

Abstract
Antimicrobial resistance (AMR) is a threat to global public health and the identification of genetic determinants of AMR is a critical component to epidemiological investigations. High-throughput sequencing (HTS) provides opportunities for investigation of AMR across all microbial genomes in a sample (i.e. the metagenome). Previously, we presented MEGARes, a hand-curated AMR database and annotation structure developed to facilitate the analysis of AMR within metagenomic samples (i.e. the resistome). Along with MEGARes, we released AmrPlusPlus, a bioinformatics pipeline that interfaces with MEGARes to identify and quantify AMR gene accessions contained within a metagenomic sequence dataset. Here, we present MEGARes 2.0 (https://megares.meglab.org), which incorporates previously published resistance sequences for antimicrobial drugs, while also expanding to include published sequences for metal and biocide resistance determinants. In MEGARes 2.0, the nodes of the acyclic hierarchical ontology include four antimicrobial compound types, 57 classes, 220 mechanisms of resistance, and 1,345 gene groups that classify the 7,868 accessions. In addition, we present an updated version of AmrPlusPlus (AMR ++ version 2.0), which improves accuracy of classifications, as well as expanding scalability and usability.
INTRODUCTION
Antimicrobial resistance (AMR) is considered one of the foremost threats to the health of humans and animals (1–4). Accordingly, investigating the emergence and dissemination of AMR genetic determinants has become a priority (5–7). While antimicrobial drugs (AMDs) are the most studied and commonly discussed antimicrobial compounds, bacteria can also harbor genes for resistance to biocides that are important as disinfectants and sanitizing agents (e.g. peroxide and acetate) as well as genes that encode resistance to metals that have antimicrobial activity (e.g. copper and zinc). Many governments, including the United States and European Union, as well as public health organizations like the Food and Agricultural Organization of the United Nations and the World Health Organization have programs for addressing AMR, all of which identify comprehensive surveillance as a critical component of control efforts (5,8). However, a unique challenge for control as well as for investigation of AMR is that many genetic determinants can be transferred and spread among different types of bacteria. Understanding—and eventually predicting—the emergence and transmission of AMR requires characterization of resistance determinants across pathogens and non-pathogens (9–11). This microbiome-wide characterization is now possible through use of high-throughput genetic sequencing (HTS), which provides access to the genetic determinants for AMR across all microbial genomes in a sample (i.e. the metagenome). This approach has highlighted the importance of microbial ecology in AMR emergence and persistence, including interaction between processes such as horizontal gene transfer and cross-selection. For example, biocide and metal resistances are now known to play an important role in AMR dynamics under some circumstances, and can promote emergence or dissemination of AMR genes through either co-localization on linked DNA or co-occurrence within positively-selected bacterial taxa (12–14). Hence, there is great potential for advancing our understanding of AMR through studying entire ecologies of resistance determinants (i.e. the resistome) (15–19). To address this need, we previously presented MEGARes, a comprehensive database of AMR genes (20), and AmrPlusPlus (AMR++), which interfaced with MEGARes to identify and quantify the AMR genes contained in a metagenomic dataset.
To-date, MEGARes 1.0 and AMR++ 1.0 have been used and cited over 85 different times to analyze sequence data for a variety of applications, ranging from agricultural studies (21,22), to public health studies, to ecological surveys (23–27). Most recently, it was used by the MetaSub Consortium to identify AMR genes in samples collected from subway systems and urban sites across the world (28). Yet, detection and characterization of genetic elements that mediate AMR continues to be problematic for multiple reasons, many related to challenges with AMR databases, as noted by Danko et al. (28). First, the evolutionary dynamics of AMR can result in rapidly-accumulating sequence variation, meaning that AMR databases must be continually updated to include novel variants (29–32). Moreover, the genetic mechanisms of AMR are varied, meaning AMR gene identification within metagenomic datasets must consider AMR genes on a mechanism-by-mechanism basis. For example, databases and corresponding ontologies must differentiate between point mutations that modify antibiotic target sites (e.g. site mutations in gyrA and rpoB genes that mediate resistance to quinolone and rifamycin antibiotics, respectively), versus full-length gene sequences that confer resistance when expressed as functional proteins (e.g. Qnr or Tet[M], which mediate resistance to quinolones and tetracyclines, respectively). These nuances must be accounted for within the bioinformatic approach for identifying and counting resistance determinants within metagenomic data—and this often means that databases must be structured accordingly. In another example of such nuance, there are genes that confer resistance to multiple compounds (including biocides, metals and antibiotic drugs), while other genes confer resistance to only one compound. The ontology used to classify metagenomic sequence reads that align to resistance determinants must accommodate these scenarios, while also supporting efficient, accurate bioinformatic analysis and proper downstream statistical analyses.
Given the diversity of AMR mechanisms, AMR databases often aggregate subsets of resistance genes based on mechanism or class of resistance. For example, the Lahey Clinic database (33)—which is now maintained by the National Center for Biotechnology Information (NCBI) as part of the Bacterial Antimicrobial Resistance Reference Gene Database (34)—characterizes genes that confer resistance to betalactam compounds; the Antibacterial Biocide and Metal Resistance Genes (BacMet) database targets genes that confer resistance to biocide and metal compounds (35); and ResFinder focuses on acquired antimicrobial resistance genes (36). In some cases, databases are accompanied by analytical tools to support use of the database itself. For example, ResFinder includes a web-based portal and Python script for uploading assembled genome or contig sequences and identifying AMR genes (36). The Comprehensive Antibiotic Resistance Database (CARD) represents one of the most encompassing ongoing efforts for curation of AMR genes, providing detailed annotations through the Antibiotic Resistance Ontology (ARO) (37); this ontology was developed to provide rich metadata about each accession, and the structure is correspondingly complex; however, when sketched as a graph, there are multiple cycles and incomplete levels, which presents challenges for short-read metagenomic data (20). Therefore, while each of these active and widely utilized AMR databases represent an important resource for AMR research and surveillance, no single resource currently enables structured, comprehensive and statistically appropriate analysis of metagenomic data for all types of antimicrobial compounds, including biocides and metals (19,38).
