


Abstract
Multiplex genetic assays can simultaneously test thousands of genetic variants for a property of interest. However, limitations of existing multiplex assay methods in cultured mammalian cells hinder the breadth, speed and scale of these experiments. Here, we describe a series of improvements that greatly enhance the capabilities of a Bxb1 recombinase-based landing pad system for conducting different types of multiplex genetic assays in various mammalian cell lines. We incorporate the landing pad into a lentiviral vector, easing the process of generating new landing pad cell lines. We also develop several new landing pad versions, including one where the Bxb1 recombinase is expressed from the landing pad itself, improving recombination efficiency more than 2-fold and permitting rapid prototyping of transgenic constructs. Other versions incorporate positive and negative selection markers that enable drug-based enrichment of recombinant cells, enabling the use of larger libraries and reducing costs. A version with dual convergent promoters allows enrichment of recombinant cells independent of transgene expression, permitting the assessment of libraries of transgenes that perturb cell growth and survival. Lastly, we demonstrate these improvements by assessing the effects of a combinatorial library of oncogenes and tumor suppressors on cell growth. Collectively, these advancements make multiplex genetic assays in diverse cultured cell lines easier, cheaper and more effective, facilitating future studies probing how proteins impact cell function, using transgenic variant libraries tested individually or in combination.
INTRODUCTION
Advancements in DNA sequencing technology have enabled multiplex genetic assays that harness sequencing as the readout. For example, multiplex assays of variant effects (MAVEs) interrogate thousands to millions of protein or nucleotide variants in the same pooled experiment (1). A key requirement of MAVEs is the physical linkage of genotype and phenotype, usually by requiring that each cell expresses one and only one variant. Additional characteristics such as stable variant expression over the course of the experiment, similar expression of variants in different cells and accurate quantification of variants by high-throughput sequencing, are other important considerations. However, existing strategies for protein variant expression, particularly in cultured mammalian cell lines, have limitations that hinder the widespread adoption of MAVEs and other multiplex genetic assays.
Current strategies for multiplex expression of protein variant libraries fall into four categories. Transient expression from plasmid dilution (2,3) is cumbersome and can yield noisy results. Low MOI lentiviral transduction is the most popular method for transgenic library expression (4,5), but pseudo-random genomic integration of lentiviral vectors introduces variability in transgene expression, and template switching during reverse transcription can scramble libraries (6,7). Saturation Genome Editing, which uses a pool of programmed oligonucleotides to introduce variant libraries through Cas9-triggered homology directed repair, enables evaluation of variants in their endogenous genomic context (8), but is currently limited to growth-based assays in the haploid chronic myeloid leukemia derived HAP-1 cell line. Moreover, variant expression cannot be temporally controlled. Notably, neither lentiviral transgene integration nor saturation genome editing are compatible with variant barcoding where short nucleotide barcodes mark each member of the variant library (9) to reduce sequencing costs.
A fourth strategy relies on directional DNA integration using the Bxb1 bacteriophage recombinase to introduce variants into a genomically integrated ‘landing pad’ locus. This landing pad approach has been used to facilitate transgene expression for cell reprogramming (10), synthetic biology circuit design (11), high titer antibody expression (12) and variant library expression (13–15). Despite these successes, the landing pad approach remains largely unoptimized for multiplex assays, limiting the types of experiments that can be performed. Thus, we present a series of improvements to the landing pad approach focused on increasing its utility for multiplex genetic assays. We incorporated the landing pad into a lentiviral vector, speeding up the process of generating landing pad cell lines. We increased the recombination rate, which can limit the library size, by including the Bxb1 recombinase in the landing pad. We replaced cumbersome and slow fluorescence-activated cell sorting (FACS)-based recombined cell enrichment using positive or negative drug selections. Lastly, because recombined cell enrichment and transgene expression were linked in the first generation landing pad, toxic transgenes rendered library-scale experiments intractable. We overcame this limitation by incorporating a second promoter, thereby unlinking transgene expression from recombined cell enrichment. We showcase these landing pad improvements by expressing a combinatorial library of oncogenes and tumor suppressors and evaluating their effects on cell proliferation. The improvements we present broaden the range of multiplex genetic assays that are possible with cultured mammalian cell lines, increasing the scope of biological problems that can be studied at high-throughput.
MATERIALS AND METHODS
Plasmid construction
dAAVS1-TetBxb1BFP_rtTA3G-2A-Blast, attB_3kGFP-IRES-mCherry (attB_3kGiM), attB_EGFP, attB-mCherry, attB-PTEN(WT)-IRES-mCherry, attB-PTEN(Y68H)-IRES-mCherry, attB-PTEN(C124S)-IRES-mCherry, attB_GFP-PTEN(WT)-IRES-mCherry-BlastR and attB-sGFP-PTEN-IRES-mCherry-P2A-bGFP were described previously (13,14). All plasmids were created using Gibson Assembly (16). pLenti-CMV-rtTA3G_Puro was created by combining amplicons from pLenti_CMV-rtTA3G_Blast (Addgene # 26429) amplified with KAM1285//KAM1288, and pCW-Cas9 (Addgene # 50661) amplified with KAM1284//KAM1287. dAAVS1_TetBxb1BFP_rtTA3G-2A-BxInt was created by combining amplicons from pCAG-NLS-Bxb1 (Addgene # 51271) amplified with KAM467//KAM469, and dAAVS1_TetBxb1BFP_rtTA3G amplified with KAM468//KAM466. pLenti_TetBxb1-BFP_rtTA3G-2A-Blast (LLP) was created by combining amplicons from dAAVS1-TetBxb1BFP_rtTA3G-2A-Blast amplified with KAM1163//KAM1166 and pLenti_CMV-rtTA3_Blast amplified with KAM1165//KAM1164. pLenti_TetBxb1-BFP-2A-BxInt_rtTA3G-2A-Blast (LLP-Int) was created by combining amplicons from LLP amplified with KAM1208//KAM1209 and dAAVS1_TetBxb1BFP_rtTA3G-2A-BxInt amplified with KAM1207//KAM1210. pLenti_TetBxb1-BFP-2A-BxInt-2A-Blast_rtTA3G (LLP-Int-Blast) was created by combining amplicons produced by LLP-Int amplified with KAM1935//KAM1936, KAM1937//KAM1938 and KAM1939//KAM1940. pLenti_TetBxb1-BFP-2A-BxInt-2A-Blast_rtTA3G (LLP-iCasp9) was created by combining amplicons produced by pMSCV-F-del Casp9.IRES.GFP (Addgene # 15567) amplified with KAM2217//KAM2218 and LLP-Int-Blast amplified with KAM2219//KAM2220. pLenti_TetBxb1-BFP_rEF1a_rtTA3G (LLP-Growth) was created by combining amplicons produced by cEF.tk-GFP (Addgene # 33308) amplified with KAM2153//KAM2154 and LLP amplified with KAM2155//KAM2156.
