBH3 catalysed repair, we quantified 3meC levels following transient overexpression of exogenous, shRNA-resistant, wild-type RAD51C or mutant RAD51CΔ42–52 in stable RAD51C knockdown cells. Alkylation damage was introduced by treating the cells with MMS. When wild-type RAD51C was overexpressed, no apparent changes in the level of 3meC signal was observed in the nucleus following MMS treatment (Figure 10A and C), suggesting complete repair of the MMS-induced 3meC. Surprisingly, in cells overexpressing mutant RAD51CΔ42–52, 3meC signal was at least 50% higher, suggesting a noticeable impact on ALKBH3 activity (Figure 10B and D). This data strongly argues in favour of the notion that efficient repair of some DNA alkylation damage requires RAD51C–ALKBH3 interaction.
Figure 10.

RAD51C-ALKBH3 interaction promotes repair of 3-methylcytosine damage following MMS treatment. PC3 cells (A) or NCI-H23 cells (C) with stable RAD51C knockdown was transiently transfected with RNAi-resistant wild-type Flag-HA-RAD51C or mutant RAD51CΔ42–52 for 24 h followed by treatment with MMS (400 μM) for 48 h and analysed using fluorescent microscopy. Representative images of fluorescence cells stained with anti-HA (for recombinant RAD51C) (green) and 3meC (red). Nuclear DNA was counterstained with DAPI (blue). The graph shows the quantification of anti-3meC antibody signal PC3 cells (B) and NCI-H23 cells (D). For each cell line at least 50 cells were counted and signals were quantified.
DISCUSSION
A new role for RAD51C in DNA alkylation repair
Here, we attempted to understand how ALKBH3 could be recruited to alkyl-adducts located within ssDNA. We observed that ALKBH3 physically interacts with RAD51C and enhances ALKBH3-mediated repair of alkyl-adducts from the 3′-tailed DNA substrate in an ATP-independent manner. RAD51C is known to be part of at least three stable complexes; a dimeric complex of RAD51C–XRCC3 and a larger complex composed of RAD51B–C–D–XRCC2 (54). RAD51C is also a member of another complex that consists of Fanconi proteins PALB2 (FANCN) (55). We report here the fourth interaction partner of RAD51C being ALKBH3. We observed that this interaction is relevant for survival when cancer cells were exposed to genotoxic DNA alkylating agent MMS. Knockdown RAD51C in prostate cancer cell line PC3 and lung cancer cell line NCI-H23 caused accumulation of 3meC in the genome almost akin to what was observed for the ALKBH3 knockdown cells. Interestingly, mutant RAD51C lacking only the ALKBH3 interaction domain failed to remove 3meC from the genome. HR pathway can repair MMS-induced DSBs and rescue stalled replication forks but it is not involved in the enzymatic reversal of 3meC. Because HR pathway does not participate in demethylation of MMS-induced methyl-adducts, accumulation of 3meC in RAD51C knockdown cells are not expected, unless RAD51C plays a role in promoting ALKBH3 activity. As expected, accumulation of 3meC was found to be little higher in the ALKBH3 knockdown cells than in RAD51C knockdown cells. This elevated level of 3meC is probably indicative of the role of ASCC3–ALKBH3 complex in the repair of 3meC from genomic DNA (56). Consistent with this, no enhancement of 3meC level was observed in RAD51C–ALKBH3 double knockdown cell line compared to ALKBH3 knockdown cells. It would be important to understand the context of this ALKBH3–RAD51C interaction. Replication fork stalling generates single-stranded gap (57). Similarly, during the commitment of a DSB to HR, resection of the DNA from 5′ to 3′ end create ssDNA of length up to 3.5 kb (58). Based on the previous report and our observation, we propose that when ALKBH3 repair DNA alkyl-adducts from the genomic DNA, it probably requires 3′-5′ helicase ASCC3 for the generation of single-stranded DNA. Whereas, when ALKBH3 repairs lesions from resected ssDNA or single-stranded gaps behind the stalled replication forks, RAD51C-ALKBH3 association might become important.