To fill this resource gap, we update our previous effort and present MEGARes 2.0, a database that incorporates standardized accessions for a comprehensive set of previously published resistance determinants for antimicrobial drugs, biocides and metals. By utilizing the same acyclical, hierarchical ontology as presented in MEGARes 1.0 (20), we are able to accommodate the unique characteristics of short-read metagenomic data and comprehensive resistome analysis, while reflecting the complex and sometimes interconnected genetic relationships between resistance mechanisms for antibiotic drug, metal and biocide compounds. As with MEGARes 1.0, this structure allows for binning of alignment counts into mutually-exclusive categories, which can then be aggregated to each of the complete levels of the hierarchical ontology. As described in the original publication for MEGARes 1.0, this type of structure prevents multiple counting of alignments and thus accommodates downstream statistical analyses. New to MEGARes 2.0, we also enable secondary bioinformatic analyses for groups of AMR genes that require special consideration, i.e. confirmation of point mutations or prediction of gene overexpression. Additionally, we present AMR++ 2.0, a bioinformatics pipeline that addresses many of the expanding use scenarios of metagenomic data, while also building capacity to support additional AMR-related analyses.
UPDATES TO MEGARES AND AMR++
Incorporating the antibacterial biocide and metal resistance genes (BacMet) into MEGARes 2.0
BacMet aggregates protein sequences that have been experimentally confirmed to confer resistance to biocides and metals (35). BacMet compiled metadata for genetic determinants from publicly available resources such as UniProtKB and NCBI’s GenBank to identify gene accessions that had been experimentally shown to confer phenotypic resistance to biocides and metal compounds through removal, mutation, insertion or overexpression of those genetic determinants. BacMet's content and accession criteria are unique, making it an important resource for investigations of resistance. However, the native annotation structure constrains BacMet's use for metagenomic analyses. Specifically, BacMet separately classified protein sequences with respect to each of 111 different compounds; this multi-classification scheme could lead to false inflation of sequence classifications in the context of a metagenomic dataset (39).
Method for determining nucleotide sequences from amino acid sequences contained in BacMet
The 753 accessions for resistance proteins contained in version 2.0 of the database ‘BacMet Experimentally Confirmed Resistance Genes’ were downloaded on 6 December 2018, from the hosting website: http://bacmet.biomedicine.gu.se/download/BacMet2_EXP_database.fasta. The protein accession ID for each of the 753 accessions were used to conduct a manual search on the UniProtKB website: https://www.uniprot.org/. Then, links in the ‘Cross-references’ section were followed and all corresponding nucleotide sequences were downloaded from the European Bioinformatics Institute. In total, 1,642 nucleotide sequences were identified and collated. Sequences were clustered by homology and redundant accessions were removed using CD-HIT-EST with the following parameters: -G 0 -c 1.0 –AS 0 –AL 0 –AI 1.0 –aS 1.0. A single representative sequence for each cluster (n = 959) was saved and aggregated for all gene accessions in a single fasta file.
Annotation of BacMet genes
BacMet's original classification scheme for each accession included the protein family, the bacterial species in which it was originally described, the gene coordinates within the source genome, the compounds to which it can confer resistance, a short text description, and a PubMed reference. For comparison, the hierarchical classification ontology developed for MEGARes 1.0 included (from highest to lowest levels) the ‘Class’ of antimicrobial compounds to which a gene confers resistance (e.g. betalactams), the ‘Mechanism’ by which this resistance is conferred (e.g. betalactamases), the ‘Group’ name of the genes (e.g. Group A betalactamases), and the ID for each individual gene accession. In order to classify BacMet accessions for inclusion in the MEGARes 2.0 ontology, we used the metadata information provided by BacMet: accessions associated with resistance to a single compound were placed in an antimicrobial class specific to that compound (e.g. Copper resistance), whereas genes associated with resistance to multiple compounds were placed in a more general classification taxon (e.g. Multi-metal resistance). This resulted in the addition of a fifth, highest hierarchical level within the MEGARes 2.0 ontology; we termed this level the ‘Type’ of compound to which the accession confers resistance (e.g. drug, biocide, metal, multi-compound). For example, the yfeABCD accessions are associated with resistance to both iron and manganese, and thus would be categorized as Type: ‘Metals’. Resistance determinants associated with multiple compound types (e.g. antimicrobial drugs, metals, and biocides) were classified as Type: ‘Multi-compound’; this is exemplified by cmeABC genes, which confer resistance to a variety of drugs, metals and biocides. For the final set of accessions added from BacMet, a total of 959 accessions were binned into three types, 32 classes, 85 mechanisms and 595 gene Groups. As part of our curation, we also added published references for each nucleotide sequence, when available.
