

Abstract
To date, large amounts of genomic and phenotypic data have been accumulated in the fields of crop genetics and genomic research, and the data are increasing very quickly. However, the bottleneck to using big data in breeding is integrating the data and developing tools for revealing the relationship between genotypes and phenotypes. Here, we report a rice sub-database of an integrated omics knowledgebase (MBKbase-rice, www.mbkbase.org/rice), which integrates rice germplasm information, multiple reference genomes with a united set of gene loci, population sequencing data, phenotypic data, known alleles and gene expression data. In addition to basic data search functions, MBKbase provides advanced web tools for genotype searches at the population level and for visually displaying the relationship between genotypes and phenotypes. Furthermore, the database also provides online tools for comparing two samples by their genotypes and finding target germplasms by genotype or phenotype information, as well as for analyzing the user submitted SNP or sequence data to find important alleles in the germplasm. A soybean sub-database is planned for release in 3 months and wheat and maize will be added in 1–2 years. The data and tools integrated in MBKbase will facilitate research in crop functional genomics and molecular breeding.
INTRODUCTION
Germplasm resources provide the genetic basis for crop improvement (1). In the past, many international organizations and research centers have been engaged in crop germplasm collection and preservation and have generated a large amount of germplasm phenotype data in field tests. These organizations include EURISCO-ECPGR (www.ecpgr.cgiar.org) (2), GRIN-NGRP (www.ars-grin.gov), NBRP (https://nbrp.jp/) (3), IRGCIS-IRRI (http://irgcis.irri.org:81/grc/IRGCISHome.html) (4), CIMMYT (www.cimmyt.org), CGRIS (www.cgris.net), Genesys (www.genesys-pgr.org) and others (5). Recently, high-throughput phenotyping technologies have enabled the collection of large-scale germplasm phenomic data under different experimental designs or environments (6).
Over the last decade, next generation sequencing (NGS) technologies have deeply impacted crop breeding. NGS has been widely used for linkage mapping, genome wide association studies (GWAS), marker-assisted selection (MAS), genomic selection (GS), domestication and population structure analysis in crops (7–12). Benefitting from the application of these technologies, a large number of important genes/QTLs have been mapped and cloned (10,13–20). These genes provide abundant operational targets for molecular design breeding and gene modification (21). Along with reduced sequencing cost, high coverage whole‐genome sequencing (WGS) of natural crop populations have opened the door to exploring genetic diversity among individuals, which results in a large amount of genomic data. More recently, single-molecule DNA sequencing of long reads has been employed in high-quality assembly with genomes of various sizes and levels of complexity (22–29). These high-quality genomes are very important for expanding variant calling, which makes it possible to obtain all alleles of all genes in WGS populations using high-throughput methods. For efficient usage, those public raw datasets need to be integrated into a big data platform where functional associations between heterogeneous datasets can be explored further. More importantly, the central challenge of integration is to establish genotype to phenotype (G2P) links and develop a user-friendly and highly flexible web tool for data visualization and mining.
Here, we describe an integrated omics knowledgebase for molecular breeding (MBKbase, www.mbkbase.org), specifically the rice sub-database, which integrates multiple reference genomes, population WGS data, germplasm information, phenotype, known genes and RNA-seq data. A soybean sub-database will be released in 3 months and wheat and maize will be added later. MBKbase aims to integrate multiple high-quality reference genomes, and reveal the relationship among germplasm, phenotype and genotype by providing genotypes for each gene locus and their associated phenotypes in a population. In this respect, MBKbase is very different from other breeding data management systems, such as GOBII (http://gobiiproject.org), NRSP10 (www.nrsp10.org), and AgBioData (www.agbiodata.org) (30) projects, as well as those mainly focusing on nucleotide variants such as GVM (31), RiceVarMap (32), and SNP-Seek (33), or the genomic or comparative genomic databases such as Ensembl (34) and Gramene (35).
