


Abstract
RNA:5-methylcytosine (m5C) methyltransferases are currently the focus of intense research following a series of high-profile reports documenting their physiological links to several diseases. However, no methods exist which permit the specific analysis of RNA:m5C methyltransferases in cells. Herein, we described how a combination of biophysical studies led us to identify distinct duplex-remodelling effects of m5C on RNA and DNA duplexes. Specifically, m5C induces a C3′-endo to C2′-endo sugar-pucker switch in CpG RNA duplex but triggers a B-to-Z transformation in CpG DNA duplex. Inspired by these different ‘structural signatures’, we developed a m5C-sensitive probe which fluoresces spontaneously in response to m5C-induced sugar-pucker switch, hence useful for sensing RNA:m5C methyltransferase activity. Through the use of this probe, we achieved real-time imaging and flow cytometry analysis of NOP2/Sun RNA methyltransferase 2 (NSUN2) activity in HeLa cells. We further applied the probe to the cell-based screening of NSUN2 inhibitors. The developed strategy could also be adapted for the detection of DNA:m5C methyltransferases. This was demonstrated by the development of DNA m5C-probe which permits the screening of DNA methyltransferase 3A inhibitors. To our knowledge, this study represents not only the first examples of m5C-responsive probes, but also a new strategy for discriminating RNA and DNA m5C methyltransferase activity in cells.
INTRODUCTION
The DNA and RNA of all living organisms, as well as that of viruses, mitochondria and chloroplasts, undergo a wide range of modifications (1–2). These modifications not only expand the structural diversity of nucleic acids, but also provide an epigenetic mechanism to fine-tune their biological functions (3–4). To date, at least 160 naturally-occurring chemical modifications have been identified, amongst which 5-methylcytosine (m5C) is currently one of the most intensively studied epigenetic modifications. The m5C modification is prevalent in DNA and multiple RNA classes (5–6). Its biological functions are best understood in DNA, where it is involved in the regulation of gene expression, genome reprogramming, organismal development and cellular differentiation (5,7,8). In contrast, there has been comparatively less studies on the biological roles of m5C in RNA. Indeed the significance of m5C modification in mRNA was not fully appreciated until recently, following the landmark discovery of widespread m5C sites in the transcriptomes of diverse organisms (9–16), suggesting that the m5C modification is far more pervasive in human mRNA than previously realised. This has reignited intense interest in the study of this epitranscriptomic mark. At present, the exact biological function of m5C modification in mRNA remains elusive although it has been linked to many cellular processes, such as nuclear export regulation (14), modulation of protein translation (17–19), and stress response (20).
The m5C methylation landscape is regulated by a complex array of m5C methyltransferases (MTases) and m5C demethylases, which specifically add and remove a 5-methyl mark from cytosine base, respectively (6,21–23). In humans, C-5 cytosine methylation of DNA is catalysed by at least three DNA:m5C MTases (i.e. DNMT1, 3A and 3B), whereas m5C methylation of RNA is catalysed by the NOL1/NOP2/Sun domain (NSUN) RNA methyltransferase family, which includes NSUN1-7, as well as the DNA MTase homologue TRDMT1 (formerly DNMT2). Notably, both the DNA and RNA m5C MTases employ a common S-adenosyl-l-methionine (SAM) dependent mechanism for methyl group transfer, although different active-site cysteine residues are likely involved in the catalysis (24).
In recent years, it has become increasingly clear that aberrant expression of m5C MTases may underlie the pathogenesis of several diseases. For instance, DNMT3A, NSUN1, NSUN2 and NSUN4 were found to be overexpressed in a number of human cancers, including breast, prostate, cervical, and colorectal cancers (25–29). NSUN3 is linked to mitochondrial diseases (30,31), whilst NSUN5 and NSUN7 are likely implicated in Williams-Beuren syndrome (a neurodevelopmental disorder) (32) and male infertility (33), respectively. Given the strong physiological links, there is currently immense interest in studying the biological roles of these enzymes and their potentials as therapeutic targets. However, at present, no methods exist which permit the direct analysis of specific m5C MTase in living cells. There are also no reports of cell-based assays capable of discriminating between DNA and RNA MTase activities. This severely impedes our understanding of these medically-important enzymes.
It is known that several nucleic acid base methylations can directly impact the secondary structure of DNA and RNA. For instance, we (34–36), and others (37–40), have recently shown that the presence of a single N6-methyladenosine (m6A) or N1-methyladenosine (m1A) modification can trigger a duplex-hairpin transformation in certain RNA sequence contexts. It is also clear from a number of NMR and X-ray crystallographic studies that m5C modification is able to induce a major B–Z structural change in some DNA sequences. This phenomenon was first reported by Behe and Felsenfeld (41), and subsequently by others (42–44), for a number of DNA duplexes with alternating CpG sequences, such as d(m5CGCGm5CG), poly d(m5CG)n and poly d(Gm5C)n. The structural influence of m5C on RNA duplexes, however, is significantly less-studied. One early study on a poly(CG) RNA sequence suggests that m5C methylation likely elicit a conformational change from an A-type duplex to an atypical duplex structure (45), whereas a separate study using a different sequence context indicates no significant structural alteration (46). To the best of our knowledge, these two reports provide the only experimental data available in this regard and, to date, it remains unclear whether m5C modification has any major impact on the secondary structures of RNA duplexes.
Herein we demonstrated, through a combination of NMR, circular dichroism (CD), and thermodynamic studies, that m5C has different conformational effects on DNA and RNA duplexes, even for duplexes with identical CpG sequence. In the case of CpG RNA duplex, it induces a considerable distortion of the phosphate backbone and a C3′-endo to C2′-endo sugar pucker switch in the terminal residues whereas, in the case of CpG DNA duplex, m5C triggered a remarkable B-to-Z structural transformation. m5C therefore produces distinctly different ‘structural signatures’ on CpG RNA and DNA duplexes under the same physiologically-relevant conditions.
Inspired by these interesting findings, we envisioned that the different duplex-remodelling effects of m5C could provide a basis for the selective detection of RNA and DNA m5C MTase activity in cells. This was demonstrated by the development of a novel m5C-sensitive nucleic acid probe which, by design, is capable of switching its terminal sugar pucker conformation spontaneously in response to m5C methylation (Figure 1). When coupled with an environment-sensitive fluorophore, it provides a powerful visual tool for sensing RNA:m5C methylation changes in cells. Prior to this work, we are not aware of any assay methods which are based on m5C-induced conformational change.
Figure 1.

The m5C-switchable probe strategy. (A) The m5C-probe 8a contains a 5′-terminal fluorescent nucleotide PC (2′-O-methyl 6-phenylpyrrolocytidine), which lights up spontaneously in response to m5C-induced terminal sugar pucker switch. (B) When the probe is unmethylated, pC is able to base-pair with guanine in the complementary strand and stack strongly with its adjacent base. This results in efficient quenching of pC fluorescence through photoinduced electron transfer. m5C methylation of the probe by RNA:m5C MTase (e.g. NSUN2), however, is expected to trigger a C2′-endo to C3′-endo sugar pucker switch in PC and, since the sugar ring pucker defines the glycosidic bond angle, such a change in sugar puckering will also convert the orientation of PC base from axial to equatorial. This, in turn, disrupts its base-pairing and base-stacking interactions, leading to fluorescence activation. (C) Schematic representation of 2′-OMe RNA probes and their methylated counterparts. The composition of the probes was confirmed through MALDI-TOF mass spectrometric analysis (see Supplementary Table S2). The structure of ‘locked 6-phenylpyrrolocytidine’ (LC) is shown. The C2′−C4′ covalent link is geometrically incompatible with C2′-endo pucker mode, it therefore locks the nucleotide into the C3′-endo conformation.
As we shall demonstrate, the m5C-probe is highly-selective and could specifically target NSUN2 over other RNA:m5C MTases (including structurally-related subfamily members NSUN3, NSUN5A, NSUN6) and DNA:m5C MTases (including DNMT1 and DNMT3A). Through the m5C-probe approach, we achieved live cell imaging and flow cytometry analyses of NSUN2 activity in HeLa cells. We also successfully applied the probe to the high-throughput cell-based screening of NSUN2 inhibitors. The discovery of such highly selective probes is rarely achieved and may provide insights on the functions of NSUN2 in m5C-regulated processes. Although this study focus on the sensing of RNA:m5C MTase activity, the m5C-switchable probe strategy outlined here could also be adapted for the study of DNA:m5C MTase. This was demonstrated by the successful development of DNA m5C-probe which is useful for the in vitro screening of DNMT3A inhibitors.
To the best of our knowledge, this study represents not only the first examples of m5C-responsive fluorescent probes, but also a new strategy for the discrimination of RNA and DNA m5C MTases activity in cells, which hopefully will inspire the development of epigenetic biosensors and diagnostics tools.
MATERIALS AND METHODS
Preparation of m5C-probes
All oligonucleotide probes investigated in this study were synthesized using standard β-cyanoethyl phosphoramidite chemistry on an automated DNA/RNA synthesiser (Applied Biosystems 394). All synthesiser reagents were purchased from Glen Research; 2′-O-methyl 6-phenylpyrrolocytidine phosphoramidite (47,48) and pyrene deoxynucleoside phosphoramidite (49) were synthesised according to literature procedure. For details of synthesis, see Supplementary Data. All oligonucleotides have a purity of at least 95% (Supplementary Tables S1-S3).