Updating MEGARes resistance determinants for AMDs from publicly available databases
The first version of MEGARes included resistance determinants for AMDs obtained from ResFinder (downloaded November 2015), ARG-ANNOT (downloaded November 2015), the Comprehensive Antibiotic Resistance Database (downloaded December 2015), and the National Center for Biotechnology Information (NCBI) Lahey Clinic beta-lactamase archive (downloaded December 2015). For MEGARes 2.0, these same sources were interrogated for the addition of new sequences since the release of MEGARes 1.0. The ARG-ANNOT database had not been updated and the website was no longer supported, and therefore no additional accessions from ARG-ANNOT were included in the update. The Lahey Clinic beta-lactamase archive had been consolidated into NCBI’s Bacterial Antimicrobial Resistance Reference Gene Database and new sequences were obtained from that source. ResFinder, a repository for acquired AMR determinants, has been updated frequently and was considered for inclusion in our update. Currently, CARD provides monthly updates driven by manual literature curation, computational text mining, and genome analysis. Importantly, CARD amends AMR gene annotations to reflect the latest published data available regarding their resistance phenotype. Therefore, we evaluated all accessions in the databases above and hand-curated these new AMR genes to incorporate them into the MEGARes 2.0 database using our updated hierarchical acyclic classification scheme.
Method for incorporating new sequences into MEGARes
Additions to be included in MEGARes 2.0 were obtained by downloading and incorporating new sequences from the latest available versions of CARD, the Bacterial Antimicrobial Resistance Reference Gene Database, and ResFinder, respectively. Specifically, CARD version 3.0.3 (21 August 2019 update) was downloaded in a single compressed document (https://card.mcmaster.ca/download/card-data.tar.bz2±) that contained AMR accessions in nucleotide and amino acid format. Each accession was categorized into one of five models: protein homolog (n = 2,404), knockout (n = 17), over-expression (n = 14), variant (n = 152) or rRNA gene variant (n = 80). Due to the sparse sequencing coverage that characterizes many metagenomic datasets (and the resulting inability to rule out false-negative classification), the knockout models were not included in MEGARes 2.0. CARD’s updates also included corrections for sequences that had been previously included in MEGARes 1.0, as well as modified header formats for each accession. Thus, to maintain harmony with CARD, we removed 2,275 CARD sequences that had been previously included in MEGARes 1.0, leaving 1,549 accessions obtained from other sources. Then, the 2,866 accessions contained in CARD version 3.0.3 were concatenated with remaining MEGARes 1.0 accessions in a single fasta file. CD-HIT was then used to remove sequences with 100% homology, resulting in a file with 4,172 unique sequences. Next, we integrated updates from NCBI’s Bacterial Antimicrobial Resistance Reference Gene Database, which contained 5,782 sequences (downloaded from: ftp://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinderPlus/data/2019-07-10.1/). Since MEGARes 2.0 focuses on resistance mechanisms for antibacterial compounds (i.e. drugs, metals and biocides), we removed sequences in the Bacterial Antimicrobial Resistance Reference Gene Database associated with ‘Heat’ and ‘Virulence’ (n = 573 accessions). The nine fasta files associated with ResFinder version 3.2 (each corresponding to accessions that confer resistance to different drug classes) were downloaded on 19 June 2019 and concatenated, providing an additional 3,095 accessions. The combined accessions from MEGARes 1.0, CARD, ResFinder, and the Bacterial Antimicrobial Resistance Reference Gene Database were again clustered by sequence homology using CD-HIT and 100% redundant sequences were removed using the parameters listed above. The final resulting MEGARes 2.0 database contained 7,868 unique AMR resistance sequences.
Another important aspect of the updated MEGARes 2.0 database is the specific annotation of genes that require the presence of single nucleotide polymorphisms (SNPs) at specific loci in order to confer resistance. To annotate and allow these genes to be identified for further analysis, we modified their sequence headers to include the label ‘RequiresSNPConfirmation’. This header is used to flag these accessions for input into a new secondary analysis component of the AMR++ 2.0 bioinformatic pipeline. This new component integrates the Resistance Gene Identifier (RGI) to confirm the presence of amino acid residues required to confer resistance (37). By pairing the MEGARes 2.0 header format with RGI and wrapping this into the AMR++ 2.0 pipeline, we thus enable automated processing of these sequences for confirmatory in silico testing to confirm the presence of resistance-conferring SNPs. This removes the burden of manual SNP analysis for these genes, while improving the accuracy of AMR gene identification from metagenomic data.
Updates to the MEGARes classification and annotation scheme
Following the addition of biocide and metal resistance accessions to MEGARes 2.0, all sequences were analyzed for homology using CD-HIT with a clustering threshold of 80% sequence homology using the following parameters: -c 0.8 -g 1. Each cluster of sequences was inspected and compared to published reference sequences using BLAST to confirm MEGARes 2.0 classifications and annotations. No discrepancies were identified between annotations from different source databases for AMR determinants associated with resistance to a single compound class. However, analysis of sequence homology in our comprehensive compilation of resistance determinants to different types of compounds (i.e. drugs, biocides and metals) allowed us to identify genetic sequences that had not previously been identified as causing resistance to multiple types of compounds. For example, we identified accessions that were classified in the BacMet database as causing resistance to biocides but that contained the same sequence as accessions previously identified in MEGARes 1.0 and other AMD resistance databases as encoding for multi-drug efflux pumps. The presence of these multi-compound cross-resistance mechanisms necessitated the expansion of the MEGARes classification scheme and the partitioning of annotation files. As noted previously, a new highest hierarchical level for compound ‘ type’ was created in the MEGARes ontology to distinguish between accessions that confer resistance to compounds of the following types: drugs, metals, biocides or genes that act on multiple types of compounds (Figure 1). In MEGARes 2.0, the nodes of the acyclic hierarchical ontology include four antimicrobial compound types, 57 classes, 220 mechanisms of resistance and 1,345 gene groups that classify the 7,868 accessions. In addition, we partitioned the annotations into two different annotation files in order to support varying resistome analysis scenarios: the file ‘megares_annotations_v2.0.csv’ contains annotations for resistance determinants related to all types of antimicrobial compounds (i.e. drugs, biocides and metals), while the annotation file ‘megares_drug_annotations_v2.0.csv’ contains only annotations for accessions that confer resistance to AMDs. Both annotation files contain a new metadata column (labeled ‘Notes’), which provides details about annotation decisions, thus increasing the transparency regarding especially the more complicated resistance classifications. As with any classification schema, the MEGARes ontology attempts to strike a balance between useful aggregation of accessions into meaningful (and interpretable) categories while avoiding potential oversimplification of important biological details of microbial genetic dynamics.