MBKbase is designed for multi-species data management and display. Each species will share the same functional framework but have an independent set of databases, web home page and a set of menu items to go to subcategories or modules (Figure 1). The basic architecture of MBKbase consists of an Apache web server, MongoDB NoSQL data management system, Pyramid web framework, Beaker web library and JQuery library. By using multiple application servers, distributed databases and efficient database structure for standardized data storage, as well as a data caching function in middleware, MBKbase ensures high-speed data access, integration analysis and visualization. We adopt a modular approach for front-end web development, which allows multiple types of data to be associated flexibly and displayed in various combinations on different web pages (Figure 1).
Figure 1.

Schematic of data, page and function modules in MKBbase. Each data module has many linked pages and functional units for displaying the information related to the module, as exemplified by the Germplasm module in the figure.
The Asian cultivated rice (Oryza sativa L.) is one of the most important crops, and the accumulated data related to rice population genomics is also the most abundant among all crops. Because of this, we first developed the sub-database for rice (http://www.mbkbase.org/rice) and only describe the rice data in this publication. Asian rice consists of two subspecies, indica and japonica, with high-quality reference genomes of the japonica rice Nipponbare (Nip) and the indica rice Shuhui498 (R498) having been released (22,36). R498 is more useful than Nip as a reference for mapping sequences of indica rice, which consists of more varieties than japonica rice and has more genetic diversity. Therefore, besides Nip, we also provide genomic variation based on the R498 reference genome, which is not found in any other databases. In addition, we provide a unified set of gene loci as a pan-genome based on the annotations on both Nip and R498 genomes. We collected publicly available rice germplasm information, population WGS data and phenotype data (Table 1 and Supplementary Tables S1–3). In particular, the data include >1000 Chinese rice accessions with both WGS data and phenotype data not available in other databases, which have been deposited into BIG-BioProject for download. MBKbase will continue to integrate all published WGS projects, phenomic data, known genes and published data related to molecular breeding to develop advanced analytical and data mining tools, as well as add more crop species.
Table 1.
The summary of rice data in MBKbase
| Phenotype | |||||
|---|---|---|---|---|---|
| Total num | With pedigree | With source locations | Traits | Value Num | |
| Germplasm | 137 769 | 24 462 | 134 626 | 130 | 4 786 640 |
| WGS sample | 7010 | 1153 | 6351 | 122 | 207 158 |
| Named Gene num | Trait gene num | Verified alleles num | |||
| Known genes | 13 219 | 4821 | 91 | ||
| Run num | Tissue | Stage or tissue | |||
| RNA-seq | 175 | 20 | 45 | ||
| MBK Allele Num | SNP num (AF ≥ 0.01) | Indel num (AF > 0.005) | |||
| Reference | Nip | 51 722 | 14 850 931 | 2 250 804 | |
| R498 | 54 973 | 13 278 107 | 2 387 538 | ||
TOP-LEVEL DATABASE MODULES
MBKbase is divided into several different top-level modules for organizing various data types and applications (Figure 1). For each data module, we provide a data description and basic search functions as described below.
The Germplasm module (www.mbkbase.org/rice/germplasm) provides basic information for rice germplasm collections as well as links to other related information of each germplasm such as genotype or phenotype. Germplasm can be searched by name or ID, which supports regular expressions. A germplasm can either have an associated sequence or not. Because a cultivar or an accession sharing the same germplasm name may have different origins, we assign a different sample ID to each instance of the same germplasm which is associated with WGS data or other omics data. The rice data include 7010 samples with WGS data, and 137 769 germplasms for reference purposes.