ALKBH3 and RAD51C are known to be silenced in breast and ovarian cancers by CpG promoter methylation (53,59). Germline mutations in RAD51C cause a severe form of Fanconi anaemia (60), whereas sporadic mutations in RAD51C predispose individuals to breast and ovarian cancers (61). It will be of interest to determine whether there is an increased burden of unrepaired DNA alkylation damage in RAD51C mutant cells. ALKBH3 overexpression is found in prostate cancer and other cancer types (20,49,62–64). RAD51C is localized to chromosome 17q23, a region frequently amplified in breast tumours (65). In this context, whether RAD51C overexpression facilitates the adaptation of cancer cells to tolerate endogenous alkylation damage to DNA remains to be determined.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: M.M., D.A. and A.D, performed experiment and analyses; A.R. and A.G. supervised their work. A.R. supervised the project, written and revised the manuscript; all authors edited the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
IIT Hyderabad and Science and Engineering Research Board (SERB), Govt. of India [EMR/2016/005135/BBM, in part]. Funding for open access charge: IIT Hyderabad Intramural funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Keppler F., Eiden R., Niedan V., Pracht J., Scholer H.F.. Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature. 2000; 403:298–301. [DOI] [PubMed] [Google Scholar]
- 2. Redeker K.R., Wang N., Low J.C., McMillan A., Tyler S.C., Cicerone R.J.. Emissions of methyl halides and methane from rice paddies. Science. 2000; 290:966–969. [DOI] [PubMed] [Google Scholar]
- 3. Marsden D.A., Jones D.J., Lamb J.H., Tompkins E.M., Farmer P.B., Brown K.. Determination of endogenous and exogenously derived N7-(2-hydroxyethyl)guanine adducts in ethylene oxide-treated rats. Chem. Res. Toxicol. 2007; 20:290–299. [DOI] [PubMed] [Google Scholar]
- 4. De Bont R., van Larebeke N.. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004; 19:169–185. [DOI] [PubMed] [Google Scholar]
- 5. Lundin C., North M., Erixon K., Walters K., Jenssen D., Goldman A.S., Helleday T.. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005; 33:3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shrivastav N., Li D., Essigmann J.M.. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010; 31:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O’Brien P.J., Ellenberger T.. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem. 2004; 279:9750–9757. [DOI] [PubMed] [Google Scholar]
- 8. O’Brien P.J., Ellenberger T.. The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J. Biol. Chem. 2004; 279:26876–26884. [DOI] [PubMed] [Google Scholar]
- 9. Sedgwick B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004; 5:148–157. [DOI] [PubMed] [Google Scholar]
- 10. Wyatt M.D., Pittman D.L.. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006; 19:1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Falnes P.O., Johansen R.F., Seeberg E.. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002; 419:178–182. [DOI] [PubMed] [Google Scholar]
- 12. Trewick S.C., Henshaw T.F., Hausinger R.P., Lindahl T., Sedgwick B.. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002; 419:174–178. [DOI] [PubMed] [Google Scholar]
- 13. Lee D.H., Jin S.G., Cai S., Chen Y., Pfeifer G.P., O’Connor T.R.. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J. Biol. Chem. 2005; 280:39448–39459. [DOI] [PubMed] [Google Scholar]
- 14. Ringvoll J., Nordstrand L.M., Vagbo C.B., Talstad V., Reite K., Aas P.A., Lauritzen K.H., Liabakk N.B., Bjork A., Doughty R.W. et al.. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 2006; 25:2189–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nay S.L., Lee D.H., Bates S.E., O’Connor T.R.. Alkbh2 protects against lethality and mutation in primary mouse embryonic fibroblasts. DNA Repair (Amst.). 2012; 11:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aas P.A., Otterlei M., Falnes P.O., Vagbo C.B., Skorpen F., Akbari M., Sundheim O., Bjoras M., Slupphaug G., Seeberg E. et al.. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003; 421:859–863. [DOI] [PubMed] [Google Scholar]
- 17. Chen B., Liu H., Sun X., Yang C.G.. Mechanistic insight into the recognition of single-stranded and double-stranded DNA substrates by ABH2 and ABH3. Mol. Biosyst. 2010; 6:2143–2149. [DOI] [PubMed] [Google Scholar]
- 18. Dango S., Mosammaparast N., Sowa M.E., Xiong L.J., Wu F., Park K., Rubin M., Gygi S., Harper J.W., Shi Y.. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol. Cell. 2011; 44:373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu D., Samson L.D., Hubscher U., van Loon B.. The interaction between ALKBH2 DNA repair enzyme and PCNA is direct, mediated by the hydrophobic pocket of PCNA and perturbed in naturally-occurring ALKBH2 variants. DNA Repair (Amst.). 2015; 35:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Konishi N., Nakamura M., Ishida E., Shimada K., Mitsui E., Yoshikawa R., Yamamoto H., Tsujikawa K.. High expression of a new marker PCA-1 in human prostate carcinoma. Clin. Cancer Res. 2005; 11:5090–5097. [DOI] [PubMed] [Google Scholar]
