


Abstract
Mechanisms that ensure repair of double-strand DNA breaks (DSBs) are instrumental in the integration of foreign DNA into the genome of transgenic organisms. After pronuclear microinjection, exogenous DNA is usually found as a concatemer comprising multiple co-integrated transgene copies. Here, we investigated the contribution of various DSB repair pathways to the concatemer formation. We injected mouse zygotes with a pool of linear DNA molecules carrying unique barcodes at both ends and obtained 10 transgenic embryos with 1–300 transgene copies. Sequencing the barcodes allowed us to assign relative positions to the copies in concatemers and detect recombination events that occurred during integration. Cumulative analysis of approximately 1,000 integrated copies reveals that over 80% of them underwent recombination when their linear ends were processed by synthesis-dependent strand annealing (SDSA) or double-strand break repair (DSBR). We also observed evidence of double Holliday junction (dHJ) formation and crossing over during the concatemer formations. Sequencing indels at the junctions between copies shows that at least 10% of DNA molecules introduced into the zygotes are ligated by non-homologous end joining (NHEJ). Our barcoding approach, verified with Pacific Biosciences Single Molecule Real-Time (SMRT) long-range sequencing, documents high activity of homologous recombination after DNA microinjection.
INTRODUCTION
Genetically modified organisms have become an important element of biomedical research (1), of production of pharmaceutical proteins (2) and in agriculture (3). Despite the widespread use of transgenic organisms, many long-standing questions remain unanswered, especially concerning molecular mechanisms involved in exogenous DNA processing. It is known that the repair of double-strand breaks (DSBs) plays an important role during genome editing and integration of foreign DNA into the genome. Homologous recombination (HR) and non-homologous end-joining (NHEJ) are the two major pathways responsible for DSB repair in eukaryotic cells. The majority of random DSBs in somatic cells are repaired by NHEJ (4), while HR is necessary to resolve more specific problems, such as rescuing a stalled replication fork or providing recombination in meiosis (5). Homology-based pathways involve invasion of single-stranded DNA filaments emerging from DSB ends into homologous template region, thus resulting in the formation of a D-loop and DNA synthesis. Different HR outcomes are possible, depending on the D-loop processing (6). In synthesis-dependent strand annealing (SDSA), the restored DNA end is released after the D-loop disruption, and anneals with the second DSB end. Break-induced replication (BIR) initiates long-range DNA synthesis in the absence of a second DSB end (replication fork collapse or telomere shortening). Double-strand break repair (DSBR) occurs when the displaced strand from a template anneals to the second broken DNA end. This way, both invading ends become physically linked in a double Holliday junction (dHJ) that could be resolved with or without crossing over (6). With the development of CRISPR-based genome editing, the focus has shifted to other roles of DNA repair pathways: HR is exploited for precise genetic modifications, such as transgene knock-ins, and NHEJ is used to knock-out genes (7,8). The obvious importance of these pathways for the field of genome editing serves as a driver for studying the activity of these pathways in different cells (9,10); understanding crosstalk between NHEJ and HR (11); and for developing new methods for the preferential activation of a particular pathway (12). Modification of the genome at the zygote stage is an advantageous method for obtaining genetically modified animals. However, in the literature there are few quantitative estimates of the activity of these DNA repair pathways in the early mammalian embryogenesis (13).
We decided to test the activity of various ways of processing DSBs in mouse zygotes after pronuclear microinjection of genetic constructs. In this method, exogenous DNA solution is injected inside the pronucleus that contains genetic material of the sperm or egg prior to fertilization (14). Usually, hundreds to thousands of DNA molecules are injected and processed subsequently by cellular DNA repair machinery, resulting in a stable DNA integration. The extensive work of many pioneer groups in the early years of transgenesis revealed several prominent features of pronuclear microinjection (15,16). For instance, transgenes always integrate at one or a few sites in the host genome and most of the transgene copies are prevalently arranged into head-to-tail tandemly oriented copies (concatemers) (data aggregated in Supplementary Table S6). Although homologous recombination was recognized as the culprit, the exact cause of concatemer formation remained unknown. Here we investigated mechanisms of exogenous DNA molecule processing by combining the classical approach of pronuclear microinjection with transgene barcoding technique and next-generation sequencing (NGS). Barcoding strategy was initially applied to address individual cell fates among heterogeneous cell population (17,18) and could be extended to trace transgene copies as well. Ten transgenic mouse embryos with varying amounts of barcoded transgenes were analyzed with NGS to read the terminal barcodes of each integrated transgene copy. Knowing the initial barcode tags of the injected transgenes, we were able to reconstitute the connections of most of the transgene copies in the concatemers and found various signatures of homology-based DNA repair pathways.
MATERIALS AND METHODS
Cloning of the barcoded vector library
Plasmid pcDNA3-Clover (Addgene #40259) was used as a base vector for cloning barcodes. The vector was digested with PciI, dephosphorylated and ligated with a short adapter fragment, introducing SbfI and NheI recognition sites. These sites were used to insert 500 bp of polymerase chain reaction (PCR) product amplified from human genome with primers carrying barcode sequences. The human region was selected to avoid potential recombination of the transgene ends with mouse genome. Sequences of the barcoded primers were as follows: 5′-CCTGCAGGNNCGANNGCANNTGCNNCTTGAATGACAACTAGTGCTCCAGG-3′ (primer with Tail barcode), 5′-GCTAGCNNACTNNGATNNGGTNNCTATCCTGACCCTGCTTGGCT-3′ (primer with Head barcode) (Supplementary Figure S1). A barcoded plasmid library was electroporated into the Top10 cells, plated and extracted using GeneJET Plasmid Midiprep Kit (Thermo Fisher Scientific, USA). The number of colonies was estimated to be ∼10 000 individual clones.
Generation of the transgenic embryos by pronuclear microinjection and PCR genotyping
The barcoded plasmid library was linearized with type IIS BsmBI restriction enzyme to provide incompatible 4 bp overhangs (∼250 bp distance from each barcode). Digested plasmid DNA was gel purified and eluted from the agarose gel, using Diagene columns (Dia-M, Russia) according to the manufacturer's recommendations. The eluate was purified further with AMPure XP magnetic beads (Beckman Coulter, USA) and finally dissolved in TE microinjection buffer (0.01 M Tris–HCl, 0.25 mM EDTA, pH 7.4). Fertilized oocytes were collected from superovulated F1 (CBA × C57BL/6) female mice crossed with C57BL/6 male mice. DNA was injected into the male pronuclei (1–2 pl ∼1000–2000 copies) as described earlier (19). Microinjected zygotes were transferred into the oviducts of the recipient pseudopregnant CD-1 females. The embryos were extracted on day 13.5 of development and PCR genotyped with primers for Clover backbone or transgene-transgene junctions (the same primers that we used for generating NGS PCR products) (Supplementary Table S5). Thermal Asymmetric Interlaced PCR (TAIL-PCR) (20) was performed as described earlier (21) with primers complementary to the 5′- or 3′-ends of the transgene (Supplementary Table S5). Conventional PCR reactions with Taq, Q5 or LongAmp polymerases (NEB, USA) were set up to amplify various rearrangements in concatemer structure, study copy order with barcode-specific primers and validate transgene-genomic borders (Supplementary Table S5).
All experiments were conducted at the Centre for Genetic Resources of Laboratory Animals at the Institute of Cytology and Genetics, SB RAS (RFMEFI61914 × 0005 and RFMEFI61914 × 0010). Animal manipulations were performed in accordance with protocols and guidelines approved by the Animal Care and Use Committee Federal Research Centre of the Institute of Cytology and Genetics, SB RAS, operating under standards set by regulatory documents of Federal Health Ministry (2010/708n/RF), and NRC and FELASA recommendations. Experimental protocols were approved by the Bioethics Review Committee of the Institute of Cytology and Genetics.
Copy number quantification by ddPCR
Droplet Digital PCR (ddPCR) was performed using ddPCR Supermix for Probes (No dUTP) and QX100 ddPCR Systems (Bio-Rad, USA) according to the manufacturer's recommendations. The 20 μl reaction contained 1× ddPCR Supermix, 900 nM primers, 250 nM probes and 3–60 ng digested genomic DNA. We adjusted the input DNA quantity for each embryo to account for tissue mosaicism and transgene copy number variation which affected transgene/control relative dilutions in every embryo. We tested various restriction digestion conditions in order to separate reliably all transgene copies (Supplementary Figure S3) and decided to perform overnight digestions with HindIII-HF or DpnII (NEB, USA) in a CutSmart buffer. Transgene copy number (Clover gene) was normalized to Emid1 control gene (23) or the unique transgene-genome border region (identified for four embryos). PCR was conducted according to the following program: 95°C for 10 min, then 40 cycles of 95°C for 30 s and 61°C for 1 min, with a final step of 98°C for 7 min and 20°C for 30 min. All steps had a ramp rate of 2°C/s. ddPCR was performed in two independent technical replicates. Sequences for primers and probes are available in Supplementary Table S5. Data were analyzed using QuantaSoft (Bio-Rad, USA).
DNA library preparation for NGS
NGS library 1 (original barcoded plasmids): The barcoded plasmid library was digested with NheI and SbfI (Figure 1A), 640 bp DNA fragment with a pair of barcodes was gel purified, eluted in TE buffer, and sequenced.
