


Abstract
Bacteria coordinate cellular behaviors using a cell–cell communication system termed quorum sensing. In Vibrio harveyi, the master quorum sensing transcription factor LuxR directly regulates >100 genes in response to changes in population density. Here, we show that LuxR derepresses quorum sensing loci by competing with H-NS, a global transcriptional repressor that oligomerizes on DNA to form filaments and bridges. We first identified H-NS as a repressor of bioluminescence gene expression, for which LuxR is a required activator. In an hns deletion strain, LuxR is no longer necessary for transcription activation of the bioluminescence genes, suggesting that the primary role of LuxR is to displace H-NS to derepress gene expression. Using RNA-seq and ChIP-seq, we determined that H-NS and LuxR co-regulate and co-occupy 28 promoters driving expression of 63 genes across the genome. ChIP-PCR assays show that as autoinducer concentration increases, LuxR protein accumulates at co-occupied promoters while H-NS protein disperses. LuxR is sufficient to evict H-NS from promoter DNA in vitro, which is dependent on LuxR DNA binding activity. From these findings, we propose a model in which LuxR serves as a counter-silencer at H-NS-repressed quorum sensing loci by disrupting H-NS nucleoprotein complexes that block transcription.
INTRODUCTION
In nature, bacteria live in complex communities of microbes where competition for resources is constant. Thus, detection and identification of neighboring cells in the community is important for survival in many niches. To achieve this feat, bacteria use cell–cell communication called quorum sensing (QS) to detect and differentiate both the number and type of cells in the local vicinity (1). QS employs small signaling molecules called autoinducers (AIs). Because AIs are constitutively produced and released, their local extracellular concentration functions as a proxy for the number of neighboring cells. The marine pathogen Vibrio harveyi produces three AIs: HAI-1 (Harveyi autoinducer 1), CAI-1 (Cholerae autoinducer 1) and AI-2 (autoinducer 2) (reviewed in (2,3)). Each of these AI molecules are sensed and bound by a cognate membrane-bound histidine-kinase receptor: HAI-1 is detected by LuxN, CAI-1 is detected by CqsS, and AI-2 is detected by LuxPQ. At low cell density (LCD), when the cellular concentration of a population is low, AI concentration is relatively low, and the receptors remain unbound and thereby function as kinases. The phosphorylation cascade is propagated through a response regulator, LuxO. When LuxO is phosphorylated at LCD, it activates the expression of the quorum regulatory RNAs (Qrrs); the Qrrs activate and repress the production of the two master QS transcription factors, AphA and LuxR, respectively. Thus, at LCD, AphA is maximally produced and LuxR is expressed at its lowest level (4). As the population grows and transitions to high cell density (HCD), the AI concentration exceeds a threshold in which the receptor proteins are saturated by AI molecules. In the ligand-bound state, the receptor proteins change from kinases to phosphatases, switching the flow of phosphate. LuxO is dephosphorylated, and the Qrrs are not expressed. Thus, at HCD, LuxR is produced maximally, and AphA protein production is inhibited. This regulatory network results in the activation and repression of hundreds of genes in response to changes in population density (5,6). The core of the QS signal transduction network architecture and the LuxR global regulator are conserved in Vibrio species, though the signaling molecules and/or receptors vary (6). Thus, in response to increases in population density and accumulating AIs, Vibrio cells increase production of LuxR protein, which results in a corresponding change in gene expression and behavior (e.g. bioluminescence, competence, and secretion of virulence factors).
LuxR is a global regulator that controls the expression of >400 genes (5–8). This family of LuxR-type proteins is conserved across vibrios (e.g. HapR in Vibrio cholerae, SmcR in Vibrio vulnificus), but does not bind AI as in the LuxI/LuxR systems widely found in Gram-negative bacteria. Rather, LuxR is classified as a TetR-type transcriptional regulator and can activate as well as repress gene expression. Our previous study showed that LuxR functions synergistically alongside the nucleoid-organizing protein called integration host factor (IHF) to activate transcription of the bioluminescence operon luxCDABE (7). Another study from our lab showed that LuxR interacts directly with the alpha subunit of RNA polymerase (RNAP) and that this interaction is required for activation of a subset of QS genes (9). These findings suggest that LuxR-dependent transcriptional activation requires the use of accessory proteins to remodel DNA structure and position RNAP at QS promoters. IHF and RNAP-interactions play an important role in LuxR-type regulation in V. vulnificus as well (10), suggesting that these mechanisms of gene regulation are conserved across the genus.
Histone-like nucleoid structuring protein (H-NS), which is another nucleoid-organizing protein, functions to directly repress transcription across the genome. H-NS has been best studied in Escherichia coli and Salmonella enterica (11). At the biophysical level, H-NS is capable of oligomerizing on DNA to form extended filaments and/or DNA–H-NS–DNA bridges (12–14). These nucleoprotein complexes function to impede the activity of RNAP, either by blocking transcription initiation or by inhibiting elongation via topological constraint of the DNA, thereby silencing gene expression from H-NS-bound loci (15,16). To counter-silence these loci and activate gene expression, bacteria employ transcription factors that are capable of displacing H-NS from promoter DNA. In V. cholerae, H-NS modulates the expression of 701 genes (17), and QS-regulated proteins have been postulated to be capable of counter-silencing activities. For example, the QS-controlled protein ToxT is required to activate the tcpA and ctx promoters, and it is hypothesized that it accomplishes this by displacing H-NS to allow transcription (18).
Here, we show that V. harveyi LuxR activates transcription of QS genes through anti-repression via H-NS remodeling and/or displacement from QS promoter DNA. RNA-seq and ChIP-seq analyses show that the regulatory overlap between LuxR and H-NS is widespread across the genome. Furthermore, ChIP-qPCR analyses show that H-NS is evicted from QS promoter DNA in vivo in a LuxR-dependent fashion. Electrophoretic mobility shift assays coupled with western blots show that LuxR is competent to displace H-NS from promoter DNA in vitro. Together, these findings expand on the growing model of LuxR-mediated transcriptional activation to include counter-silencing as a means of activation at H-NS-occupied loci.
MATERIALS AND METHODS
Bacterial strains and media
The E. coli S17-1λpir strain was used for cloning purposes, and the E. coli BL21 (DE3) strain was used for overexpression and purification of all proteins (Supplemental information Table S2). Escherichia coli strains were cultured at 37°C with shaking (250–275 RPM) in Lysogeny Broth (LB) medium with 40 μg/ml kanamycin, 100 μg/ml ampicillin, and/or 10 μg/ml chloramphenicol when selection was required. V. harveyi BB120 was recently reclassified as Vibrio campbellii BB120 (a.k.a., ATCC BAA-1116) (19), but for consistency in the literature, we refer to it as V. harveyi. BB120 and derivatives were cultured at 30°C with shaking (250–275 RPM) in Luria Marine (LM) medium with 250 μg/ml kanamycin, 5/10 μg/ml chloramphenicol, and/or 50 μg/ml polymyxin B when selection was required. Plasmids were transformed into electrocompetent E. coli S17-1λpir cells and subsequently conjugated into V. harveyi strains. V. harveyi exconjugants were selected using polymyxin B (50 U/ml).
Bioluminescence assays
Bacterial cultures were back-diluted to OD600 = 0.0005 in 50 ml LM in flasks and grown shaking at 275 RPM. For the standard assay (Figure 1A), optical density (OD600) was measured using a spectrophotometer and a Biotek Cytation3 plate reader was used to measure light units per 200 μL. For the high-throughput assay, cells were diluted 30 000×-fold into 200 μl LM and incubated at 30°C shaking overnight in the Biotek plate reader with readings every 30 min. Bioluminescence per cell was determined by dividing the relative light units (RLU) by the OD600. For Figure 1B–D and Supplementary Figure S2, the plate reader was used to determine bioluminescence and optical density.
Figure 1.
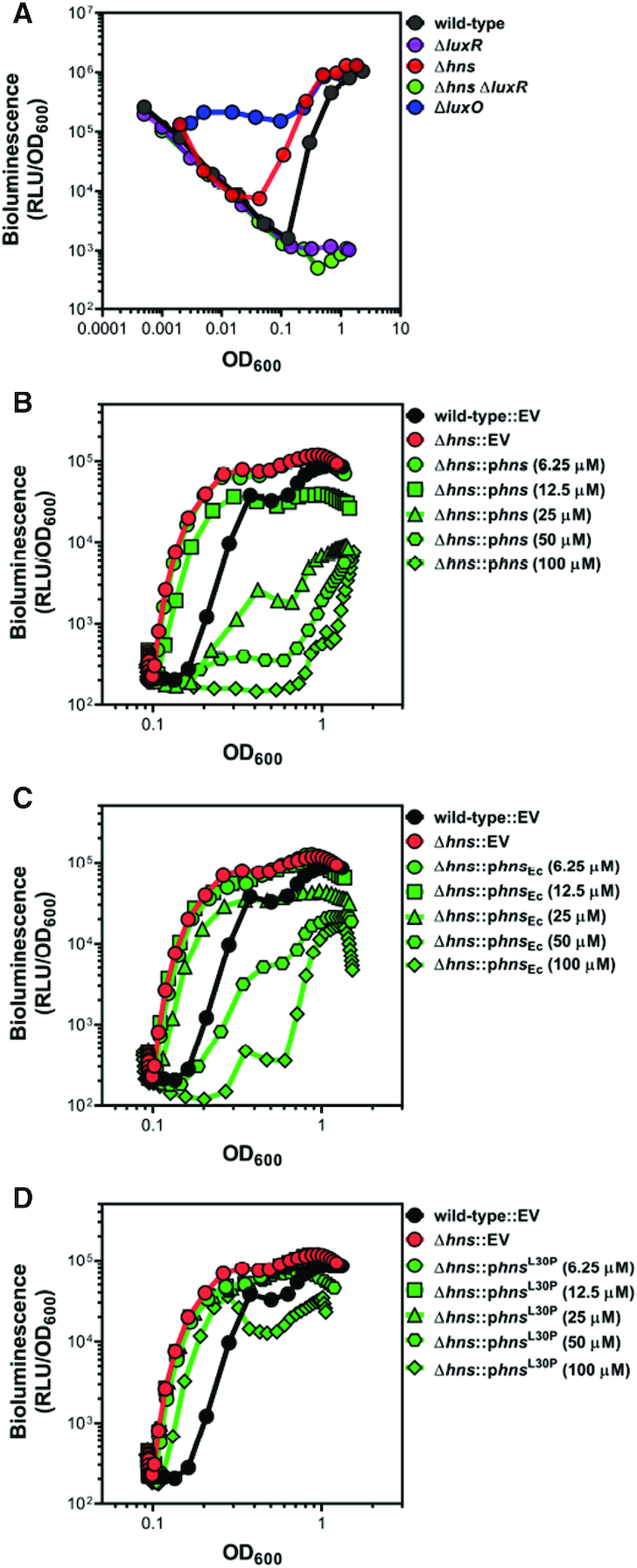
H-NS controls bioluminescence in V. harveyi and is functionally similar to H-NS in E. coli. (A) Bioluminescence (relative light units, RLU; Lux/OD600) was measured for V. harveyi wild-type (BB120), ΔluxR (KM669), Δhns (RRC045), Δhns ΔluxR (RRC168) and ΔluxO (JAF78). Data shown are three independent biological experiments. (B–D) Bioluminescence (relative light units, RLU; Lux/OD600) was measured for V. harveyi wild-type (BB120) and Δhns (RRC045) strains. Strains contained either an empty vector (EV, pMMB67EH-kanR), a plasmid expressing V. harveyi hns under an IPTG-inducible promoter (pRC053; panel B), a plasmid expressing E. coli hns under an IPTG-inducible promoter (pRC054; panel C), or a plasmid expressing V. harveyi hns L30P under an IPTG-inducible promoter (pRC055; panel D). IPTG was added at concentrations of 6.25 μM, 12.5 μM, 25 μM, or 50 μM; IPTG concentration had no effect on the curve for the wild-type EV or Δhns EV strains (data not shown). Data shown are averages of two technical replicates and are representative of five independent biological experiments.