The Phenotype module (www.mbkbase.org/rice/phenotype) provides information on traits and phenotypes of a germplasm or a sample. Phenotype can be searched by trait symbol or name or browsed by trait class. We used standard terms for trait description and phenotype classification when they were available. The standard terms come from the standard evaluation system for Rice (37), the Chinese Crop Germplasm Information System (CGRIS, www.cgris.net/cgris_english.html) and Plant Trait Ontology (TO) (38). Due to complexity and inconsistency of trait terms in real data, we roughly classified the rice phenotypes into five top-level classes. The trait classes were further divided into subclasses manually based on the TO terms. We will update the trait definitions, classifications and phenotypes continually in MBKbase and connect the phenotype data explicitly to TO accessions in the future. The phenotype data in MBKbase come from two different sources: (a) evaluation data from resource databases or books for general descriptions of germplasms or cultivars that are not necessarily sequenced or genotyped, and (b) population phenotypes collected for GWAS along with sequences or genotypes. The distribution of phenotype values can be found in the trait detail page (Supplementary Figure S1). The rice data include 122 traits with phenotype data for WGS samples and 130 traits with phenotype data for un-sequenced germplasms.
The Genotype module (www.mbkbase.org/rice/genotype) provides genotype information of all sequenced germplasms or samples. The WGS data come from published projects that were collected from NCBI-SRA (www.ncbi.nlm.nih.gov/sra) and BIG-BioProject (https://bigd.big.ac.cn/bioproject/). Sequence variation including both single-nucleotide polymorphisms (SNPs) and insertions-deletions (indels) in MBKbase are called based on multiple reference genomes. The WGS data were mapped to reference genomes using the BWA-MEM (39) tool, multiple mapping reads filtered using samtools (v1.8) (40) and SNPs and indels are called using the GATK (v3.8) UnifiedGenotyper (41) tool. We define gene locus as a conceptual gene to hold multiple alleles in both reference genomes and WGS populations. Currently the rice data include 51 722 gene alleles, 14 850 931 SNPs and 2 250 804 short indels based on Nip; and 54 973 alleles, 13 278,107 SNPs and 2 387 538 short indels based on R498. Most of the loci in rice contain the alleles of both Nip and R498, which can be found with a locus search. Genotype can be searched at a locus or a set of user-selected positions. Many functions and tools are provided based on genotypes in MBKbase and they are further described below.
The KnownGene module (www.mbkbase.org/rice/knownGene) provides information on functionally annotated genes and experimentally verified alleles. The module supports gene search by symbol, name or related traits, and provides gene browsing by trait categories. Currently there are 13 219 rice genes with assigned symbols and functional annotations downloaded from Q-TARO, GenBank, funRiceGenes and Oryzabase. Particularly, 63 experimental rice genes with verified variation sites were curated from the literature and associated with their corresponding phenotypes in populations.
The Population module (www.mbkbase.org/rice/population) provides information on the collected WGS populations and their phylogenetic trees. Most of the rice populations were collected from external data sources and one was from an internally sequenced population. A germplasm sample can be searched to show its position in the tree with its closely related germplasms/samples. Clicking on a node in the tree will show a sub-tree structure of nodes, and the geographic distribution of the germplasm/samples is also displayed (Supplementary Figure S2).
UNITED GENE SET AS PAN-GENOME
A pan-genome consists of the genes of all individuals in a species. In MBKbase, we defined the concept of a virtual locus corresponding to a gene in a pan-genome based on reference genomes only. The genes from multiple reference genomes are united into a single gene set consisting of virtual loci. A locus can contain a single allele if the gene is present in only one reference genome, or multiple alleles if these alleles are in a collinear position in multiple reference genomes. From Nip and R498, there are a total of 95 325 loci at present. For example, under locus OsG00067204 (Figure 2A), there are alleles from both Nip (with multiple annotations based on different annotation methods) and R498. The different gene annotation sets are usually complementary to each other, providing more useful genes than a single annotation. As more reference genomes are added in MBKbase, the locus set will also be expanded with more loci as well as more alleles.
Figure 2.