attB_HygroR-2A-mCherry, attB_PuroR-2A-mCherry, attB_Blast-2A-mCherry were created by combining an amplicon from attB_mCherry amplified with KAM1585//KAM1586, combined with pMK290_mAID-Clover-HygroR (Addgene # 72828) amplified with KAM1587//KAM1588, pLenti_CMV-rtTA3G_Puro amplified with KAM1589//KAM1590 and pLenti_CMV-rtTA3G_Blast amplified with KAM1591//KAM1592, respectively. attB_sGFP-PTEN-IRES-mCherry-P2A-HygroR, attB_sGFP-PTEN-IRES-mCherry-P2A-PuroR and attB_sGFP-PTEN-IRES-mCherry-P2A-BlastR were created by combining an amplicon from attB-sGFP-PTEN-HA-IRES-mCherry-P2A-bGFP amplified with KAM1646//KAM1647, combined with pMK290_mAID-Clover-HygroR amplified with KAM1648//KAM1649, pLenti_CMV-rtTA3G_Puro amplified with KAM1650//KAM1651 and pLenti_CMV-rtTA3G_Blast amplified with KAM1652//KAM1653, respectively. revIRES-mCherry_attB_3kGFP (rIM_attB_GFP) was created by combining amplicons produced by attB_3kGiM amplified with KAM2179//KAM2180 and attB-EGFP amplified with KAM2181//KAM2182. revIRES-PuroR-2A-mCherry_attB_3kGFP (rIP2M_attB_GFP) was created by combining amplicons from rIM_attB_GFP amplified with KAM1477//KAM2227, and attB_PuroR-2A-mCherry amplified with KAM2226//KAM010.
attB_sGFP-VHL-IRES-mCherry-2A-bGFP (attB_sViM2b) was created by amplifying the VHL open reading from HA-VHL wt-pBabe-puro (Addgene # 19234) using KAM799//KAM802, and combining it with attB_sGFP-PTEN-IRES-mCherry-2A-bGFP amplified with KAM800//KAM801, and attB_sViM2b-L188Q was subsequently created by amplifying with KAM938//KAM939. attB_VHL-sGFP-IRES-mCherry-2A-bGFP (attB_VsiM2b) was created by amplifying the VHL open reading from HA-VHL wt-pBabe-puro using KAM885//KAM886, and combining it with attB_PTEN-SGFP-IRES-mCherry-2A-bGFP amplified with KAM883//KAM884, and attB_VsiM2b-L188Q was subsequently created by amplifying with KAM938//KAM939. attB_TP53-IRES-mCherry was created by amplifying the TP53 open reading frame from the pcDNA3 p53 WT plasmid (Addgene # 69003) using primers KAM1318//KAM1319, and inserting it into attB_3kGiM N-terminal of GFP using primers KAM1316//KAM1317. The GFP fusion was then removed to create attB_TP53-IRES-mCherry using primers KAM1963//KAM1964, and the TP53-R273H variant was created using primers KAM1963, KAM1964, KAM1965 and KAM1966. attB-RB1-IRES-mCherry was created by amplifying the RB1 open reading frame from plasmid pSG5L_HA_RB (Addgene # 10720) using KAM1320//KAM1321, and inserting it into attB_3kGiM N-terminal of GFP using primers KAM1316//KAM1317. The GFP fusion was then removed to create attB_RB1-IRES-mCherry using primers KAM1967//KAM1968. attB_EGFP-p21 was created by amplifying the p21 open reading frame from a gBlock (IDT; KAM1703) using KAM1740//KAM1741, and adding it into attB_EGFP-Bgl2 (13) which was amplified with KAM1742//KAM1743. attB_ecDHFR-3kGFP-IRES-mCherry was created by amplifying the ecDHFR open reading frame from a gBlock (IDT; KAM1704) using KAM1736//KAM1737, and combining it with attB_3kGiM amplified with KAM1738//KAM1739. attB_p21-IRES-mCherry was created by amplifying attB_EGFP-p21 with KAM1793//KAM1794, and combining it with attB_ecDHFR-3kGFP-IRES-mCherry amplified with KAM1795//KAM1889. attB_BAX-IRES-mCherry and attB_AKT(E17K)-IRES-mCherry were created by amplifying attB_PTEN(WT)-IRES-mCherry with KAM2068//KAM2069, and combined with hBax_C3-EGFP (Addgene # 19741) and pDONR223_AKT1_p.E17K (Addgene # 81459), respectively. attB_p16(INK4A)-IRES-mCherry and attB_p14(ARF)-IRES-mCherry were created by amplifying the open reading frames out of a gBlock (IDT; KAM1971) using KAM2014//KAM2015 and KAM2016//KAM2017, respectively, and combining it with an amplicon from attB_PTEN(WT)-IRES-mCherry using KAM2012 and KAM2013. attB_HygroR-P2A-Puro-3kGFP-TEVcs-PTEN(Y68H)-IRES-mCherry was created by combining amplicons from pMK290_mAID-Clover-HygroR amplified with KAM1550//KAM1553, attB-PTEN(Y68H)-IRES-mCherry amplified with KAM1556//KAM1551, pLenti-TetBxb1BFP-rtTA3G-2A-Puro amplified with KAM1552//KAM1555, and attB_3kGiM amplified with KAM1554//KAM1557. attB_mAID-GFP-PTEN(WT)-IRES-mCherry-BlastR was created by combining amplicons from pMK290_mAID-Clover-HygroR amplified with KAM1713//KAM1714 and attB_GFP-PTEN(WT)-IRES-mCherry-BlastR amplified with KAM1715//KAM1716. All plasmids were extracted using a Qiagen miniprep kit, though libraries and plasmids that were repeatedly used were extracted with a Qiagen midiprep kit. Primers used for molecular cloning are listed in Supplementary Table S1.
Plasmid library construction and high-throughput sequencing
In the first step, the positions 1 and 2 transgenes were barcoded through amplification, with the termini of the position 1 amplicons harboring identical sequences to the termini of the position 2 amplicons, permitting Gibson assembly of the fragments (Supplementary Figure S1a, top). A total of 40 ng of plasmid DNA was mixed with 0.333 μM of forward primer, 0.333 μM of reverse primer and 2× Kapa Hifi added as half the volume (Supplementary Table S2). The mixtures were cycled 12 times in a thermocycler. A total of 1 μl of DPNI enzyme (20 units) was added to each tube, and incubated at 37°C for 2 h. A total of 15 μl of product was mixed with 3 μl of (6× gel loading dye), and run on a 0.8% agarose gel in Tris-borate-EDTA buffer. Bands were cut, position 1 bands and position 2 bands were pooled separately, and extracted using the Qiagen Gel Extraction kit. Position 1 and position 2 DNA were eluted in 10 μl nuclease-free water. A total of 5 μl of each eluate were mixed together with 10 μl 2× Gibson mix and incubated at 50°C for 30 min. After incubation, the product was eluted in 5 μl, and 4 μl of library was electroporated into 10-beta cells (NEB) using 0.1 μm cuvettes (Bio-Rad) according to manufacturer instructions. Approximately 2.16 million colony forming units of transformants were grown in 200 ml Luria Broth overnight, and plasmid DNA was extracted with a Qiagen Midiprep Kit (Qiagen). In the second step, polymerase chain reaction (PCR) and Gibson assembly was used to insert an IRES-Puromcyin-2A-mCherry cassette in the reverse orientation 5′ of the Bxb1 attB site, so the library could be selected in the absence of Dox induction (Supplementary Figure S1a, bottom). A total of 320 ng of library DNA was mixed with 2× Kapa Hifi Readymix and a final concentration of 0.333 μM KAM2311 and 0.333 μM KAM2312, with 30 μl distributed into eight replicate tubes. A total of 160 ng of rIP2M_attB_GFP plasmid was mixed with 2× Kapa Hifi and a final concentration of 0.333 μM KAM2313 and 0.333 μM KAM2314, with 30 μl distributed into four replicate tubes. Both sets of tubes were amplified over 18 cycles. A total of 1 μl (20 units) of DPNI enzyme was added to each tube, and incubated at 37°C for 2 h. After incubation, the rIP2M_attB_GFP amplicon was gel extracted from a 0.8% agarose gel and eluted in 10 μl, whereas the library amplicon was cleaned and concentrated (Zymo), and eluted in 5 μl. A total of 4 μl of each were mixed together with 8 μl of 2× Gibson mix and incubated at 50°C for 30 min. The product was clean-and-concentrated, eluted in 5 μl, and electroporated in 10-beta cells using 0.1 μm cuvettes (Bio-Rad) according to manufacturer's instructions. Approximately 0.5 million colony forming units were amplified in 200 ml Luria Broth overnight, and extracted using the Qiagen Midiprep kit. Sanger sequencing of eight colonies revealed six to be intended library members. This library was called attB-TSPCR.