Molecular and chemical methods
PCR was performed using Phusion HF polymerase (New England Biolabs). All restriction enzymes and T4 polynucleotide kinase were purchased from New England Biolabs and used according to the manufacturer's protocol. Site-directed mutagenesis was performed using Phusion HF polymerase (New England Biolabs) following the QuikChange mutagenesis protocol (Stragene). All oligonucleotides were ordered from Integrated DNA Technologies (IDT). Oligonucleotides used for EMSAs and qPCR are listed in Supplemental Information Table S4. PCR products and plasmids were sequenced using Eurofins Genomics. All plasmids used in this study are listed in Supporting Information Table S3, and cloning procedures are available upon request. DNA samples were resolved using 1% agarose (1× TBE). V. harveyi autoinducer 1 (HAI-1) was synthesized using a previously published protocol (20).
Construction of deletion/epitope-tagged strains
All V. harveyi BB120 and KM669 derivative strains in this study were constructed following a previously published technique (7). Briefly, the pRE112 suicide vector was used to construct unmarked deletions or insertions of epitopes in which 1000 bp of upstream and downstream flanking sequence was cloned into pRE112. The pRE112 derivatives were conjugated into V. harveyi and selected on chloramphenicol to induce chromosomal recombination of the plasmid. Subsequently, the plasmid was excised via counterselection on 20–25% sucrose. Cells in which the plasmid excision yielded a non-WT locus were detected via colony PCR. All gene deletions were confirmed by DNA sequencing.
RNA-seq
To isolate RNA, cells were back-diluted to OD600 = 0.005 and grown shaking at 30°C until OD600 = 1.0. RNA was collected, extracted, DNase-treated, and purified as described previously (7). RNA-seq was performed as described previously (7). Sequence data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (NCBI GEO: GSE136191).
Chromatin immunoprecipitation
Cells were grown to an OD600 of 1.0 and protein/nucleic acids were crosslinked using 1% formaldehyde at 30°C for 30 min. Crosslinking was quenched with the addition of 0.42 M glycine and incubated at 30°C for 5 min. 10 ODs of cells were harvested for each replicate and cell pellets were stored at −80°C. Cells were lysed using lysis buffer (1× Bugbuster (Milipore), 1% Triton X-100, 1 mM PMSF, 50 μg/ml lysozyme and 1× protease inhibitors) at room temperature for 30 min on a nutator. Protease inhibitor cocktail (100× stock) contained the following: 0.07 mg/ml phosphoramidon (Santa Cruz), 0.006 mg/ml bestatin (MPbiomedicals/Fisher Scientific), 1.67 mg/ml AEBSF (DOT Scientific), 0.07 mg/ml pepstatin A (Gold Bio), 0.07 mg/ml E64 (Gold Bio). For ChIP-qPCR experiments, cell lysates were sonicated using a Branson SFX 150 sonicator (30% amplitude, 10’ pulse, 30’ rest on ice, 4 cycles). For ChIP-seq experiments, cell lysates were sonicated using a Covaris S220 sonicator (175 W peak incident power, 10% duty factor, 200 cycles per burst, 15-minute treatment time, 7.5°C). After sonication, lysates were clarified via centrifugation at 15 000 RPM for 15 min at 4°C. FLAG-tagged proteins (LuxR was tagged N-terminally and H-NS was tagged C-terminally) were captured from clarified lysates using anti-FLAG agarose M2 beads (Sigma). Lysates were incubated with the anti-FLAG resin for 90 min at room temperature in immunoprecipitation buffer (50 mM HEPES–NaOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF) on a nutator. After incubation with lysate, the beads were washed with immunoprecipitation buffer, wash buffer 1 (50 mM HEPES–NaOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, 0.1% SDS), wash buffer 2 (10 mM Tris–HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% deoxycholate) and 10 mM TE. Nucleoprotein complexes were eluted from the beads using elution buffer (50 mM Tris–HCl pH 7.5, 10 mM EDTA, 1% SDS) at 65°C for 30 min. Subsequently, nucleic acids were freed from crosslinked protein via proteinase K digestion (1.8 mg/ml proteinase K, 2 h at 42°C, 16 h at 65°C). ChIPed DNA was purified using a PCR clean up kit (Qiagen). For input samples (an aliquot of the sonicated, clarified lysate prior to incubation with beads), 1:1 ratio of lysate:10 mM Tris–HCl pH 8.0 and 1.8 mg/ml RNase A was added and incubated at room temperature for 10 min. Proteinase K treatment of input DNA was performed as described above. All ChIP-seq experiments were performed using three biological replicates. Immunoprecipitated DNA concentration/quality was determined using an Agilent 2200 TapeStation and the DNA was subsequently stored at −20°C.
ChIP-seq and analyses
Chromatin immunoprecipitation for ChIP-seq experiments was performed as described above. Sonication was performed using a Covaris S220 water bath sonicator; manufacturers’ protocol was followed to achieve ∼150 bp DNA fragments. Immunoprecipitated chromatic DNA was subjected to library preparation using NEBNext Ultra II DNA Library kit (NEB cat# E7645S). Briefly, DNA fragments were end repaired and 5′ phosphorylated, and a 3′ end Adenosine-tail was added. The products were ligated to a NEB hairpin adapter which contains an uracil at the loop region. The adapter loop was opened by Uracil-Specific Excision Reagent (NEB) to excise the uracil. The products were cleaned with Agencourt AMPure XP beads (Beckman Coulter cat# A63882) but no size selection was applied. Ten or 12 PCR cycles were run using 8-nt barcoded oligos from NEBNext Multiplex Oligos barcode kit (NEB cat# 6609S). The libraries were purified with Agencourt AMPure XP beads, multiplexed and sequenced on NextSeq 500 (Illumina) with NextSeq75 High Output v2.5 kit (Illumina cat# 20024906) to generate paired-end 2 × 42 bp reads. The read sequences were de-multiplexed using bcl2fastq (software versions 2.1.0.31). ChIP-seq datasets were trimmed for quality and adapters using Trimmomatic (v0.33 for all samples except the H-NS ΔluxR ChIP H-NS samples which used v0.38) with the following parameters: ILLUMINACLIP:adapters.fa:3:20:6 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:30. Reads were mapped to the V. harveyi ATCC BAA-1116 genome using bwa (v0.7.17). Peaks were identified using macs2 (v2.1.2) call peak algorithm. To compare H-NS peak heights in the wildtype and ΔluxR mutant backgrounds we had to normalize the data. Naively, we would just normalize each sample based on the number of ChIP-seq reads mapped to the genome but it was unclear whether this would be accurate given the potential interaction between H-NS and luxR. To validate this approach, we identified 98 H-NS peaks that were not associated with luxR binding sites. The coverage underlying the peak in each replicate was normalized to the number of reads mapped to the genome. The mean, calculated across replicates, normalized peak coverage for these 98 luxR independent peaks in the wildtype and ΔluxR strains was plotted. Linear regression shows that there is a significant correlation between these points and that these points generates a line with a slope close to 1 that passes close to the origin. Based on this result we proceeded to normalize H-NS peak heights after normalizing by mapped reads. Distances between LuxR/H-NS and H-NS/H-NS peaks were performed using a custom script. Points were plotted as a function of distance from LuxR peak to the nearest H-NS peak (x-axis) against distance from nearest H-NS peak to the next closest H-NS peak (i.e. doublet, y-axis). H-NS doublet peaks were identified using a custom script that searched for H-NS peaks within 2 kb of each other that had a trough ≤80% of the lower of the two peaks. Sequence data were deposited in the NCBI GEO database (accession number: GSE136050). Custom scripts are available on GitHub (https://github.com/Juliacvk/LuxR_H-NS).