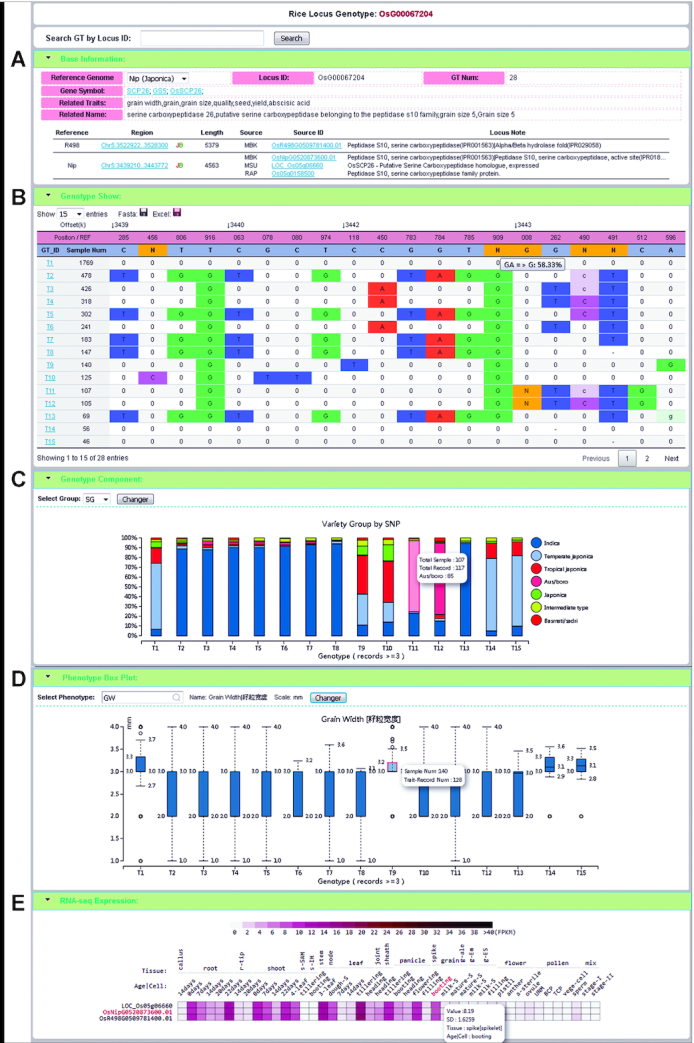
An example of a locus page showing multiple types of information associated with the locus. Rice gene GS5 controlling grain size is on locus OsG00067204. (A) Two reference genomes contain different alleles at the locus. (B) Genotype table showing all genotypes in the rice population, with reference genome positions in the first row of the table and reference bases in the second row. Genotype IDs are enumerated from T1 as the most frequent genotype (allele or haplotype for homozygous genome). ‘0’ indicates that the genotype at this position is the same as in the reference genome. Capitalized bases indicate homozygous variants, lowercase bases indicate heterozygous variants and ‘-’ indicates missing information. In the REF row, the letter ‘N’ represents a short insertion in the reference genome compared with some other samples. In the GT row, the ‘N’ represents insertion of bases in the samples compared with the reference genome. (C) Distribution of each genotype (allele) in a rice population. For example, the majority of genotype (allele) T1 is found in temperate japonica lines (1251 out of 1769). (D) Boxplot showing the relationship between genotypes and phenotypes. The phenotype can be selected by users from all collected traits in the database. In this case, the locus OsG0067204 is known to be associated with grain width (GW) traits. (E) Expression profile at the locus in different reference genomes in different tissues.
GENOTYPES IN MBKBASE
Genotype is one of the key concepts in MBKbase on which many applications are built. We define a genotype to be a group of SNPs and short indels, which are either adjacent or non-adjacent on a chromosome. A genotype is equivalent to a haplotype when the identified SNP and indel are homozygous in a genome. We define four types of genotypes: (i) positional genotype at a single SNP or indel position, (ii) locus genotype at a gene locus (i.e. an allele in a homozygous genome) (Figure 2), (iii) customized genotype for a group of user-selected SNPs and indels at any positions throughout the genome (Supplementary Figure S3) and (iv) window genotype for a group of core SNPs and indels in 20-kb windows. We developed a set of web tools for searching and displaying genotypes.