The barcode region within the 10 ng of plasmid DNA was amplified with 14 cycles of PCR in a 50 μl NEB Q5 polymerase reaction (New England Biolabs) using a Bio Rad mini opticon and Sybr Green I. Cycling was stopped at 14 cycles during low to mid log phase amplification. The amplicons were mixed equally based on their final relative fluorescence units as measured by the mini opticon and run on a 1% TBE-agarose gel. The product band was excised and extracted using the Freeze and Squeeze Kit according to the manufacturer's instructions (Bio-Rad). Gel-purified amplicons were quantified by qPCR using the Illumina Library Quantification Kit (Kapa Biosystems). Then, indexed amplicons were pooled and sequenced on a Nextseq (NextSeq 75 High V2 kit, Illumina, for primers see Supplementary Data Set 1).
The library contained ∼400 000 unique plasmid barcodes, which we were able to quantitate with reasonable accuracy across technical replicate amplification and sequencing reactions (Pearson's r: 0.9; Supplementary Figure S1b). When plasmid identifiers were combined for each of the 216 observed combinations out of the 224 possible (Supplementary Table S3), we observed precise quantitation across the technical replicates (Pearson's r: 1; Supplementary Figure S1b). Notably, there was a strong correlation between the frequency of each transgene identifier combination within the plasmid library with the number of times that combination was associated with a unique plasmid identifier combination (Supplementary Figure S1d; Pearson's r: 0.99), showing there was little selection of each plasmid combination during growth in bacteria.
Cell culture, plasmid transfection and landing pad clone generation
All cell culture reagents were purchased from ThermoFisher Scientific unless otherwise noted. All cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (D10). Cells were passaged by detachment with Trypsin-Ethylenediaminetetraacetic acid 0.25%.
Lentiviral vectors were produced by transfection of 250 000 HEK 293T cells in a 6-well with 500 ng pMD-VSV-G (Addgene #12259), 1750 ng PsPax2 (Addgene # 12260) and 1750 ng of the LLP or derivative vector template, using 6 μg of Fugene 6 (Promega). Media was changed the next day, and the supernatant collected over the next 72 h. The collected media was pelleted at 300 × g for 4 min, and the supernatant was passed through a 0.45 μm filter. HEK293T cells, A549 cells, NIH3T3 cells and HT-29 cells were incubated with various dilutions of lentiviral supernatant ranging from 100 μl to 4 ml, and transduced cultures were visually confirmed to have been transduced at MOI clearly less than one. Cells were then incubated with 10 μg/ml of Blasticidin. After a week of selection, cells were assessed with a BD FACS Aria II for fluorescence, and individual BFP+ cells were sorted into separate wells of 96-well plates. Cell clones that grew out were transferred into a 24-well plate and subsequently transferred to 6-well plates for analysis. Cells were induced to express off of the Tet-inducible promoter by adding doxycycline to a final concentration of 2 μg/ml (D10-dox). Cells were also maintained in D10-dox unless otherwise noted.
HEK 293T-based cells were recombined by transfecting 250 000 HEK 293T cells in a 6-well plate, with 1500 ng of pCAG-NLS-Bxb1, and 1500 ng of either individual or a mixture of attB recombination plasmids, incubated with 6 μg of Fugene 6 reagent in doxycycline-free media. Two or more days following transfection, the media was changed to D10-dox media, and cells were assessed for recombination at least 2 days after. For recombination of A549 cells, 150 000 cells plated in a 6-well with 2 ml of media were transfected with a mixture of 3.75 μl Lipofectamine 3000 diluted in 125 μl OPTI-MEM, and 5 μg DNA with 10 μl P3000 reagent diluted in 125 μl OPTI-MEM. For recombination of 3T3 cells, 150 000 cells plated in a 6-well with 2 ml of media were transfected with a mixture of 7.5 μl Lipofectamine 3000 diluted in 125 μl OPTI-MEM and 5 μg DNA with 10 μl P3000 reagent diluted in 125 μl OPTI-MEM. Recombined cells were positively selected by growing the cells for a week in D10-dox media supplemented with the indicated amounts of hygromycin, puromycin or blasticidin (Invivogen). Cells were split and media replenished every 3 days. Recombined cells were negatively selected with the addition of 10 nM AP1903 / Rimiducid (MedChemExpress). Cells were maintained in AP1903 for 2 days. Sodium Butyrate (Sigma) was dissolved as a 0.5 M stock solution in H2O.
Flow cytometry and fluorescence-activated cell sorting (FACS)
Cells were detached with trypsin, and resuspended in PBS containing 5% serum. Analytical flow cytometry was performed with a BD LSRII flow cytometer. Blue fluorescent protein (BFP) was excited with a 405 nm laser, and emitted light was collected after passing through a 450/50 nm band pass filter. EGFP was excited with a 488 nm laser, and emitted light was collected after passing through 505 nm long pass and 530/30 nm band pass filters. mCherry was excited with a 561 nm laser, and emitted light was collected after passing through 595 nm long pass and 610/20 nm band pass filters. Cells were sorted using a BD Aria III FACS machine equipped with either an 85 or 100 μm nozzle. Filter sets were identical to those described for the LRSII, with the exception of mCherry emission which was detected using 600 nm long pass and 610/20 band pass filters. Before analysis of fluorescence, live, single cells were gated using FSC-A and SSC-A (for live cells) and FSC-A and FSC-H (for single cells). For landing pad clone isolation, individual BFP+ cells were directly sorted into wells of 96-well plates.
Library recombination and high-throughput sequencing
Four million HEK 293T LLP-rEF1α Clone 21 cells were transfected with a 1:1 mixture of pCAG-NLS-Bxb1 and the attB-TSPCR plasmid using Fugene 6, as described above, in the absence of doxycycline. The cells were then passaged for 2 weeks in the absence of doxycycline but in the presence of 0.5 ng/μl puromycin. Cells were then induced through the addition of 2 μg/μl doxycycline, and cells were collected the next day to serve as the initial timepoint. Cells were maintained in dox-containing media for up to 2 weeks, with additional cells collected, pelleted at 300 × g for 4 min and scored at −20*C.
Genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer's instructions with the following modification: proteinase K digestion was amended with a final concentration of 0.1 mU RNaseA (Sigma-Aldrich) extended to 30 min at 56°C. Upon DNA extraction, PCR was used to amplify the region of interest using 20 μl of DNeasy eluate (∼1000–3000 ng template) in a 50 μl NEB Q5 polymerase reaction (New England Biolabs) using a Bio Rad mini opticon and Sybr Green I. Cycling was stopped at 25 cycles during low to mid log phase amplification. The amplicons were mixed equally based on their final relative fluorescence units as measured by the mini opticon and run on a 1% TBE-agarose gel. The product band was excised and extracted using the Freeze and Squeeze Kit according to the manufacturer's instructions (Bio-Rad). Gel-purified amplicons were quantified by qPCR using the Illumina Library Quantification Kit (Kapa Biosystems). Then, indexed amplicons were pooled and sequenced on a Nextseq (NextSeq 75 High V2 kit, Illumina, for primers see Supplementary Table S2).
Plasmid identifier combinations were assessed at the initial time point to determine library diversity in the enriched recombined cells. We used a frequency filter of 1 × 10−5 to separate bonafide recombined barcodes from sequencing errors. We observed as many as 8741 unique barcodes at the initial time point (median 2820). However, these barcodes corresponded to 202 of the 216 possible transgene combinations observed in the plasmid library. Notably, technical replicates were precise at this combinatorial transgene identifier level, with a Pearson's r of 0.99 at each of four time points taken within the first replicate. Fastq sequencing files containing only the transgene identifier combinations from the various time points across the eight replicate experiments were entered into Enrich2, and slopes for the enrichment or depletion of each combined transgene was calculated for each replicate experiment (17).
Data analysis
Data analysis was performed using version 1.2.1335 of RStudio, with the exception of flow cytometry data, which was analyzed using version 10.5.3 of FlowJo. An R Markdown file containing code for the analyses is provided as a Supplementary File, and the data and analysis files have been uploaded to the Fowler Lab GitHub repository.
RESULTS
Lentiviral landing pads expedite cell engineering
We previously developed the landing pad system for large scale, single-copy expression of variant libraries in human-derived cultured cells (13). Homology directed repair was used to insert a synthetic landing pad sequence encoding a promoter preceding a Bxb1 attP recombination sequence at the AAVS1 locus. These landing pad cells can be used to recombine in plasmids encoding the complementary Bxb1 attB site (Figure 1A). Successful recombination switches expression from the initial transgene encoded by the landing pad to a new transgene encoded by the recombination plasmid (Figure 1A). This process occurs independently in each cell, allowing expression of a large library of protein variants when performed en masse.
Figure 1.
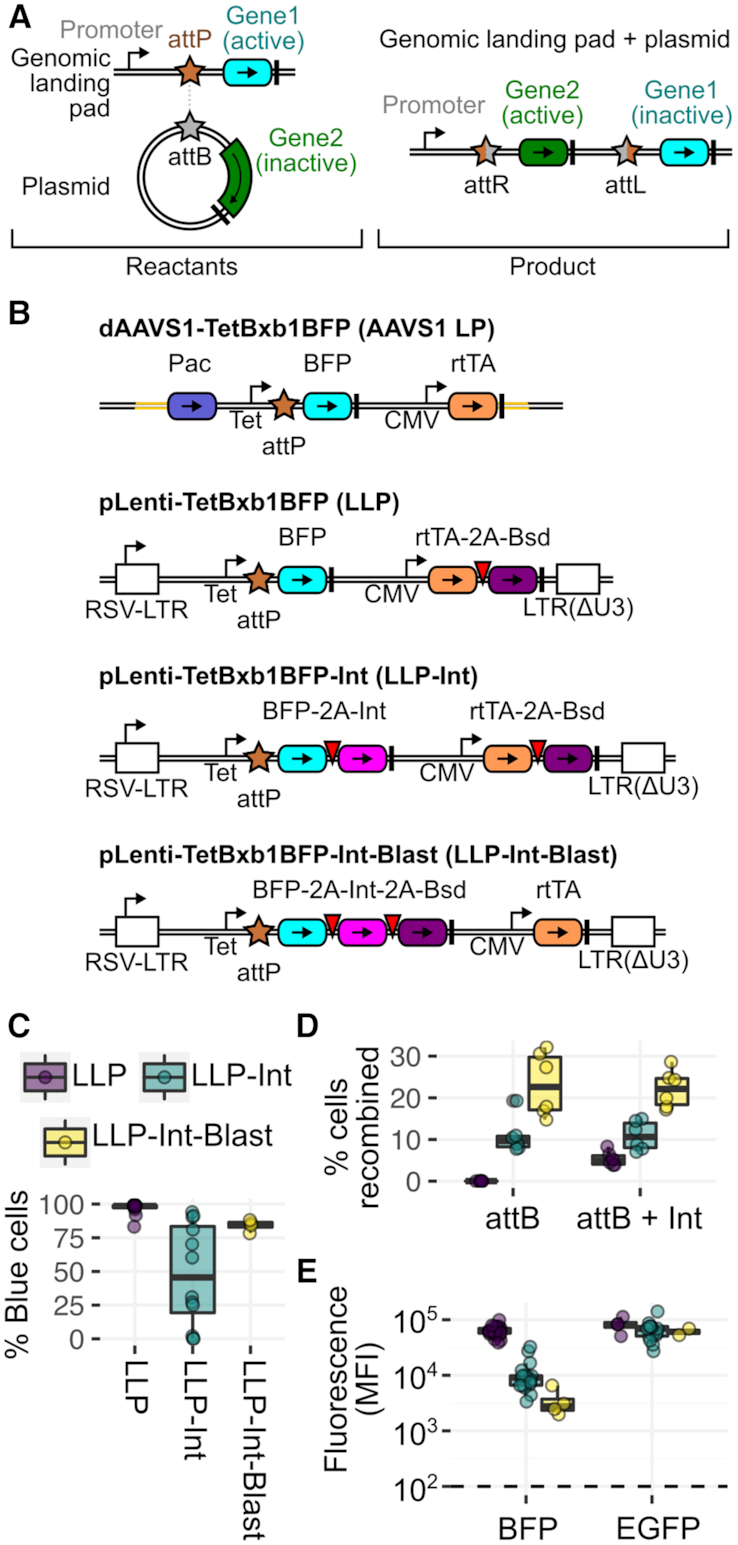
Lentiviral Landing Pad (LLP) Derivatives. (A) Schematic of the integration of an attB recombination plasmid into a genomically integrated landing pad. Coding regions are colored boxes, promoters are stemmed arrows, terminator sequences are thick black lines, Bxb1 recombination sequences are stars. (B) LLP constructs, with the original landing pad (AAVS1 LP) for comparison. Components are colored according to panel A, and viral 2A-like sequences are red inverted triangles. Pac is the puromycin resistance gene. Bsd is the blasticidin resistance gene. rtTA is the reverse Tet transactivator. LTR is a lentiviral long terminal repeat. (C) Percentage of BFP positive cells in 293T clones generated with each LLP construct. (D) Recombination rate comparisons of 293T AAVS1 LP clone 4, 293T LLP-Int clone 6 and 293T LLP-Int-Blast clone 2, with a mixture of attB-EGFP and attB-mCherry recombination plasmids without exogenous Bxb1 recombinase expression plasmid (left), or with Bxb1 recombinase expression plasmid included (right). (E) Median fluorescence intensity of each clone from panel C. Left panel shows blue fluorescence before recombination. Right panel shows green fluorescence after recombination of an attB-EGFP recombination plasmid (right). The dotted line represents background fluorescence in parental HEK-293T cells. Panels D and E are a result of at least six replicates.