Figure 1.

Schematic overview of the experimental design. (A) The main stages of the generation of the barcoded plasmid library (NGS library 1) and pronuclear microinjection of the barcoded molecules. (B) Determining the barcodes at the ends of the concatenated copies by inverse PCR. DNA was digested with PciI and ligated in conditions favoring self-ligation (NGS library 3). (C) Determining the barcodes of the adjacent copies (NGS library 2). (D) Element of a transgene ‘subway map’—our visualization approach that combined the NGS data from three sequencing experiments. Colors indicate different barcode connections: green—actual transgene copy in the concatemer from the inverse PCR data; red—transgene-transgene junctions; blue—copies that retained the combinations of the barcodes, which were present in the injected plasmids.
NGS library 2 (PCR of the barcoded transgene-transgene junctions): Concatemer junctions containing barcodes were PCR amplified using Q5 polymerase (Figure 1C). PCR conditions were tested to avoid accumulation of PCR artifacts in late cycles and to account for copy number variation between embryos (range of 0.2 copies to 300 copies). PCR program: 98°C for 30 s, then 25–30 cycles of 98°C for 15 s, 64°C for 30 s, 72°C for 1 min, with 3 min of final extension at 72°C. PCR reactions (25 μl) were purified using AMPure XP magnetic beads (Beckman Coulter, USA), eluted in TE buffer and sequenced. To obtain additional information about transgene–transgene junctions, we also sought to sequence PCR products corresponding to the internal junctions (100 bp from BsmBI cut site) in three multicopy embryos (#2, 3, 7). To stay within the range of an acceptable read length (∼150 bp), we used one primer close to the junction and another one flanking the barcode (primer pairs 2+14 and 1+13 at Supplementary Figure S6A, B). PCR products were generated for both orientations. This way, a unique signature of the trimmed junction could be assigned to specific barcodes (Figure 6).
Figure 6.
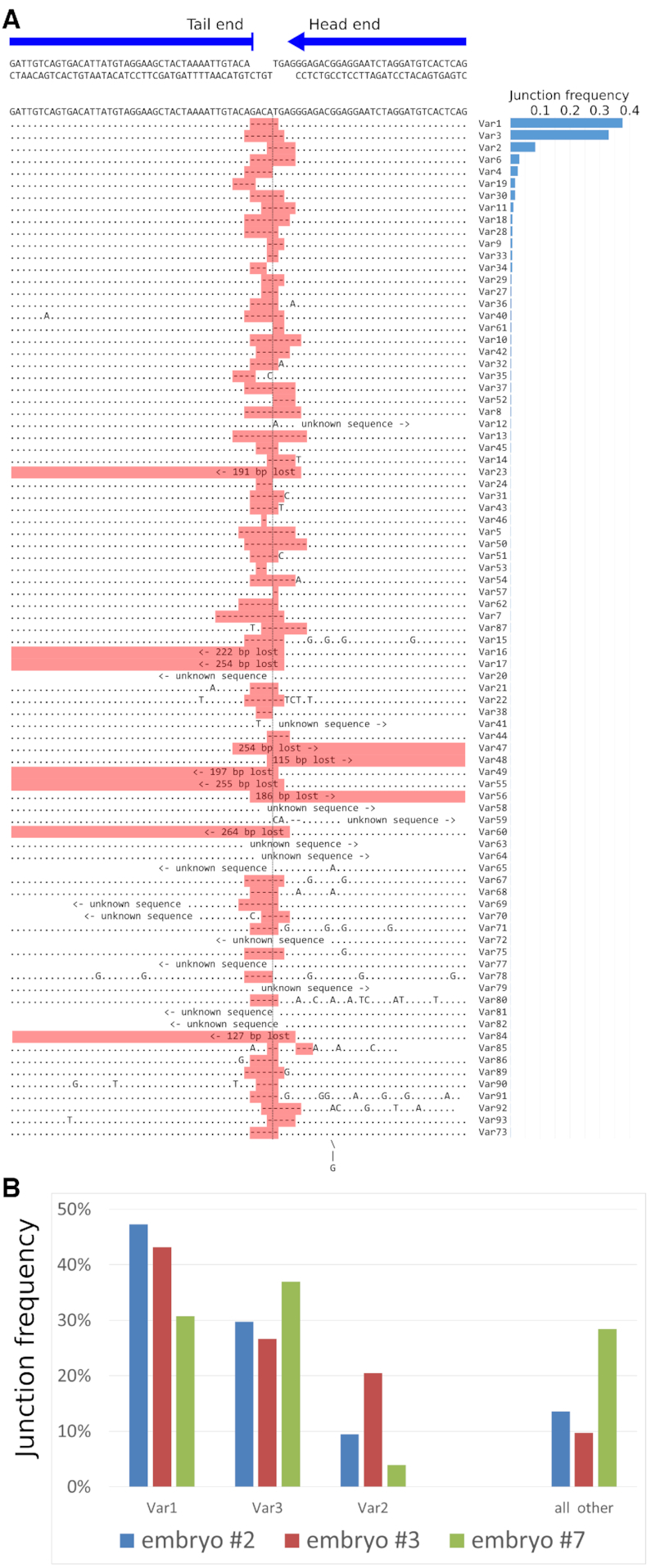
Sequence variants of indels at the junctions. (A) 4 bp 5′-protruding ends were generated by BsmBI digestion prior to microinjection. Sequence variants of the transgene–transgene head-to-tail junction region (Var1-Var93) are aligned below the original sequence. In most of the junctions, end processing removed only 5′-overhangs. (B) We calculated junction variant frequency for all detected mutations. The diagram shows the distribution of the top three variants (Var1, 2, 3) and remaining variants (‘all other’) in analyzed embryos.
NGS library 3 (inverse PCR of the barcoded transgene ends): Genomic DNA from transgenic embryos was digested overnight with an excess of PciI enzyme that cuts 87 bp away from one of the barcodes, inside the junction (Figure 1B). Digested DNA was purified with AMPure XP magnetic beads. To avoid incomplete digestion, the sticky ends generated with PciI were filled-in with Klenow enzyme to produce blunt ends. This signature was later used for filtering during sequence analysis. After heat inactivation of Klenow enzyme (20 min at 75°C), 300 ng of digested DNA was ligated overnight at 16°C in a large reaction volume (100 μl) to facilitate self-ligation of transgene monomers. Terminal barcodes of self-ligated DNA fragments were PCR amplified with Q5 polymerase by means of the same primers and conditions as were used for NGS library 2 generation. To remove PCR fragments resulting from undigested DNA, Illumina-prepared adapter-ligated PCR products were additionally treated with PciI, gel purified, and used for sequencing. We prepared 10 libraries corresponding to 10 embryos and one control library representing a 1:1 mix of DNA from embryos #1 and #4 (150 ng + 150 ng of digested genomic DNA in 100 μl ligation). This control library was used to estimate the level of random intermolecular transgene ligation, which is reflected by the proportion of chimeric PCR products containing barcodes from both embryos #1 and #4. After filtering with our standard threshold levels (see Pair filtering), the control library had zero chimeric sequence reads (Supplementary Figure S7); therefore, it is safe to assume that our inverse PCR libraries contain mostly self-ligated copies.
PCR artifacts, such as barcode mutations or polymerase template switching, are considered a common source of noise in the NGS data (22), and, theoretically, could produce unrealistic barcode combinations in our investigation. All of the above experiments were performed using Q5 polymerase (NEB) as it was shown that other polymerases have a much higher rate of strand conversion (20).
DNA fragments from three libraries were prepared with NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, USA), pooled together, and sequenced on the Illumina HiSeq 2500 platform (Illumina, USA). Libraries were assessed using an Agilent 2100 bioanalyzer (Agilent, USA) and a Qubit dsDNA HS assay kit (Life Technologies, USA).
PacBio SMRT sequencing
High molecular weight DNA extraction from fresh frozen tissues of transgenic embryo #8, PacBio DNA library preparation, and sequencing on PacBio Sequel platform was performed by Novogene (Hong-Kong).
PacBio data were converted to FASTA format, using bam2fasta tool and used to build a Basic Local Alignment Search Tool (BLAST) database. The obtained database was queried for the transgene sequence, using a National Center for Biotechnology Information (NCBI) megablast tool, which allowed identification of 113 reads containing transgene sequences. Since some of these sequences were redundant, that is, resulting from the same circular consensus sequence, we merged the sequences with the same identifier using a ccs tool with parameters –maxLength = 80 000 –minPasses = 0 –force –noPolish. The 76 unique sequences obtained were again compared to the transgene by the use of a megablast. These final BLAST results were manually analyzed to identify barcode sequences and junctions between transgene molecules.
To determine the average genomic coverage of PacBio reads, we aligned all data to the mm10 mouse genome using BLAST with default parameters and then computed the coverage by using GATK DepthOfCoverage.