A genotype can be the same as or different from the reference sequence. The genotypes based on WGS data of a population are affected by two major factors: variation type and variation number, which are further influenced by many factors, such as the size of the population, the length of selected chromosomal regions, the allele frequency, missing allele frequency, sequencing depth and mapping quality. In the customized genotype page, all related parameters can be customized by users (Supplementary Figure S3). This function is useful for users to examine the type of variations in a set of target genome regions in a subpopulation.
INTEGRATION OF MULTI-OMICS DATA
In MBKbase, we used known genes controlling important agronomic traits as a bridge between genotype and phenotype. The experimentally verified functional alleles controlling phenotypic traits were manually annotated with trait class and description. In the locus genotype or custom genotype query page, all genotypes are listed in one genotype table. Samples with the same genotype are merged and assigned a genotype ID and sorted according to the number of samples sharing a genotype in descending order (Figure 2B). This provides a convenient base for revealing the relationship among germplasm, genotypes and phenotypes. The percent of ecotype or originating country of each genotype are displayed in a separate panel (Figure 2C), with the boxplots of G2P relationships generated online (Figure 2D). Users can choose which phenotype is associated for display with the genotypes based on their knowledge of known genes or variation. The phenotypes directly associated with genotypes in published GWAS projects are used for default plotting. If this type of phenotype value is missing, other phenotype data under the same germplasm name will be retrieved automatically for comparison purpose.
In the genotype table, clicking any of the genotype IDs in the first column (GT_ID) leads to the genotype detail page, which contains the sample information list, the sample geographical distribution and the traits with phenotypic value (Supplementary Figure S4). If the chromosomal region of the genotype contains genes, each gene's mRNA expression profile is also shown in the expression panel (Figure 2E).
ADVANCED GENOTYPE-BASED FUNCTIONS
We provide several advanced data analytical or mining tools that are based on genotypes. They are described as follows.
Germplasm search
MBKbase provides advanced search methods for germplasm by genotype or by sequence. In the former case, a user can set up logical queries using a different combination of positional genotypes to identify the germplasm containing the genotypes. For example, a user can set up the following logical query: (Chr1:11663:CCA and Chr1:26254:T) and (Chr2:30096330:A or Chr6:1767006:G) (Figure 3A), to find and display all germplasms passing the filter. Clicking the filtered germplasm number will display the germplasm information and their geographical distribution in a result table and Google Maps. A known allele can also be used to search for germplasm that contains the allele. In the latter case, a user can submit a short DNA sequence as a query and find out which germplasms contain the same DNA sequence. For example, 142 germplasms were found that contain the same sequence as BD295334.1 (Figure 3B).
Figure 3.

Advanced functions based on genotype. (A) Advanced germplasm query by genotype (GT), the options in the drop-down list of genotype source include position GT, Locus GT and Verified GT. Users can set up complex logical queries by selecting different combinations of variants. In this example, 177 samples passed the filter with the same genotype. (B) Advanced germplasm query by sequence. Users can submit a genome sequence and identify the germplasms containing the sequence. (C) Comparison of two germplasm samples (A and B) based on Locus GT. X-axis: window position along the chromosome; Y-axis: locus number in the window, positive for sample A, negative for sample B. A red line represents the number of identical genotypes between A and B. A green line represents the number of specific genotypes of sample A. A teal line represents the number of specific genotypes of sample B. A purple line represents the number of missing or low frequency genotypes (containing sample number <10). (D) Identification of the known alleles in a WGS sample. This can be used for WGS samples either stored in the database or submitted by users through the web interface. (E) Tools for viewing the effect of variation. Clicking on any variation in a reference row of genotype table, for example, clicking ‘T’ (at position 30,315,214 bp) in the example will result in the sequence view panel automatically jumping to the corresponding position. As shown, this mutation results in the substitution of amino acids (K→M).
Germplasm comparison
A user can compare two samples in MBKbase to show the genetic variations between them at the chromosome level (Figure 3C) to find out how similar they are. Furthermore, global comparisons between three samples are also available for comparing breeding parents and offspring to identify the chromosomal segments of the offspring from each of its parents. In the genotype home page of MBKbase, clicking the ‘sample compare’ link will open the variant genotype comparison home page and clicking the ‘pedigree compare’ link will enter the pedigree genotype comparison home page.