Quantitative PCR
ChIP input and elution samples were diluted 1:100 and 1:4, respectively, using nuclease free H2O. qPCR reactions were performed using SensiFAST SYBR Hi-ROX (Bioline) according to the manufacturer's guidelines. qPCR primers were designed to fit the following parameters: amplicon size: 100–105 bp, primer size: 18–22 nt, and primer melting temperature: 56–60°C. Each reaction contained 2 μl of diluted DNA template (input or elution) and 0.4 μM of each primer (total volume was 10 μl). qPCR reactions were incubated and monitored using a LightCycler 4800 II (Roche). The clpP gene (VIBHAR_01417) was chosen as a negative control for ChIP-qPCR experiments because it was devoid of binding peaks for LuxR and H-NS. For quantification of relative enrichment, the following equation was utilized: 2CTinput – CTelution. All ChIP-qPCR experiments were performed with three biological replicates.
Purification of LuxR, H-NS, and RNAP proteins
Recombinant, untagged LuxR and E. coli 6xHis-RNAP proteins were purified using protocols previously reported (7,9). H-NS was purified by cloning the hns ORF into the pET28B expression vector (Novagen) to generate a 6xHis tag on the C-terminus. The pET28B-hns-6xHis plasmid (pRC049) was transformed into NiCo21 E. coli (New England Biolabs), which contains lacks several genes encoding proteins that readily contaminate immobilized metal affinity chromatography purifications. The resulting strain, RRC226 (NiCo21-pRC049), was grown at 30°C to an OD600 = 0.6–0.8 and hns overexpression was induced using 1 mM IPTG for 3 h. Induced cell pellets were resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole, 1× protease inhibitors, 1 mM PMSF, 0.2 mg/ml DNaseI (GoldBio), 1× FastBreak (Millipore)) and incubated at room temperature for 35 min. The protease inhibitor mix included the following: 0.07 mg/ml phosphoramidon (Santa Cruz), 1.67 mg/ml AEBSF (DOT Scientific), 0.07 mg/ml pepstatin A (DOT Scientific), 0.07 mg/ml E-64 (Gold Bio), and 0.06 mg/ml bestatin (MPbiomedicals/Fisher). Clarified lysate was loaded onto a 5 ml His-Trap Ni-NTA column (GE Healthcare Life Sciences) using an Akta Pure (GE Healthcare Life Sciences). Protein was eluted from the column using a linear gradient of elution buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 M imidazole). Fractions were analyzed by SDS-PAGE to confirm the presence of 6xHis-H-NS and pooled together. Pooled fractions were concentrated using 10 kDa cutoff centrifugal filters (Sartorius) and dialyzed overnight in against storage buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 5% glycerol). Dialyzed protein was aliquoted, snap frozen in liquid N2, and stored at −80°C.
Electrophoretic mobility shift assays
PCR was used to produce promoter substrates; PluxC, P02100 and P03626 were amplified using RC003/042, RC549/606 and RC604/605, respectively. Binding reactions were performed using either 1 nM radiolabeled DNA or 8.5 nM non-labeled DNA in EMSA binding buffer (50 mM HEPES–NaOH pH 7.5, 50 mM NaCl, 5% glycerol). Poly(dI-dC) (10 ng/μl, Sigma) was used as non-specific competitor DNA when noted. Reactions were incubated at room temperature for 15 min. For competitive binding experiments, reactions were supplemented with the secondary protein in binding buffer and incubated at room temperature for an additional 15 min. Reactions containing small substrates were resolved on 6% polyacrylamide (19:1) 1× TBE (100 mM Tris, 100 mM boric acid, 2 mM EDTA) gels using 25 mA constant current for 30–40 min at room temperature. These gels were dried down and visualized using a phosphor storage screen (imaged with a GE Life Technologies Typhoon 9500). Reactions containing large substrates were resolved on 6% polyacrylamide (37.5:1) 1× TBE gels using 20 mA constant current for 50–70 min at 4°C. DNA was detected by staining the gel with 1 μg/ml ethidium bromide for 20 min at room temperature. To detect displaced proteins in competitive EMSAs, the ethidium bromide-stained gel was transferred to a nitrocellulose membrane. Proteins were detected using anti-His-HRP (Sigma) and anti-FLAG-HRP (Sigma) antibodies.
RESULTS
H-NS is a repressor of bioluminescence in V. harveyi
We previously identified a protein bound to the promoter of the luxCDABE bioluminescence genes using a promoter pull-down assay that is predicted to be H-NS (7). The protein encoded by the hns gene in V. harveyi (VIBHAR_01827) shares 53% amino acid identity and 68% amino acid similarity to E. coli H-NS (Supplementary Figure S1A). DNA binding motifs in the linker domain (charged residues) and C-terminal domain (AT-hook) are highly conserved between E. coli and V. harveyi H-NS (21,22). Additionally, homology is maintained throughout the first three α-helices in the N-terminal domain, which are important for oligomerization (22). Based on the literature that defines H-NS as a transcriptional repressor, we hypothesized that H-NS functions as a repressor of luxCDABE at LCD. To test this hypothesis, we constructed a Δhns strain and monitored bioluminescence throughout a growth curve. A typical wild-type bioluminescence growth curve is a ‘U-shape’ (Figure 1A). At the start of the experiment, stationary HCD cells are diluted into fresh media to LCD. Early in the growth curve, because the cells are at LCD, bioluminescence gene expression is off, due to lack of LuxR, a required activator. The calculation to determine approximate bioluminescence per cell is performed by dividing the bioluminescence by the optical density (OD600). Because the bioluminescence number remains constant and low at LCD and the optical density is increasing, the early LCD part of the growth curve has a negative slope. As the population density increases, LuxR expression begins to increase indicating quorum has been reached (approximately at OD600 = 0.1), and bioluminescence production increases until stationary phase is reached. A ΔluxR strain does not produce bioluminescence and exhibits the negative slope of the data as described throughout the plot (Figure 1A). Conversely, the ΔluxO strain lacks the ability to repress LuxR production via the Qrrs and exhibits constitutive bioluminescence (Figure 1A). The Δhns strain exhibits several important characteristics: (i) the levels of bioluminescence at LCD mirror wild-type, (ii) the initial production of bioluminescence appears earlier in growth compared to wild-type and (iii) bioluminescence levels are similar to wild-type at late stationary phase (Figure 1A). Furthermore, the ΔluxR Δhns strain phenocopies a ΔluxR strain, indicating that LuxR is epistatic to H-NS. To examine the effect of H-NS copy number on bioluminescence, we developed a high-throughput bioluminescence assay using a plate reader that monitors bioluminescence during the latter part of the ‘U-shape’ curve. We complemented the hns deletion using an ectopic IPTG-inducible expression vector and observed bioluminescence levels comparable to wild-type (Figure 1B). Using an array of IPTG concentrations, we observed dose-responsiveness between H-NS expression and bioluminescence; at 100 μM IPTG, bioluminescence is severely decreased (Figure 1B). To determine whether V. harveyi H-NS functions similarly to E. coli H-NS, we complemented the Δhns V. harveyi strain with the hns gene from E. coli (hnsEc). Expression of hnsEc complemented the bioluminescence phenotype in the Δhns strain to produce approximately wild-type levels of bioluminescence (Figure 1C). At 100 μM IPTG, expression of E. coli H-NS results in bioluminescence levels that are drastically reduced compared to wild-type. In addition, the L30P allele of E. coli H-NS is able to bind DNA but unable to oligomerize, which is essential for its repressor functionality in vivo (23,24). We therefore complemented the Δhns strain with V. harveyi hns L30P allele to test its activity. As expected, the Δhns strain expressing H-NSL30P phenocopied the Δhns strain and produced more bioluminescence than wild-type during exponential phase (Figure 1D). Even at high concentrations of IPTG, this mutant is unable to complement the Δhns strain. Importantly, using native PAGE, we show that purified V. harveyi H-NS is capable of oligomerizing into higher order structures similar to E. coli H-NS (Supplementary Figure S1B). From these data, we conclude that V. harveyi H-NS represses bioluminescence in a concentration dependent manner and is a functional homologue of H-NS from E. coli. Further, the role of H-NS in repression of luxCDABE occurs at HCD; at LCD, deletion of hns has no impact on bioluminescence.
H-NS and LuxR co-regulate 124 genes
To determine whether other promoters are regulated by H-NS and LuxR in a similar manner to PluxCDABE, we used RNA-seq to define the regulons for LuxR and H-NS. For the RNA-seq, we compared transcripts in a Δhns strain to a wild-type strain. Conventionally, we have performed QS-related RNA-seq analyses at OD600 = 1.0 because this is when HCD is reached and QS genes are maximally expressed (specifically in LM medium; this may differ in other media). Therefore, we also performed the wild-type vs. Δhns RNA-seq at this point in the growth curve. We observed that H-NS regulates 726 genes (>2-fold, P < 0.05; Figure 2A, dataset S1) in V. harveyi, which is strikingly comparable to the 701 genes regulated by H-NS in V. cholerae (17). Among the 726 genes, 596 are repressed by H-NS. Because H-NS is a global repressor and thus functions in the opposing manner to LuxR at luxCDABE, we hypothesized that LuxR acts to derepress H-NS-bound loci. Indeed, the luxCDABE genes were absent from the H-NS regulon in the wild-type background, which supports this hypothesis. To determine if the presence of LuxR affects H-NS gene regulation at other loci, we next performed RNA-seq comparing a ΔluxR strain to a ΔluxR Δhns strain. Under these conditions, H-NS regulates 824 genes (>2-fold, P < 0.05; Figure 2A, dataset S1) and, as expected, the luxCDABE genes were present. However, only 430 genes are repressed by H-NS in this comparison. Together, these RNA-seq data show that H-NS regulates a similar number of genes in the presence or absence of LuxR, though the scale and direction of regulation varies widely. Among the combined genes in the H-NS regulons (wild-type and ΔluxR backgrounds combined), 178 genes overlapped with the previously identified 308-gene LuxR regulon (>2-fold, P < 0.05; Figure 2A; (7)). These genes include many systems previously shown to be highly regulated by LuxR, including the luxCDABE genes, the type III secretion operons, the proXWV glycine betaine transport system, and several ABC transport systems, among others. From these data, we conclude that H-NS and LuxR are relevant global co-regulators in V. harveyi.
Figure 2.