Southern blot DNA analysis
Genomic DNA samples (∼10 μg each) obtained from mouse embryos were digested with the BamHI restriction endonuclease, fractionated on 0.8% agarose gels, and transferred to GeneScreen nylon membranes (NEN DuPont). The membrane was hybridized with a 32P-labeled DNA probe obtained from BsmBI digested Clover-barcoded plasmid, using random prime DNA labeling kit (Roche), and [α-32P] dCTP (PerkinElmer). Membrane was washed with 0.5× SSPE (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA, pH 7.7) and 0.5% SDS at 65°C and exposed to MS-film (Kodak) at −80°C.
Computational data analysis
Overview of data processing pipeline
NGS data processing contained four steps. First, the reads were trimmed by using cutadapt to remove constant sequences flanking barcodes. Second, read pairs were examined for a complete or partial match of barcode patterns (NN CGA NN GCA NN TGC NN for tail barcode, NN ACT NN GAT NN GGT NN for head barcode), and pairs sharing identical barcodes were merged. This results in the initial set of barcode pairs, which was further filtered at the third step of the pipeline to produce the final set of pairs (Supplementary Figure S24) (Supplementary Tables S2–S4). These resulting pairs were visualized using the Network module of the vis.js framework (http://visjs.org/). All computations were performed using nodes of Novosibirsk State University high-throughput computational cluster. A detailed description of the analysis of NGS data is presented in the supplementary notes.
RESULTS
Generation of the barcoded concatemers by pronuclear microinjection
We decided to replicate the conditions of a typical microinjection experiment. To investigate the mechanisms leading to the formation of concatemers, we injected the zygotes with a library of transgene molecules labeled with unique DNA barcodes. The strategy for introducing barcodes is shown in Figure 1A. A 7 kb plasmid vector expressing Clover was tagged with two barcodes placed 280 bp from the future ends (Supplementary Figure S1). We sought to reach a compromise between the length of the DNA ends precluding barcodes, as longer fragments would preserve the barcodes from exonuclease trimming, while shorter ends are more suitable for generating PCR products for NGS sequencing (∼700 bp in our case). Thus, we sequenced the DNA of the barcodes in the plasmids and estimated that our library consists of 12 657 different molecules (Figure 1A). This barcoded plasmid library was linearized with BsmBI, which cuts between the two barcodes to generate incompatible 4 bp 5′-overhangs. Linear DNA (6719 bp including plasmid backbone) was subsequently injected into pronuclei following standard protocol (each zygote received around 1000–2000 DNA molecules), and the zygotes were transferred into the pseudopregnant foster mothers. Embryos were collected at day E13.5 of development and their DNA analyzed by PCR genotyping (Supplementary Figure S2). Out of 50 embryos, 10 turned out to be positive for the transgene integration (20%), constituting a normal outcome for this method.
Determination of transgene copy number
First, we quantified the transgene copy number using droplet digital PCR (ddPCR). A pair of probes was designed for the multiplex ddPCR: (i) transgene-specific probe for the Clover gene in the middle of the vector, and (ii) a standard reference probe for the gene Emid1 at chromosome 11 as control (tested in (23)). As seen in Supplementary Figure S4, the transgene copy numbers varied greatly between embryos. In some cases, this number was less than one copy owing to mosaicism of the embryo tissues (embryos #5 and #6). Mosaicism is frequently observed in transgenic animals because transgene integration could occur after zygote division. Integration at the two-cell stage, for example, will result in 50% of the embryo cells’ bearing transgene and, consequently, in a 50% reduction of signal from the transgene and underestimation of the transgene copy number. Fortunately, we managed to obtain genomic localization information for some embryos, using Thermal Asymmetric Interlaced PCR (TAIL-PCR), а simple method based on the annealing of random primers close to the transgene-genome border and amplification of transgene flanking sequences (20). Transgene-genomic border is a unique site that could be used as a probe target region for the ddPCR to implement mosaicism correction (Supplementary Figure S3). Thus, replacing the standard reference Emid1 gene with a transgene-genome border-specific probe allowed us to clarify the copy number for four of the embryos (Supplementary Figure S4). For example, embryo #4 had 23 copies corrected to 45 (∼50% mosaicism), embryo #5: 0.4 to 1, embryo #9: 5 to 22, embryo #10: 3.5 to 4. Our copy number estimates were also confirmed by Southern blot analysis in six embryos (Supplementary Figure S25). In summary, we obtained 10 transgenic embryos with a broad distribution of transgene copy numbers, ranging from 1 to ∼300 copies, as expected in typical pronuclear microinjection experiments (24).
NGS of DNA barcodes in the concatemers
To understand the internal structure and origin of the concatemers, we sequenced the barcodes at the ends of the individual transgene copies. We performed two alternative sequencing experiments. First, we applied the inverse PCR method to determine the head and tail barcodes for each of the molecules in the concatemer (Figure 1B). This was important, considering the transgene recombination that may take place prior to integration. Genomic DNA from the transgenic embryos was digested with PciI endonuclease that makes a single cut inside the transgene-transgene junctions (Figure 1B, Supplementary Figure S1). Ligation was performed in a highly diluted solution of the digested DNA. Such conditions favor the self-ligation of individual transgenes—as a result, terminal barcodes come close at a distance of about 700 bp, and they can be PCR amplified and sequenced using paired-end NGS. The DNA sequencing of the inverse PCR library made it possible to establish genuine pairs of barcodes at the ends of each transgene that constitute concatemers. Additionally, we PCR amplified and sequenced barcodes directly at the transgene-transgene head-to-tail junctions to get information about the relative positions of molecules in the concatemers (barcodes of adjacent copies) (Figure 1C).
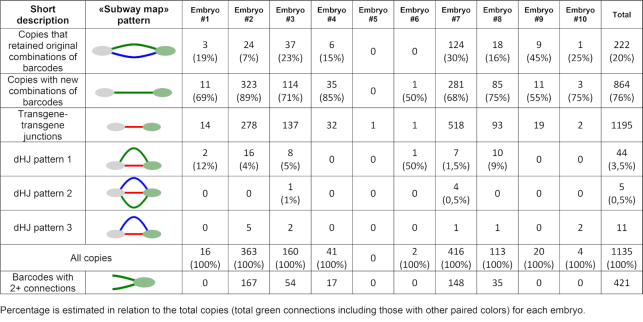
Combining NGS information from all the sequencing experiments (initial plasmid library + inverse PCR + junction PCR) allowed us to collect comprehensive data on which the transgene molecules were injected into the pronuclei, on how each molecule changed during the end processing, and on their relative positions inside concatemers. We visualized the structure of the concatemers in a graphical format (Figure 2A) (Supplementary Figures S8–S19) (Sup. Files 1–11). The barcodes from the NGS data were represented as nodes of two colors (green or gray for the head or tail barcodes, respectively). The adjacent barcodes that were discovered in the same PCR fragment were joined with a connection. Figure 2A illustrates the organization of all the connections between the barcodes of embryo #9, taken as an example. For clarity, each type of barcode connection was assigned one of three colors, based on the PCR library where they originate. Blue connections correspond to the transgene copies that retained the terminal barcodes that they had in the injected plasmid library (‘expected’ barcodes). The green connections correspond to the transgene copies that were observed in the embryos by inverse PCR (‘actual’ barcodes). These two connection types are not mutually exclusive, and, normally, one would expect that most of the observed copies in the concatemers would have green + blue double connections. However, as we discuss further in the text, most of the observed transgene copies switched their initial barcode tags owing to recombination. These transgene copies, which have head and tail barcodes from different molecules, no longer have a blue connection. Finally, transgene copies were linked together with short red connections that correspond to the transgene-transgene junctions in the concatemers. Altogether, these connections combine into continuous ‘connection chains’ that accurately reflect the barcode order in a concatemer. We named these colored schemes ‘transgene subway maps’ (Figure 1D). A count of the unique barcode connections (green) in 10 embryos revealed >1000 individual copies of barcoded transgenes (summed up in Table 1 and Supplementary Figure S4).
Figure 2.

Concatemer structure. (A) Transgene ‘subway map’ for embryo #9. Transgenes are oriented in head-to-tail fashion: green and gray colored ellipses designate the head and tail barcodes, respectively. Gaps between transgene chains are due to either deletions or palindromic junction orientations. (B) Box plots represent the distributions of the relative copy number of each of the terminal barcode combinations (green connections) in transgenic embryos. Relative copy numbers were calculated as NGS read counts divided by the median. Most of the outliers (relative copy number >1) were tied to deletions that create a shorter PCR product. N indicates the copy number (green connections) analyzed for each embryo. (C) Verification of the transgene amplification case with PacBio long-range sequencing. Transgene copy with barcodes #6037-#152462 was observed as direct repeat in PacBio read (top). On the subway map (bottom), these barcodes are linked by red + green connection with increased read count. (D) Verification of barcode copying (‘branching’) with PacBio long-range sequencing. Barcode #3680 was detected in three different regions of the concatemer (top). Like many other barcodes, barcode #3680 has multiple connection partners on the subway map (a fragment of such map for embryo #8 is shown at the bottom). As seen from PacBio data, this connection ‘branching’ is caused by iterative copying of a barcode during recombination. dHJ pattern 1—a signature of HR that is discussed further in the text.
Table 1.