Figure 1.

Diagram representing nodes in the five levels of the acyclic hierarchical ontology for MEGARes 2.0 (compound type, classes, mechanisms of resistance and gene groups that classify accessions).
Future updates
MEGARes 2.0 pulls accessions from multiple source databases, each with their own update schedule. To maintain congruity with these resources, MEGARes 2.0 will be updated at least yearly. In cases where new accessions are found to have 100% nucleotide sequence homology to an accession already in MEGARes, we will maintain the MEGARes accession as the single representative of that gene, with links to the original database from which the accession was sourced. To annotate new, non-homologous sequences, we will cluster all such sequences with existing MEGARes sequences using CD-HIT as described above (i.e. at 80% homology). Sequences that cluster with existing MEGARes accessions will be automatically assigned the same annotation. Sequences that do not cluster with any existing MEGARes accessions will be manually handled in the following manner: first, the annotations from the original databases will be identified; second, primary literature describing the accessions will be identified; third, the cluster with the highest homology to each new accession will be identified, and the annotation of this ‘nearest cluster’ will be compared to the annotation from the original database and the primary literature. In cases where annotations from the original database, the ‘nearest cluster’ and the primary literature disagree, we will initiate a discussion with the curators of the originating database to resolve these discrepancies. If a new annotation label must be created at any level of the annotation hierarchy, it will be integrated into the existing MEGARes ontology such that its acyclic, hierarchical and full-level classification scheme is maintained. Finally, the annotation decision process will be summarized in the ‘Notes’ column of the annotation file (see above) and the new accession will be assigned a header that conforms to the MEGARes style (see below).
Standardized headers and database table relationships
MEGARes is structured as a relational database in which the fasta header of each accession is the primary key to attributes stored in additional tables that are formatted as comma-delimited files. In MEGARes 1.0, we used headers exactly as they appeared in the original source databases; however, this resulted in inconsistent header formats and content across different accessions. For MEGARes 2.0, we improved accession headers by establishing a standardized format to include a unique accession ID, compound type, resistance class, resistance mechanism, resistance group, source database, and an optional label to signify that the accession requires further analysis to confirm presence of resistance-conferring SNPs. These standardized headers are machine-readable and easily parse-able aiding the ability to perform analysis on various subsets of classified sequencing reads. Importantly, MEGARes accession headers can still be linked to the originating source (i.e. BacMet, CARD, Resfinder, ARG-ANNOT or Lahey/NCBI) through our relational database. For extended browsing of the MEGARes annotation and sequences, we offer a web interface (http://megares.meglab.org). The content of MEGARes is summarized on the home page as a D3 interactive graphic (40) and users can ‘Browse’ database features or ‘Search’ for specific genes. All database files are available in the ‘Download’ section, and users can restrict downloads to include only the results of their search term.
AMR++ 2.0: bioinformatic pipeline update
The AMR++ bioinformatic pipeline was designed to analyze paired-end, short-read sequencing data (with support for multiple samples), interfacing with MEGARes to produce a count matrix of alignments to AMR gene accessions contained in each sample. AMR++ 1.0 was implemented via nextflow scripting language (41) and made publicly available via Galaxy (42), an easy-to-use web-based platform. Although the Galaxy-centered approach offers relatively high usability, this platform is unreasonably slow for large-scale projects. Therefore, with the release of AMR++ 2.0, we provide support for both proprietary and non-proprietary computing clusters, facilitating large-scale resistome data analysis. Access to the necessary bioinformatic tools is also facilitated through the use of Singularity containers (43), which eliminates the need for ‘sudo’ access in computing clusters. Extended descriptions of each step of the pipeline have been previously reported (20) and updated documentation is available on the MEGARes website (http://megares.meglab.org). Here, we provide an overview of AMR++ 2.0 and its new features.
Quality trimming and host filtering
AMR++ processes raw sequencing reads from metagenomic samples through a series of steps to characterize the resistome and microbiome (Figure 2). Briefly, reads are trimmed for quality and adapter contamination using Trimmomatic (44), after which reads are aligned to the presumptive host genome using Burrows-Wheeler-Aligner (BWA) (45); reads identified as host are removed using BEDTools (46). Trimming and filtering statistics for these processes are automatically summarized in tab-delimited text files using customized python scripts.
Figure 2.