However, our initial approach requires months to isolate, expand and characterize each new clonal landing pad line. Many cell types are poorly transfectable, greatly hindering the already inefficient process of homology-directed repair to install the landing pad (13). Additionally, the cell engineering and clonal expansion process took one to two months, largely waiting for the dilution of the homology repair template plasmid. Moreover tedious genotyping of a large number of candidate cell lines was required to identify lines with a single integrated landing pad (13). Both problems can be circumvented by using low multiplicity of infection transduction with VSV-G pseudotyped lentiviral vectors, which pseudo-randomly integrate recombinant DNA into the genomes of almost any cultured cell line. Thus, we developed a series of lentiviral landing pad (LLP) constructs (Figure 1B).
Similar to a landing pad format recently used for multi-copy antibody overexpression (12), we transferred the sequence encoding our original landing pad (AAVS1 LP) into a plasmid encoding a self-inactivating lentiviral vector to make our initial LLP construct (Figure 1B). LLP virus was transduced into the human HEK 293T cell line, murine NIH3T3 fibroblast cell line, A549 lung cancer cell line, HT-29 colon cancer cell line, and PC-3 prostate cancer cell line at low multiplicity of infection. Cells successfully transduced by the LLP lentiviral vector, identifiable owing to a BFP marker, were sorted by FACS and single cells were grown out over a period of ∼2 weeks. For each cell line, up to 12 clones were assessed for a single, high-intensity blue fluorescence peak using flow cytometry, and at least three clones were selected for downstream analysis for recombination. To ensure that the resulting candidate landing pad clonal cell lines did not contain multiple integrated copies of the landing pad, each line was transfected with a mixture of attB-EGFP and attB-mCherry recombination plasmids, along with a plasmid encoding the Bxb1 recombinase. Here, cell lines with multiple landing pads would yield GFP/mCherry double positive recombinant cells (13). Clonal lines harboring a single landing pad were derived for each parental cell line, with the HEK 293T, NIH 3T3 and A549 lines yielding appreciable recombinant cells (Supplementary Figure S2a), while no recombinants were observed with the HT-29 or PC-3 clones (data not shown). The fraction of blue fluorescent A549 landing pad cells diminished with continued passaging, though this apparent silencing of the landing pad could be partially reversed by growth in blasticidin or addition of the histone deacetylase inhibitor sodium butyrate (18) (Supplementary Figure S2b).
The rapid cell engineering possible with LLPs allowed us to design and test derivatives of the original landing pad format with additional features (Figure 1B). Previously, we demonstrated that expression of the Bxb1 recombinase in cells prior to transfection with the Bxb1 attB-containing recombination plasmid boosted recombination rates (13). Thus, we created LLP-Int, a landing pad encoding human codon-optimized Bxb1 recombinase C-terminally linked to BFP with a parechovirus 2A-like translational stop-start sequence (19) (Figure 1B). LLP-Int allows for doxycycline (Dox) inducible expression of the Bxb1 recombinase until it catalyzes integration of a transfected Bxb1 attB-containing recombination plasmid, terminating Bxb1 expression. To minimize landing pad silencing, we also developed LLP-Int-Blast, where the Bxb1 recombinase is also 2A-linked with the blasticidin resistance gene, Bsd, allowing selection of Bxb1 recombinase-retaining cells during clonal expansion in the presence of the blasticidin antibiotic (LLP-Int-Blast; Figure 1B). HEK293T cells transduced by LLP-Int or LLP-Int-Blast were compared to those previously transduced by LLP, which were included as a control. All transductions yielded polyclonal populations of BFP+ cells, which were sorted and expanded to obtain clonal lines. Notably, all twelve LLP lines were highly BFP positive following expansion, whereas many LLP-Int lines exhibited appreciable loss of BFP expression (Figure 1C). However, growth of LLP-Int-Blast cells in 10 ug/mL blasticidin enabled effective selection against silenced cells and all four expanded clones were BFP positive (Figure 1C).
To determine whether Bxb1 integrase expression from the landing pad enhanced recombination efficiency, we compared the original 293T AAVS1 LP Clone 4 cells (13), 293T LLP-Int Clone 6 cells, and 293T LLP-Int-Blast Clone 2 cells. As expected, 293T AAVS1 LP Clone 4 cells did not recombine in the absence of transfected Bxb1 integrase expression vector, while the Bxb1 expressed from the landing pad in 293T LLP-Int and LLP-Int-Blast cell lines drove recombination (Figure 1D). In fact, landing pad-based Bxb1 expression enhanced recombination by 2- and 4-fold compared to transfection of a Bxb1 expression plasmid (Figure 1D).
LLP-Int or LLP-Int-Blast cells also sped the recovery of recombined cells by FACS. Both LLP and LLP-Int cells recombined with an mCherry marker rapidly become positive for red fluorescence. However, the corresponding decrease in blue fluorescence, also used to isolate recombined cells, is much more rapid in LLP-Int cells (Supplementary Figure S2c and d). This is because the 293T LLP-Int and LLP-Int-Blast cell lines exhibited decreased steady-state blue fluorescence (Figure 1E), likely due to decreased overall translation of their longer BFP-2A-Bxb1 transgenes. Expression of recombined EGFP in these cell lines was the same as in LLP cells, supporting this notion (Figure 1E). Owing to this lower steady-state BFP expression, 293T LLP-Int-Blast cells became BFP negative after only 7 days whereas LLP cells required more than 11 days to become BFP negative (Supplementary Figure S2c and d). Thus, compared to our original landing pad configuration, the LLP-Int-Blast landing pad exhibited a much higher recombination rate and faster BFP decay.
Positive and negative drug selection to enrich recombinant cells
Despite improved recombination rates, library-based experiments with the landing pad still require enrichment of recombined cells. The first version of the landing pad used FACS to enrich for recombined cells, which was time and resource intensive. For example, adequate coverage of an ∼10 000 member library may require isolating ∼400 000 recombined cells. At an ∼5% recombination rate and sorting ∼2 000 cells per second, ∼2 h of sorting time is required. Larger libraries or cell lines with poor viability require isolating even more recombined cells, which is impractical by FACS. Positive antibiotic selection can enrich for recombined stem cells (10), synthetic circuits (11) or variant libraries (15) in related systems. We engineered a panel of both positive and negative drug selectable markers into the landing pad to facilitate enrichment of recombined cells without using FACS.
For positive selection of recombined cells, we developed a series of attB recombination plasmids that, upon recombination into the landing pad, express the Escherichia coli-derived hph transgene conferring hygromycin resistance (20) the Aspergillus terreus-derived bsd transgene conferring blasticidin resistance (21), or the Streptomyces alboniger-derived pac transgene conferring puromycin resistance (22) (Figure 2A). These attB recombination plasmids were transfected into 293T AAVS1 LP Clone 4 cells or 293T LLP-Int Clone 6 cells. The transfected cells were divided into wells with a range of concentrations of the appropriate antibiotic, and the enrichment of recombined cells in each population was ascertained using flow cytometry. Unrecombined 293T AAVS1 LP Clone 4 cells were still largely puromycin sensitive despite the presence of the pac puromycin resistance gene driven by the endogenous PPP1R12C promoter at the AAVS1 locus (23) (Figure 2B, top and Supplementary Figure S3a, top). In contrast, unrecombined 293T LLP-Int Clone 6 cells were strongly blasticidin resistant as these cells already contain a blasticidin resistance cassette driven by a CMV promoter in the landing pad (Figure 2B, bottom and Supplementary Figure S3a, bottom). Aside from this exception, strong enrichment of recombined cells was observed with all three antibiotics. Puromycin selection was most effective in both cases (Figure 2B and Supplementary Figure S3a).