Frequencies of various connection patterns in transgenic embryos
|
Amongst noteworthy map features are gaps in connection chains (embryos #1 and #9 are prominent examples) (Supplementary Figures S8 and S18). These cases obviously indicate a lack of PCR products connecting the barcodes in our sequence reads. This could happen for two reasons. First, transgene–genome borders are not subjects for PCR with our NGS primers; thus, at least two detached barcodes are ensured for any map. Additionally, we cannot exclude multiple integration events that will increase the number of connection gaps. Partial transgene deletions and inversions are another source of discontinuity: most of the gaps are certainly caused by NGS primer site loss at the concatemer junctions. As expected, we detected many transgene deletions and complex rearrangements with conventional PCR, TAIL-PCR, and long-range sequencing (Supplementary Figure S16). The number of transgene rearrangements correlated with the copy number of each embryo. Embryos #2, 3, 7 and 8 have hundreds of copies and a plethora of rearrangements (Supplementary Figure S5). Conversely, the remaining embryos had few (#4, 6, 9) or zero (#1, 5, 10) abnormal transgene junctions. The list of some sequenced deletions and rearrangements is available in the Supplementary material next to the corresponding concatemer maps. Some interesting cases (embryos #8, #9, #10) are highlighted in the Discussion section (Supplementary Figures S16, S18, S19).
Verification of transgene subway maps
To confirm that our transgene subway maps reflect the composition of the concatemers, we conducted two control experiments. First, we made sure that amplification during inverse PCR does not introduce artifacts due to the formation of chimeric molecules consisting of barcodes from different copies. To do this, we mixed genomic DNA from two embryos (#1 and #4) in equal proportions and performed all stages of the analysis for this sample, including amplification in inverse PCR, library preparation, sequencing, read filtering, and construction of a subway map as we did earlier for individual embryos. As can be seen in Supplementary Figure S7 (and Supplementary File 11), such joint processing of two independent concatemers did not lead to the appearance of the artifact links between the transgene maps of embryos #1 and #4. This suggests that our proposed method reproduces well the actual composition of the concatemers.
Nevertheless, we decided to complement our barcode analysis by utilizing the method without the PCR amplification step at all. Therefore, we decided to sequence the concatemer of one of the embryos by using Pacific Biosciences Single Molecule Real-Time (SMRT) technology. This technique is remarkable, in that, it allows the sequencing of single long DNA molecules, and amplification is not used at any stage in the preparation of the library for sequencing or in the sequencing itself. Furthermore, the long PacBio reads allow the sequencing of up to six linked copies in the concatemer. We performed the genomic DNA sequencing of embryo #8 with ∼3-fold genome coverage. In these data, we identified 76 reads containing a transgene totaling 1.16 Mb of sequence. Median read length was around 20 kb (N50 = 20 387 bp) with a read length spanning from 239 to 51 644 bp. Independent overlapping reads were merged into 31 contigs (Supplementary Figure S16). Unfortunately, the PacBio approach has an inherent error rate of around 15%, which almost guarantees that our 17 bp barcodes will have a mutation. This fact greatly complicates the automatic analysis of barcodes. Thus, we had to validate the barcode sequences manually. Supplementary Figure S17 shows the longest contig that we were able to assemble on the basis of PacBio data. It contains 6 copies whose sequence is fully consistent with the subway map. Moreover, the contig covers the transgene–genomic border and allows filling the gap in the subway map, the formation of which was due to the presence of a truncated copy in tandem. Thus, we have confirmed that our approach accurately reflects the barcode connections in the embryos. Next, we inspected our transgene subway maps to understand the molecular mechanisms that lead to concatemer emergence.
De novo amplifications make little if any contribution to concatemer formation
One of the motivations for our work was to test the hypothesis that rolling circle replication or analogs take part in the formation of a tandem of head-to-tail oriented copies (25). This mechanism is used by some viruses of eukaryotes to amplify their genome (26), and is suspected to participate in telomere maintenance (t-circles) (27) as well as in yeast mitochondria replication (28). It is established that after microinjection, some DNA copies can be circularized by NHEJ (29). Such circular molecule could probably undergo rolling circle replication with the involvement of a BIR-like mechanism and strand displacement, for example. Conceptually, this mechanism can be a good candidate for the role of the constructor of tandemly oriented concatemers. However, in our data, we did not find any evidence supporting this hypothesis since the number of unique barcoded molecules found in the concatemers was well in agreement with the estimates obtained by the ddPCR method (Supplementary Figure S4), even in multicopy embryos (70–300 copies). Nonetheless, to assess whether individual transgenes were amplified, we analyzed the distribution of the sequence read counts for each of the unique transgene copies (green connections) (Figure 2B). Although we found several transgenes that had increased read counts, in most of these cases, the shift was explained by deletions in the transgene-transgene junction regions, thus resulting in shorter PCR products and altered PCR kinetics. Still, there are several copies for which this technical explanation does not work. Apparently, these are genuine examples of molecules that have doubled their copy number (embryo #3 and #8 (Figure 2B)). Remarkably, we directly observed one amplification event in PacBio reads for embryo #8. In this case, the transgene junction with terminal barcodes #6037 and #152462 was repeated at least three times (transgene copy repeated at least two times) (Figure 2C) (Supplementary Figure S16S). This transgene repeat was found in two independent reads and partially in another, which spans the transgene–genome border (Supplementary Figure S16C). It should be noted that the copy lost its initial barcode combination (no blue connection); thus, it was not created through an amplification of circularly permuted original molecule but formed during linear end recombination. Although our data do not allow us to suggest a non-contradictory mechanism for this phenomenon, it is worth noting that in our analysis there were more than 1,000 molecules in the concatemers, of which only around 10 could be suspected of amplification. Therefore, we can conclude that the concatemers are generally formed by direct linkage of the injected molecules, rather than by de novo amplification mechanism.
HR is essential for concatemer formation
Our transgene subway maps illustrate high recombination activity that assembles the transgenes into the concatemers, resulting in barcode ‘switching’—the exchange of terminal barcodes between the copies (green connections without paired blue connections). We identified several typical connection patterns in the transgene ‘subway maps’ and proposed which of the known DNA repair pathways could led to their formation. Of these, we examined the NHEJ and two sub-pathways of HR, that is, SDSA and DSBR (Supplementary Figure S20). First, it is important to note that identical linear copies of DNA cannot be combined by the HR mechanism without the template region bridging two copies. Therefore, the formation of any concatemer undoubtedly begins with non-homologous end joining. However, aside from initial junction ligation, NHEJ plays a debatable role in assembling concatemers, compared to HR (see estimates in the next chapters). We discovered that most of the transgene copies in all embryos participated in barcode switching — a signature of HR. For example, as seen in Figure 2A, in embryo #9, only 9 out of 20 copies preserved the initial combination of barcodes that were observed in the injected plasmid library (coinciding blue and green connections), while the other 11 copies contain the head barcode from one molecule, and the tail barcode from the other (therefore, no blue connection). Such an exchange of genetic information between molecules is a characteristic signature of HR and strikingly differs from the simple combination of intact (with the exception of small indels at the junction) molecules produced by NHEJ mechanism. In our total sample of 1135 copies, only ∼20% (222) retained the original combination of barcodes (Table 1). Thus, we can conclude that at least 80% of the molecules in the concatemers were processed by the HR mechanism. Most likely, this is an underestimation because in our experimental system, barcodes are located almost 300 bp away from the ends and resection might not always reach the barcode sequence to change it through recombination - compare Figure 4 (barcode recombination) to Supplementary Figure S20 (transgene joining without barcode recombination).
Figure 4.

Principle of recombination between transgene copies causing barcode ‘switching’. Stages common to all pathways of homologous recombination (A) and stages characteristic of DSBR (B) and SDSA (C). The numbers denote barcodes. Outcomes of recombination are shown as elements of the transgene ‘subway map’. The mismatch repair steps are shown in the box.
Recombination mechanisms devised from connection patterns
We planned to use terminal barcodes as mere informative tags for concatenation analysis, but their location at the ends unexpectedly turned them into an indicator of recombination activity. Figure 4 shows possible mechanisms explaining the formation of recombined copies. In DSBR, after 3′-end resection and homologous duplex invasion, the D-loop synthesis reaches the barcode and forms a mismatch on one of the strands. This mismatch is a substrate for the mismatch repair system, which removes the fragment of the strand containing the mismatch and completes the gap on the template of the remaining strand. It is known that during the recombination, strand discrimination removes information from the invading strand rather than from the repair template, resulting in gene conversion (30,31). Thus, the mismatch repair leads to the copying of the donor barcode into the invading transgene. Simply put, the exchange of barcodes between the copies is a typical example of gene conversion. It is interesting that our assumption about the active participation of the mismatch repair system was confirmed by the analysis of embryo #1. Here, we found two chains of transgenes consisting of 3 and 11 copies. As can be seen in Figure 3A, some barcode connections form an unusual structure with a fork at one end (bottom part of the map). We assumed that the embryo #1 is a mosaic, whose cells contain one of two barcodes in a given position of the concatemer in equal proportions. This is possible if, for some reason, the mismatch that was formed during the recombination was not repaired before the replication, and the daughter cells received two different barcode variants (Figure 3B) (Supplementary Figure S21). These separated barcodes have about half the number of reads than the other copies in the concatemer (Figure 3C). We did not observe this pattern in any other embryo. Therefore, it must represent a unique event.