Analyzing user submitted WGS data
A user can submit WGS data of germplasm samples to MBKbase for genotype identification. After submission of WGS data, they will be processed in real time and the resulting known alleles identified in the samples are displayed in the sample detail page (Figure 3D). MBKbase also supports offline batch uploading of private raw data of WGS samples and phenotypes, and the private data can be combined with published project data for analysis and visualization. Users can decide when to share the private data.
Variant annotation
We developed an annotation tool for adding experimentally verified functional annotations to a sequence variant such as an SNP or an indel. In the custom genotyping page, for any gene, its structure, reference protein sequence and genotype protein sequence are displayed (Figure 3E). Users can select the gene model by clicking an mRNA ID in the gene structure panel, and the corresponding protein sequence will be automatically generated and displayed in the sequence panel. In the same way, users can select a genotype by clicking a genotype row in the genotype table, and the corresponding DNA and protein sequence will be displayed. Note that the protein sequence of the genotype (haplotype) comes from the resulting sequence with all the variants in the query region, including SNPs and indels. Users can click on a variant in the genotype table, which pops up a variant annotation page. If this variant has been annotated, the variant font is marked in red, as shown at position 30 315 214 bp (Figure 3E). This provides a convenient way to manually update functional annotations as more and more genes are cloned.
CONCLUSION AND FUTURE DIRECTIONS
Here, we report MBKbase, specifically the rice sub-database, for integrating germplasm information and multi-omics data to support functional genomics and molecular breeding in rice. We did not describe all features available in the database here. Users can go to tutorials on the MBKbase website (www.mbkbase.org/rice/help) for more information. Starting with the rice sub-database, we have completed the development of the functional framework and database structure of MBKbase. We have also developed a set of pipelines for genotype construction, making it possible to achieve online integration analysis and visualizations of genotype and phenotype. As population genomic and phenotypic data become available in other crops, they will be incorporated into MBKbase as independent sub-databases. Our next released crop is soybean, which will be available in 3 months. The database will be continually updated by adding more reference genomes (such as those from (42)) with an expanded pan-genome, as well as the increasing publicly available WGS data (such as from (43)) and high-throughput phenomic data. We will also add more annotations of experimentally verified alleles and trait ontology terms to improve the annotation of phenotypes; more connections to external databases such as Gramene Pathways; and more web tools to support molecular design breeding based on known gene networks and large-scale G2P associations and prediction models for major agronomic traits.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Ruaraidh Sackville Hamilton and Grace Lee Capilit at IRGCIS (International Rice Genebank Collection Information System) for their help in using the IRRI germplasm information in MBKbase.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Chinese Academy of Sciences ‘Strategic Priority Research Program’ fund [XDA08020302]. Funding for open access charge: Institutional Core Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Abelson P.H. Resources of plant germplasm. Science. 1991; 253:833. [DOI] [PubMed] [Google Scholar]