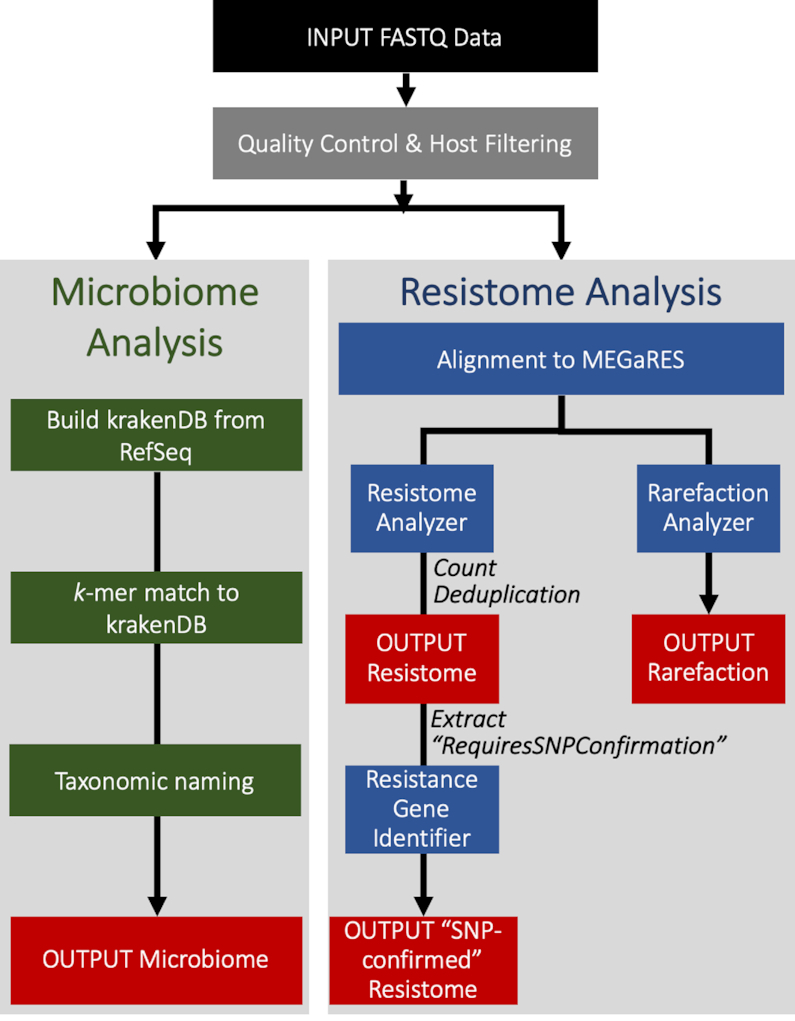
Overview of AMR++ 2.0 pipeline. Input fastq files are trimmed and aligned to host genome(s) before being analyzed for resistome and microbiome content using MEGARes and Kraken2 databases, respectively. Relevant resistome output can be piped into the Resistance Gene Identifier for secondary confirmation of specific resistance-conferring SNPs. Output content is indicated by red boxes.
Resistome analysis
High-quality, non-host reads are then aligned to the MEGARes database using BWA to produce a SAM file that is used as input for (a) resistome characterization, and (b) rarefaction analysis. For resistome analysis, the SAM file is parsed using ResistomeAnalyzer, a custom C++ program that minimizes the potential for false-positive classification by removing accessions with sparse alignments (source code available at https://github.com/cdeanj/resistomeanalyzer). The filtering criterion can be user-defined and is based on a threshold rule for the ‘gene fraction,’ defined as the minimum proportion of nucleotides in a given reference sequence that must be aligned by at least one read in order to be considered as ‘present’ in the original metagenomic sample. These filtered data are then used to produce a count of aligned reads for each accession in each sample, using the header annotations as the basis for hierarchical classifications, starting at the gene accession ID, and then aggregating upward to the group, mechanism, class, and resistance type levels. To facilitate statistical analysis of resistome results, accession-level counts for all samples are combined into a single count matrix, and then counts are aggregated up the ontological hierarchy to produce separate count matrices for each level in the MEGARes ontology (i.e. group, mechanism, class, and type).
AMR++ 2.0 also integrates Samtools (47) functionality to allow filtering of reads with identical sequence alignments, thus providing a method for controlling for potential PCR duplication; this step can be omitted from the pipeline based on user preference. In this update, we have also incorporated the Resistance Gene Identifier (RGI) tool to confirm that relevant alignments contain the specific SNPs required to confer resistance (37). This secondary analysis is crucial for mechanisms of resistance that involve single nucleotide changes in genes that otherwise perform non-resistance-associated functions within bacteria, e.g. rpoB or gyrA. Likewise, multi-drug efflux pumps can require the presence of mutations that promote overexpression to confer resistance to AMDs, e.g. acrAB or mexAB. The biological relevance of these SNPs and the expression of efflux pumps can be context-dependent, i.e. can be contingent upon the bacterial species carrying the mutation, and interpretation of relevance is incumbent upon users. These important complexities are well characterized but difficult to confirm within short-read metagenomic data, therefore, MEGARes users must use their discretion to appropriately interpret results in the context in which they are applying this resource. To enable inclusion of these types of resistance mechanisms within resistome analysis while also guarding against false positive AMR gene identification, AMR++ integrates the ‘Perfect’ algorithm implemented in RGI. This integration is enabled through the standardized MEGARes 2.0 headers, which contain the label ‘RequiresSNPConfirmation’ for accessions requiring this additional analysis. The AMR++ 2.0 pipeline will extract relevant alignments from the SAM file into a single fasta file for analysis with RGI. Using Prodigal (48), RGI translates sequences in all six reading frames to predict open reading frames, and then detects homologs to proteins in CARD’s database using DIAMOND (49). Results are then filtered with significance thresholds based on bitscore cut-offs with three different algorithms: Perfect, Strict, and Loose. AMR++ 2.0 utilizes RGI’s ‘Perfect’ detection algorithm, which requires that the relevant reads match the reference sequence with 100% homology, and therefore contain the resistance-conferring SNP(s). The AMR++ pipeline then combines the RGI-confirmed read counts with the alignment counts for accessions that did not require SNP confirmation, to produce a comprehensive count matrix with the final results for all samples. Lastly, the RarefactionAnalyzer program provides an assessment of sequencing depth through rarefaction analysis of the alignment SAM files at the group, mechanism, and class levels. Further details about use of RarefactionAnalyzer, including various user-defined parameters, are available at https://github.com/cdeanj/rarefactionanalyzer).