Figure 3.
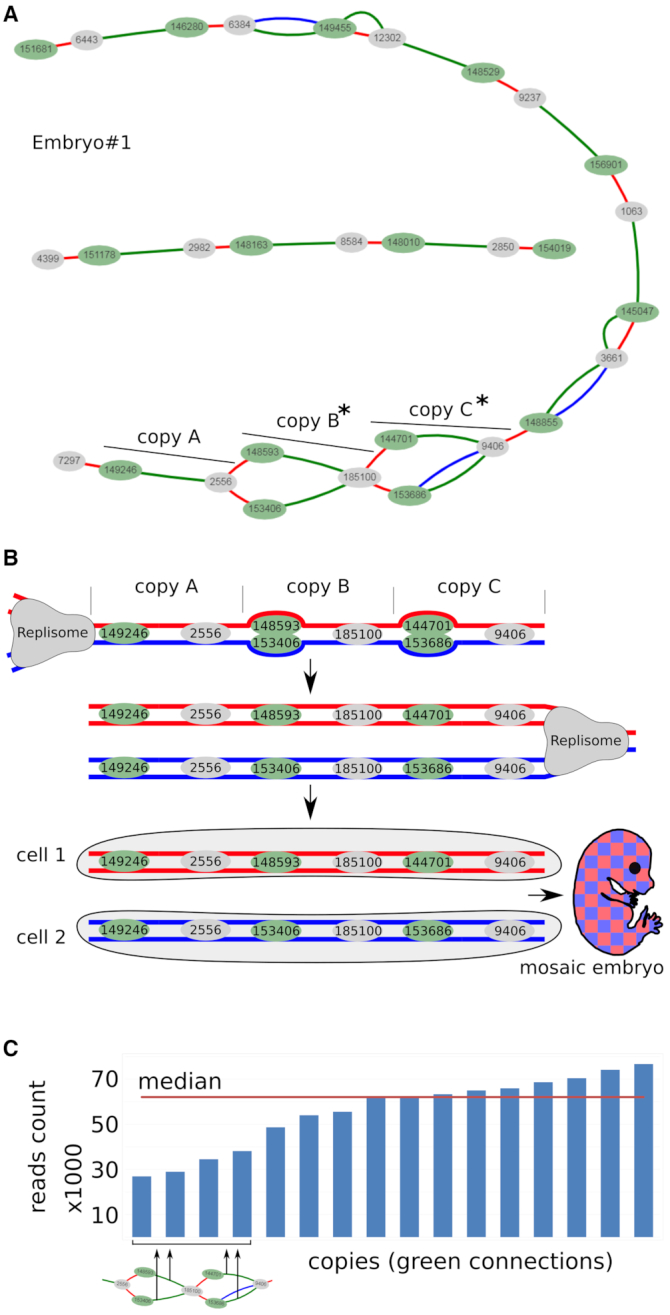
(A) Transgene ‘subway map’ for embryo #1. Copies B and C have two alternative head barcodes. (B) The scheme explaining the emergence of mosaic embryo consisting of two cell populations in case the barcode mismatches were not repaired in time before DNA replication (more details in Supplementary Figure S21). (C) Copies B and C have roughly half as many reads as the other copies in the embryo #1.
Copying barcodes from a donor template explains why in many cases concatemer maps look like webs of branched nodes (embryos #2, 3, 4, 7, 8) (Supplementary Figures S9, S10, S11, S14, S15). Note that the graphical term ‘branching’ is equal to barcode copying from a molecular perspective. Whenever the junctions that were originally linked together by HR or NHEJ serve as a donor template, they can transfer their barcode (one or both, depending on the extent of the resection of the invading end) to the invading copy, thus causing it to switch barcodes (Figure 4). This way, one barcode will be connected to two partners at independent junctions even if these regions lie at distant positions in the concatemer. For example, a head-to-tail junction between barcodes A and B can be invaded and partially copied by three transgene molecules that have X, Y and Z barcodes on their 3′-invading ends. If all these copies were to be incorporated in the final concatemer array, we would detect four junctions: A-B, A-X, A-Y and A-Z that share barcode A. Hence, the node of barcode A would have four connections (branching) on the subway map. An abundance of these nodes demonstrates intensive HR activity that sometimes copies one junction 3–5 times with different invading ends (404 counts overall, Table 1) but also greatly complicates the ‘subway map’ for visual inspection. It is worth mentioning that, as stated earlier, most of the original transgenes (blue connections) fail to be incorporated into the concatemers even if they provide recombination templates for other copies. Analysis of long PacBio sequence reads confirmed barcode copying (Figure 2D). For instance, barcode #3680 was detected at least 3 times at the independent junctions (Figure 2D); many barcodes and barcode pairs (#147233–#3447, #150859–#3172, #149759–#13397) could be observed at least twice in our PacBio reads (Supplementary Figure S16).
Evidence of dHJ formation
In the schemes described above, SDSA products are indistinguishable from non-crossover DSBR products (Figure 4 and Supplementary Figure S20). However, we found three connection patterns that strongly support the fact of dHJ resolution with crossing over. dHJpattern 1: If both ends of a single transgene molecule invade one junction, dHJ is formed and can be resolved with the formation of crossover products. This leads to the integration of the ‘attacking’ copy between the two original ones, and both of the ‘attacking’ copies’ barcodes are overwritten by those in the junction (Figure 5A). On our concatemer maps, this is represented by paired green and red connections. For example, there are two such connections in embryo #1 (Supplementary Figure S8) and 44 in total (Table 1). It is interesting that if dHJ is resolved without formation of crossover products, then the attacking molecule becomes circularized. Such circles are either lost during cell division or possibly serve as templates for other linear transgene copies.
Figure 5.

Resolution of dHJs during DSBR leads to characteristic connection patterns. (A) Crossing over between copies could result in assimilation of another copy (‘attacker’) into the junction with the loss of the ‘attacker’’s barcodes (green + red pattern). (B) DSBR between linear ends and a circular copy can have two detectable outcomes: the circular copy donates barcodes without crossing over (red + blue pattern) or gets incorporated into the ‘attacking’ molecule while also donating barcodes (green + red + blue pattern). Outcomes of recombination are shown as elements of the transgene ‘subway map’.
dHJpattern 2: Another crossover scenario occurs when a single circular copy is attacked by the ends of two other molecules (Figure 5B). In this case, dHJ resolution with the formation of crossover products leads to the joining of all three copies, while the ‘attacking’ molecules copy barcodes from the circular template. This outcome appears as two barcodes linked by three connections at once. There are five such structures in our maps (Table 1). Besides, even if the ‘attacking’ molecules copied barcodes from the template without crossing over and physical integration of the circular copy, we would still see such events (Figure 5B). On our maps, such barcodes are connected by red and blue connections (dHJpattern 3). This pattern is characteristic of a circularized copy as well. However, these are only a few (11 of 1135 total molecules in our analysis), which says that the closure of a single molecule in a ring is a rare event (Table 1). This is important because, according to the initial theories, circularization and subsequent random breakage were considered one of the key stages of the formation of concatemers (32).
We would like to emphasize that although we found only sparse evidence of crossing over (<5% of the copies) (dHJ patterns in Table 1), all of these were simple, categorizable cases that are just the tip of the iceberg, as many simultaneous recombination events must have created higher order patterns. For instance, crossovers could be formed by multicopy tandems that are incorporated into junctions (this would result in a side loop on the transgene ‘subway map’). As many of the individual transgenes also switch barcodes by junction invasions, this side loop would be connected to multiple nodes in other concatemer regions, thus vanishing in the complex ‘subway map’ (as in embryos #2, 3, 4, 7, 8) (Supplementary Figures S9, S10, S11, S14, S15).
Role of NHEJ in concatemer formation
Our data suggest that HR plays a leading role in the formation of tandemly oriented copies. NHEJ pathway facilitates concatemer formation by creating template transgene-transgene junctions for initial HR invasions and by ligating truncated copies. NHEJ contribution is thought to be proportional to DNA concentration. In the concatemer of the multicopy embryo #8 sequenced with PacBio, we observed ∼20 deletions, 8 palindromic orientations (head-to-head or tail-to-tail) (most emerged from truncated copies) (Supplementary Figure S16, orange labels) and 4 ‘elongation beyond original broken end’ (EBOBE) patterns (Supplementary Figure S16A, H, K), which are distributed between at least 56 copies (Supplementary Figure S16). That is much more of a random ligation than in low-medium copy lines (5–20 copies) (Supplementary Figure S8) that tend to have fewer gaps and a higher proportion of head-to-tail junctions.