- 2. Weise S., Oppermann M., Maggioni L., van Hintum T., Knupffer H.. EURISCO: the European search catalogue for plant genetic resources. Nucleic Acids Res. 2017; 45:D1003–D1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamazaki Y., Akashi R., Banno Y., Endo T., Ezura H., Fukami-Kobayashi K., Inaba K., Isa T., Kamei K., Kasai F. et al.. NBRP databases: databases of biological resources in Japan. Nucleic Acids Res. 2010; 38:D26–D32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackson M.T. Conservation of rice genetic resources: the role of the International Rice Genebank at IRRI. Plant Mol. Biol. 1997; 35:61–67. [PubMed] [Google Scholar]
- 5. Sachs M.M. Cereal germplasm resources. Plant Physiol. 2009; 149:148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mir R.R., Reynolds M., Pinto F., Khan M.A., Bhat M.A.. High-throughput phenotyping for crop improvement in the genomics era. Plant Sci. 2019; 282:60–72. [DOI] [PubMed] [Google Scholar]
- 7. Varshney R.K., Singh V.K., Hickey J.M., Xun X., Marshall D.F., Wang J., Edwards D., Ribaut J.M.. Analytical and decision support tools for genomics-assisted breeding. Trends Plant Sci. 2016; 21:354–363. [DOI] [PubMed] [Google Scholar]
- 8. Bhat J.A., Ali S., Salgotra R.K., Mir Z.A., Dutta S., Jadon V., Tyagi A., Mushtaq M., Jain N., Singh P.K. et al.. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016; 7:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheben A., Batley J., Edwards D.. Genotyping-by-sequencing approaches to characterize crop genomes: choosing the right tool for the right application. Plant Biotechnol. J. 2017; 15:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jamil M., Ali A., Gul A., Ghafoor A., Napar A.A., Ibrahim A.M.H., Naveed N.H., Yasin N.A., Mujeeb-Kazi A.. Genome-wide association studies of seven agronomic traits under two sowing conditions in bread wheat. BMC Plant Biol. 2019; 19:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Z., Jiang Y., Wang Z., Gou Z., Lyu J., Li W., Yu Y., Shu L., Zhao Y., Ma Y. et al.. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015; 33:408–414. [DOI] [PubMed] [Google Scholar]
- 12. Wang W., Mauleon R., Hu Z., Chebotarov D., Tai S., Wu Z., Li M., Zheng T., Fuentes R.R., Zhang F. et al.. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018; 557:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang X., Wei X., Sang T., Zhao Q., Feng Q., Zhao Y., Li C., Zhu C., Lu T., Zhang Z. et al.. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010; 42:961–967. [DOI] [PubMed] [Google Scholar]
- 14. Miura K., Ashikari M., Matsuoka M.. The role of QTLs in the breeding of high-yielding rice. Trends Plant Sci. 2011; 16:319–326. [DOI] [PubMed] [Google Scholar]
- 15. Zuo J., Li J.. Molecular genetic dissection of quantitative trait loci regulating rice grain size. Annu. Rev. Genet. 2014; 48:99–118. [DOI] [PubMed] [Google Scholar]
- 16. Zuo J.R., Li J.Y.. Molecular dissection of complex agronomic traits of rice: a team effort by Chinese scientists in recent years. Natl. Sci. Rev. 2014; 1:253–276. [Google Scholar]
- 17. Qian Q., Guo L.B., Smith S.M., Li J.Y.. Breeding high-yield superior quality hybrid super rice by rational design. Natl. Sci. Rev. 2016; 3:283–294. [Google Scholar]
- 18. Sun C.W., Zhang F.Y., Yan X.F., Zhang X.F., Dong Z.D., Cui D.Q., Chen F.. Genome-wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the Yellow and Huai Valley of China. Plant Biotechnol. J. 2017; 15:953–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu X., Zuo J., Wang J., Liu L., Sun G., Li C., Ren X., Sun D.. Multi-locus genome-wide association studies for 14 main agronomic traits in barley. Front. Plant Sci. 2018; 9:1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu H., Yang Y., Li H., Liu Q., Zhang J., Yin J., Chu S., Zhang X., Yu K., Lv L. et al.. Genome-wide association studies of photosynthetic traits related to phosphorus efficiency in soybean. Front. Plant Sci. 2018; 9:1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peleman J.D., van der Voort J.R.. Breeding by design. Trends Plant Sci. 2003; 8:330–334. [DOI] [PubMed] [Google Scholar]
- 22. Du H., Yu Y., Ma Y., Gao Q., Cao Y., Chen Z., Ma B., Qi M., Li Y., Zhao X. et al.. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017; 8:15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jarvis D.E., Ho Y.S., Lightfoot D.J., Schmockel S.M., Li B., Borm T.J., Ohyanagi H., Mineta K., Michell C.T., Saber N. et al.. The genome of Chenopodium quinoa. Nature. 2017; 542:307–312. [DOI] [PubMed] [Google Scholar]