Figure 2.

Positive or Negative Drug-Based Selection for Recombined Cells. (A) Schematics of two recombination plasmid formats for expressing drug resistance genes. The gray box denotes the coding region for a transgene of interest (PTEN in this analysis). (B) Percentage of red fluorescent cells in mixed populations recombined with the cap-dependent antibiotic resistance vectors, after selection for four or more days in the presence of the indicated concentrations of each drug. Results are an average of three biological replicate experiments, with error bars denoting standard deviation. (C) Schematic of the iCasp9-based negative selection LLP construct. DNA elements are represented in the same manner as Figure 1A. (D) Representative flow cytometry plots of attB-EGFP recombined 293T LLP-iCasp9-Blast Clone 4 cells, before and after negative selection with AP1903. (E) Percent BFP- but GFP+ or mCherry+ positive cells, indicative of recombination, before and after selection with 10 nM AP1903, over eight replicates in 293T LLP-iCasp9-Blast Clone 12.
For negative selection of unrecombined cells, we developed LLP-iCasp9-Blast, a LLP derivative based on the inducible caspase, iCasp9 (Figure 2C). iCasp9 is a synthetic fusion between Caspase 9 and the inducible dimerization domain of FKBP1A variant F36V (24). Caspase dimerization is triggered by the small molecule AP1903 (Rimiducid), causing rapid cell death through apoptosis. Using lentiviral transduction of HEK-293T cells, we generated multiple clonal lines containing LLP-iCasp9-Blast. 293T LLP-iCasp9-Blast cells recombined with attB-EGFP were enriched to ∼75% after 24 h of AP1903 treatment (Figure 2D and E; Supplementary Figure S3b). This selection can be performed shortly after recombination, as existing iCasp9 protein degrades and cells become insensitive to AP1903 2 days after iCasp9 transcription is turned off (Supplementary Figure S3c). Thus, the iCasp9-Blast landing pad enables rapid enrichment of recombinant cells. Together, the antibiotic resistance attB recombination plasmids and LLP-iCasp9-Blast landing pad allow for either positive or negative drug selection to enrich for recombined cells.
A dual-promoter landing pad for toxic transgene expression
In our original landing pad system, expression of transgenes that negatively impact cell growth complicate isolation of recombinant cells. For example, recombined 293T AAVS1 LP Clone4 cells expressing CDKN1A, which encodes the cell-cycle arresting protein p21-WAF (25), quickly depleted from the culture upon induction of expression (half life ∼1.25 days), while cells expressing GFP or mCherry alone did not (half life ∼12 days) (Supplementary Figure S4a and b). This rapid depletion of recombined cells would make multiplex genetic assays characterizing CDKN1A, or any other toxic transgene, challenging because recombined cells would be difficult to enrich.
To surmount this problem, we designed another landing pad, LLP-rEF1α, which uses dual promoters that converge on the recombination site to unlink recombined cell selection from transgene expression. Along with the original Tet promoter, this landing pad also encodes a constitutive EF1α promoter situated in the reverse orientation (Figure 3A). This second promoter allows recombination of a promoterless rEF1α-compatible attB recombination plasmid where a transgene is integrated downstream of the Tet inducible promoter and a selection marker is integrated downstream of the EF1α promoter. Transcription of the selection marker is facilitated by an IRES sequence.
Figure 3.
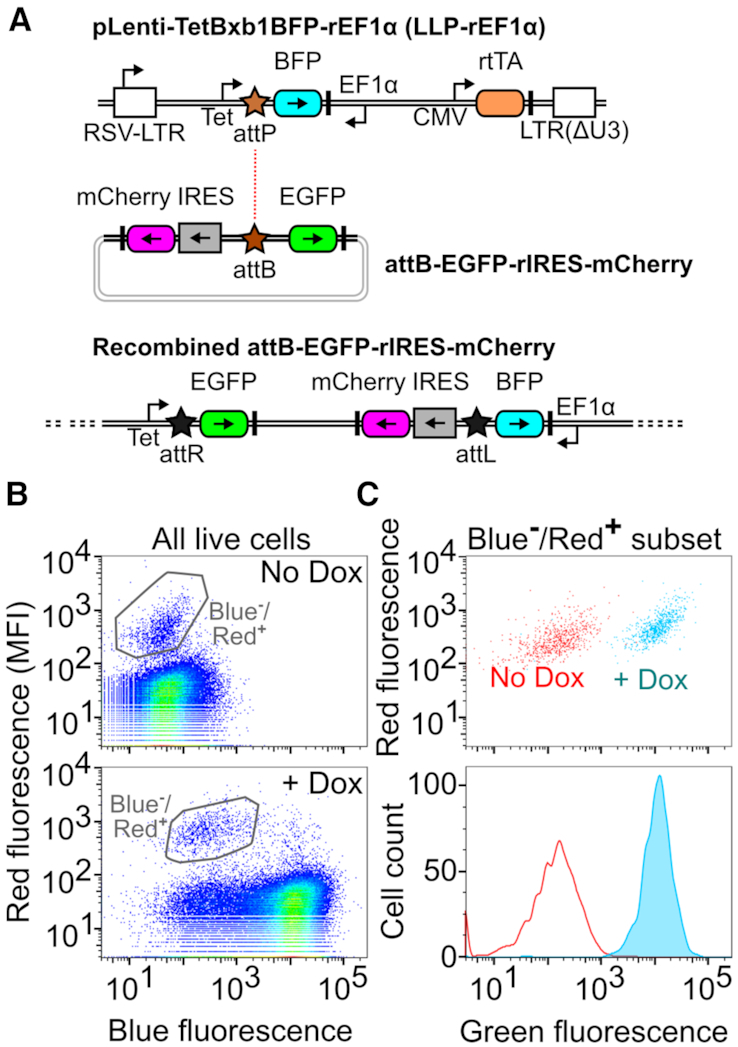
A Landing Pad Compatible with Toxic Transgenes. (A) Schematic of the toxic transgene-compatible LLP construct, the corresponding attB recombination vector and an integrated version of this landing pad after insertion of the recombination vector. DNA elements are represented in the same manner as Figure 1A. (B) Representative flow cytometry scatter plots of growth-compatible 293T LLP-rEF1α cells before (top) or after induction with dox (bottom). (C) Scatter plot (top) and smoothed histogram (bottom) of the gated populations in panel B.
Recombination of an attB rEF1α-compatible recombination plasmid encoding an EGFP transgene and a mCherry selection marker in these cells yielded a population of dim red fluorescent recombinant cells (Figure 3B). As expected, these recombinant cells could be induced to fluoresce green upon doxycycline addition (Figure 3C). We also generated a dual mCherry-puromycin selection cassette, where the puromycin resistance marker was appended to mCherry using a 2A sequence. Red fluorescence from this construct was reduced ∼1.7-fold but was still ∼10-fold higher than background. Moreover, the addition of puromycin enriched for recombined cells (Supplementary Figure S4c). Thus, by unlinking recombinant cell enrichment from transgene expression, the rEF1α landing pad enables multiplex genetic assays with growth-altering genes.