Since the typical signatures of NHEJ are small indels at the repair sites (4), we decided to explore the repertoire of indels at the transgene–transgene junctions in the multicopy embryos (#2, 3, 7). We analyzed the sequences of junctions adjacent to 1803 barcodes altogether. We found almost a hundred possible variants for the structure of ligation sites between copies (Figure 6A). However, the frequency of sequence variants was distributed very unevenly, so that the three most frequent ones were found in 80% of the junctions (Figure 6B). These top three variants were the same in all examined embryos. These variants were clippings of the protruding 4 bp 5′-ends: –5 bp (Var1), –5 bp (Var2), –7 bp (Var3). Remarkably, other deletions of the same or even a smaller size were rare (Figure 6A). In our case, the processing of the 5′-overhangs may have revealed complementary nucleotides (GA in Var1 and AG in Var3), hence favoring ligation of these variants over others. Recent rigorous analysis of DSB repair patterns in mouse ES cells (33) demonstrated that 5′-protruding ends are repaired by either NHEJ or TMEJ (polymerase theta-mediated end-joining) leading to ∼30% cases of insertions and delins (deletion with nucleotide insertion). In our case, >99% of the junctions (Var1–3 plus Var4, Var6, Var11, Var19, Var30) did not have any additional insertions or SNPs and formed uniform clusters (Figure 6B).
Unfortunately, the fact that NHEJ favored few junction variants disrupted our initial idea to estimate the total number of transgene molecules, which were independently joined by NHEJ and served as template for HR. Nevertheless, information about the sequence of the junctions made it possible to check our prediction that the result of the joining of molecules by HR mechanism would be an exact copying of the template junction. For example, different copies with the same barcodes in dHJ patterns (Figure 5) should also have the same variant of junction. We checked it, and this is true for most of the cases. Only in 8% of cases did the barcode have not one but two different variants of the junction (data not shown).
We can roughly estimate the efficiency of the NHEJ-mediated ligation: the number of independent NHEJ-ligated molecules should be no fewer than the number of unique junction variants from the embryos. According to our data, this corresponds to 1 event per 10 injected copies. Obviously, this is the lower estimate and the real value is several times higher (individual ligation events would likely produce one of the same top 3 preferred signatures; at the same time, many unique junctions are not incorporated in the concatemer and are lost from our assessment). We also did not analyze other copy orientation variants (head-to-head or tail-to-tail) because their sequencing was impossible owing to technical reasons (see Discussion section). In general, we can conclude that NHEJ plays a significant role during the initial ligation of exogenous DNA in zygotes.
DISCUSSION
Concatemers are a prominent feature of the pronuclear microinjection method (Supplementary Table S6) and have been observed for >40 years. To explain concatenation, researchers came up with many theories that could be summed up in three models.
Concatenation model #1 implies that concatemers are formed through recombination of linear transgene copies with circular intermediates and was shared by most scientists (34,35). Concatenation model #2 states that self-ligation of linear transgene ends could lead to the generation of circular molecules. Random breakage of these circles could create overlapping fragments for recombination (29,34). Lastly, concatenation model #3 is based on a rolling circle replication mechanism not necessarily involving classic origin replication but based on a homologous recombination pathway similar to a break-induced replication. This hypothesis was initially termed ‘localized replication’ (25). De novo amplification would have explained enormous processivity of concatenation that could sometimes join hundreds of copies (when only a thousand copies are injected) into tandemly oriented arrays.
Aspects of transgene concatenation could be tested conveniently in a cell culture but most of the experiments focus solely on the end-joining aspect of concatemer formation (that is, studying signatures of NHEJ/MMEJ repair at transgene-transgene junctions) (36–38). To our knowledge, the only paper that addressed the mechanism of concatemer formation directly was published by the Mario Capecchi group >35 years ago (15). In this seminal experiment, albeit performed on cultured mammalian cells, the researchers injected nuclei with 2–500 copies of DNA molecules (linear or circular). Besides clarifying various technical aspects, the group also devoted a part of the report to investigating how concatemers are formed. They injected nuclei with a mixture of two similar transgenes (A- and B-molecules)—plasmid backbones with HSV thymidine kinase gene in two orientations. Southern blot analysis with specific restriction enzymes showed that the transgene concatemers consisted of interspersed tandemly oriented A/B copies. This elegant effort challenged the transgene amplification hypothesis, but authors were cautious about low copy number integrations and random fluctuations due to the presence of only two transgene versions. Our data unequivocally confirm that concatemers are created through recombination of individual transgenes without large-scale de novo amplification; although in rare cases, transgene copies undergo expansion by unknown mechanism (Figure 2C).
We also managed to obtain decisive evidence that head-to-tail tandems are mostly formed by HR between linear copies (concatenation model #1). We base this conclusion on the lack of red + blue double connections (self-circularized copies), which discards concatenation model #2. SDSA and DSBR are two main ways of repairing double-strand DNA breaks by homologous recombination (5). These pathways have similar initial stages and usually resolve into indiscernible non-crossover products, evident in the form of barcode ‘switching’ in our investigation (green connections without blue connection from the initial plasmid library). However, DSBR sometimes manifests itself in the formation of crossover products after dHJ resolution. We found several convincing examples of crossovers leaving traces on the concatemer ‘subway map’ (<5% copies) (Table 1). Apparently, the formation of crossovers is quite dangerous for somatic cells as it can lead to the loss of heterozygosity of a large chromosome fragment (39). The fact that crossing over is not completely suppressed in early embryos is of interest and expands our scarce knowledge of DNA repair at this stage.
In addition to crossovers and barcode ‘switching’, we also noticed another indication of HR activity. Analysis of the head-to-tail junctions and transgene-genome integration sites reveals that sometimes the transgene copies contain junction sequences, corresponding to the D-loop disruption intermediates (40). We labeled this pattern ‘EBOBE’ (Supplementary Figure S22). This pattern is closely reminiscent of a non-canonical HR termination model described recently (41). One can imagine that the resected transgene's 3′-end invades the homologous template at the transgene–transgene junction, copies a portion of the junction, and following the D-loop disruption, it gets incorporated into the concatemer or genome by NHEJ or MMEJ (like in embryo #10) (Supplementary Figure S19). We sequenced 4 EBOBE junctions from embryo #8 (Supplementary Figure S16) and 1 junction from embryo #6 (Supplementary Figure S13). We also detected EBOBE fragments at the transgene–genome borders in embryos #5 and #10. A similar pattern was noticeable in some published transgene integration models (35,42). We have two reasons to suspect that the D-loop disruption intermediates in the EBOBE creation, instead of the traditionally accepted random transgene fragmentation. First of all, junctions which border these fragments lack the blue barcode connection and does not result from a self-ligated and broken copy. Second, the EBOBE fragments are terminated at the barcode sequence (∼270 bp from the end), which hints that the synthesized displaced strand probably could not reinvade the homologous duplex because of the barcode heterogeneity. Some of the EBOBE fragments have profound tracts of microhomologies at the ends (see the transgene-genome borders in embryo #10). We speculate that large transgene deletions could also be attributed to EBOBE (if the D-loop disruption took place in the backbone region), but they escape detection in our investigation. These cases demonstrate that HR intermediates could be processed by NHEJ/MMEJ at par with typical fragmented copies. We think that this finding represents considerable interest in light of new discoveries of deleterious recombination pathways, such as the microhomology-mediated BIR (MM-BIR) (43) and multi-invasion induced rearrangement (MIR) (6). We hope that this report will stimulate further research aimed at evaluating the real representation of these EBOBE cases in other transgenic lines using modern sequencing methods.
Also, it is noteworthy that we could not detect directly any activity of alternative HR pathways, such as single-strand annealing (SSA) and BIR. SSA might link randomly broken transgene circles (formed after initial circularization) and therefore result in red + blue connections, which were in fact very rare. BIR would manifest itself as the amplification of the continuous regions of the concatemers, and, indeed, we found one region encompassing three transgene molecules with double read counts (embryo #3) (Figure 2B), but this was a single event. Another potential source of recombination is the MIR pathway (6,44). It was shown that resected ssDNA regions far from the 3′-terminus are capable of invading multiple regions of homology at different loci simultaneously. Endonucleolytic processing of such recombination substrates leads to gross translocations between donor fragments (44). In our investigation, MIR could produce additional barcode recombination by translocating transgene fragments (if recombination involved the backbone region) and translocating barcode junctions (if recombination occurred inside the transgene-transgene junction). However, we presumed that the SSA, BIR or MIR pathways do not contribute much to the concatemer formation because competition with SDSA prevents long-range resection (45).
The real proportion of NHEJ-processed copies in concatemers has always remained enigmatic. NHEJ signatures from mouse embryonic stem cells (33) and zebrafish embryos (46) show that this pathway participates actively in transgene end joining, but what proportion of these joined molecules is retained in the final concatemer is unclear. Southern blot estimations and new sequencing approaches for many transgenic mouse lines (Supplementary Table S6) confirm that head-to-tail orientation is dominant (>90% of copies versus 50% in the case of random ligation). Likewise, our transgene ‘subway maps’ display continuous tandem head-to-tail chains (>10 copies) with no gaps (e.g. in embryo #1 (Supplementary Figure S8), #4 (Supplementary Figure S11A) or #9 (Supplementary Figure S18A)). Unfortunately, studying complex rearrangements in concatemers is nearly impossible at present because PCR is not suitable for detection of palindromic junctions in transgenic animals (our experience; (47); also see (48) and Figure S2 therein), and the repetitive nature of concatemers complicates NGS-based methods (49,50). However, it is well established that NHEJ often contributes to concatemer emergence with fragmented and truncated copies arranged in random orientation (49,51). We ourselves detected truncated copies in most of the transgenic embryos and their abundance correlated with a transgene copy number (Supplementary Figure S5A, B). New transgene mapping methods, such as targeted locus amplification (TLA) and long-range sequencing, will soon expand and probably reformat our view on concatemer formation and integration processes. TLA approach, which is based on DNA cross-linking and PCR enrichment of the closely positioned transgene–genome fragments (52,53), is a convenient method for large-scale examination of integration sites. This was demonstrated recently by mapping transgene integration loci in 40 transgenic mouse lines from JAX Repository (50). TLA exposed many cases of complex genomic rearrangements accompanying transgene integrations (duplications, inversions and co-integrations of chromosome fragments) (50,52). Compared to TLA, long-range sequencing could provide more information about internal concatemer junctions and rearrangements and is especially valuable to the sequencing of palindromic junctions. In a recent study, researchers applied Oxford Nanopore sequencing to investigate the transgene integration site in the popular transgenic mouse line Oct4:EGFP (54). They described the chromosome integration site and, essentially, managed to identify three palindromic junctions inside the concatemer (25 copies). In our case, Pacific Biosciences SMRT sequencing reveals 8 palindromic junctions with various degrees of ends trimming (orange connections) (Supplementary Figure S16). Thus, long-range sequencing methods are well suited for studying palindromes and their long-term stability, although very deep sequencing is required to obtain high-quality junction sequences, which is presently quite costly.