Microbiome characterization
AMR++ 2.0 also provides the ability to analyze the microbial composition of metagenomic samples using Kraken2 (50), a metagenomic classifier based on exact k-length subsequence (k-mer) matches. By comparing all k-mers found in a set of sequence reads to those from a set of reference genomes, Kraken2 achieves accurate classification and a flexible scoring system that can be used to refine the specificity of classification at lower taxonomic levels. AMR++ 2.0 provides both the standard Kraken2 output as well as the results obtained from setting the ‘–confidence’ flag with the highest possible value (i.e. ‘1’), which generates the most conservative scoring. Results for each metagenomic sample are then parsed using a custom python script included within the AMR++ 2.0 pipeline; counts for strain-level classification are aggregated to the species level and the entire taxonomic lineage of each feature is output into a matrix that contains counts for each feature within each sample. While short-read metagenomic data is not always well-suited to co-localization analyses (51), users who wish to perform microbiome-resistome co-occurrence analysis can then use these count matrices as input into separate statistical analyses [see, e.g. (52)].
Pipeline portability
AMR++ 2.0 comes pre-installed and fully integrated with all necessary bioinformatic tools and dependencies within a publicly accessible Amazon Machine Image (AMI) named Microbial_Ecology_Group_AMR_AMI, allowing users to easily employ the AMR++ 2.0 pipeline within the Amazon Web Services (AWS) ecosystem. With this approach, users pay for the cost of a suitable AWS EC2 instance without the challenge of accessing large computing clusters and individually installing each piece of software necessary to run the pipeline (including all dependencies). Integration within AWS also allows users to scale the computing resources to fit the needs of any project size.
In addition, we improved the usability of AMR++ 2.0 on non-AWS computing clusters by providing the configuration file templates that nextflow uses to interface with SLURM Workload Manager, a common job scheduler. Easy access to required bioinformatic tools is made available through the use of a Singularity container (https://singularity-hub.org/collections/3418), minimizing challenges of installation on computational servers (53).
DISCUSSION
Here we present MEGARes 2.0, a database resource developed specifically for high-throughput sequencing analysis of previously published AMR genes within metagenomic data. Our goal is to provide a single, comprehensive database that incorporates all published reference sequences for genetic determinants of resistance to antimicrobial drugs, metals, and biocides. Additionally, MEGARes 2.0 uses an acyclic hierarchical annotation structure that facilitates high-throughput classification and subsequent statistical analyses. MEGARes does not replace source databases such as CARD and ResFinder, which provide detailed descriptions of accessions, but rather offers a simpler, hierarchical ontological structure that is specially adapted for the unique characteristics of high-throughput analysis of metagenomic resistome data. In addition, MEGARes focuses on previously published sequences, rather than novel variant discovery. Metagenomic data are an important resource for novel AMR gene discovery, which requires specialized tools (e.g. fARGene (54), Meta-MARC (39) and PCM (55) and databases (e.g. Mustard (56)). In contrast, MEGARes 2.0 attempts to facilitate reproducible, systematic and statistically-appropriate bioinformatic identification of previously published sequences associated with resistance to AMDs, metals and biocides. In MEGARes 2.0, we have approximately doubled the number of accessions in comparison to the previous version. These accessions expand coverage to include genes associated with resistance to biocides and metals by incorporating accessions from BacMet (35). Similarly, we integrate a large number of additional AMD resistance genes and advance the ability to identify and confirm the presence of SNPs in genes that require specific gene variants to confer resistance by integration with CARD’s RGI tool. Confirmation of these requisite SNPs is an especially challenging task with metagenomic data, and the seamless integration of RGI into a pre-packaged resistome pipeline will greatly ease resistome analyses of these genes. Additionally, with the release of AMR++ 2.0, we increase accessibility by providing a publicly accessible Amazon Machine Image. We believe that MEGARes 2.0 integrated within AMR++ 2.0 will aid AMR research and public health epidemiology by promoting the adoption of HTS and metagenomic analysis.
DATA AVAILABILITY
The MEGARes 2.0 reference database and annotation files are available at https://megares.meglab.org. All AMR++ 2.0 pipeline source files are freely available on GitHub at https://github.com/meglab-metagenomics/AmrPlusPlus_v2.
ACKNOWLEDGEMENTS
We thank the undergraduate students who assisted with manual curation and annotation of new gene accessions. We also thank users of MEGARes and AmrPlusPlus that have provided feedback regarding this work.
FUNDING
USDA NIFA [2015-68003-23048]; College of Veterinary Medicine and Biomedical Sciences - Texas A&M University; N.N. was supported by NIH [R01 1R01AI141810-01]; Agricultural Research, Education, Extension and Technology Transfer program at the University of Minnesota; C.D. was supported by USDA NIFA [2018-51300-28563]. Funding for open access charge: Startup funding for Dr Morley's current position.
Conflict of interest statement. None declared.