- 21. Sundheim O., Vagbo C.B., Bjoras M., Sousa M.M., Talstad V., Aas P.A., Drablos F., Krokan H.E., Tainer J.A., Slupphaug G.. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006; 25:3389–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harrison L., Brame K.L., Geltz L.E., Landry A.M.. Closely opposed apurinic/apyrimidinic sites are converted to double strand breaks in Escherichia coli even in the absence of exonuclease III, endonuclease IV, nucleotide excision repair and AP lyase cleavage. DNA Repair (Amst.). 2006; 5:324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma W., Westmoreland J.W., Gordenin D.A., Resnick M.A.. Alkylation base damage is converted into repairable double-strand breaks and complex intermediates in G2 cells lacking AP endonuclease. PLos Genet. 2011; 7:e1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roca A.I., Cox M.M.. RecA protein: structure, function, and role in recombinational DNA repair. Prog. Nucleic Acid Res. Mol. Biol. 1997; 56:129–223. [DOI] [PubMed] [Google Scholar]
- 25. Lin Z., Kong H., Nei M., Ma H.. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. PNAS. 2006; 103:10328–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pittman D.L., Cobb J., Schimenti K.J., Wilson L.A., Cooper D.M., Brignull E., Handel M.A., Schimenti J.C.. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell. 1998; 1:697–705. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida K., Kondoh G., Matsuda Y., Habu T., Nishimune Y., Morita T.. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol. Cell. 1998; 1:707–718. [DOI] [PubMed] [Google Scholar]
- 28. Godin S.K., Sullivan M.R., Bernstein K.A.. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016; 94:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shivange G., Monisha M., Nigam R., Kodipelli N., Anindya R.. RecA stimulates AlkB-mediated direct repair of DNA adducts. Nucleic Acids Res. 2016; 44:8754–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nigam R., Mohan M., Shivange G., Dewangan P.K., Anindya R.. Escherichia coli AlkB interacts with single-stranded DNA binding protein SSB by an intrinsically disordered region of SSB. Mol. Biol. Rep. 2018; 45:865–870. [DOI] [PubMed] [Google Scholar]
- 31. Mazin A.V., Zaitseva E., Sung P., Kowalczykowski S.C.. Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. EMBO J. 2000; 19:1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qiu Y., Antony E., Doganay S., Koh H.R., Lohman T.M., Myong S.. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat. Commun. 2013; 4:2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shivange G., Kodipelli N., Monisha M., Anindya R.. A role for Saccharomyces cerevisiae Tpa1 protein in direct alkylation repair. J. Biol. Chem. 2014; 289:35939–35952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nigam R., Anindya R.. Escherichia coli single-stranded DNA binding protein SSB promotes AlkB-mediated DNA dealkylation repair. Biochem. Biophys. Res. Commun. 2018; 496:274–279.29326044 [Google Scholar]
- 35. Rodrigue A., Lafrance M., Gauthier M.C., McDonald D., Hendzel M., West S.C., Jasin M., Masson J.Y.. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 2006; 25:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kroetz M.B., Hochstrasser M.. Identification of SUMO-interacting proteins by yeast two-hybrid analysis. Methods Mol. Biol. 2009; 497:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller K.A., Yoshikawa D.M., McConnell I.R., Clark R., Schild D., Albala J.S.. RAD51C interacts with RAD51B and is central to a larger protein complex in vivo exclusive of RAD51. J. Biol. Chem. 2002; 277:8406–8411. [DOI] [PubMed] [Google Scholar]
- 38. Schild D., Lio Y.C., Collins D.W., Tsomondo T., Chen D.J.. Evidence for simultaneous protein interactions between human Rad51 paralogs. J. Biol. Chem. 2000; 275:16443–16449. [DOI] [PubMed] [Google Scholar]
- 39. Miller K.A., Sawicka D., Barsky D., Albala J.S.. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res. 2004; 32:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurumizaka H., Enomoto R., Nakada M., Eda K., Yokoyama S., Shibata T.. Region and amino acid residues required for Rad51C binding in the human Xrcc3 protein. Nucleic Acids Res. 2003; 31:4041–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lio Y.C., Mazin A.V., Kowalczykowski S.C., Chen D.J.. Complex formation by the human Rad51B and Rad51C DNA repair proteins and their activities in vitro. J. Biol. Chem. 2003; 278:2469–2478. [DOI] [PubMed] [Google Scholar]
- 42. Li Q., Sritharathikhun P., Motomizu S.. Development of novel reagent for Hantzsch reaction for the determination of formaldehyde by spectrophotometry and fluorometry. Anal. Sci. 2007; 23:413–417. [DOI] [PubMed] [Google Scholar]