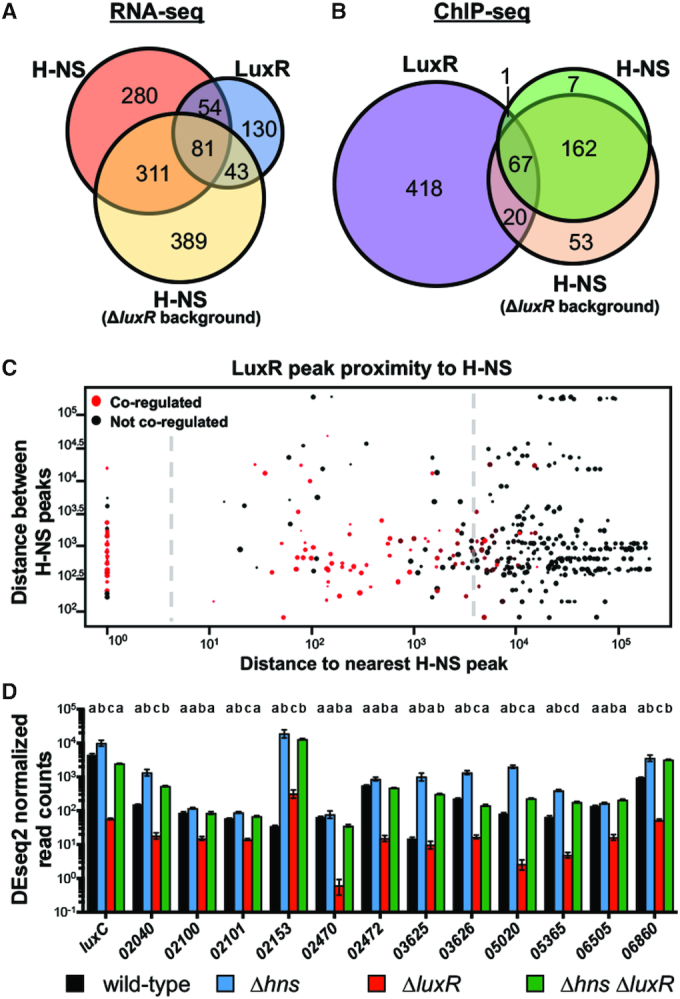
LuxR and H-NS directly co-regulate QS genes. (A) RNA-seq was used to determine genes regulated >2-fold, P < 0.05. The H-NS regulon was determined by comparing transcript levels of a ΔluxR strain (KM669) to a Δhns ΔluxR strain (RRC168). The LuxR regulon was previously determined by comparing wild-type (BB120) to a ΔluxR strain (KM669) (7). (B) FLAG-LuxR and H-NS-FLAG ChIP-seq profiles were analyzed for overlapping peaks within 500bp. The H-NS-FLAG ChIP-seq profile was determined in the ΔluxR background (RRC237), and the FLAG-LuxR ChIP-seq profile was determined in a wild-type background (JV039). (C) Each point represents a LuxR binding site with the diameter corresponding to the strength of the peak at that site; all 506 LuxR peaks are plotted. The distance between a LuxR peak and the nearest H-NS peak is plotted on the x-axis. The distance between the nearest H-NS peak and the next proximal H-NS peak is plotted on the y-axis. Red points represent LuxR peaks that are proximal to genes that are co-regulated by LuxR and H-NS while black points represent genes that are regulated by only one protein or neither. The plot is divided into three segments by the dashed line that represent LuxR peaks that overlap with H-NS peaks (left), LuxR peaks that are proximal to H-NS peaks (middle), and LuxR peaks that are not associated with H-NS peaks (right). (D) DESeq2 normalized read counts determined by RNA-seq of RNA collected from wild-type (BB120), ΔluxR (KM669), Δhns (RRC045), Δhns ΔluxR (RRC168) V. harveyi strains. V. harveyi locus tags are listed on the x-axis (VIBHAR_XXXXX). Different letters indicate significant differences between strains (P < 0.05; two-way analysis of variation (ANOVA), followed by Tukey's multiple comparison test of log-transformed data; n = 3).
LuxR and H-NS co-occupy and co-regulate 19 promoters
Based on our bioluminescence and transcriptomic data, our working hypothesis was that as LuxR protein concentration increases during QS, LuxR binds to promoters containing LuxR binding sites to displace H-NS, which alleviates promoters from H-NS-mediated repression. To test this hypothesis and identify promoters that are bound by both LuxR and H-NS, we used ChIP-seq to define global binding profiles for LuxR and H-NS individually. FLAG-luxR and hns-FLAG alleles were each introduced separately into V. harveyi BB120 replacing the native copy of the respective gene, and ChIP-seq was performed comparing each of the FLAG-tagged mutant strains to the wild-type strain. The FLAG epitope tag was chosen for ChIP-seq experiments because of the highly specific nature of the FLAG:α-FLAG antibody interaction, which produced less contaminants compared to a 6xHis tag (data not shown). We identified 506 binding peaks for LuxR and 237 binding peaks for H-NS across the genome (Figure 2B, dataset S2). We also analyzed LuxR and H-NS binding peaks to determine how many are proximal to each other and found that LuxR and H-NS peaks overlap at 68 regions (Figure 2B, dataset S2). Furthermore, we analyzed the distance between H-NS and LuxR peaks across the genome. We observed distinct types of LuxR binding site locations using this analysis: (i) LuxR binding sites that directly overlap with H-NS binding sites, (ii) LuxR binding sites that are proximal to H-NS binding sites (within ∼1000 bp) and (iii) LuxR binding sites that are distant from H-NS peaks (>5000 bp away) (Figure 2C). Interestingly, most LuxR peaks within 1000 bp of an H-NS peak are proximal to genes that are co-regulated by both LuxR and H-NS (red dots, Figure 2C). As expected, we observed LuxR and H-NS peaks at the luxCDABE promoter (Figure 3A), and no peaks at a control locus (clpP; Figure 3B).
Figure 3.
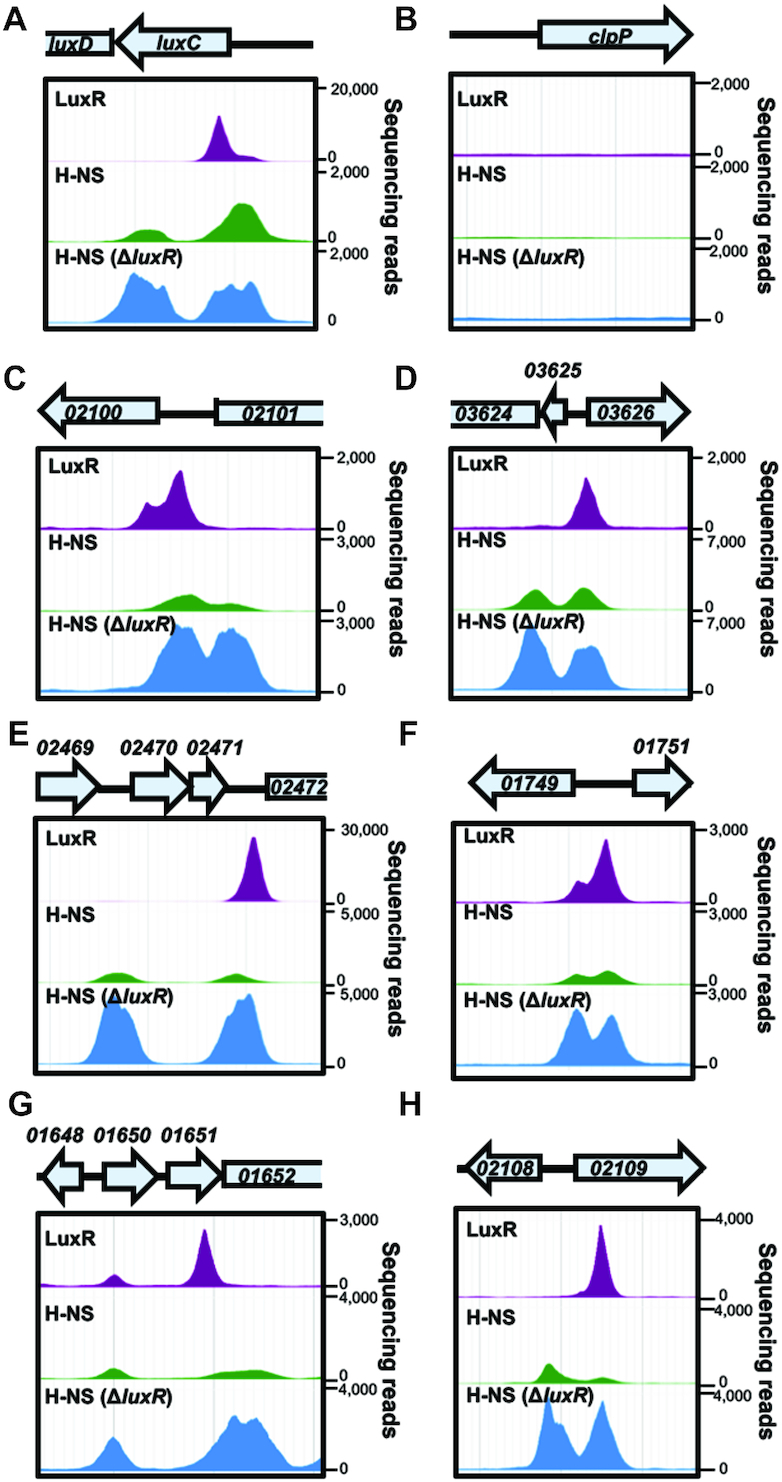
LuxR and H-NS binding peak distribution. (A–H) LuxR and H-NS ChIP-seq peaks at QS loci luxC, VIBHAR_02100, 03626, 02472, 01749, 01652and 02108 are shown for experiments in which LuxR (JV039) or H-NS (RRC169) were ChIPed in the wild-type background (top two rows for each panel) or in the ΔluxR background (RRC237, bottom row for each panel). ChIP-seq profiles were visualized using JBrowse 1.12.3.