Probing the combinatorial effects of tumor suppressor and oncogene expression on cell growth
To demonstrate the expanded scope of biological problems that can be addressed using the improved landing pad system, we studied the effects of overexpressing 216 pairwise combinations of a set of cancer-related proteins. First, we created a molecular cloning framework that allows for combinatorial barcoding, expression, and sequencing of two distinct transgenes separated by a 2A element (Figure 4A). Similar to the Combigem approach (26), barcodes identifying transcribed sequence on the same DNA fragment were concatenated so that the combined transcript combinations could be identified using high-throughput sequencing. For simplicity, we refer to the transgene N-terminal to the 2A sequence as position 1, and the transgene C-terminal to the 2A sequence as position 2 (Figure 4A).
Figure 4.
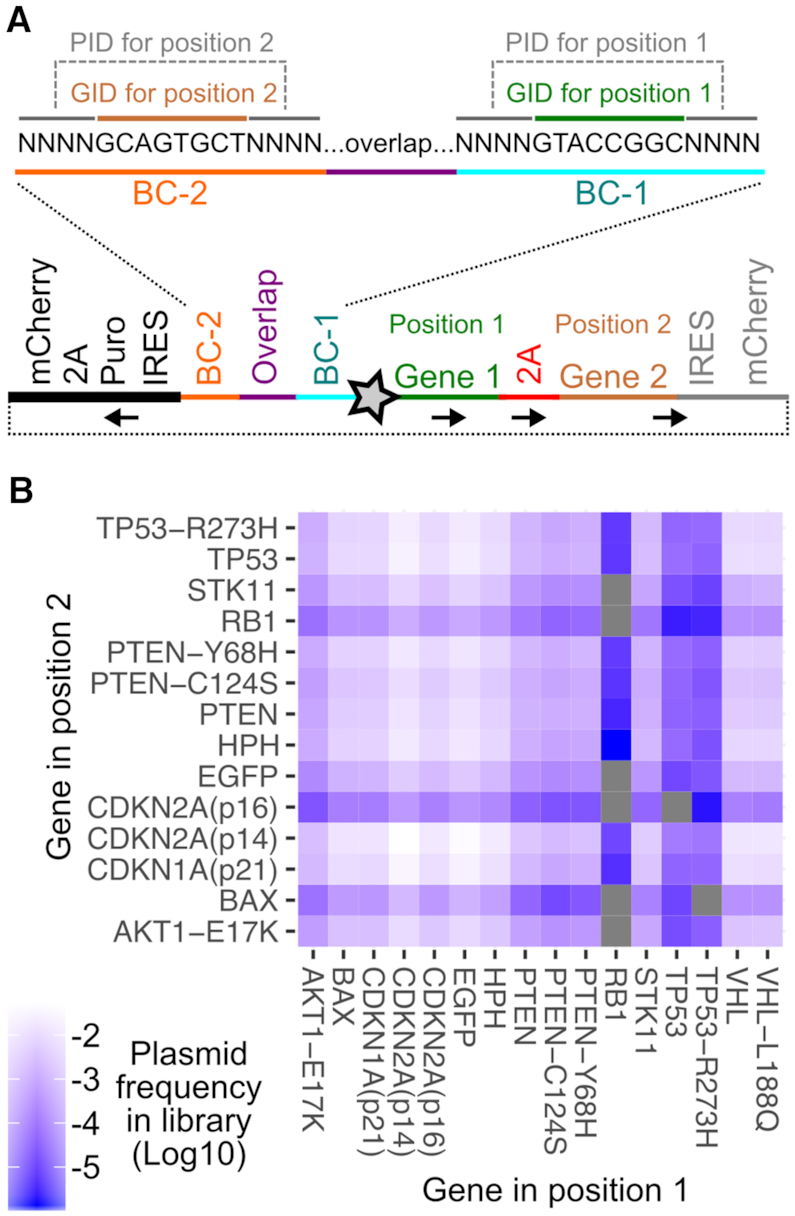
Combinatorial library of tumor suppressors and oncogenes. (A) Schematic showing the combinatorial library construct. The unique plasmid identifier (PID) sequences and representative transgene identifier sequences (GID) are shown. The sequence indicated by the black, bold line was added in a second step after the initial amplification and circularization step, to drive puromycin resistance from the reverse EF1α promoter. The star is the Bxb1 attB site. Black arrows denote direction of coding sequences. (B) Heatmap showing the frequencies of each transgene combination in the final plasmid library.
Using this approach, we created a combinatorial library of oncogenes and tumor suppressors. Each transgene in each position was given a 16 nt identifier sequence, separated by a 20 nt constant region. Each 16 nt identifier was composed of 8 nt of known sequence serving as a transgene identifier (GID) (Supplementary Table S3), flanked on either side by four degenerate nucleotides serving as unique plasmid identifier (PID) sequences (Figure 4A, top). This combination of identifier sequences served as the full plasmid ‘barcode’. EGFP and the hygromycin resistance transgene hph were included to serve as inert controls. Of the 224 possible transgene combinations, 216 were observed in the final plasmid library (Figure 4B). Some combinations, particularly involving RB1 and TP53, were observed at low frequencies likely due to inefficient amplification before pooled Gibson assembly (Figure 4B).
We recombined the library into LLP-rEF1α cells (Figure 3A) and grew the cells for 2 weeks in the absence of doxycycline but in the presence of puromycin, which enriched recombined cells to ∼40%. Transgene expression was then induced with doxycycline, and cells were collected at various points during up to 2 weeks of outgrowth. Genomic DNA was extracted from each cell pellet, and recombined plasmid barcodes were amplified and sequenced at high-throughput. We performed a total of eight replicate experiments from two independent recombinations.
The relative frequency of each transgene combination did not substantially differ between the recombined cells and the plasmid library, except for combinations where RB1, TP53 or TP53-R273H were present in position 1. Although these three transgenes were the least frequent in position 1 of the plasmid library (Figure 4B), they were enriched in the recombined pool by one to two orders of magnitude (Supplementary Figure S5a, cyan bars). Resequencing of the plasmid library did not alter this result (Supplementary Figure S5a, orange bars). Similarly, the number of unique plasmid identifiers associated with these position 1 combinations increased in the recombined cells (Figure 5A). The underlying causes of these observations remain to be elucidated.
Figure 5.

Effects of combinatorial library expression on cell proliferation in a mixed culture. (A) Log10-transformed ratio of the frequency of each unique PID sequence associated with each transgene combination in recombined cells divided by the frequency of that PID in the plasmid library. (B) Heatmap showing the mean enrichment or depletion scores of each combination of transgenes after outgrowth. Combinations scored in fewer than 4 replicates are indicated by smaller tiles. Missing combinations are indicated by gray tiles. (C) Histograms of scores from replicate experiments for specific transgene combinations. (D) Enrichment scores of each transgene with all partners. Color indicates the position of the indicated transgene within the 2A-linked translation product. Large hollow points denote the median score for each position, and the black line indicates the mean of the two median points. (E) Heatmap of interaction scores, computed by subtracting each gene's individual median score from the observed growth score for the pair.