Most of the head-to-tail junctions that we sequenced (Figure 6) had little or no deletions (5–7 bp). In theory, some palindromic head-to-head or tail-to-tail junctions should have similar junctions, but most of them harbor asymmetric deletions. We found only one ‘perfect’ palindromic (tail-to-tail) junction that lost 4 bp (Supplementary Figure S16J). What is the mechanism to break the central symmetry? Palindromic sequences are quite stable in mammalian zygotes and are inherited by their offspring (55). Thus, palindrome symmetry is likely disrupted in the initial step of concatenation (during extrachromosomal recombination). We documented high activity of HR recombination between ends (>80% of copies), and it made us believe that frequent strand invasion and D-loop formation could provoke secondary structures, such as hairpins and cruciforms in template palindromic junctions (Supplementary Figure S23). Another possible HR-related mechanism is the folding back of the single-stranded resected end of a linear transgene after copying a fragment of the palindromic junction (56). Subsequently, cell repair systems recognize and remove these hairpin structures
Ultimately, we aimed to utilize barcode connections to figure out unequivocal transgene order in concatemers. The closest we achieved this goal was in embryo #1 (only 1 gap unresolved) (Supplementary Figure S8), embryo #9 (Supplementary Figure S18A), and embryo #10 (Supplementary Figure S19). PacBio sequencing, coupled with manual inspection of the barcodes from the NGS data for embryo #8, helped us to reconstitute long tandemly oriented chains interspersed with broken fragments and palindromic junctions (Supplementary Figure S16). In embryo #9, inverted copies (full-sized or truncated) presumably separate tandemly oriented chains of variable lengths. We sequenced some of them (Supplementary Figure S18B). Lastly, embryo #10 puzzled us with a complex barcode distribution pattern. As seen in Supplementary Figure S19, the transgene ‘subway map’ in this embryo has quite a peculiar plan with four transgene copies but only 2 unique barcode pairs (8 barcodes in total). We validated the duplication of each barcode with ddPCR (Supplementary Table S1). We employed long-distance PCR with barcode-specific primers to position all copies and their respective barcodes in the four-copy concatemer (Supplementary Figure S19). It appears that two initially ligated copies with unique barcode pairs underwent recombination and assimilated two other copies. It definitely was not a simple duplication event because the barcodes were shuffled between the copies.
Concluding remarks
Using DNA barcodes helped us to explain some of the long-standing questions in the field of transgenesis. First of all, we showed that hundreds of copies are joined together independently without the contribution of long range de novo synthesis. Terminal barcodes were also useful for tracking self-ligated copies and we found no concatemer contribution from such rings, although it had been frequently proposed that concatemers are formed by recombination of the overlapping fragments of broken circular copies. In theory, injection of circular copies that go through random breakage and recombine with backbone regions must lead to complete disappearance of barcode ‘switching’ in our investigation. Clearly, understanding HR regulation in zygotes will be of great importance for the implementation of revolutionary transgenesis methods, such as newly designed gene drivers (57) or continuous DNA assembly by overlaps (58), and might help fine-tune HR to prevent unwanted recombination that complicates CRISPR/Cas9 knock-in experiments (59).
DATA AVAILABILITY
Raw sequence reads have been deposited at the Sequence Read Archive (SRA) under accession number PRJNA533172. The processed data (transgene ‘subway maps’ in web-browser file format) are available as supplementary (Sup. Files 1–11).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank L. Gubar for technical assistance with Southern blot analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Science Foundation [16-14-00095]; NGS libraries preparation was partially performed using experimental equipment of the Resource Center of the Institute of Cytology and Genetics SB RAS [0324-2019-0041]; Pacbio sequencing was performed with support from Novosibirsk State University. Funding for open access charge: Russian Science Foundation [16-14-00095].
Conflict of interest statement. None declared.
REFERENCES
- 1. Savić N., Schwank G.. Advances in therapeutic CRISPR/Cas9 genome editing. Transl. Res. 2016; 168:15–21. [DOI] [PubMed] [Google Scholar]
- 2. Bertolini L.R., Meade H., Lazzarotto C.R., Martins L.T., Tavares K.C., Bertolini M., Murray J.D.. The transgenic animal platform for biopharmaceutical production. Transgenic Res. 2016; 25:329–343. [DOI] [PubMed] [Google Scholar]
- 3. Kamthan A., Chaudhuri A., Kamthan M., Datta A.. Genetically modified (GM) crops: milestones and new advances in crop improvement. Theor. Appl. Genet. 2016; 129:1639–1655. [DOI] [PubMed] [Google Scholar]
- 4. Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R.. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017; 18:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ranjha L., Howard S.M., Cejka P.. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma. 2018; 127:187–214. [DOI] [PubMed] [Google Scholar]
- 6. Piazza A., Heyer W.-D.. Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol. 2019; 29:135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Low B.E., Kutny P.M., Wiles M.V.. Simple, efficient CRISPR-Cas9-Mediated gene editing in mice: Strategies and methods. Methods Mol. Biol. 2016; 1438:19–53. [DOI] [PubMed] [Google Scholar]
- 8. Jiang F., Doudna J.A.. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017; 46:505–529. [DOI] [PubMed] [Google Scholar]
- 9. Cheng L.-T., Sun L.-T., Tada T.. Genome editing in induced pluripotent stem cells. Genes Cells. 2012; 17:431–438. [DOI] [PubMed] [Google Scholar]
- 10. Nishiyama J. Genome editing in the mammalian brain using the CRISPR–Cas system. Neurosci. Res. 2019; 141:4–12. [DOI] [PubMed] [Google Scholar]
- 11. Pawelczak K.S., Gavande N.S., VanderVere-Carozza P.S., Turchi J.J.. Modulating DNA repair pathways to improve Precision genome engineering. ACS Chem. Biol. 2018; 13:389–396. [DOI] [PubMed] [Google Scholar]
- 12. Charpentier M., Khedher A.H.Y., Menoret S., Brion A., Lamribet K., Dardillac E., Boix C., Perrouault L., Tesson L., Geny S. et al.. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018; 9:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Derijck A., van der Heijden G., Giele M., Philippens M., de Boer P.. DNA double-strand break repair in parental chromatin of mouse zygotes, the first cell cycle as an origin of de novo mutation. Hum. Mol. Genet. 2008; 17:1922–1937. [DOI] [PubMed] [Google Scholar]
- 14. Pu X., Young A.P., Kubisch H.M.. Production of transgenic mice by pronuclear microinjection. Methods Mol. Biol. 2019; 1874:17–41. [DOI] [PubMed] [Google Scholar]
- 15. Folger K.R., Wong E.A., Wahl G., Capecchi M.R.. Patterns of integration of DNA microinjected into cultured mammalian cells: evidence for homologous recombination between injected plasmid DNA molecules. Mol. Cell Biol. 1982; 2:1372–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brinster R.L., Chen H.Y., Trumbauer M.E., Yagle M.K., Palmiter R.D.. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc. Natl. Acad. Sci. 1985; 82:4438–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu R., Neff N.F., Quake S.R., Weissman I.L.. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 2011; 29:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yunusova A.M., Fishman V.S., Vasiliev G.V., Battulin N.R.. Deterministic versus stochastic model of reprogramming: new evidence from cellular barcoding technique. Open Biol. 2017; 7:160311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown G.A.J., Corbin T.J.. Oocyte injection in the mouse. Transgenesis Techniques. 2002; 180:New Jersey: Humana Press; 39–70. [DOI] [PubMed] [Google Scholar]
- 20. Liu Y.-G., Chen Y.. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. BioTechniques. 2007; 43:649–656. [DOI] [PubMed] [Google Scholar]
- 21. Serova I.A., Dvoryanchikov G.A., Andreeva L.E., Burkov I.A., Dias L.P.B., Battulin N.R., Smirnov A.V., Serov O.L.. A 3, 387 bp 5′-flanking sequence of the goat alpha-S1-casein gene provides correct tissue-specific expression of human granulocyte colony-stimulating factor (hG-CSF) in the mammary gland of transgenic mice. Transgenic Res. 2012; 21:485–498. [DOI] [PubMed] [Google Scholar]