The ChIP-seq experiments described above were performed with cells collected at HCD (OD600 = 1.0), similar to our RNA-seq analyses because this is the time point is when LuxR protein is maximally produced prior to stationary phase (5). Based on our hypothesis that LuxR displaces H-NS at HCD, we reasoned that our experimental ChIP-seq setup may have missed H-NS binding sites that were obscured by competition with LuxR because they were collected at the HCD state. Therefore, to observe all H-NS-bound sites in the genome, we performed ChIP-seq with H-NS-FLAG in a ΔluxR mutant strain background. As we predicted, the number of H-NS binding peaks increased from 237 to 302 in the absence of LuxR (dataset S2). The number of co-occupied H-NS and LuxR loci increased from 68 to 88 in the absence of LuxR (dataset S2). By examining these 88 regions further in the context of transcriptomic data and gene organization, we narrowed this list down to 65 regions that are co-occupied by LuxR and H-NS (dataset S2). Among these are 28 promoters controlling expression of 63 genes that are also co-regulated by LuxR and H-NS (dataset S2). Examples of binding peaks at co-regulated and/or co-occupied regions are shown in Figure 3. The remaining co-occupied loci are not present in the LuxR/H-NS co-regulon. These genes may be regulated <2-fold, regulated by only one of these proteins, or regulated only under specific conditions that we did not test. From these data, we conclude that H-NS and LuxR co-localize throughout the genome to co-regulate genes.
LuxR binding alters H-NS binding profiles in vivo
From the ChIP-seq data, we also observed substantial differences in H-NS peak heights in the presence or absence of LuxR. We hypothesized that these profile changes are due to binding of LuxR and displacement of H-NS. To ensure peak height differences were not due to differences in sample preparation (i.e. library preparation/number of sequencing reads), we identified 98 loci that contain H-NS peaks but are devoid of any LuxR peaks. We reasoned that these peaks would not be directly influenced by LuxR and thus we could normalize the H-NS sequencing read distribution to these peaks (normalization procedure detailed in methods). Following normalization, we re-compared these 98 peaks and found that the loci remained significantly correlated between wild-type and ΔluxR backgrounds (Supplementary Figure S2). With the read counts normalized, we then analyzed the H-NS peak heights at select LuxR-bound loci that showed strong ChIP-seq peak signals for both proteins. Each of the loci which are directly activated by LuxR/repressed H-NS showed an increase in H-NS peak height in the absence of LuxR (Figure 3A, C, D–F), indicating increased binding of H-NS at these loci. Interestingly, when we analyzed the difference in H-NS peak heights between the wild-type and ΔluxR backgrounds, we noticed a single LuxR binding site often influenced both H-NS peaks, even if separated by 2 kb as is the case with VIBHAR_02472 (Figure 3E). Even at two loci that LuxR does not regulate under our tested conditions, there is a measurable H-NS peak height difference when LuxR is bound compared to the ΔluxR background (Figure 3G, H).
Another intriguing property of the H-NS ChIP-seq profile is the frequent occurrence of doublets in which two peaks of similar height are proximal to one another. This phenomenon is also detectable in H-NS ChIP-seq experiments performed in V. cholerae (25). We suspect that these peak profiles represent bridges formed by H-NS. While the peak height is generally similar within doublets, the distance between peaks is highly variable ranging from ∼400 bp to 2 kb. We analyzed the H-NS ChIP-seq data for binding sites that matched these parameters (two similarly sized peaks within 2 kb of each other). We identified 52 H-NS doublets throughout the genome, and 32 are proximal to a LuxR binding site (within 1 kb). The notable disruption of these H-NS doublets by the presence of LuxR (e.g. Figure 3A, C, H) suggests that LuxR influences H-NS binding distribution, and thus H-NS bridging. Collectively, these data suggest that LuxR binding displaces H-NS, decreasing H-NS binding and/or bridging at co-regulated and co-bound loci.
LuxR is not required for transcription in the absence of H-NS
Based on our observations of LuxR activation and H-NS repression activities at the luxCDABE promoter, our working hypothesis was that as LuxR protein concentration increases during QS, LuxR binds to PluxCDABE and displaces H-NS, which alleviates promoters from H-NS-mediated repression. As a control, we examined gene expression of the luxCDABE genes to compare to our bioluminescence assays and previously published expression data. The RNA-seq data for the luxC gene indicate that Δhns cells express 1.3-fold more luxC transcripts than wild-type cells at OD600 = 1.0 (Figure 2D). As expected, the ΔluxR strain exhibits very low levels of luxC transcript (Figure 2D), which has been shown previously (26) and is evidenced by lack of bioluminescence production (Figure 1A). We were therefore surprised to observe that the ΔluxR Δhns strain restored luxC transcripts to near wild-type levels (Figure 2D). This result suggests that LuxR is only required for luxC transcription in the presence of H-NS. However, the bioluminescence production of the ΔluxR Δhns strain remains very low (Figure 2D). Importantly, the regulatory pattern of luxDABE in the four strain backgrounds tested is identical to luxC, (dataset S1), which suggests that a posttranscriptional mechanism of regulation, coordinated via LuxR, is required for bioluminescence production.
To test whether this occurs at other LuxR/H-NS co-regulated and co-occupied promoters, we examined the other nine loci in addition to luxCDABE that are activated by LuxR and repressed by H-NS (dataset S2). Each of the 37 genes under control of the 13 promoters show the same pattern of gene expression as luxC: transcript levels are lowest in the ΔluxR strain for all genes, suggesting that LuxR is required for activation of these genes (Figure 2D). However, when hns is deleted in the ΔluxR background, transcript levels are completely or partially restored to wild-type levels (Figure 2D). From these data, we conclude that LuxR is not required for transcription activation of these promoters in the absence of H-NS.
H-NS is evicted from promoter regions as cells transition from LCD to HCD
To investigate the role of LuxR in H-NS localization at promoters, we developed a ChIP-qPCR assay that mimics various QS states to quantify relative amounts of LuxR and H-NS protein occupancy at promoters. These experiments were performed using a strain background in which the genes encoding the HAI-1 autoinducer synthase and CAI-1 and AI-2 receptors were deleted (TL25: ΔluxPQ, ΔluxM, ΔcqsS) (5). This strain responds solely to exogenously supplied HAI-1. Based on previously established concentrations of HAI-1 that mimic the transition states between LCD and HCD, we tested a range of synthetic HAI-1 from 0 to 1000 nM. When either no HAI-1 or low concentrations of HAI-1 are added to this strain, both aphA/luxR transcripts and AphA/LuxR proteins are detected (5). As the HAI-1 concentration added increases to 1000 nM, aphA transcript sharply decreases and AphA protein is no longer detected, whereas luxR transcript and LuxR protein increase (5). To pinpoint AI concentrations that mimic LCD, mid cell density (MCD), and HCD, a bioluminescence induction assay was used. Based on the bioluminescent output, we found that addition of 0 (or 4), 40 and 1000 nM were sufficient to replicate light production levels present at LCD, MCD and HCD (Supplementary Figure S3). The FLAG-LuxR and H-NS-FLAG alleles were individually introduced into the TL25 background, and ChIP was performed using these strains grown with the above HAI-1 concentrations. The immunoprecipitated DNA was analyzed via qPCR directed at seven promoters that showed strong, overlapping peak signals for LuxR and H-NS. The promoter region upstream of clpP was used a negative control for this experiment because no ChIP-seq peaks were detected for LuxR/H-NS (Figure 3B), and similarly, this promoter is not captured by LuxR or H-NS with the ChIP-qPCR assay (Figure 4B). At promoters in which LuxR and H-NS are both present, such as PluxC, LuxR protein occupancy increases at the promoter while H-NS occupancy decreases as cells transition from LCD to HCD (Figure 4A). This trend was conserved at six additional promoters in which we observed a marked increase in LuxR occupancy with a simultaneous decrease in H-NS occupancy (Figure 4C–H). While the overall trend of decreasing H-NS occupancy is consistent across promoters, the amount of HAI-1 required to reach maximum H-NS displacement is variable. Some promoters, such as PluxC and P02100, require 1000 nM HAI-1, whereas others, such as P01749 and P02472, require ≤40 nM HAI-1. Taken together, these data support the hypothesis that LuxR accumulation coincides with the displacement of H-NS from QS promoters.
Figure 4.
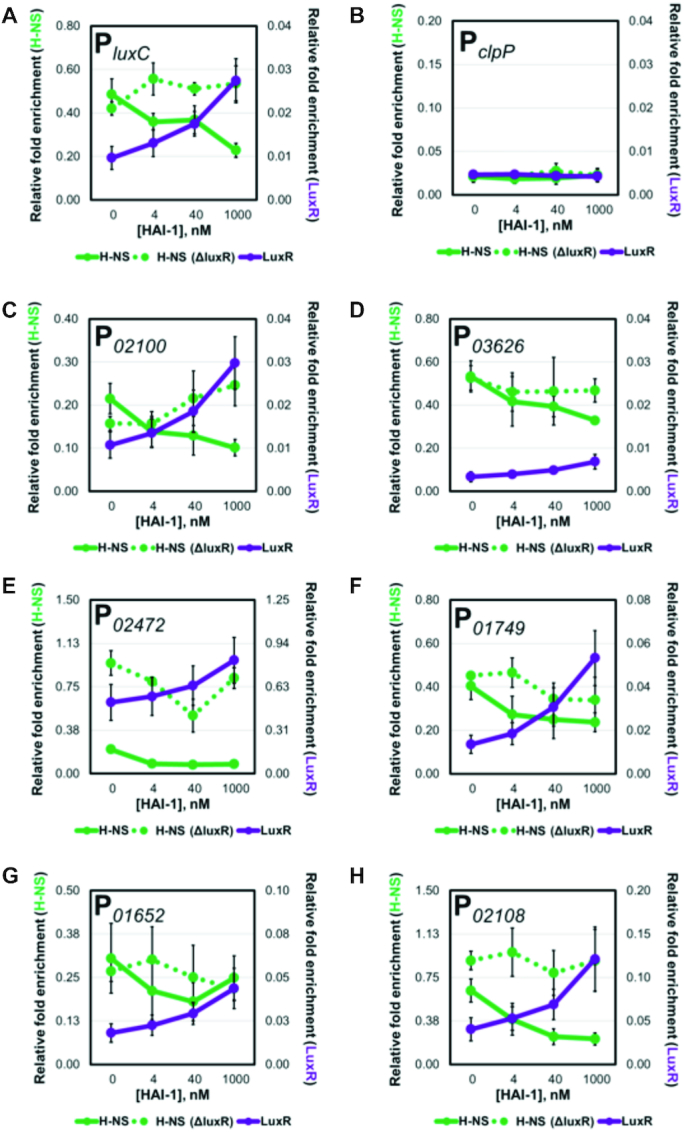
LuxR and H-NS occupancy is inversely related at QS loci. (A–H) The relative enrichment of DNA from various promoters analyzed by ChIP-qPCR experiments in the presence of 0, 4 nM, 40 nM, or 1000 nM HAI-1 in hns-FLAG (RRC169), FLAG-luxR (JV039) and hns-FLAG ΔluxR (RRC237) strains.