Studies on the duplex-remodelling effects of m5C using NMR, CD and UV-based thermodynamic analyses
All 1H NMR experiments were recorded on a Bruker DRX-500 spectrometer operating at 500.23 MHz and the data processed using the standard Bruker processing software TopSpin. Estimated accuracy for protons is within 0.02 ppm. The oligos were dissolved in either D2O (99.98%) or buffer/D2O mix; the buffer used was 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2. Final concentrations were 3 mM for the unmethylated duplexes (1a, 7a) and 2 mM for the m5C-methylated duplexes (1b, 7b). The proton chemical shifts were referenced relative to residual H2O signal (4.75 ppm at 25°C). The 1H imino spectra of duplexes in 9:1 buffer/D2O solvent mix (pH 7.4) were acquired at 4°C and 10°C with suppression of water signal by gradient-tailored excitation (WATERGATE) sequence, as previously described (34,50); at these temperatures, the oligos exist in the duplex state, as verified by UV-melting and CD analyses. Two-dimensional 1H NMR spectra of duplexes in D2O and buffer/D2O were acquired in the phase-sensitive mode using previously described method (51). Two-dimensional NOESY spectra of duplexes in D2O (52–54) were recorded at 25°C with a spectral width of 4000 Hz using 1000 t2 complex points, 1000 t1 (real) points, pulse repetition delay of 2.5 s and mixing times (τm) of 80, 150 and 300 ms. Two-dimensional NOESY spectra of duplexes in 9:1 buffer/D2O were acquired at 10°C with water suppression by WATERGATE sequence (34,50). Spectra were recorded using 2048 t2 complex points, 256 t1 (real) points, pulse repetition delay of 1.7 s and mixing time of 200 ms. Proton-decoupled 31P NMR spectra of duplexes in 9:1 buffer/D2O were acquired at 10°C on a Bruker AV300 spectrometer equipped with an autotune 5 mm QNP probe. Spectra were recorded at 121.4 MHz, with a spectral width of 10 kHz. The 31P chemical shifts were externally referenced to 85% phosphoric acid (H3PO4). Estimated accuracy for phosphorus is within 0.03 ppm. For CD spectroscopy and UV-based thermal denaturation experiments, see Supplementary Data.
Fluorescence analyses of m5C-probes
Fluorescence analysis was performed using a Cary Eclipse fluorescence spectrophotometer, as previously reported with modifications (36). The fluorescence emission spectra (λem = 400–600 nm) of the probes were recorded in a quartz cuvette at 37°C, at a total stand concentration 5 μM in a buffer of 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2. An average of five scans was recorded. Background fluorescence spectra were acquired after the probe has been incubated for 30 min at 37°C. For determination of fluorescence quantum yields of the m5C-probes, see Supplementary Data.
In vitro m5C-probe methyltransferase assay
RNA methyltransferase assay: the assay was performed at 37°C in triplicate in a Corning Costar 96-well flat bottom black plate. Reaction consisted of enzyme (0.5 μM), methyl donor S-adenosyl-l-methionine (SAM; 200 μM), m5C-probe 9a (substrate; 5 μM) or locked-probe 10a (control; 5 μM) in a 50 mM HEPES buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2. The final reaction volume is 25 μl. The time course of fluorescence activation was recorded immediately after the addition of enzyme with a Tecan ultra microplate reader (λex 360 nm; λem 465 nm). An average of five scans was recorded. DNA methyltransferase assay: the assay was performed as described above. Reaction consisted of enzyme (NSUN2 or DNMT3A; 0.5 μM), methyl donor S-adenosyl-l-methionine (SAM; 200 μM), DNA m5C-probe 11a (substrate; 5 μM) in a 50 mM HEPES buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2. The fluorescence of the reaction was monitored at λem 400 nm (λex 340 nm).
Steady-state kinetic analyses of the methylation of m5C-probe, tRNA and ssRNA by NSUN2
The substrates investigated include m5C-probe 9a, human tRNALeu(CAA) (5′-CCAGACUCAAGUUCUGG-3′), and CpG-rich ssRNA (5′-CGCGCGCGCGCG-3′). The kinetic constants (Vmax, Km,kcat) of the substrate were determined using an NSUN2 concentration of 0.1 μM and substrate concentrations of 1, 2, 3, 5, 8 and 10 μM. The amount of m5C-methylated product formed at different substrate concentrations was determined based on the relative intensities of the substrate and the product peaks observed in MALDI-TOF mass spectra. All reactions were performed at 37°C in triplicate.
Fluorescence microscopy
The HeLa cells were grown in 35 mm glass bottom culture dishes (Thermo Scientific). The cells were transfected with either m5C-probe 9a (10 μM) or locked-probe 10a (10 μM; control) using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocols. Fluorescence images were then acquired at 37°C in the presence of 5% CO2 using a Nikon BioStation IM-Q live cell imaging system equipped with 20× and 63× oil immersion objective lens. The fluorescence images were captured using the DAPI filter setting (i.e. λex 340–380 nm; λem 435–485 nm) and the fluorescence intensity quantified using Biostation IM multichannel software by collecting mean gray values for regions exhibiting fluorescence. All experiments were performed in triplicates and the mean fluorescence intensities were normalised to that of the control. The corresponding transmitted light image for each fluorescence image was also acquired.
Single-cell flow cytometry analysis
The HeLa cells were transfected with m5C-probe 9a (10 μM) or control probe 10a (10 μM) using Lipofectamine 2000. They were then collected by trypsinization at 37°C with 5% CO2, washed with phosphate buffered saline (PBS) media, pH 7.4, resuspended in PBS media, and analysed using a BD LSRFortessa™ flow cytometer (BD Bioscience). Fluorescence was measured using an excitation laser of 355 nm, and a 450/50 bandpass emission filter. Acquisition was stopped when 20, 000 events per sample were acquired. The fluorescence data were then analysed using BD FACS software. Violin plot was produced using the Origin software.
Cell-based m5C-probe methyltransferase assay
The HeLa cells were pre-incubation with various concentrations of non-specific MTase inhibitors, including sinefungin, S-adenosyl-l-homocysteine (SAH), adenosine, and homocysteine at 37°C for 30 min. Nine different concentrations of inhibitors were used (0.1, 0.3, 1, 3, 10, 50, 100, 250, 1000 μM) and the final DMSO concentration was kept at less than 1% (v/v) of the assay mix. These concentrations of inhibitors were shown to be non-toxic to HeLa cells in MTT cytotoxicity assays. This was followed by the delivery of either the m5C-probe 9a (10 μM) or control probe 10a (10 μM) into the cells via Lipofectamine 2000. Aliquots were then collected for flow cytometry measurements (λex 355 nm; λem 425–475 nm) at the indicated time points. The mean fluorescence intensity of at least 20 000 live cells was determined. IC50 determination: the IC50 values were then calculated from the variation in fluorescence at different inhibitor concentrations using nonlinear regression, with normalised dose-response fit on GraphPad Prism 6.0™. The assay was performed in triplicate for each inhibitor concentration. Z’ factor determination: the Z’ factor for our m5C-probe assay was calculated using previously described method (55). For details, see Supplementary Data.
Cell culture, gene silencing and gene overexpression
HeLa cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin). Cells were grown at 37°C in a 5% CO2 humidified incubator. To silence the expression of specific gene transiently, the cells were first plated in a 24-well plate (0.5 × 105 cells/well). The next day, the cells were transfected with either siNSUN2, siNSUN6, siTRDMT1, siDNMT3A or siControl using Lipofectamine RNAiMAX (Invitrogen), according to manufacturer's protocols. The sequences used for siRNAs and siControls are provided in the Supplementary Data. The protein levels were analysed by western blot using specific antibodies. Overexpression of specific gene. The construction of pcDNA3-Flag vectors expressing NSUN2, TRDMT1 and DNMT3A was previously described (56–58). Full length NSUN2, TRDMT1 and DNMT3A genes were amplified by PCR using human HeLa cells cDNA, then subcloned into the EcoRI-BamHI sites of mammalian pcDNA3-Flag vector (Invitrogen) to obtain pcDNA3-Flag-NSUN2, pcDNA3-Flag-TRDMT1, and pcDNA3-Flag-DNMT3A plasmids, respectively. The plasmids were then transfected into HeLa cells using the calcium chloride transfection method. For construction of pcDNA3-Flag-NSUN6 plasmid (59), the cDNA encoding NSUN6 was PCR-amplified using Phusion DNA polymerase and subcloned into the KpnI-XhoI sites of pcDNA3.1-Flag with CloneExpress II One step Cloning Kit (Vazyme). The primer sequences used for vector construction are given in Supplementary Data.
Western blot analysis
The HeLa cells were first lysed using radioimmunoprecipitation assay (RIPA) buffer (i.e. 25 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate, 1% NP-40 and 1× protease inhibitors), followed by immunoblotting using standard protocol. In brief, the extract was fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto nitrocellulose filter membranes (Whatman), then incubation with the respective antibodies. This was followed by incubation with either horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (Bio-Rad) or HRP-conjugated anti-mouse IgG antibody (Bio-Rad) for 2 h at room temperature. Enhanced chemiluminescence substrates (Luminata Crescendo, EMD Millipore) were then applied and the signals exposed to autoradiography film. The immunoblots were then quantified by densitometric analyses using ImageJ software. All antibodies used were purchased from commercial sources (for details, see Supplementary Data).