- 43. Roy T.W., Bhagwat A.S.. Kinetic studies of Escherichia coli AlkB using a new fluorescence-based assay for DNA demethylation. Nucleic Acids Res. 2007; 35:e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tran T.Q., Ishak Gabra M.B., Lowman X.H., Yang Y., Reid M.A., Pan M., O’Connor T.R., Kong M.. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017; 15:e2002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. French C.A., Tambini C.E., Thacker J.. Identification of functional domains in the RAD51L2 (RAD51C) protein and its requirement for gene conversion. J. Biol. Chem. 2003; 278:45445–45450. [DOI] [PubMed] [Google Scholar]
- 46. Sigurdsson S., Van Komen S., Bussen W., Schild D., Albala J.S., Sung P.. Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev. 2001; 15:3308–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen F., Bian K., Tang Q., Fedeles B.I., Singh V., Humulock Z.T., Essigmann J.M., Li D.. Oncometabolites d- and l-2-hydroxyglutarate nhibit the AlkB family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 2017; 30:1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Badie S., Liao C., Thanasoula M., Barber P., Hill M.A., Tarsounas M.. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J. Cell Biol. 2009; 185:587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamato I., Sho M., Shimada K., Hotta K., Ueda Y., Yasuda S., Shigi N., Konishi N., Tsujikawa K., Nakajima Y.. PCA-1/ALKBH3 contributes to pancreatic cancer by supporting apoptotic resistance and angiogenesis. Cancer Res. 2012; 72:4829–4839. [DOI] [PubMed] [Google Scholar]
- 50. Deans A.J., West S.C.. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011; 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuznetsov S.G., Haines D.C., Martin B.K., Sharan S.K.. Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res. 2009; 69:863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sullivan M.R., Bernstein K.A.. RAD-ical New Insights into RAD51 Regulation. Genes. 2018; 9:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stefansson O.A., Hermanowicz S., van der Horst J., Hilmarsdottir H., Staszczak Z., Jonasson J.G., Tryggvadottir L., Gudjonsson T., Sigurdsson S.. CpG promoter methylation of the ALKBH3 alkylation repair gene in breast cancer. BMC Cancer. 2017; 17:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Masson J.Y., Tarsounas M.C., Stasiak A.Z., Stasiak A., Shah R., McIlwraith M.J., Benson F.E., West S.C.. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001; 15:3296–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park J.Y., Singh T.R., Nassar N., Zhang F., Freund M., Hanenberg H., Meetei A.R., Andreassen P.R.. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. 2014; 33:4803–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yi C., He C.. DNA repair by reversal of DNA damage. Cold Spring Harb. Perspect. Biol. 2013; 5:a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petermann E., Helleday T.. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010; 11:683–687. [DOI] [PubMed] [Google Scholar]
- 58. Zhou Y., Caron P., Legube G., Paull T.T.. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014; 42:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cunningham J.M., Cicek M.S., Larson N.B., Davila J., Wang C., Larson M.C., Song H., Dicks E.M., Harrington P., Wick M. et al.. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014; 4:4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vaz F., Hanenberg H., Schuster B., Barker K., Wiek C., Erven V., Neveling K., Endt D., Kesterton I., Autore F. et al.. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010; 42:406–409. [DOI] [PubMed] [Google Scholar]
- 61. Meindl A., Hellebrand H., Wiek C., Erven V., Wappenschmidt B., Niederacher D., Freund M., Lichtner P., Hartmann L., Schaal H. et al.. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010; 42:410–414. [DOI] [PubMed] [Google Scholar]
- 62. Choi S.Y., Jang J.H., Kim K.R.. Analysis of differentially expressed genes in human rectal carcinoma using suppression subtractive hybridization. Clin. Exp. Med. 2011; 11:219–226. [DOI] [PubMed] [Google Scholar]
- 63. Shimada K., Nakamura M., Anai S., De Velasco M., Tanaka M., Tsujikawa K., Ouji Y., Konishi N.. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009; 69:3157–3164. [DOI] [PubMed] [Google Scholar]
- 64. Tasaki M., Shimada K., Kimura H., Tsujikawa K., Konishi N.. ALKBH3, a human AlkB homologue, contributes to cell survival in human non-small-cell lung cancer. Br. J. Cancer. 2011; 104:700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barlund M., Monni O., Kononen J., Cornelison R., Torhorst J., Sauter G., Kallioniemi O.-P., Kallioniemi A.. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res. 2000; 60:5340–5344. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.