To test if LuxR is necessary and/or sufficient to displace H-NS from QS loci in vivo, we constructed the TL25 FLAG-hns ΔluxR strain. We performed our ChIP-qPCR analysis to monitor changes in H-NS occupancy in the absence of LuxR. We observed that the relative amount of H-NS present at a promoter is higher in the ΔluxR background compared to wild-type at some or all concentrations of HAI-1 (Figure 4B–H). While overall H-NS occupancy is higher in these profiles, we do observe H-NS displacement in the absence of LuxR at certain promoters. For instance, at P02472 H-NS displacement is observed between 0 and 40 nM HAI-1 and at P01749 H-NS is displaced between 4 and 40 nM HAI-1. These results are not surprising as LuxR likely works alongside other transcription factors to activate transcription; thus, the observation of H-NS displacement in the absence of LuxR can be attributed to the action of other regulators. The results of these experiments provide evidence that LuxR is required for maximal H-NS displacement at QS promoters.
LuxR displaces H-NS from promoter DNA in vitro
The results of the ChIP-qPCR experiments showed that increasing concentrations of AI and LuxR-binding results in lower H-NS binding at QS promoters in vivo. To determine whether LuxR is able to displace H-NS from DNA in vitro, we first used a bioinformatics approach (FIMO software, (27)) to identify putative H-NS binding sites in the luxCDABE promoter. Using the high-affinity H-NS nucleation site identified in E. coli (TCGATAAATT) (28), we identified 13 potential sites in PluxCDABE, many of which overlap with the seven established LuxR binding sites (Figure 5A, Supplementary Table S1). Next, we used electrophoretic mobility gel shift assays (EMSAs) and performed titration curves with purified 6xHis-H-NS (referred to as H-NS) and LuxR individually to determine the binding kinetics for each protein with luxCDABE promoter DNA. For both sets of reactions, we observed the DNA shift to a larger molecular weight, suggesting that all the DNA was bound (Figure 5B). For H-NS, a steep binding curve was observed, which likely reflects the cooperative DNA binding activities of H-NS (29,30). For LuxR, multiple intermediate LuxR-DNA bands were observed, as expected (7).
Figure 5.
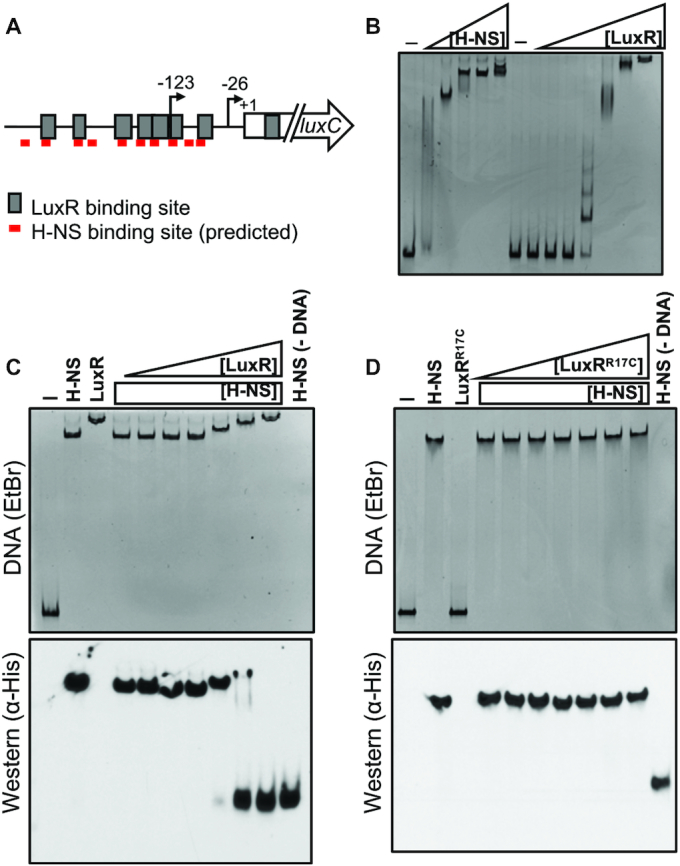
H-NS is displaced from PluxC DNA by LuxR in vitro. (A) Diagram of luxCDABE promoter. Gray boxes indicate LuxR binding sites, and red lines indicate FIMO-predicted H-NS binding sites. Arrows indicate transcription start sites relative to the start codon (+1). (B) EMSA reactions containing 8.8 nM PluxC DNA and either H-NS (100, 150, 200, 250, 300 nM) or LuxR (31.25, 62.5, 125, 250, 500, 1000, 2000 nM) purified protein. Reactions denoted ‘–’ contained no protein. (C) Competitive EMSA reactions consisting of 8.8 nM PluxC DNA and 200 nM purified H-NS and either 31.25, 62.5, 125, 250, 500, 1000 or 2000 nM purified LuxR. Lanes labelled ‘H-NS’, ‘LuxR’, ‘–’, and H-NS (–DNA) contained 200 nM H-NS, 2000 nM LuxR, no protein, and 200 nM H-NS without DNA, respectively. The reactions were run on polyacrylamide gels and stained with ethidium bromide (top) and then transferred to a nitrocellulose membrane and probed for H-NS using α-His-HRP antibodies (bottom). (D) The same experimental procedures were used as described in panel C but instead using the DNA-binding deficient LuxRR17C purified protein.
To examine competition between LuxR and H-NS, we generated reactions with both H-NS and LuxR added to luxCDABE promoter DNA. We added a constant amount of H-NS and varying concentrations of LuxR. We observed that increasing concentrations of LuxR incrementally shifted the migration of the nucleoprotein complex to mirror that of the LuxR-PluxCDABE complex (Figure 5C). This result suggests that LuxR evicts H-NS from this promoter. To determine whether any H-NS protein remained bound to the shifted DNA substrate, EMSA reactions were then transferred to a nitrocellulose membrane such that the proteins associated with bound DNA could be monitored via western blot. We observed that as LuxR concentration increases across reactions, the H-NS protein signal corresponding to the H-NS-PluxCDABE bound complex decreases. In parallel, the signal corresponding to unbound H-NS accumulates (Figure 5C).
The experiment described above was performed in the absence of competitor DNA because H-NS can bind to DNA non-specifically. To verify that the displacement of H-NS was dependent on LuxR associating with specific binding sites rather than non-specific interactions with the DNA, we performed the same competition experiment in the presence of competitor DNA (poly[dI-dC]) to limit the amount non-specific protein-DNA interactions. Under these conditions, LuxR remains able to displace H-NS from PluxCDABE DNA (Supplementary Figure S4C). Importantly, we tested a LuxRR17C DNA binding mutant (8) and found that it was unable to displace H-NS from the luxCDABE promoter (Figure 5D). This result confirms that LuxR eviction of H-NS relies on LuxR DNA binding activity. Next, we extended these experiments to two additional promoters that are repressed by H-NS and activated by LuxR (P02100 and P03626). Competition between H-NS and LuxR was observed using these promoters as well (Supplementary Figure S4A, B), indicating that LuxR eviction of H-NS is a conserved mechanism at multiple loci.
Lastly, we wanted to examine the influence of H-NS on the binding of RNAP to PluxCDABE promoter DNA. Using the same competitive EMSA experimental design as described above, we pre-bound the PluxCDABE substrate with H-NS and titrated in various amounts of purified E. coli 10xHis-RNAP (referred to as RNAP). With increasing concentrations of RNAP, the H-NS–DNA complex showed slower migration through the gel (Supplementary Figure S5). This observation indicates that RNAP is capable of binding H-NS-bound PluxCDABE promoter DNA. However, although RNAP can bind the H-NS-bound DNA, it is not sufficient to displace detectable amounts of H-NS (Supplementary Figure S5). Together, these results indicate two important properties of H-NS: 1) LuxR is capable of displacing H-NS through competitive DNA binding, and 2) RNAP can bind to H-NS-bound DNA but cannot displace H-NS at the luxCDABE promoter.
DISCUSSION
LuxR/HapR have been extensively studied for decades as key regulators of QS genes in vibrios, yet our collective understanding of the molecular workings of these unique transcriptional regulators has remained limited. In the present study, we show that LuxR engages in a functional relationship with H-NS in addition to IHF and RNA polymerase. We show that the actions of LuxR and H-NS are fundamentally opposed in order to coordinate LCD/HCD gene expression programs in V. harveyi.
Derepression describes a transcriptional regulatory mechanism in which a negative regulator is cleared from a promoter to enable the recruitment and/or stabilization of RNAP and thereby stimulate transcription. H-NS has been the subject of studies investigating derepression mechanisms in Gram-negative bacteria. Similarly to V. cholerae ToxT, the positive regulator LeuO evicts H-NS from the DNA at the ompS1 promoter in Salmonella enterica (31). The work presented in this study demonstrates that LuxR is also capable of derepression of H-NS to directly activate gene expression. At one example promoter, the luxCDABE bioluminescence promoter, our experiments provide compelling evidence for a model in which H-NS oligomerizes to form a filament and/or bridge that represses gene expression. The V. harveyi H-NS L30P mutant allele fails to complement bioluminescence in the Δhns strain in V. harveyi, which strongly suggests that oligomerization (i.e. filament formation) is essential for H-NS to serve as a negative regulator at the luxCDABE promoter. Because the DNA-binding deficient LuxRR17C mutant was unable to displace H-NS in vitro, this suggests that LuxR evicts H-NS from DNA through competition for the same binding sites. Notably, because both LuxR and H-NS DNA binding sites are AT-rich sequences, it is unsurprising that they may compete for similar sites at a promoter.