RESULTS AND DISCUSSION
m5C induces a local distortion of the phosphate backbone and a C3′-endo to C2′-endo sugar pucker switch in CpG RNA duplex
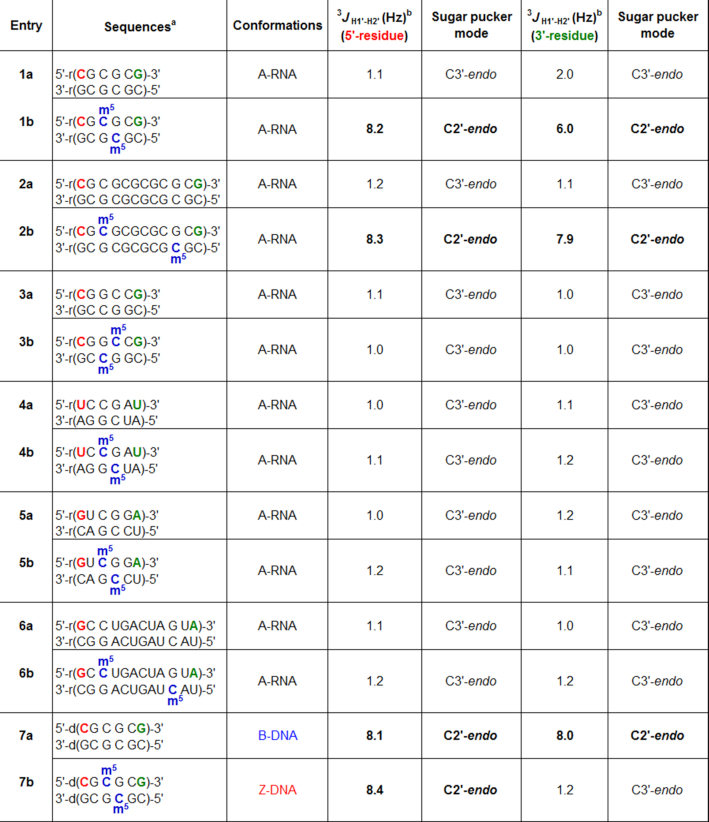
To date, there has not been a systematic study directly comparing the structural impact of m5C methylation on CpG RNA duplexes and CpG DNA duplexes. Such an analysis might provide insights into the respective conformational and/or mechanistic roles of m5C in DNA and RNA, hence is of special interest. To investigate this question in multiple sequence contexts, we prepared a set of 6-mer and 12-mer RNA duplexes containing either alternating CpG sequences (1a–2a), out-of-alternation CpG sequence 3a or random sequences (4a–6a), as well as their corresponding m5C-methylated counterparts (1b–6b; Table 1). To facilitate comparison, the CpG DNA duplexes 7a and 7b were also generated. For details of their chemical synthesis, see Supplementary Data.
Table 1.
Sequences of RNA and DNA duplexes investigated in this study. The 3JH1′−H2′ values for the 5′- and 3′-terminal residues of each sequence are indicated in red and green, respectively. m5C-induced terminal sugar pucker switch was observed in 6-mer and 12-mer CpG duplexes (1b and 2b) but not in out-of-alternation CpG duplex 3b and random duplexes 4b–6b, suggesting that the structural effect of m5C may be unique to alternating CpG RNA sequences. m5C methylation of 7a i.e. the DNA equivalent of 1a caused a C2′-endo to C3′-endo sugar pucker switch in all guanosine residues, whilst all cytosine residues preserved their C2′-endo orientation, consistent with a B-Z structural conversion
|
aThe composition of the oligos was confirmed through MALDI-TOF mass spectrometric analysis (see Supplementary Table S1).
bObserved at 25°C in D2O.
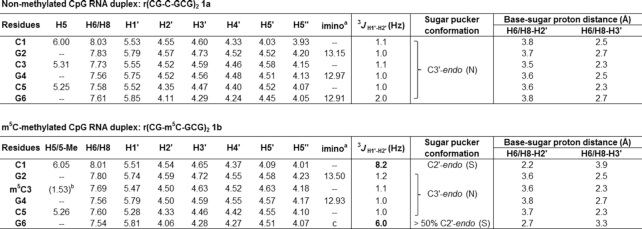
We began by analysing the secondary structure of CpG RNA hexamer r(CG-C-GCG)21a and its methylated equivalent r(CG-m5C-GCG)21b using 1H and 31P NMR. Assignment of the proton resonances was accomplished by 2D NOESY experiments and by comparison with reported NMR data for 1a (60). The results are summarised in Figure 2 and Table 2. Consistent with previous NMR studies (60), the imino proton spectrum of 1a exhibited three NH signals at 10°C, which is expected for a palindromic duplex with six base-pairs (Figure 2A). Upon m5C methylation (1b), we observed a downfield shift in G2 resonance by ∼0.35 ppm and a disappearance of the terminal G6 resonance. The latter is likely due to ‘fraying’ of the terminal base-pairs as the G6 signal could again be detected at a lower temperature of 4°C (Supplementary Figure S1). This result suggests that m5C modification likely promotes the disruption of base-pairing interactions at duplex terminal ends. As anticipated, there is little or no change in the imino resonance of G4 which is involved in base-pairing with m5C, confirming that C5-cytosine methylation does not hinder C:G Watson–Crick base-pairing interactions. Nevertheless, we did observe considerable variations in 31P resonances between the methylated and unmethylated duplexes, indicating a significant distortion of the phosphate backbone (Figure 2B).
Figure 2.

m5C methylation directly alters the phosphate backbone in CpG RNA duplex, leading to a C3′-endo to C2′-endo sugar pucker switch of the terminal residues under physiologically-relevant conditions. (A) Representative 1H imino NMR spectra of r(CG-C-GCG)21a (top) and its methylated counterpart r(CG-m5C-GCG)21b (bottom) in 9:1 buffer/D2O solvent mix acquired at 10°C. The buffer used was 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2. The G6 imino resonance disappeared upon m5C methylation, presumably due to ‘fraying’ of the terminal base-pairs. (B) Proton-decoupled 31P NMR spectra of 1a (top) and 1b (bottom) in 9:1 buffer/D2O solvent mix acquired at 10°C. There is considerable variations in 31P resonances between the methylated and unmethylated duplexes, suggesting a significant distortion of the phosphate backbone. Representative 2D NOESY spectra of the H2′/H3′-aromatic region of (C) 1a and (D) 1b in D2O at 25°C (pH 7.4; τm = 150 ms) showing the H6/8-H2′ NOE connectivity pathways (blue dashed-line) and the H6/8-H3′ NOE connectivity pathways (black solid line). The schematic illustrations (left) showed the likely sugar pucker mode for terminal residues C1 and G6, and their calculated intranucleotide base-sugar distances (labelled in red; determined based on the intensity of NOEs between the base and sugar protons).
Table 2.
1H NMR chemical shifts δH (ppm) for CpG RNA duplex 1a (top) and its m5C-methylated counterpart 1b (bottom) observed at 25°C in D2O with WATERGATE suppression (τm 150 ms). Assignment of the proton resonances was accomplished by 2D NOESY experiments, and by comparison with reported NMR data for 1a (60). The representative NMR spectra for the determination of 3JH1′−H2′ coupling constants are given in Supplementary Figure S2. For calculation of sugar-base proton distances, see Supplementary Data
|
aObserved at 10°C in 9:1 buffer/D2O solvent mix. The buffer used was 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2.
bChemical shift for 5-methyl group on m5C.
cNot observed even at 4°C.
Prompted by this finding, we proceeded to examine whether changes in phosphate backbone also alter the sugar ring geometry. To this end, we determined the coupling constant between sugar proton H1′ and H2′ (3JH1′−H2′), which is highly sensitive to the dihedral angle and, therefore, provides a direct indication of the sugar pucker conformation. In general, a 3JH1′−H2′ value <2 Hz is suggestive of a C3′-endo sugar pucker (North type), whereas 3JH1′−H2′ value >8 Hz is characteristic of a C2′-endo pucker (South type) (61). The 3JH1′−H2′ values for 1a and 1b are summarized in Table 2 and the representative spectra are shown in Supplementary Figure S2. In the absence of cytosine methylation, all ribose rings in 1a exhibit a C3′-endo conformation (3JH1′−H2′ ∼1–2 Hz), which is the preferred sugar puckering mode for A-form RNA duplex. Upon m5C methylation (1b), however, the 5′-terminal cytosine (C1) undergoes a sugar pucker switch from C3′-endo to C2′-endo, as evidenced by a large increase in 3JH1′−H2′ from 1.1 Hz to 8.2 Hz. This is accompanied with a switch in sugar puckering of the 3′-terminal guanosine (G6) to an intermediate conformation between C3′-endo and C2′-endo (3JH1′−H2′ = 6.0 Hz). No changes in sugar pucker mode are observed for the four internal residues G2 to C5, which continue to adopt a C3′-endo orientation. Our data support the findings of an earlier NMR study (45) and, together, they demonstrate that m5C can alter the sugar pucker conformation of the 5′- and 3′-terminal residues in CpG RNA duplexes.
To confirm our results, we further analysed the intensity of intranucleotide NOEs between the base protons (H6 in cytosine; H8 in guanine) and the sugar protons (H2′ and H3′), which correlates with the distance between the protons (Figures 2C, D and Table 2) (62,63). Consistent with our 3JH1′−H2′ data, the C1 residue of 1b displays a strong NOE between its base proton H6 and sugar proton H2′ (∼2.2 Å) and a weak NOE between H6 and H3′ (∼3.9 Å), which is indicative of a C2′-endo conformation, and the G6 residue of 1b shows approximately similar NOE intensities for H8-H2′ (∼2.7 Å) and H8-H3′ (∼3.3 Å), suggesting an intermediate C2′-endo/C3′-endo conformation. In comparison, both the C1 and G6 residues of unmethylated 1a exhibit a C3′-endo sugar pucker, thus m5C methylation indeed produces a switch in the terminal sugar pucker orientation.