REFERENCES
- 1. Huttner A., Harbarth S., Carlet J., Cosgrove S., Goossens H., Holmes A., Jarlier V., Voss A., Pittet D. for the World Healthcare-Associated Infections Forum participants Antimicrobial resistance: a global view from the 2013 World Healthcare-Associated Infections Forum. Antimicrob. Resist. Infect. Control. 2013; 2:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jasovský D., Littmann J., Zorzet A., Cars O. Antimicrobial resistance—a threat to the world's sustainable development. Ups. J. Med. Sci. 2016; 121:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prestinaci F., Pezzotti P., Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Glob. Health. 2015; 109:309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roca I., Akova M., Baquero F., Carlet J., Cavaleri M., Coenen S., Cohen J., Findlay D., Gyssens I., Heure O.E. et al. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 2015; 6:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Humphreys G., Fleck F. United Nations meeting on antimicrobial resistance. Bulletin of the World Health Organization. 2016; 94.9:638–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cecchini M., Langer J., Slawomirski L. Antimicrobial resistance in G7 countries and beyond: economic issues, policies and options for action. Organization for Economic Co-operation and Development. 2015; 1:1–75. [Google Scholar]
- 7. Metcalfe S., Baker M.G., Freeman J., Wilson N., Murray P. Combating antimicrobial resistance demands nation-wide action and global governance. N. Z. Med. J. 2016; 129:8–14. [PubMed] [Google Scholar]
- 8. Abu Sin M., Nahrgang S., Ziegelmann A., Clarici A., Matz S., Tenhagen B.-A., Eckmanns T. [Global and national strategies against antibiotic resistance]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2018; 61:507–514. [DOI] [PubMed] [Google Scholar]
- 9. Soucy S.M., Huang J., Gogarten J.P. Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 2015; 16:472–482. [DOI] [PubMed] [Google Scholar]
- 10. von Wintersdorff C.J.H., Penders J., van Niekerk J.M., Mills N.D., Majumder S., van Alphen L.B., Savelkoul P.H.M., Wolffs P.F.G. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol. 2016; 7:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall James P. J., Brockhurst Michael A., Ellie Harrison Sampling the mobile gene pool: innovation via horizontal gene transfer in bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2017; 372:20160424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pal C., Bengtsson-Palme J., Kristiansson E., Larsson D.G.J. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics. 2015; 16:964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li B., Yang Y., Ma L., Ju F., Guo F., Tiedje J.M., Zhang T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015; 9:2490–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma L., Xia Y., Li B., Yang Y., Li L.-G., Tiedje J.M., Zhang T. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ. Sci. Technol. 2016; 50:420–427. [DOI] [PubMed] [Google Scholar]
- 15. Francis S.S., Riley L.W. Metagenomic epidemiology: a new frontier. J. Epidemiol. Community Health. 2015; 69:306–308. [DOI] [PubMed] [Google Scholar]
- 16. Hu Y., Gao G.F., Zhu B. The antibiotic resistome: gene flow in environments, animals and human beings. Front. Med. 2017; 11:161–168. [DOI] [PubMed] [Google Scholar]
- 17. Baquero F. Metagenomic epidemiology: a public health need for the control of antimicrobial resistance. Clin. Microbiol. Infect. 2012; 18:67–73. [DOI] [PubMed] [Google Scholar]
- 18. Port J.A., Cullen A.C., Wallace J.C., Smith M.N., Faustman E.M. Metagenomic frameworks for monitoring antibiotic resistance in aquatic environments. Environ. Health Perspect. 2014; 122:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McArthur A.G., Tsang K.K. Antimicrobial resistance surveillance in the genomic age. Ann. N. Y. Acad. Sci. 2017; 1388:78–91. [DOI] [PubMed] [Google Scholar]
- 20. Lakin S.M., Dean C., Noyes N.R., Dettenwanger A., Ross A.S., Doster E., Rovira P., Abdo Z., Jones K.L., Ruiz J. et al. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017; 45:D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doster E., Rovira P., Noyes N.R., Burgess B.A., Yang X., Weinroth M.D., Lakin S.M., Dean C.J., Linke L., Magnuson R. et al. Investigating effects of tulathromycin metaphylaxis on the fecal resistome and microbiome of commercial feedlot cattle early in the feeding period. Front. Microbiol. 2018; 9:1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huebner K.L., Martin J.N., Weissend C.J., Holzer K.L., Parker J.K., Lakin S.M., Doster E., Weinroth M.D., Abdo Z., Woerner D.R. et al. Effects of a Saccharomyces cerevisiae fermentation product on liver abscesses, fecal microbiome, and resistome in feedlot cattle raised without antibiotics. Sci. Rep. 2019; 9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doan T., Arzika A.M., Hinterwirth A., Maliki R., Zhong L., Cummings S., Sarkar S., Chen C., Porco T.C., Keenan J.D. et al. Macrolide resistance in MORDOR I — A cluster-randomized trial in niger. N. Engl. J. Med. 2019; 380:2271–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruppé E., Cherkaoui A., Lazarevic V., Emonet S., Schrenzel J. Establishing Genotype-to-Phenotype relationships in bacteria causing hospital-acquired pneumonia: a prelude to the application of clinical metagenomics. Antibiotics. 2017; 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grall N., Lazarevic V., Gaïa N., Couffignal C., Laouénan C., Ilic-Habensus E., Wieder I., Plesiat P., Angebault C., Bougnoux M.E. et al. Unexpected persistence of extended-spectrum β-lactamase-producing Enterobacteriaceae in the faecal microbiota of hospitalised patients treated with imipenem. Int. J. Antimicrob. Agents. 2017; 50:81–87. [DOI] [PubMed] [Google Scholar]