- 22. Omelina E.S., Ivankin A.V., Letiagina A.E., Pindyurin A.V.. Optimized PCR conditions minimizing the formation of chimeric DNA molecules from MPRA plasmid libraries. BMC Genomics. 2019; 20:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Codner G.F., Lindner L., Caulder A., Wattenhofer-Donzé M., Radage A., Mertz A., Eisenmann B., Mianné J., Evans E.P., Beechey C.V. et al.. Aneuploidy screening of embryonic stem cell clones by metaphase karyotyping and droplet digital polymerase chain reaction. BMC Cell Biol. 2016; 17:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burkov I.A., Serova I.A., Battulin N.R., Smirnov A.V., Babkin I.V., Andreeva L.E., Dvoryanchikov G.A., Serov O.L.. Expression of the human granulocyte–macrophage colony stimulating factor (hGM-CSF) gene under control of the 5′-regulatory sequence of the goat alpha-S1-casein gene with and without a MAR element in transgenic mice. Transgenic Res. 2013; 22:949–964. [DOI] [PubMed] [Google Scholar]
- 25. Rohan R.M., King D., Frels W.I.. Direct sequencing of PCR-amplified junction fragments from tandemly repeated transgenes. Nucleic Acids Res. 1990; 18:6089–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krupovic M., Forterre P.. Single-stranded DNA viruses employ a variety of mechanisms for integration into host genomes. Ann. N. Y. Acad. Sci. 2015; 1341:41–53. [DOI] [PubMed] [Google Scholar]
- 27. Tomaska L., Nosek J., Kramara J., Griffith J.D.. Telomeric circles: universal players in telomere maintenance. Nat. Struct. Mol. Biol. 2009; 16:1010–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X.J., Clark-Walker G.D.. Unveiling the mystery of mitochondrial DNA replication in yeasts. Mitochondrion. 2018; 38:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bishop J.O. Chromosomal insertion of foreign DNA. Reprod. Nutr. Dev. 1996; 36:607–618. [PubMed] [Google Scholar]
- 30. Stone J.E., Ozbirn R.G., Petes T.D., Jinks-Robertson S.. Role of proliferating cell nuclear antigen interactions in the mismatch Repair-Dependent processing of mitotic and meiotic recombination intermediates in yeast. Genetics. 2008; 178:1221–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakraborty U., George C.M., Lyndaker A.M., Alani E.. A delicate balance between repair and replication factors regulates recombination between divergent DNA Sequences in saccharomyces cerevisiae. Genetics. 2016; 202:525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Auerbach A.B. Production of functional transgenic mice by DNApronuclear microinjection. Acta Biochim. Pol. 2004; 51:9–31. [PubMed] [Google Scholar]
- 33. Schimmel J., Kool H., Schendel R., Tijsterman M.. Mutational signatures of non‐homologous and polymerase theta‐mediated end‐joining in embryonic stem cells. EMBO J. 2017; 36:3634–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pawlik K.M., Sun C.-W., Higgins N.P., Townes T.M.. End joining of genomic DNA and transgene DNA in fertilized mouse eggs. Gene. 1995; 165:173–181. [DOI] [PubMed] [Google Scholar]
- 35. Hamada T., Sasaki H., Seki R., Sakaki Y.. Mechanism of chromosomal integration of transgenes in microinjected mouse eggs: sequence analysis of genome-transgene and transgene-transgene junctions at two loci. Gene. 1993; 128:197–202. [DOI] [PubMed] [Google Scholar]
- 36. Kostyrko K., Neuenschwander S., Junier T., Regamey A., Iseli C., Schmid-Siegert E., Bosshard S., Majocchi S., Le Fourn V., Girod P.-A. et al.. MAR-Mediated transgene integration into permissive chromatin and increased expression by recombination pathway engineering. Biotechnol. Bioeng. 2017; 114:384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grandjean M., Girod P.-A., Calabrese D., Kostyrko K., Wicht M., Yerly F., Mazza C., Beckmann J.S., Martinet D., Mermod N.. High-level transgene expression by homologous recombination-mediated gene transfer. Nucleic Acids Res. 2011; 39:e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sasaki S., Sato M., Katsura Y., Kurimasa A., Chen D.J., Takeda S., Kuwano H., Yokota J., Kohno T.. Rapid assessment of two major repair activities against DNA double-strand breaks in vertebrate cells. Biochem. Biophys. Res. Commun. 2006; 339:583–590. [DOI] [PubMed] [Google Scholar]
- 39. Daley J.M., Gaines W.A., Kwon Y., Sung P.. Regulation of DNA pairing in homologous recombination. Cold Spring Harb. Perspect. Biol. 2014; 6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wright W.D., Shah S.S., Heyer W.-D.. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018; 293:10524–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hartlerode A.J., Willis N.A., Rajendran A., Manis J.P., Scully R.. Complex breakpoints and template switching associated with Non-canonical termination of homologous recombination in mammalian cells. PLos Genet. 2016; 12:e1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan B., Li D., Gou K.. Homologous illegitimate random integration of foreign DNA into the X chromosome of a transgenic mouse line. BMC Mol. Biol. 2010; 11:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kramara J., Osia B., Malkova A.. Break-Induced replication: the where, the why, and the how. Trends Genet. 2018; 34:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piazza A., Wright W.D., Heyer W.-D.. Multi-invasions are recombination byproducts that induce chromosomal rearrangements. Cell. 2017; 170:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verma P., Greenberg R.A.. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016; 30:1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dai J., Cui X., Zhu Z., Hu W.. Non-homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryos. Int. J. Biol. Sci. 2010; 6:756–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Masumura K., Sakamoto Y., Kumita W., Honma M., Nishikawa A., Nohmi T.. Genomic integration of lambda EG10 transgene in gpt delta transgenic rodents. Genes Environ. 2015; 37:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mikhailov K.V., Efeykin B.D., Panchin A.Y., Knorre D.A., Logacheva M.D., Penin A.A., Muntyan M.S., Nikitin M.A., Popova O.V., Zanegina O.N. et al.. Coding palindromes in mitochondrial genes of Nematomorpha. Nucleic Acids Res. 2019; 47:6858–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiang C., Jacobsen J.C., Ernst C., Hanscom C., Heilbut A., Blumenthal I., Mills R.E., Kirby A., Lindgren A.M., Rudiger S.R. et al.. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 2012; 44:390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goodwin L.O., Splinter E., Davis T.L., Urban R., He H., Braun R.E., Chesler E.J., Kumar V., van Min M., Ndukum J. et al.. Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 2019; 29:494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suzuki O., Hata T., Takekawa N., Koura M., Takano K., Yamamoto Y., Noguchi Y., Uchio-Yamada K., Matsuda J.. Transgene insertion pattern analysis using genomic walking in a transgenic mouse line. Exp. Anim. 2006; 55:65–69. [DOI] [PubMed] [Google Scholar]
- 52. Cain-Hom C., Splinter E., van Min M., Simonis M., van de Heijning M., Martinez M., Asghari V., Cox J.C., Warming S.. Efficient mapping of transgene integration sites and local structural changes in Cre transgenic mice using targeted locus amplification. Nucleic Acids Res. 2017; 45:gkw1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laboulaye M.A., Duan X., Qiao M., Whitney I.E., Sanes J.R.. Mapping transgene insertion sites reveals complex interactions between mouse transgenes and neighboring endogenous genes. Front. Mol. Neurosci. 2018; 11:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nicholls P.K., Bellott D.W., Cho T.-J., Pyntikova T., Page D.C.. Locating and characterizing a transgene integration site by nanopore sequencing. G3; Genes|Genomes|Genet. 2019; 9:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Akgün E., Zahn J., Baumes S., Brown G., Liang F., Romanienko P.J., Lewis S., Jasin M.. Palindrome resolution and recombination in the mammalian germ line. Mol. Cell Biol. 1997; 17:5559–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen H., Lisby M., Symington L.. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell. 2013; 50:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grunwald H.A., Gantz V.M., Poplawski G., Xu X.-R.S., Bier E., Cooper K.L.. Super-Mendelian inheritance mediated by CRISPR–Cas9 in the female mouse germline. Nature. 2019; 566:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tacken P.J., Zee A.V.D., Beumer T.L., Florijn R.J., Gijpels M.J.J., Havekes L.M., Frants R.R., Dijk K.W.V., Hofker M.H.. Effective generation of very low density lipoprotein receptor transgenic mice by overlapping genomic DNA fragments: high testis expression and disturbed spermatogenesis. Transgenic Res. 2001; 10:211–221. [DOI] [PubMed] [Google Scholar]
- 59. Skryabin B.V., Gubar L., Seeger B., Kaiser H., Stegemann A., Roth J., Meuth S.G., Pavenstädt H., Sherwood J., Pap T. et al.. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR/Cas9-mediated genome editing events. 2019; bioRxiv doi:08 March 2019, preprint: not peer reviewed 10.1101/570739. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence reads have been deposited at the Sequence Read Archive (SRA) under accession number PRJNA533172. The processed data (transgene ‘subway maps’ in web-browser file format) are available as supplementary (Sup. Files 1–11).