Notably, however, these structural perturbations appear to have little or no effect on the overall secondary structure of the RNA duplex. This is apparent from CD analysis where the spectra of 1a and 1b exhibit characteristics which are typical of a right-handed A-form duplex, and they superimpose almost perfectly with each other (Figure 3A). Hence, m5C modification likely only affects the local nucleotide geometry without altering the overall conformation of CpG RNA duplexes.
Figure 3.
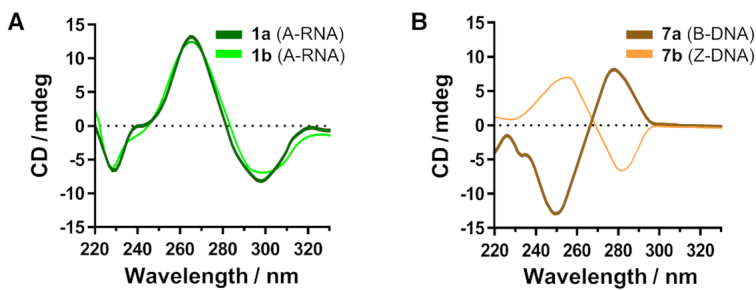
m5C methylation induces distinctly different conformational effects on RNA and DNA duplexes. Superimposition of the CD spectra of (A) CpG RNA duplex 1a with that of its m5C-methylated counterpart 1b, and (B) CpG DNA duplex 7a with that of its m5C-methylated equivalent 7b. Under physiologically-relevant salt and pH conditions, m5C methylation has negligible effect on the overall conformation of 1a. In sharp contrast, methylation of 7a triggered a transformation from right-handed B-DNA to left-handed Z-DNA, as apparent from the characteristic inversion of its CD spectrum.
m5C-induced terminal sugar pucker switch is likely unique to alternating CpG RNA sequences
We next examined whether m5C could also elicit terminal sugar pucker switch in other RNA sequence contexts. To this end, we determined the 3JH1′−H2′ coupling constant values for the 5′- and 3′-terminal residues of each sequence (Table 1). Our results revealed a similar C2′-endo to C3′-endo switch in the longer 12-mer CpG duplex 2b, but not in the out-of-alternation CpG sequence 3a and random sequences 4a–6a. On the basis of these results, we performed further NMR experiments to investigate how the position of m5C methylation affects terminal sugar pucker conformation in alternating CpG duplexes 1a and 2a. The 3JH1′−H2′ values are summarised in Supplementary Table S4. For 6-mer CpG duplexes, terminal sugar switch occurred only when the third cytosine residue (C3) was methylated (i.e. 1b), and not when the first (1c) or second (1d) cytosine residue was methylated. Similarly for 12-mer CpG duplexes, m5C-induced sugar switch was observed only when methylation was on C3 (2b), and not on C1 (2c) or C5 (2d), thus the structural effect of m5C is highly-dependent on the position of m5C methylation.
The sequence requirements and mechanisms by which m5C induces terminal sugar pucker switch are unclear at present and require further investigations. Nevertheless, the present study, the first to investigate the structural influence of m5C on multiple RNA sequence contexts, suggests that this conformational effect might be unique to alternating CpG RNA sequences.
m5C triggers a B-to-Z DNA transformation in CpG DNA duplex
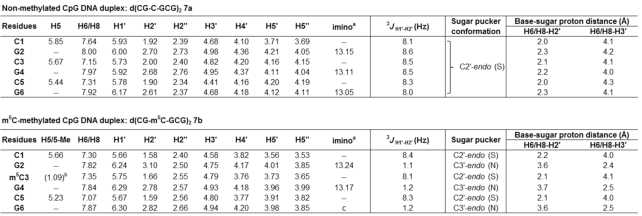
We next investigated the structural influence of m5C on CpG DNA duplex 7a, i.e. the DNA equivalent of 1a under the same physiologically-relevant conditions. A preliminary analysis of the 3JH1′−H2′ values and sugar-base proton distances of 7a and 7b revealed a switch in sugar puckering of all guanosine residues from C2′-endo to C3′-endo mode, whilst all cytosine residues preserve their C2′-endo orientation, which is highly suggestive of a B-to-Z structural transformation (Table 3; the representative 2D NOESY spectrum of 7b is shown in Supplementary Figure S3). As further evidence, CD analysis showed a characteristic inversion of the CD spectrum upon m5C methylation, confirming that m5C does indeed triggers a major B-Z structural transformation in CpG DNA duplex 7a (Figure 3B). This result is notable because it demonstrates that m5C produces different structural outcomes on RNA and DNA duplexes.
Table 3.
1H NMR chemical shifts δH (ppm) for CpG DNA duplex 7a (top) and its m5C-methylated counterpart 7b (bottom) observed at 25°C in D2O with WATERGATE suppression. The representative 2D NOESY spectrum is given in Supplementary Figure S3. For calculation of sugar-base proton distances, see Supplementary Data
|
aObserved at 10°C in 9:1 buffer/D2O solvent mix. The buffer used was 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2.
bChemical shift for 5-methyl group on m5C.
cNot observed even at 4°C.
The observed B-Z conversion is likely attributed to the ability of m5C to stabilize the Z-form DNA over its B-form structure (42,64). Indeed, UV-based thermodynamic analysis of 7a showed that m5C methylation increases the stability of its Z-duplex by ∼2.5 kcal/mol relative to its B-duplex (compare ΔG°310 of 7a with 7b; Supplementary Table S3 and Supplementary Figure S4). Moreover, m5C-induced B-Z helicity change is primarily an entropy-driven process, as judged by the more favourable entropic contributions (ΔΔS°7a→7b = 31.6 cal/mol/K) which counteracts the enthalpy cost (ΔΔH°7a→7b = 7.3 kcal/mol). Further thermodynamic analysis of 7b in the absence of MgCl2 (a known Z-conformation inducer) revealed a free energy of B-to-Z transition (ΔΔG°T) of ∼1.5 kcal/mol (compare ΔG°310 of 7b with 7b*); since 7b does not undergo B-Z conversion in the absence of MgCl2, any free energy differences observed at these two salt conditions, i.e. between 7b and 7b* is largely ascribed to conformational transition (Supplementary Table S3 and Supplementary Figure S5).
m5C does not promote A–Z transition in CpG RNA duplex
To date, the effects of m5C methylation on the stability of Z-form RNA is unclear. We are also not aware of any prior studies investigating whether m5C could promote an analogous A–Z transition in RNA duplex. Therefore, we performed further CD experiments to monitor the conformation of CpG RNA duplex 1a and its methylated equivalent 1b at a range of MgCl2 concentrations (1–300 mM). The results showed that both 1a and 1b maintain an A-type double helical structure at all MgCl2 concentrations investigated (Supplementary Figure S6). In fact, the methylated RNA 1b could not be induced to form Z-RNA even at MgCl2 concentration as high as 300 mM. Consistent with this observation, the presence of m5C does not significantly improve the stability of 1a (ΔΔG°310 ∼0.8 kcal/mol; Supplementary Table S3). Thus m5C methylation favours B–Z conversion in CpG DNA duplex, but not A–Z transition in CpG RNA duplex under the same physiological salt and pH conditions.
Overall, our results demonstrated that m5C methylation can directly influence the conformation of RNA and DNA duplexes. In the case of CpG RNA hexamer, m5C modification induces a local distortion of the phosphate backbone and a C3′-endo to C2′-endo terminal sugar pucker switch whereas, in CpG DNA hexamer, m5C triggers a transformation from a right-handed B-DNA to a left-handed Z-DNA. We appreciate that detailed crystallographic studies are required to fully characterise these structural changes, nevertheless, our available data clearly demonstrated that m5C modification produces distinctly different ‘structural signatures’ on RNA and DNA duplexes, even for duplexes with identical CpG sequence. The significance of this finding is unclear at present, however it might infer a possible involvement of m5C-induced conformational change in biological regulatory processes.
Design principle of m5C-switchable probes
Inspired by these interesting findings, we envisaged that the different structure-remodelling effects of m5C on RNA and DNA duplexes could be exploited for the design of ‘methylation-switchable probe’ useful for studying m5C methylation in DNA and RNA. Notably, at present, no methods exist which permit the direct analysis of m5C MTase activity in living cells. There are also no reports of assay methods that are able to distinguish between the activities of DNA m5C MTases and RNA m5C MTases.
As a proof-of-principle study, we developed a novel m5C-probe which is able to fluoresce spontaneously in response to m5C-induced terminal sugar pucker switch, hence useful for sensing RNA:m5C MTase activity. The probe (8a; Figure 1) is modelled after the CpG RNA duplex 1a but contains two important modifications. First, in order to visualise m5C-induced sugar pucker switch, the 5′-cytosine residues on the forward strand was replaced with the fluorescent cytosine analogue 6-phenylpyrrolocytosine (pC) (47,48). pC is well-suited as reporter for our probe because it is highly fluorescence (quantum yield (ΦF) 0.31) and its emission is extremely sensitive to microenvironment changes (65–67). In particular, previous photophysical studies showed that pC emits brightly when in a single-stranded environment, but becomes significantly quenched when hybridised with its complementary strand (65–67). Second, to render the m5C-probe sufficiently stable for live-cell applications, the RNA backbone was replaced with a 2′-O-methyl backbone, which is reasonably resistant to cellular nuclease degradation.