- 24. Deschamps S., Zhang Y., Llaca V., Ye L., Sanyal A., King M., May G., Lin H.. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 2018; 9:4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. International Wheat Genome Sequencing, C., investigators, I.R.p. Appels R., Eversole K., Feuillet C., Keller B., Rogers J., Stein N. International Wheat Genome Sequencing, C., investigators, I.R.p. investigators, I.w.-g.a.p. Pozniak C.J. et al.. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018; 361:eaar7191. [DOI] [PubMed] [Google Scholar]
- 26. Shen Y., Liu J., Geng H., Zhang J., Liu Y., Zhang H., Xing S., Du J., Ma S., Tian Z.. De novo assembly of a Chinese soybean genome. Sci. China Life Sci. 2018; 61:871–884. [DOI] [PubMed] [Google Scholar]
- 27. Sun S., Zhou Y., Chen J., Shi J., Zhao H., Zhao H., Song W., Zhang M., Cui Y., Dong X. et al.. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018; 50:1289–1295. [DOI] [PubMed] [Google Scholar]
- 28. Shi J., Ma X., Zhang J., Zhou Y., Liu M., Huang L., Sun S., Zhang X., Gao X., Zhan W. et al.. Chromosome conformation capture resolved near complete genome assembly of broomcorn millet. Nat. Commun. 2019; 10:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang M., Tu L., Yuan D., Zhu, Shen C., Li J., Liu F., Pei L., Wang P., Zhao G. et al.. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019; 51:224–229. [DOI] [PubMed] [Google Scholar]
- 30. Harper L., Campbell J., Cannon E.K.S., Jung S., Poelchau M., Walls R., Andorf C., Arnaud E., Berardini T.Z., Birkett C. et al.. AgBioData consortium recommendations for sustainable genomics and genetics databases for agriculture. Database. 2018; 2018:doi:10.1093/database/bay088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song S., Tian D., Li C., Tang B., Dong L., Xiao J., Bao Y., Zhao W., He H., Zhang Z.. Genome Variation Map: a data repository of genome variations in BIG Data Center. Nucleic Acids Res. 2018; 46:D944–D949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao H., Yao W., Ouyang Y., Yang W., Wang G., Lian X., Xing Y., Chen L., Xie W.. RiceVarMap: a comprehensive database of rice genomic variations. Nucleic Acids Res. 2015; 43:D1018–D1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mansueto L., Fuentes R.R., Borja F.N., Detras J., Abriol-Santos J.M., Chebotarov D., Sanciangco M., Palis K., Copetti D., Poliakov A. et al.. Rice SNP-seek database update: new SNPs, indels, and queries. Nucleic Acids Res. 2017; 45:D1075–D1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tello-Ruiz M.K., Naithani S., Stein J.C., Gupta P., Campbell M., Olson A., Wei S., Preece J., Geniza M.J., Jiao Y. et al.. Gramene 2018: unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res. 2018; 46:D1181–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawahara Y., de la Bastide M., Hamilton J.P., Kanamori H., McCombie W.R., Ouyang S., Schwartz D.C., Tanaka T., Wu J., Zhou S. et al.. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. International Rice Research Institute Standard Evaluation System (SES) for Rice. 2013; Manila: International Rice Research Institute. [Google Scholar]
- 38. Cooper L., Meier A., Laporte M.A., Elser J.L., Mungall C., Sinn B.T., Cavaliere D., Carbon S., Dunn N.A., Smith B. et al.. The Planteome database: an integrated resource for reference ontologies, plant genomics and phenomics. Nucleic Acids Res. 2018; 46:D1168–D1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stein J.C., Yu Y., Copetti D., Zwickl D.J., Zhang L., Zhang C., Chougule K., Gao D., Iwata A., Goicoechea J.L. et al.. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018; 50:285–296. [DOI] [PubMed] [Google Scholar]
- 43. Zhao Q., Feng Q., Lu H., Li Y., Wang A., Tian Q., Zhan Q., Lu Y., Zhang L., Huang T. et al.. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018; 50:278–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.