- 26. Karkman A., Do T.T., Walsh F., Virta M.P.J. Antibiotic-resistance genes in waste water. Trends Microbiol. 2018; 26:220–228. [DOI] [PubMed] [Google Scholar]
- 27. Ziegler M., Jang H., Gopinath G., Horlbog J.A., Stephan R., Guldimann C. Whole-Genome shotgun sequencing of three listeria monocytogenes strains isolated from a ready-to-eat salad-producing facility in Switzerland. Genome Announc. 2018; 6:e00547-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danko D.C., Bezdan D., Afshinnekoo E., Ahsanuddin S., Alicea J., Bhattacharya C., Bhattacharyya M., Blekhman R., Butler D.J., Castro-Nallar E. et al. Global genetic cartography of urban metagenomes and anti-microbial resistance. 2019; bioRxiv doi:05 August 2019, preprint: not peer reviewed 10.1101/724526. [DOI]
- 29. Baquero F., Alvarez-Ortega C., Martinez J.L. Ecology and evolution of antibiotic resistance. Environ. Microbiol. Rep. 2009; 1:469–476. [DOI] [PubMed] [Google Scholar]
- 30. Bengtsson-Palme J., Kristiansson E., Larsson D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018; 42:doi:10.1093/femsre/fux053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bengtsson-Palme J. The diversity of uncharacterized antibiotic resistance genes can be predicted from known gene variants—but not always. Microbiome. 2018; 6:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allen H.K., Donato J., Wang H.H., Cloud-Hansen K.A., Davies J., Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010; 8:251–259. [DOI] [PubMed] [Google Scholar]
- 33. Bush K., Jacoby G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010; 54:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feldgarden M., Brover V., Haft D.H., Prasad A.B., Slotta D.J., Tolstoy I., Tyson G.H., Zhao S., Hsu C.-H., McDermott P.F. et al. Using the NCBI AMRFinder tool to determine antimicrobial resistance Genotype-Phenotype correlations within a collection of NARMS isolates. 2019; bioRxiv doi:15 February 2019, preprint: not peer reviewed 10.1101/550707. [DOI] [PMC free article] [PubMed]
- 35. Pal C., Bengtsson-Palme J., Rensing C., Kristiansson E., Larsson D.G.J. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014; 42:D737–D743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zankari E., Hasman H., Cosentino S., Vestergaard M., Rasmussen S., Lund O., Aarestrup F.M., Larsen M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012; 67:2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia B., Raphenya A.R., Alcock B., Waglechner N., Guo P., Tsang K.K., Lago B.A., Dave B.M., Pereira S., Sharma A.N. et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017; 45:D566–D573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xavier B.B., Das A.J., Cochrane G., De Ganck S., Kumar-Singh S., Aarestrup F.M., Goossens H., Malhotra-Kumar S. Consolidating and exploring antibiotic resistance gene data resources. J. Clin. Microbiol. 2016; 54:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lakin S.M., Kuhnle A., Alipanahi B., Noyes N.R., Dean C., Muggli M., Raymond R., Abdo Z., Prosperi M., Belk K.E. et al. Hierarchical Hidden Markov models enable accurate and diverse detection of antimicrobial resistance sequences. Commun. Biol. 2019; 2:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teller S. Data Visualization with D3.Js. 2013; Birmingham: Packt Publishing Ltd; 1–180. [Google Scholar]
- 41. Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017; 35:316–319. [DOI] [PubMed] [Google Scholar]
- 42. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kurtzer G.M., Sochat V., Bauer M.W. Singularity: scientific containers for mobility of compute. PLoS ONE. 2017; 12:e0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:16 March 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 46. Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup The sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hyatt D., Chen G.-L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 50. Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. 2019; bioRxiv doi:07 September 2019, preprint: not peer reviewed 10.1101/762302. [DOI] [PMC free article] [PubMed]
- 51. Martínez J.L., Coque T.M., Baquero F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015; 13:116–123. [DOI] [PubMed] [Google Scholar]
- 52. Feng J., Li B., Jiang X., Yang Y., Wells G.F., Zhang T., Li X. Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ. Microbiol. 2018; 20:355–368. [DOI] [PubMed] [Google Scholar]
- 53. Singularity: Scientific containers for mobility of compute. [DOI] [PMC free article] [PubMed]
- 54. Berglund F., Österlund T., Boulund F., Marathe N.P., Larsson D.G.J., Kristiansson E. Identification and reconstruction of novel antibiotic resistance genes from metagenomes. Microbiome. 2019; 7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cortés-Ciriano I., Ain Ul Q., Subramanian V., Lenselink E.B., Méndez-Lucio O., IJzerman A.P., Wohlfahrt G., Prusis P., Malliavin T.E., van Westen G.J.P. et al. Polypharmacology modelling using proteochemometrics (PCM): recent methodological developments, applications to target families, and future prospects. MedChemComm. 2015; 6:24–50. [Google Scholar]
- 56. Ruppé E., Ghozlane A., Tap J., Pons N., Alvarez A.-S., Maziers N., Cuesta T., Hernando-Amado S., Clares I., Martínez J.L. et al. Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat. Microbiol. 2019; 4:112–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MEGARes 2.0 reference database and annotation files are available at https://megares.meglab.org. All AMR++ 2.0 pipeline source files are freely available on GitHub at https://github.com/meglab-metagenomics/AmrPlusPlus_v2.