The analytical principle of the m5C-probe is illustrated in Figures 1A and B. By design, when the probe is unmethylated, the pC fluorophore is able to base-pair with guanine and stack strongly with its adjacent base. This results in efficient quenching of pC fluorescence through photoinduced electron transfer (PET). m5C methylation of the probe by RNA:m5C MTases, however, is expected to trigger a spontaneous C3′-endo to C2′-endo sugar pucker switch in pC. Since the sugar ring pucker defines the glycosidic bond angle, a change in sugar puckering mode will also convert the orientation of PC base from axial to equatorial, thereby disrupting its base-pairing and base-stacking interactions, leading to fluorescence activation.
The m5C-probe has the sensitivity to detect a single m5C mark change
To verify our probe design, we first compared the fluorescence emission of the probes at different m5C methylation levels (λex 360 nm; λem 465 nm; Supplementary Figure S7). Consistent with our detection strategy, the starting unmethylated probe 8a displayed weak fluorescence, with a relatively low fluorescence quantum yield (ΦF) of ∼0.03, whilst its methylated counterpart 8b yielded a 2-fold increase in fluorescence intensity (ΦF ∼0.06; Supplemental Figure S7). Similarly the unmethylated probe 8c, which was designed to simultaneously exploit sugar pucker switch at both 5′-ends, also gave negligible fluorescence response (ΦF ∼0.04; Supplementary Figure S7). This result is notable because most DNA/RNA duplexes are known to undergo transient opening of the terminal base-pairs in solution. Conceivable, this might cause auto-activation of pC fluorescence, which will severely limit the applications of these probes. Nevertheless, our fluorescence data from starting probes 8a and 8c suggests that emission arising from such transient end-fraying event is relatively insignificant. Indeed, with the improved probe 8c, we are able to clearly detect a 2.8-fold increase in fluorescence intensity upon m5C methylation (8d; ΦF ∼ 0.11; Supplemental Figure S7), which is remarkable in light of only a single m5C mark change. Thus our m5C-probe has the sensitivity to detect subtle methylation changes. Moreover, the introduction of a second m5C modification, i.e. 8e led to approximately a doubling of fluorescence emission (5.3-fold), hence the fluorescence intensity of the probe increases proportionately with m5C methylation level.
Interestingly, the fluorescence output of the probe could be further increased by incorporating additional pC fluorophores, as demonstrated by probe 9a (Figure 4A) where the concurrent replacement of cytosine 1 and 5 in both strands with pC led to significant improvements in fluorescence light-up response (ΔΦF (9b) 3.6-fold; (9c) 7.2-fold). Despite the introduction of 2′-O-Me backbone and four relatively bulky pC residues, NMR data indicate that both the 5′- and 3′-terminal residues of probe 9a continue to adopt a C3′-endo pucker conformation (3JH1′−H2′ ∼1–2 Hz; Supplementary Table S4), and they spontaneously assume a C2′-endo pucker orientation (3JH1′−H2′ ∼8 Hz) upon m5C methylation (9c), thus these modifications do not interfere with fluorescence activation of the probe. There is also minimal disturbance to the overall duplex structure of probe 9a relative to parent probes 8a and 1a, as verified by CD analysis (Supplementary Figure S8). Amongst the probes investigated, 9a gives the greatest fluorescence response, it was therefore selected as representative m5C-probe for subsequent evaluations.
Figure 4.

Verification of the m5C-probe design. (A) The fluorescence emission spectra of probe 9a (λex 360 nm; λem 465 nm) were recorded at 5 μM strand concentration in 10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2, 37°C. The probe is highly responsive to its m5C methylation level; introduction of one and two m5C modifications led to a considerable 3.6-fold (9b) and 7.2-fold (9c) increase in fluorescence intensity, respectively. (B) Time-course fluorescence analysis of 9a (5 μM). Significant fluorescence could be detected after a short 30-minute incubation with NSUN2 (0.5 μM; blue line); maximum emission was reached after ∼3 h, giving ∼8 fold increase in fluorescence intensity. Probe fluorescence reduces significantly in the presence of generic MTase inhibitor sinefungin (10 μM; grey line), thus the probe is specifically activated by NSUN2 MTase activity. Incubation of NSUN2 with locked-probe 10a (5 μM; black line) gave negligible fluorescence response, confirming that probe activation is dependent on a change in sugar puckering of PC. (C) The fluorescence intensity increases linearly with the concentration of methylated probe, this enables a direct read-out of NSUN2 MTase activity. Data are expressed as mean ± SD of three replicates. (D, E) Steady-state kinetics analyses of the methylation of m5C-probe, human tRNALeu(CAA) (5′-CCAGACUCAAGUUCUGG-3′) and CpG-rich ssRNA (5′-CGCGCGCGCGCG-3′) by NSUN2. Data are expressed as mean ± SD of three replicates.
The m5C-probe is highly-selective for NSUN2 over other RNA/DNA:m5C MTases
We next examined whether m5C-probe 9a could function as fluorogenic substrate for RNA:m5C MTase by testing it against NSUN2, which is of particular interest in light of emerging evidence linking NSUN2 with m5C methylation of human mRNA (14) and its association with human cancers (25–29). In a typical assay, 5 μM of 9a was incubated with NSUN2 (0.5 μM) and methyl donor S-adenosyl-l-methionine (SAM; 200 μM) under physiologically-relevant conditions (at 37°C in 50 mM HEPES buffer containing 150 mM NaCl and 20 mM MgCl2, pH 7.4). The formation of methylated probe was measured by an increase in fluorescence signal at 465 nm (λex 360 nm). Our result showed that the probe could be readily methylated by NSUN2, yielding ∼8-fold increase in fluorescence signal after 3 h (Figure 4B).
To rule out fluorescence activation due to other non-specific mechanisms, we repeated the assay in the absence of NSUN2 or methyl donor SAM; in both cases, there was no detectable fluorescence light-up response (Supplementary Figure S9). Furthermore, pre-incubation of NSUN2 with 10 μM of sinefungin (a non-specific DNA/RNA MTase inhibitor) (25,68,69) resulted in a significant reduction in fluorescence intensity, confirming that fluorescence activation was specifically mediated by NSUN2 MTase activity (Figure 4B). Notably, the fluorescence intensity increases linearly with the concentration of methylated probe, thus providing a direct read-out of NSUN2 MTase activity (Figure 4C).
We next verified whether m5C-induced sugar pucker switch in PC was indeed responsible for fluorescence turn-on. For this purpose, we synthesised ‘locked-probe’ 10a (Figure 1C) wherein the pC fluorophore was conformationally restricted in a C3′-endo sugar pucker orientation via a methylene bridge connecting O2′ with C4′ (LC; Figure 1C). As a result of this modification, the locked-probe will no longer be able to undergo sugar pucker switch upon m5C methylation. Consistent with our hypothesis, 10a gave negligible fluorescence light-up response when exposed to NSUN2 (Figure 4B), even though MALDI-TOF MS assay clearly showed that this probe can be methylated by NSUN2 (Supplementary Figure S10). Thus probe activation mechanism is strictly dependent on a change in sugar pucker mode of the 5′-pC residues.
We then evaluated the specificity of probe 9a by testing it against a panel of human RNA:m5C MTases, including three structurally-related homologues NSUN3, NSUN5A, and NSUN6, as well as DNMT homologue TRDMT1 (formerly DNMT2) (6,22). In all cases, there was no significant fluorescence activation (Supplementary Figure S9). Furthermore, MALDI-TOF MS assay showed no methylated product formation even after a prolonged 8 h-incubation with these enzymes, suggesting that 9a is highly selective for NSUN2 over other RNA:m5C MTases (Supplementary Figure S10). Remarkably, 9a also gave no appreciable fluorescence response and methylated product when exposed to key human DNA:m5C MTases, namely DNMT1 and DNMT3A (21,22), hence the probe is also able to discriminate against DNA:m5C MTases (Supplementary Figures S9 and S10). Steady-state kinetic analysis using MALDI-TOF MS assay indicated that the m5C-probe is a relatively good substrate for NSUN2 (kcat/Km = 0.3 min-1μM-1), and is only ∼2-fold less efficiently methylated compared with its native tRNA substrate, tRNALeu(CAA), and CpG-rich ssRNA (Figures 4D and E).
The m5C-probe is applicable for live-cell imaging, single-cell flow cytometry analysis and cell-based inhibitor screening
On the basis of these promising data, we proceeded to investigate whether the m5C-probe strategy could provide direct visualisation of NSUN2 activity in living cells. Accordingly, HeLa cells, which constitutively express NSUN2, were transfected with either 9a (10 μM) or the locked-probe 10a (control) via Lipofectamine 2000, and then imaged using fluorescence microscopy (λex 340–380 nm, λem 435–485 nm). Cells treated with probe 9a gave off bright blue fluorescence after 1 h; this is ∼7-fold higher intensity compared with control (Figures 5A–C).
Figure 5.

The m5C-probe provides real-time visualisation of cellular NSUN2 MTase activity. Live cell imaging of HeLa cells following treatment with m5C-probe 9a (10 μM) using (A) bright field and (B) fluorescence microscopy. (C) Fluorescence image of HeLa cells treated with locked-probe 10a (10 μM) obtained from a separate experiment than the correlated images in panels A and B. It is displayed using the same brightness and contrast settings as that in panel B. Scale bar, 20 μm. (D) Violin plots showing the flow cytometry analysis of HeLa cells transfected with probe 9a (green), and with NSUN2 knockdown (orange) or NSUN2 overexpression (red) (λex 355 nm; λem 425–475 nm). The boxes in violin plots show the median (white dot), 25–75 percentile (black box) and 5–95 percentile values (black bar). (E) The fluorescence intensity of m5C-probe changes with the expression levels of NSUN2 in HeLa cells, but is unresponsive towards NSUN6, TRDMT1 and DNMT3A. A minimum of 20 000 live cells were analysed for determination of mean fluorescence intensity (MFI). Data are expressed as mean ± SD of three biological replicates. **P < 0.01; n.s. = not significant.