Within the 506 and 302 peaks identified for LuxR and H-NS, respectively, 88 peaks overlapped and were proximal to an ORF (≤500 bp). The LuxR ChIP-seq profile previously published identified 1165 LuxR binding peaks (8). However, that previous experiment was performed with FLAG-luxR expressed from a multi-copy plasmid in at ΔluxR background, which is the likely reason for the increased number of LuxR binding peaks identified. Overall, the ChIP-seq data show that QS loci are regulated via the competitive binding dynamics of LuxR and H-NS (and likely other regulators). The function of LuxR at 13 of these promoters appears to be eviction of H-NS, and this results in activation of gene expression. The nucleoprotein bridges formed by H-NS may inhibit transcription initiation, trap elongating RNAP, and/or affect the topology of the DNA such that RNAP elongation is inhibited. Importantly, we observed that at many promoters, the two H-NS peaks were positioned such that the promoter was between the peaks. If these peaks indeed represent DNA–H-NS–DNA bridges, it is likely that even if RNAP can initiate transcription effectively, the bridge would block elongation. In addition, our finding that several H-NS peaks colocalize near genes repressed by LuxR presents the question: does LuxR also function alongside H-NS to repress gene expression? Additional experimentation will be necessary to properly address this question, but we speculate that LuxR may be sufficient to displace transcriptional activators that prevent H-NS from binding to these loci. In support of this theory is the finding that HapR and H-NS function together to ‘double-lock’ the vieSAB promoter in V. cholerae (32).
Perhaps the most surprising evidence that LuxR functions to oppose the action of H-NS globally is the observation that transcript levels of many QS genes are restored to wild-type levels in the ΔluxR Δhns strain. This finding implies that, at HCD, LuxR becomes dispensable when H-NS is not present for a subset of genes. It is important to note that, for at least luxCDABE, while transcript levels are restored to near wild-type levels in the ΔluxR Δhns strain, the bioluminescence phenotypic output remains very low. This result suggests that the bioluminescence genes are controlled both transcriptionally and post-transcriptionally. One possibility is that LuxR regulates the expression of a sRNA or an RNA-binding protein that is necessary for translation of these genes. Alternatively, a cis-acting RNA regulatory element such as a riboswitch or RNA thermometer may be regulating the translation of the luxCDABE mRNA. Further analyses will be required to test these possibilities.
From the collective work on the luxCDABE promoter, it is clear that these genes are highly regulated by multiple mechanisms, likely due to the cost of expressing these proteins at such high levels. At LCD, the ChIP-seq H-NS peak profile suggests that H-NS is bound to the luxCDABE promoter in a nucleoprotein bridge. LuxR protein levels are insufficient to stimulate transcription of the luxCDABE genes, even in the absence of hns. Thus, there are likely other regulatory mechanisms acting at the transcriptional and/or posttranscriptonal levels to repress expression at LCD. As population density increases and LuxR protein levels rise, LuxR binds to the binding sites in the luxCDABE promoter, and the H-NS nucleoprotein filament is destabilized resulting in the mass eviction of H-NS monomers/dimers from the DNA. Once the promoter has been purged of H-NS, it is probable that LuxR and other transcription factors, such as IHF, remodel promoter architecture to poise RNAP for transcription of the luxCDABE genes. LuxR interaction with RNAP has been established at the luxCDABE promoter (9), and likely is responsible for the ∼4-fold increase in transcription in the presence of LuxR. The observation that RNAP can bind to H-NS-bound DNA but is unable to displace H-NS is suggestive that RNAP recruitment is not the rate-limiting step for expression of the luxCDABE operon. Instead, it is likely that RNAP can bind to the luxCDABE promoter but remains incompetent for transcription. This hypothesis aligns well with the observation that RNAP can partially elongate through H-NS-DNA bridges but inevitably pauses due to the topological constraints of the H-NS–DNA bridge (13). Additionally, H-NS filaments can interact with promoter-bound RNAP to inhibit open complex formation (33). While the experiments performed in this study are insufficient to identify which mechanism(s) occurs at LuxR-dependent promoters, we speculate that the mechanism is probably promoter dependent.
We therefore propose an updated model for the activation of QS genes (such as the luxCDABE operon) in V. harveyi (Figure 6). At LCD, the amount of LuxR in the cell is inadequate to compete against H-NS for binding of QS promoter DNA. Under these conditions, H-NS remains bound to QS promoters as a nucleoprotein bridge, thereby silencing gene expression. As the population transitions to HCD, the intracellular concentration of LuxR increases ∼10-fold (34). Given the gradual accumulation of LuxR as well as the variation of binding site affinities, LuxR likely associates with high-affinity binding sites followed by moderate to low affinity sites within promoter regions. We propose that this incremental binding of LuxR to the promoter is part of a stepwise mechanism that allows for the systematic displacement of H-NS nucleoprotein bridges and filaments. Once H-NS bridges have been remodeled and H-NS displaced from the promoter, LuxR aids in the recruitment and/or stabilization of RNA polymerase to increase transcription levels (9). Thus, in a Δhns strain, LuxR is not required for transcription, but the presence of LuxR increases transcription by interaction with RNA polymerase.
Figure 6.
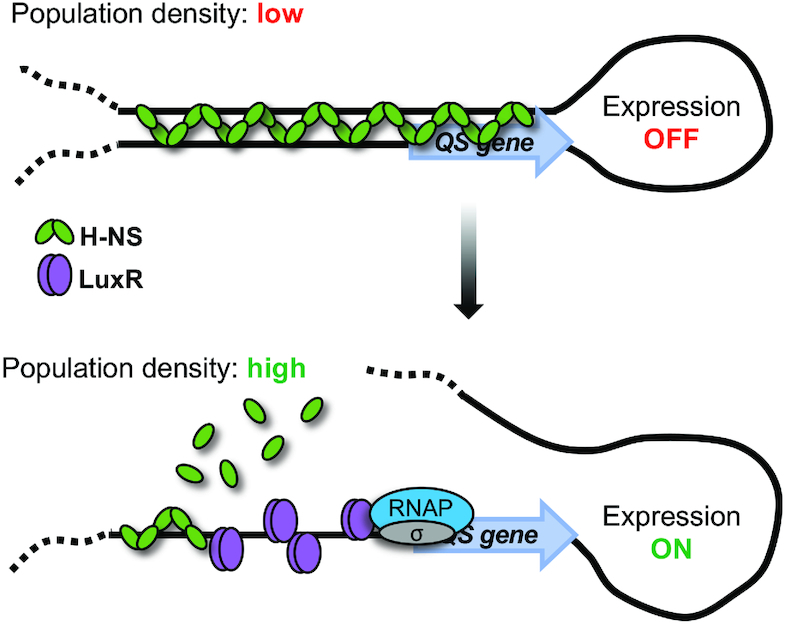
Proposed model for activation of H-NS-repressed QS loci. At low cell density, the levels of LuxR are low and thus H-NS outcompetes LuxR for binding DNA at QS promoters. Presumably in this state, the DNA-H-NS-DNA bridge blocks transcription and the gene is not expressed. As cells transition to high cell density, LuxR concentration in the cell increases, which enables LuxR to outcompete H-NS for binding of QS promoter DNA and disrupt inhibitory H-NS bridges and filaments. These events together with LuxR interaction with RNA polymerase alpha drives maximal transcription of QS genes.
It is well established that H-NS functions to silence genes that have been acquired through horizontal gene transfer (HGT) (35). Inappropriate expression of these genes leads to severe fitness disadvantages and thus H-NS is employed to buffer the transition of laterally acquired genes into proper regulatory networks (36). Although the luxCDABE (or other H-NS/LuxR co-regulated genes) do not show clear signs of originating from HGT based on GC-content analyses, it is possible that, at one point in the evolutionary past of V. harveyi, they were acquired via HGT and have since been fully incorporated into host regulatory circuits. It is interesting to speculate about nature of the interplay between LuxR and H-NS, which poses a slew of new questions. Over the course of the evolutionary timeline of V. harveyi, how many QS genes were acquired via HGT? DNA uptake is linked to QS in other Vibrios—is the regulatory scheme we present in this paper a conserved mechanism for incorporating exogenous DNA into the host genome once quorum has been achieved? Answers to these questions could explain the global extent of H-NS/LuxR co-regulation and shed light on its physiological significance.
Collectively, biochemical and genetic studies of LuxR/HapR proteins have made significant progress in determining the mechanism of transcriptional activation and repression by these master QS regulators. LuxR-type proteins function alongside IHF, directly oppose H-NS, associate with RNAP, and stimulate and/or block activities of other transcription factors (4,7,9,10,37–39). Importantly, though many of these mechanisms are conserved in Vibrio species and at multiple promoters, we surmise that the transcriptional control of each QS promoter is likely different. The luxCDABE promoter provides an easily monitored system for examining possible LuxR functions that can be tested at other promoters. Our observation that LuxR counter-silencing of H-NS is globally functional in V. harveyi and that similar observations have been reported in other QS bacteria alludes that H-NS counter-silencing via QS regulators could represent a conserved QS regulatory mechanism.