Further characterisation of the probe by time course fluorescence analysis showed that the probe remained strongly emissive in HeLa cell lysate for at least 3 h, suggesting good photostability (Supplementary Figure S11A). Moreover, there was no obvious increase in probe fluorescence in cell lysate which had been spiked with 1 mM sinefungin, suggesting no significant fluorescence arising from NSUN2-independent mechanisms (Supplementary Figure S11A). The lack of fluorescence increase further suggests that the probe is reasonably resistant to cellular degradation, which would otherwise release unstacked fluorophore. Consistent with this result, a repeat of the cell lysate experiment using locked-probe 10a gave very little or no increase in fluorescence in HeLa cell lysate, both in the presence and absence of 1 mM sinefungin (Supplementary Figure S11B). These data suggest that probe degradation by cellular nucleases and other nucleic acid modifying enzymes do not interfere with the performance of the m5C-probe 9a significantly, at least not within the time frame of our cell-based assay (typically 1 h). Furthermore, the probe is relatively non-cytotoxic, as evidenced by MTT toxicity assay and cell morphological assessment, where >80% of the cells remained viable after being exposed to 40 μM probe for 24 h (Supplementary Figure S11C).
Besides live-cell imaging, the m5C-probe is also suitable for use in single-cell flow cytometry analysis (λex 355 nm; λem 425–475 nm). In particular, NSUN2 knockdown in HeLa cells caused a marked reduction in probe fluorescence, whereas overexpression of NSUN2 led to significant increase in fluorescence signal (Figures 5D and E). Remarkably, the fluorescence intensity was unchanged with NSUN6, TRDMT1, and DNMT3A knockdown and overexpression, thus, consistent with our in vitro data, the probe is highly selective for NSUN2 over other m5C MTases. Prior to this work, we are not aware of any assay method that is able to discriminate between DNA and RNA m5C MTase activities in cells.
Finally, the developed m5C-probe could also be used to directly measure NSUN2 inhibition in cells. Treatment of HeLa cells with increasing concentrations of known MTase inhibitors, sinefungin or S-adenosyl-L-homocysteine (SAH), for 30 min prior to the addition of probe 9a (10 μM) led to a concentration-dependent decrease in fluorescence signal (Figures 6A–C). The determined IC50 values of sinefungin (8.9 μM) and SAH (3.1 μM) against NSUN2 are comparable with their inhibitory activities against other RNA MTases (IC50 values typically range between 0.1 μM to 20 μM (25,68); Figure 6D). Importantly, the negative controls, adenosine and homocysteine, showed poor inhibition towards NSUN2, as anticipated (IC50s >200 μM and >400 μM, respectively). Thus our assay is able to identify known NSUN2 inhibitors from amongst structurally-related analogues. We further performed Z’ factor analysis (55) to evaluate the reliability of our assay for high-throughput screening of inhibitor (for experimental details, see Supplementary Data). Our assay produces an average Z’ factor of 0.73 in a 96-well plate format, indicating that it has the required statistical reproducibility for cell-based screening of NSUN2 inhibitors (Figure 6E).
Figure 6.

Application of the m5C-probe assay in cell-based screening of NSUN2 inhibitors. (A) IC50 values of non-specific MTase inhibitors against NSUN2. Results are from m5C-probe assay. (B) Analysis of NSUN2 inhibition in live HeLa cells by real-time flow cytometry (λex 355 nm; λem 425–475 nm). Cells were pre-incubated with sinefungin (0, 10 and 1000 μM; colour coded) for 30 min prior to treatment with m5C-probe 9a (10 μM) or locked-probe 10a (10 μM; control). (C) The flow cytometry profile at 1 h showed a concentration-dependent decrease in mean fluorescence intensity (MFI) in cells treated with 9a. (D) Inhibition of NSUN2 by selected MTase inhibitors. The m5C-probe assay could distinguish known inhibitors from structurally-related negative controls. Data are expressed as mean ± SD of three replicates. (E) Screening validation of the m5C-probe assay in a 96-well format. The assay has a Z’ factor of 0.73, which demonstrates excellent reproducibility.
The m5C-switchable probe strategy could be adapted for the study of DNA:m5C MTase activity
In light of present finding that m5C methylation can induce a spontaneous B–Z conformational change in CpG DNA duplex 7a (Figure 3B and Supplementary Figure S6), our m5C-probe strategy may, in principle, also be adapted for the study of DNA:m5C MTase activity. To examine this possibility, we prepared DNA probe 11a, which is an analogue of 7a containing a 5′-overhang pyrene deoxyriboside (Py) in the forward strand (Figure 7). The design of this probe exploits differences in the ability of Py to participate in end-stacking interactions in the B-DNA and Z-DNA forms. It is clear, from a number of studies, that whereas the B-DNA (diameter of helix ∼20 Å) is able to adopt a continuous base-stacking arrangement, the more compact Z-DNA (diameter of helix ∼18 Å) only permits discontinuous series of four-base stack (70). Because of these inherent differences in base-stacking pattern, the unpaired Py will only be able to end-stack on adjacent G:C base pair when it is in a B-DNA environment, and not in the Z-DNA environment. Accordingly, conversion of probe 11a from B-DNA to Z-DNA form upon m5C methylation is expected to cause a destacking of Py fluorophores and, consequently, fluorescence activation.
Figure 7.

The m5C-probe approach could be adapted for the detection of DNA:m5C MTases activity. (A) The DNA m5C-probe 11a is conformationally-responsive to m5C methylation. It exists as a right handed B-DNA structure when unmethylated, but undergoes spontaneous and rapid transformation to the more compact, left-handed Z-DNA structure upon m5C methylation by DNA:m5C MTases (e.g. DNMT3A). Such a major B-Z conversion severely disrupts end-stacking interaction of the fluorophore, pyrene deoxyriboside (Py), leading to fluorescence activation. (B) Schematic representation of DNA m5C-probe 11a and its methylated counterpart 11b. The structure of Py monomer is shown.
Consistent with our probe detection strategy, m5C methylation readily triggered a B-Z transformation in probe 11a, with concomitant increase in fluorescence emission (ΔΦF ∼5.5-fold; λex 340 nm; λem 380 nm and 400 nm; Figures 8A and B). Notably, there was no fluorescence activation in the absence of MgCl2 where 11a* did not undergo B-to-Z conversion, thus the observed fluorescence response was primarily mediated by a B–Z conversion of the probe (Figure 8B). Moreover, our thermodynamic analysis supports the notion that B-Z transition triggers a destacking of the dangling Py (Supplementary Table S3). In particular, in the absence of m5C methylation, Py is able to stack readily at the 5′-end of probe 11a, giving an end-stacking stabilisation of ∼2.8 kcal/mol (compare the difference in duplex stability between 11a and 7a; Supplementary Table S3 and Supplementary Figures S12 and S13). Upon m5C methylation, however, end-stacking interaction is completely abolished (negligible difference in stability between 11b and 7b). This suggests that the end-stacking property of Py and, hence, its fluorescence output is sensitive to m5C methylation status.
Figure 8.

DNA probe 11a provides highly sensitive detection of DNA:m5C methylation. (A) Fluorescence measurement of probe 11a (5 μM) showed two distinct pyrene emission peaks at 380 nm and 400 nm (λex 340 nm). The spectra were recorded at 37°C under physiologically-relevant conditions (10 mM sodium phosphate buffer (pH 7.4) containing 150 mM NaCl and 20 mM MgCl2). The probe exhibited a significant 5.5-fold increase in fluorescence intensity upon m5C methylation (11b), but gave no appreciable light-up response in the absence of MgCl2 (11b*), implying that fluorescence switch-on was primarily mediated by B–Z structural conversion of the probe. (B) m5C methylation triggered a major inversion of the CD spectrum of probe 11a under physiologically-relevant conditions. There was little or no conformational change in the absence of MgCl2 (11b*). (C) Time-course fluorescence analysis (λex 340 nm; λem 400 nm) of probe 11a (5 μM) in the presence of DNMT3A (0.5 μM; red line) or NSUN2 (0.5 μM; black line). The results demonstrate that probe 11a is selectively activated by DNMT3A, and not by NSUN2. (D) Probe 11a could be used for in vitro inhibition assay of DNMT3A. Both sinefungin (known DNMT3A inhibitor) and adenosine (negative control) displayed clear concentration-dependent decrease in fluorescence, with determined IC50 values of 0.7 μM and 106.3 μM, respectively. Data are expressed as mean ± SD of three replicates.
Preliminary biochemical analysis further demonstrated that probe fluorescence is only activated by DNA:m5C MTases DNMT3A, and not by RNA:m5C MTases NSUN2 (Figure 8C). Importantly, m5C-probe 11a may also be applied to the inhibition study of DNMT3A. As shown in Figure 8D, incubation of probe 11a (5 μM) and DNMT3A (0.5 μM) with increasing concentrations of sinefungin (known DNMT3A inhibitor) or adenosine (negative control) for 30 min led to clear concentration-dependent decrease in fluorescence intensity. The determined IC50 values of sinefungin (0.7 μM) and adenosine (106.3 μM) are comparable with previously reported values (71), thus the developed m5C-probe strategy could be used for in vitro screening of DNMT3A inhibitors. Work is currently underway to explore the possibility of employing both probes concurrently for the simultaneous detection of RNA and DNA m5C MTase activity.
CONCLUSION
Overall, we use a combination of NMR, CD and thermodynamic analyses to investigate the structural impact of m5C methylation on RNA and DNA duplexes. Our results revealed that, although m5C does not hinder canonical Watson–Crick base-pairing interactions, this modification can, in fact, elicits significant conformational change in certain RNA and DNA sequence contexts. We further demonstrated that m5C produces distinctly different ‘structural signatures’ on RNA and DNA duplexes, including duplexes with identical CpG sequences. In the case of CpG RNA duplex (as exemplified by 1a), m5C methylation induces a local distortion of the phosphate backbone and a C3′-endo to C2′-endo sugar pucker switch in the terminal residues. However, in the case of CpG DNA duplex (as exemplified by 7a), m5C triggers a remarkable B-Z structural transformation. The significance of this result is unclear at present nevertheless, given that majority of the m5C modifications in DNA and mRNA are distributed within the CpG consensus motif, sequence information alone is insufficient to discriminate m5C marks on RNA and DNA CpG sites. Therefore, one question which arises from this finding is whether the duplex-remodelling property of m5C might provide a mechanism for its specific recognition by RNA/DNA:m5C binding proteins.
On the basis of this interesting finding, we further provided proof-of-principle that m5C-induced conformational change can be exploited for direct detection of m5C MTase activity in cells. This was demonstrated by the development of the first m5C-responsive probe 9a, which switches sugar pucker conformation spontaneously according to its m5C methylation status, hence useful for sensing RNA:m5C MTase activity.
The m5C-probe is simple, inexpensive and highly selective for NSUN2 over other RNA:m5C MTases (including NSUN3, NSUN5A, NSUN6 and TRDMT1) and DNA:m5C MTases (including DNMT1 and DNMT3A). Through the use of this probe, we achieved fluorescence imaging and real-time flow cytometry analysis of NSUN2 activity in live HeLa cells. We further demonstrated the utility of the probe in cell-based screening of NSUN2 inhibitors. Prior to this study, we are not aware of any methods that permit direct sensing of RNA:m5C MTase activity in cells. There are also no reports of assays that selectively target NSUN2 over other RNA/DNA:m5C MTases. The discovery of such highly selective probes is rarely achieved and may prove valuable in advancing our knowledge of NSUN2 in m5C-regulated processes. Importantly, our m5C-probe approach could also be adapted for the analysis of DNA:m5C MTase activity. This was demonstrated by the development of DNA m5C-probe 11a, which is useful for in vitro screening of DNMT3A inhibitors.
We appreciate that m5C-induced terminal sugar pucker switch is likely an interesting anomaly that is unique to alternating CpG RNA duplexes, since this phenomenon was not observed in majority of other sequences investigated in this study. Therefore, one limitation of the proposed approach is that it is not applicable to RNA:m5C methyltransferases that do not methylate CpG substrates. Nevertheless, given the importance of NSUN2, both as a key regulator of m5C marks in mRNA and a potential therapeutic target, we envisaged that the proposed NSUN2 assay strategy would be of broad scientific interest.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the Chemical, Molecular and Materials Analysis Centre at the National University of Singapore for their support in NMR measurements and helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Singapore Ministry of Health's National Medical Research Council [NMRC/BNIG/2008/2013]; Singapore Ministry of Education [AcRF Tier 1 Grant R148-000-231-114 and R148-000-238-114]. Funding for open access charge: Singapore Ministry of Education [AcRF Tier 1 Grant R148-000-238-114].
Conflict of interest statement. None declared.
REFERENCES
- 1. Boccaletto P., Machnicka M.A., Purta E., Piatkowski P., Baginski B., Wirecki T.K., de Crecy-Lagard V., Ross R., Limbach P.A., Kotter A. et al.. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic. Acids. Res. 2018; 46:D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sood A.J., Viner C., Hoffman M.M.. DNAmod: the DNA modification database. J. Cheminform. 2019; 11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grosjean H. Fine-Tuning of RNA Functions by Modification and Editing. 2005; Berlin, Heidelberg: Springer. [Google Scholar]
- 4. Motorin Y., Helm M.. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA. 2011; 2:611–631. [DOI] [PubMed] [Google Scholar]
- 5. Breiling A., Lyko F.. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenet. Chromatin. 2015; 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Motorin Y., Lyko F., Helm M.. 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 2010; 38:1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith Z.D., Meissner A.. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013; 14:204–220. [DOI] [PubMed] [Google Scholar]
- 8. Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. [DOI] [PubMed] [Google Scholar]
- 9. Squires J.E., Patel H.R., Nousch M., Sibbritt T., Humphreys D.T., Parker B.J., Suter C.M., Preiss T.. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012; 40:5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edelheit S., Schwartz S., Mumbach M.R., Wurtzel O., Sorek R.. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013; 9:e1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khoddami V., Cairns B.R.. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013; 31:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Legrand C., Tuorto F., Hartmann M., Liebers R., Jacob D., Helm M., Lyko F.. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 2017; 27:1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amort T., Rieder D., Wille A., Khokhlova-Cubberley D., Riml C., Trixl L., Jia X.-Y., Micura R., Lusser A.. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017; 18:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X., Yang Y., Sun B.-F., Chen Y.-S., Xu J.-W., Lai W.-Y., Li A., Wang X., Bhattarai D.P., Xiao W. et al.. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017; 27:606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. David R., Burgess A., Parker B., Li J., Pulsford K., Sibbritt T., Preiss T., Searle I.R.. Transcriptome-wide mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and non-coding RNAs. Plant Cell. 2017; 29:445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui X., Liang Z., Shen L., Zhang Q., Bao S., Geng Y., Zhang B., Leo V., Vardy L.A., Lu T. et al.. 5-Methylcytosine RNA methylation in Arabidopsis thaliana. Mol. Plant. 2017; 10:1387–1399. [DOI] [PubMed] [Google Scholar]
- 17. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang H., Fan X., Xing J., Liu Z., Jiang B., Dou Y., Gorospe M., Wang W.. NSun2 delays replicative senescence by repressing p27 (KIP1) translation and elevating CDK1 translation. Aging. 2015; 7:1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xing J., Yi J., Cai X., Tang H., Liu Z., Zhang X., Martindale J.L., Yang X., Jiang B., Gorospe M. et al.. NSun2 promotes cell growth via elevating cyclin-dependent kinase 1 translation. Mol. Cell. Biol. 2015; 35:4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aguilo F., Li S., Balasubramaniyan N., Sancho A., Benko S., Zhang F., Vashisht A., Rengasamy M., Andino B., Chen C.H. et al.. Deposition of 5-Methylcytosine on enhancer RNAs enables the coactivator function of PGC-1α. Cell Rep. 2016; 14:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018; 19:81–92. [DOI] [PubMed] [Google Scholar]
- 22. Grosjean H. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. 2009; Austin: Landes Bioscience. [Google Scholar]
- 23. Kohli R.M., Zhang Y.. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013; 502:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y., Santi D.V.. m5C RNA and m5C DNA methyl transferases use different cysteine residues as catalysts. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:8263–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boriack-Sjodin P.A., Ribich S., Copeland R.A.. RNA-modifying proteins as anticancer drug targets. Nat. Rev. Drug Discov. 2018; 17:435–453. [DOI] [PubMed] [Google Scholar]
- 26. Freeman J.W., Hazlewood J.E., Auerbach P., Busch H.. Optimal loading of scraped HeLa cells with monoclonal antibodies to the proliferation associated Mr 120, 000 Nucleolar Antigen. Cancer Res. 1988; 48:5246–5250. [PubMed] [Google Scholar]
- 27. Frye M., Watt F.M.. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr Biol. 2006; 16:971–981. [DOI] [PubMed] [Google Scholar]