DATA AVAILABILITY
Datasets for RNA-seq and ChIP-seq have been deposited into NCBI GEO: GSE136050 and GSE136191
Token: izutkkqgvdkftcf
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136050
The link for a Jbrowse session (instead of UCSC) containing all uploaded sequences is: https://jbrowse.cgb.indiana.edu/index.html?data=vanKessel_GSF2248&loc=NC_009784.1%3A528561.565749&tracks=DNA%2CGenes%2CGSF2248-1_S19.Plus%2CGSF2248-2_S20.Plus%2CGSF2248-3_S21.Plus%2CGSF1697-WT1-input.Plus%2CGSF1697-WT2-ChIPed.Plus%2CGSF1697-WT3-ChIPed.Plus%2CGSF1697-HNS1-ChIPed.Plus%2CGSF1697-HNS2-ChIPed.Plus&highlight=
Github: All Perl scripts have been deposited in Github: https://github.com/Juliacvk/LuxR_H-NS
Experiments have all been conducted in accordance with ENCODE to the best of our ability given that no guidelines are listed for bacterial samples. ChIP-seq was validated by qPCR.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Chelsea Simpson, Evan Schneider, and Victoria Lydick for excellent technical assistance. We thank Jun Liu and Dave Miller at the Indiana University Center for Bioinformatics and Genomics for assistance with ChIP-seq and RNA-seq processing and analyses.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R35GM124698 to J.V.K., SC3GM118199 to L.C.M.C.]; National Science Foundation [DUE-1258366 to M.L.N.T.]. Funding for open access charge: NIH [R35GM124698].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mukherjee S., Bassler B.L.. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019; 17:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papenfort K., Bassler B.L.. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016; 14:576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ng W.L., Bassler B.L.. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009; 43:197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rutherford S.T., van Kessel J.C., Shao Y., Bassler B.L.. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 2011; 25:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Kessel J.C., Rutherford S.T., Shao Y., Utria A.F., Bassler B.L.. Individual and combined roles of the master regulators AphA and LuxR in control of the vibrio harveyi Quorum-Sensing regulon. J. Bacteriol. 2013; 195:436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ball A.S., Chaparian R.R., van Kessel J.C.. Quorum sensing gene regulation by LuxR/HapR Master regulators in vibrios. J. Bacteriol. 2017; 199:e00105-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaparian R.R., Olney S.G., Hustmyer C.M., Rowe-Magnus D.A., van Kessel J.C.. Integration host factor and LuxR synergistically bind DNA to coactivate quorum-sensing genes in Vibrio harveyi. Mol. Microbiol. 2016; 101:823–840. [DOI] [PubMed] [Google Scholar]
- 8. van Kessel J.C., Ulrich L.E., Zhulin I.B., Bassler B.L.. Analysis of activator and repressor functions reveals the requirements for transcriptional control by LuxR, the master regulator of quorum sensing in Vibrio harveyi. mBio. 2013; 4:e00378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ball A.S., van Kessel J.C.. The master quorum-sensing regulators LuxR/HapR directly interact with the alpha subunit of RNA polymerase to drive transcription activation in Vibrio harveyi and Vibrio cholerae. Mol. Microbiol. 2019; 111:1317–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeong H.S., Kim S.M., Lim M.S., Kim K.S., Choi S.H.. Direct interaction between quorum-sensing regulator SmcR and RNA polymerase is mediated by integration host factor to activate vvpE encoding elastase in Vibrio vulnificus. J. Biol. Chem. 2010; 285:9357–9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dorman C.J. H-NS, the genome sentinel. Nat. Rev. Microbiol. 2007; 5:157–161. [DOI] [PubMed] [Google Scholar]
- 12. Dame R.T., Noom M.C., Wuite G.J.L.. Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature. 2006; 444:387–390. [DOI] [PubMed] [Google Scholar]
- 13. Kotlajich M.V., Hron D.R., Boudreau B.A., Sun Z., Lyubchenko Y.L., Landick R.. Bridged filaments of histone-like nucleoid structuring protein pause RNA polymerase and aid termination in bacteria. eLife. 2015; 4:e04970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Valk R.A.d., Vreede J., Qin L., Moolenaar G.F., Hofmann A., Goosen N., Dame R.T.. Mechanism of environmentally driven conformational changes that modulate H-NS DNA bridging activity. eLife. 2017; 6:e27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Landick R., Wade J.T., Grainger D.C.. H-NS and RNA polymerase: a love-hate relationship. Curr. Opin. Microbiol. 2015; 24:53–59. [DOI] [PubMed] [Google Scholar]
- 16. Shen B.A., Landick R.. Transcription of bacterial chromatin. J. Mol. Biol. 2019; doi:10.1016/j.jmb.2019.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., Ayala J.C., Benitez J.A., Silva A.J.. RNA-seq analysis identifies new genes regulated by the histone-like nucleoid structuring protein (H-NS) affecting Vibrio cholerae virulence, stress response and chemotaxis. PLoS One. 2015; 10:e0118295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu R.R., DiRita V.J.. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 2002; 43:119–134. [DOI] [PubMed] [Google Scholar]
- 19. Lin B., Wang Z., Malanoski A.P., O’Grady E.A., Wimpee C.F., Vuddhakul V., Alves N. Jr, Thompson F.L., Gomez-Gil B., Vora G.J.. Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ. Microbiol. Rep. 2010; 2:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ke X., Miller L.C., Bassler B.L.. Determinants governing ligand specificity of the Vibrio harveyi LuxN quorum‐sensing receptor. Mol. Microbiol. 2015; 95:127–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Y., Foo Y.H., Winardhi R.S., Tang Q., Yan J., Kenney L.J.. Charged residues in the H-NS linker drive DNA binding and gene silencing in single cells. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grainger D.C. Structure and function of bacterial H-NS protein. Biochem. Soc. Trans. 2016; 44:1561–1569. [DOI] [PubMed] [Google Scholar]
- 23. Lim C.J., Lee S.Y., Kenney L.J., Yan J.. Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci. Rep. 2012; 2:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ueguchi C., Seto C., Suzuki T., Mizuno T.. Clarification of the dimerization domain and its functional significance for the Escherichia coli nucleoid protein H-NS. J. Mol. Biol. 1997; 274:145–151. [DOI] [PubMed] [Google Scholar]
- 25. Ayala J.C., Wang H., Silva A.J., Benitez J.A.. Repression by H-NS of genes required for the biosynthesis of the Vibrio cholerae biofilm matrix is modulated by the second messenger cyclic diguanylic acid. Mol. Microbiol. 2015; 97:630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swartzman E., Silverman M., Meighen E.A.. The luxR gene product of Vibrio harveyi is a transcriptional activator of the lux promoter. J. Bacteriol. 1992; 174:7490–7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang B., Blot N., Bouffartigues E., Buckle M., Geertz M., Gualerzi C.O., Mavathur R., Muskhelishvili G., Pon C.L., Rimsky S. et al.. High-affinity DNA binding sites for H-NS provide a molecular basis for selective silencing within proteobacterial genomes. Nucleic Acids Res. 2007; 35:6330–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouffartigues E., Buckle M., Badaut C., Travers A., Rimsky S.. H-NS cooperative binding to high-affinity sites in a regulatory element results in transcriptional silencing. Nat Struct Mol Biol. 2007; 14:441–448. [DOI] [PubMed] [Google Scholar]
- 30. Liu Y., Chen H., Kenney L.J., Yan J.. A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev. 2010; 24:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. la Cruz M., Fernández‐Mora M., Guadarrama C., Flores‐Valdez M., Bustamante V.H., Vázquez A., Calva E.. LeuO antagonizes H‐NS and StpA‐dependent repression in Salmonella enterica ompS1. Mol. Microbiol. 2007; 66:727–743. [DOI] [PubMed] [Google Scholar]
- 32. Ayala J.C., Wang H., Benitez J.A., Silva A.J.. Molecular basis for the differential expression of the global regulator VieA in Vibrio cholerae biotypes directed by H‐NS, LeuO and quorum sensing. Mol. Microbiol. 2018; 107:330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shin M., Lagda A.C., Lee J.W., Bhat A., Rhee J.H., Kim J.S., Takeyasu K., Choy H.E.. Gene silencing by H‐NS from distal DNA site. Mol. Microbiol. 2012; 86:707–719. [DOI] [PubMed] [Google Scholar]
- 34. Teng S.W., Wang Y., Tu K.C., Long T., Mehta P., Wingreen N.S., Bassler B.L., Ong N.P.. Measurement of the copy number of the master quorum-sensing regulator of a bacterial cell. Biophys. J. 2010; 98:2024–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dorman C.J. H-NS-like nucleoid-associated proteins, mobile genetic elements and horizontal gene transfer in bacteria. Plasmid. 2014; 75:1–11. [DOI] [PubMed] [Google Scholar]
- 36. Ali S.S., Soo J., Rao C., Leung A.S., Ngai D.H., Ensminger A.W., Navarre W.W.. Silencing by H-NS potentiated the evolution of Salmonella. PLoS Pathog. 2014; 10:e1004500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeong H.S., Lee M.H., Lee K.H., Park S.J., Choi S.H.. SmcR and cyclic AMP receptor protein coactivate Vibrio vulnificus vvpE encoding elastase through the RpoS-dependent promoter in a synergistic manner. J. Biol. Chem. 2003; 278:45072–45081. [DOI] [PubMed] [Google Scholar]
- 38. Kim H., Wen Y., Son J.S., Lee K.H., Kim K.S.. The Fur-Iron complex modulates the expression of the Quorum-Sensing master regulator SmcR to control the expression of virulence factors in vibrio vulnificus. Infect. Immun. 2013; 81:2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shao C.P., Lo H.R., Lin J.H., Hor L.I.. Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU. J. Bacteriol. 2011; 193:2557–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets for RNA-seq and ChIP-seq have been deposited into NCBI GEO: GSE136050 and GSE136191
Token: izutkkqgvdkftcf
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136050
The link for a Jbrowse session (instead of UCSC) containing all uploaded sequences is: https://jbrowse.cgb.indiana.edu/index.html?data=vanKessel_GSF2248&loc=NC_009784.1%3A528561.565749&tracks=DNA%2CGenes%2CGSF2248-1_S19.Plus%2CGSF2248-2_S20.Plus%2CGSF2248-3_S21.Plus%2CGSF1697-WT1-input.Plus%2CGSF1697-WT2-ChIPed.Plus%2CGSF1697-WT3-ChIPed.Plus%2CGSF1697-HNS1-ChIPed.Plus%2CGSF1697-HNS2-ChIPed.Plus&highlight=
Github: All Perl scripts have been deposited in Github: https://github.com/Juliacvk/LuxR_H-NS
Experiments have all been conducted in accordance with ENCODE to the best of our ability given that no guidelines are listed for bacterial samples. ChIP-seq was validated by qPCR.