- 28. Hussain S., Benavente S.B., Nascimento E., Dragoni I., Kurowski A., Gillich A., Humphreys P., Frye M.. The nucleolar RNA methyltransferase Misu (NSun2) is required for mitotic spindle stability. J. Cell Biol. 2009; 186:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kar S.P., Beesley J., Amin A., Olama A., Michailidou K., Tyrer J., Kote-Jarai Z., Lawrenson K., Lindstrom S., Ramus S.J. et al.. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 2016; 6:1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Haute L., Dietmann S., Kremer L., Hussain S., Pearce S.F., Powell C.A., Rorbach J., Lantaff R., Blanco S., Sauer S. et al.. Deficient methylation and formylation of mt tRNA Met wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016; 7:2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haag S., Sloan K.E., Ranjan N., Warda A.S., Kretschmer J., Blessing C., Hübner B., Seikowski J., Dennerlein S., Rehling P. et al.. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016; 35:2104–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aguilo F., Li S., Balasubramaniyan N., Sancho A., Benko S., Zhang F., Vashisht A., Rengasamy M., Andino B., Chen C.H. et al.. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1α. Cell Rep. 2016; 14:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris T., Marquez B., Suarez S., Schimenti J.. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol. Reprod. 2007; 77:376–382. [DOI] [PubMed] [Google Scholar]
- 34. Yang T., Cheong A., Mai X., Zou S., Woon E.C.Y.. A methylation-switchable conformational probe for the sensitive and selective detection of RNA demethylase activity. Chem. Commun. 2016; 52:6181–6184. [DOI] [PubMed] [Google Scholar]
- 35. Zou S., Toh J.D.W., Wong K.H.Q., Gao Y.-G., Hong W., Woon E.C.Y.. N 6-Methyladenosine: a conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci. Rep. 2016; 6:25677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheong A., Low J.J.A., Lim A., Yen P.M., Woon E.C.Y.. A fluorescent methylation-switchable probe for highly sensitive analysis of FTO N6-methyladenosine demethylase activity in cells. Chem. Sci. 2018; 9:7174–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engel J.D., von Hippel P.H.J.. Effects of methylation on the stability of nucleic acid conformations. Studies at the polymer level. Biol. Chem. 1978; 253:927–934. [PubMed] [Google Scholar]
- 38. Micura R., Pils W., Höbartner C., Grubmayr K., Ebert M.O., Jaun B.. Methylation of the nucleobases in RNA oligonucleotides mediates duplex–hairpin conversion. Nucleic. Acids. Res. 2001; 29:3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kierzek E., Kierzek R.. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003; 31:4472–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roost C., Lynch S.R., Batista P.J., Qu K., Chang H.Y., Kool E.T.. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 2015; 137:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Behe M., Felsenfeld G.. Effects of methylation on a synthetic polynucleotide: the B-Z transition in poly(dG-m5dC).poly(dG-m5dC). Proc. Natl. Acad. Sci. U. S. A. 1981; 78:1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tran-Dinh S., Taboury J., Neumann J.-M., Huynh-Dinh T., Genissel B., Langlois d’Estaintot B., Igolen J.. Proton NMR and circular dichroism studies of the B and Z conformations of the self-complementary deoxyhexanucleotide d(m5C-G-C-G-m5C-G): mechanism of the Z-B-coil transitions. Biochemistry. 1984; 23:1362–1371. [DOI] [PubMed] [Google Scholar]
- 43. Butkus V., Klirnaauskas S., Petrauskiene L., Maneliene Z., Janulaitis A., Minchenkoval L.E., Schyolkinal A.K.. Synthesis and physical characterization of DNA fragments containing N4-methylcytosine and 5-methycytosine. Nucleic Acids Res. 1987; 15:8467–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Orbons L.P., Altona C.. Conformational analysis of the B and Z forms of the d(m5C-G)3 and d(br5C-G)3 hexamers in solution. A 300-MHz and 500-MHz NMR study. Eur. J. Biochem. 1986; 160:141–148. [DOI] [PubMed] [Google Scholar]
- 45. Ceolin F., Babin F., Huynh-Dinh T., Igolen J.. RNA fragment r(CGm5CGCG) that exhibits two conformations in slow exchange on the NMR time scale in low salt solution. J. Am. Chem. Soc. 1987; 109:2539–2541. [Google Scholar]
- 46. Bloch G., Neumann J.M., Babin F., Huynh-Dinh T.. Sequence-dependence of the conformational changes induced by the 5-methyl cytosine in synthetic RNA oligomers. FEBS Lett. 1987; 219:464–468. [DOI] [PubMed] [Google Scholar]
- 47. Hudson R.H.E., Ghorbani-Choghamarani A.. Selective fluorometric detection of guanosine-containing sequences by 6-phenylpyrrolocytidine in DNA. Synlett. 2007; 6:870–873. [DOI] [PubMed] [Google Scholar]
- 48. Cho S.J., Ghorbani-Choghamarani A., Saito Y., Hudson R.H.E.. 6-Phenylpyrrolocytidine: an intrinsically fluorescent, environmentally responsive nucleoside analogue. Curr. Protoc. Nucleic Acid Chem. 2019; 76:e75. [DOI] [PubMed] [Google Scholar]
- 49. Ren R.X.-F., Chaudhuri N.C., Paris P.L., Rumney S., Kool E.T.. Naphthalene, phenanthrene, and pyrene as DNA base analogues: synthesis, structure, and fluorescence in DNA. J. Am. Chem. Soc. 1996; 118:7671–7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sklénar V., Piotto M., Leppik R., Saudek V.. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. A. 1993; 102:241–250. [Google Scholar]
- 51. States D.J., Haberkorn R.A., Ruben D.J.. A two-dimensional nuclear overhauser experiment with pure absorption phase in four quadrants. J. Magn. Reson. 1982; 48:286–292. [Google Scholar]
- 52. Jeener J., Meier B.H., Bachmann P., Ernst R.R.. Investigation of exchange processes by two‐dimensional NMR spectroscopy. J. Chem. Phys. 1979; 71:4546–4593. [Google Scholar]
- 53. Barkhuisen H., de Beer R., Bovee W.M.M.J., van Ormondt K.. Retrieval of frequencies, amplitudes, damping factors, and phases from time-domain signals using a linear least-squares procedure. J. Magn. Reson. 1985; 61:465–481. [Google Scholar]
- 54. Otting G., Widmer H., Wagner G., Wuthrich K.. Origin of τ2 and τ2 ridges in 2D NMR spectra and procedures for suppression. J. Magn. Reson. 1986; 66:187–193. [Google Scholar]
- 55. Zhang J.H., Chung T.D.Y., Oldenburg K.R.. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999; 4:67–73. [DOI] [PubMed] [Google Scholar]
- 56. Zhang X., Liu Z., Yi J., Tang H., Xing J., Yu M., Tong T., Shang Y., Gorospe M., Wang W.. NSun2 stabilizes p16INK4 mRNA by methylating the 3′-untranslated region of p16. Nat. Commun. 2012; 3:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dev R.R., Ganji R., Singh S.P., Mahalingam S., Banerjee S., Khosla S.. Cytosine methylation by DNMT2 facilitates stability and survival of HIV-1 RNA in the host cell during infection. Biochem. J. 2017; 474:2009–2026. [DOI] [PubMed] [Google Scholar]
- 58. Chen Z.X., Mann J.R., Hsieh C.L., Riggs A.D., Chedin F.. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell Biochem. 2005; 95:902–917. [DOI] [PubMed] [Google Scholar]
- 59. Wang Z.-L., Li B., Luo Y.-X., Lin Q., Liu S.-R., Zhang X.-Q., Zhou H., Yang J.-H., Qu L.-H.. Comprehensive genomic characterization of RNA-binding proteins across human cancers. Cell Rep. 2018; 22:286–298. [DOI] [PubMed] [Google Scholar]
- 60. Popenda M., Biala E., Milecki J., Adamiak R.W.. Solution structure of RNA duplexes containing alternating CG base pairs: NMR study of r(CGCGCG)2 and 2′-O-Me(CGCGCG)2 under low salt conditions. Nucleic. Acids. Res. 1997; 25:4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Altona C., Sundaralingam M.. Conformational analysis of the sugar ring in nucleosides and nucleotides. Improved method for the interpretation of proton magnetic resonance coupling constants. J. Am. Chem. Soc. 1973; 95:2333–2344. [DOI] [PubMed] [Google Scholar]
- 62. Neuhaus D., Williamson M.P.. The Nuclear Overhauser Effect in Structural and Conformational Analysis. 2000; NY: John Wiley & Sons, Inc. [Google Scholar]
- 63. Butts C.P., Jones C.R., Towers E.C., Flynn J.L., Appleby L., Barron N.J.. Interproton distance determinations by NOE – surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem. 2011; 9:177–184. [DOI] [PubMed] [Google Scholar]
- 64. Fujii S., Wang A.H.-J., van der Marel G., van Boom J.H., Rich A.. Molecular structure of (m5dC-dG)3: the role of the methyl group on 5-methyl cytosine in stabilizing Z-DNA. Nucleic Acids Res. 1982; 10:7879–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wojciechowski F., Hudson R.H.E.. Fluorescence and hybridization properties of peptide nucleic acid containing a substituted phenylpyrrolocytosine designed to engage guanine with an additional H-bond. J. Am. Chem. Soc. 2008; 130:12574–12575. [DOI] [PubMed] [Google Scholar]
- 66. Wahba A.S., Esmaeili A., Damha M.J., Hudson R.H.E.. A single-label phenylpyrrolocytidine provides a molecular beacon-like response reporting HIV-1 RT RNase H activity. Nucleic Acids Res. 2010; 38:1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wahba A.S., Azizi F., Deleavey G.F., Brown C., Robert F., Carrier M., Kalota A., Gewirtz A.M., Pelletier J., Hudson R.H. et al.. Phenylpyrrolocytosine as an unobtrusive base modification for monitoring activity and cellular trafficking of siRNA. ACS Chem. Biol. 2011; 6:912–919. [DOI] [PubMed] [Google Scholar]
- 68. Yebra M.J., Sanchez J., Martin C.G., Hardisson C., Barbes C.. The effect of sinefungin and synthetic analogues on RNA and DNA methyltransferases from Streptomyces. J. Antibiot. 1991; 44:1141–1147. [DOI] [PubMed] [Google Scholar]
- 69. Woon E.C.Y., Arcieri M., Wilderspin A.F., Malkinson J.P., Searcey M.. Solid-phase synthesis of chlorofusin analogues. J. Org. Chem. 2007; 72:5146–5151. [DOI] [PubMed] [Google Scholar]
- 70. Neidle S. Oxford Handbook of Nucleic Acid Structure. 1999; NY: Oxford University Press. [Google Scholar]
- 71. Kilgore J.A., Du X., Melito L., Wei S., Wang C., Chin H.G., Posner B., Pradhan S., Ready J.M., Williams N.S.. Identification of DNMT1 selective antagonists using a novel scintillation proximity assay. J. Biol. Chem. 2013; 288:19673–19684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.