We next looked at how cells expressing each combination of transgenes enriched or depleted in the mixed population. There were 181 transgene combinations scored in four or more replicates, an additional 23 combinations that were scored in one to three replicates and 20 combinations that were not scored (Figure 5B). Replicate enrichment scores generally grouped together (Figure 5C), and were averaged to give each transgene combination a composite growth score (Figure 5B).
To capture the individual growth contributions of each transgene, we computed the median scores of all combinations involving that transgene at either positions 1 or 2 (Figure 5D, large hollow circles). Many transgenes had median scores close to zero, including EGFP and hph, neither of which were expected to impact growth (Figure 5D). Furthermore, the enrichment score of each transgene paired with EGFP or hph, correlated with the composite positions 1 or 2 median scores calculated for that transgene (Pearson's r: 0.82; Supplementary Figure S5b). This correlation suggests that the median enrichment scores capture the individual proliferation effects of each transgene expressed from each position. Other transgenes had median scores that were significantly different from zero. Of transgenes observed in 10 or more combinations at each position, TP53 exhibited the lowest median scores in the experiment, while TP53-R273H exhibited the highest. CDKN1A(p21) also exhibited consistently low scores, as expected from our preliminary experiments (Supplementary Figure S4a and b).
We next assessed whether the enrichment score of any given gene, when paired with each of its partners, exhibited consistently lower scores in one orientation over the other. CDKN2A(p14) consistently gave lower scores when expressed from position 1 rather than position 2, which remained statistically significant following correction for multiple hypothesis testing (permutation test, P = 0.04). This suggests that CDKN2A(p14) may exhibit different effects on cell growth depending on its position in the 2A-linked translation product.
Finally, we assessed whether any combinations of transgenes exhibited unexpected effects on cell growth, relative to the genes’ independent effects. Here, for each transgene pair, we computed an interaction score by subtracting each gene's individual positions 1 or 2 median enrichment score (large red and green hollow circles in Figure 5D) from the observed growth score for the pair (shown in Figure 5B). Interaction scores greater than zero indicate transgene pairs with a positive effect on growth when expressed in combination, relative to the individual effects of each gene. Interaction scores less than zero indicate transgene pairs with a negative effect on growth when expressed in combination, relative to the individual effects.
Some transgene pairs had large, potentially biologically meaningful interaction scores (Figure 5E). For example, PTEN counteracted the TP53 proliferation defect, in either orientation. VHL-L188Q also counteracted the TP53 proliferation defect, whereas WT VHL seemed to exaggerate this defect. One possible explanation for this observation is an interaction between pVHL and p53, which protects p53 and stimulates its transcriptional activity (27). STK11 in position 1 exhibited a strong negative interaction when paired with TP53, which may relate to p53-dependent STK11 induced cell cycle arrest or apoptosis (28,29). However, this interaction was absent when STK11 and TP53 were expressed from the opposite positions. This positional dependence could be an artifact of the 2A linkage, which leaves behind short peptides on the terminus of linked proteins. Regardless of these complexities, this experiment demonstrates that the LLP-rEF1α landing pad can be used to conduct multiplex genetic assays with toxic transgenes. The work also highlights the possibility of using our new combinatorial transgene expression library format to experimentally test hundreds to thousands of genetic interactions.
DISCUSSION
The Bxb1 landing pad system is useful for library-scale expression of transgenes in cultured mammalian cell lines. Here, we describe a series of advances that improve recombination rates, enable positive or negative drug selection of recombinant cells, and facilitate assays involving toxic transgenes. We also introduce landing pads into a variety of commonly used cell lines using lentiviral transduction. We demonstrate how these new tools can empower multiplex genetic assays by screening 216 combinations of cancer-related transgenes that perturb cell growth. We identified transgenes that reproducibly enhance or reduce cell growth, including transgene combinations with unexpected effects.
The basic LLP format we present is similar to the original AAVS1-based landing pad (13), and is thus a good starting point when exploring a new cell line where BFP expression levels or Bxb1 integrase toxicity has not been characterized. If recombination rates are prohibitively low, the LLP-Int or LLP-Int-Blast formats can enhance recombination rates. This may be particularly important in with additional cell lines, as the recombination rates in NIH 3T3 and A549 cells were much lower than in 293Ts. Effective drug-based selection against landing pad silencing makes LLP-Int-Blast superior to LLP-Int. However, because of the reduced BFP and bsd protein levels driven by LLP-Int-Blast, LLP-Int might be preferable in cells with low Dox-inducible promoter expression which could make the fluorescent reporter or the blasticidin resistance markers unusable. Bxb1 recombinase sequences with central GA dinucleotides enhance recombination (30). Future modification of the LLP-Int-Blast vector and its corresponding recombination vector to GA dinucleotides will likely yield the highest recombination rates yet.
Unless more efficient Bxb1 recombinase variants are discovered, enrichment of recombinant cells is required. In the past, enrichment was accomplished using FACS; here we introduce positive and negative drug selection schemes. Recombination plasmids expressing drug selection markers enable efficient enrichment for recombined cells. A landing pad expressing an inducible caspase, iCasp9, enables rapid and nearly quantitative negative selection of un-recombined cells. This iCasp9-based landing pad provides an orthogonal strategy for the selection of recombinants and is backward compatible with previous attB recombination plasmids (13,14). Combining these tools enables selection of recombinants by both positive and negative selection, which we anticipate may achieve a near-pure population of recombinants without the use of FACS.
Overexpression of disease-relevant proteins can cause drastic effects on cell growth. Problematically, in library-scale experiments, such toxic library members are rapidly lost. By unlinking expression of the recombination reporter from the expression of the transgene(s) of interest, the LLP-rEF1α landing pad enables experiments with toxic transgenes. For example, the TP53 variant-specific growth effects we observed could be used to read out the function of a library of TP53 variants bolstering data collected by other groups in different systems (5,31). One drawback of the LLP-rEF1α format is that the EF1α promoter expression is relatively weak, presenting a narrow effective window for antibiotic selection. This flaw likely contributed to the bottleneck in PID sequences observed in the recombined library. We suggest that antibiotic selections in these cell lines should be piloted at library scale to guard against bottlenecking. Alternative promoters could improve expression, alleviating this problem.
The landing pad improvements we present here enable multiplex genetic assays for transgene libraries in cultured human-derived cells. One important application of these tools will be the simultaneous evaluation of thousands of variants of disease-related proteins (13,14). Another application could be evaluation of combinations of transgenes to understand genetic interactions. Our approach could be used to study protein–protein interactions, including a set of proteins that promiscuously form heterodimers, or variants of a pair of proteins that have evolved to bind each other. Inclusion of known loss-of-function variants, such as the TP53-R273H variant, in combinatorial transgene libraries can provide clearer understanding of specific functions that may underlie certain genetic interactions. By addressing many of the key weaknesses of the original landing pad system, including the difficulty of generating new landing pad cell lines, the relatively low recombination rate, the requirement for FACS to enrich recombined cells and the incompatibility of the landing pad with toxic transgenes, we provide the tools needed to rapidly and efficiently execute such multiplex genetic assays in the future.
DATA AVAILABILITY
An R markdown script recreating the analysis from the datafiles can be found at the Fowler lab GitHub repository (https://github.com/FowlerLab/LentiLandingPad).
The Illumina raw sequencing files can be accessed at the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE135717.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Leith of the UW Foege Flow Lab and D. Prunkard of the UW Pathology Flow Cytometry Core Facility for assistance with cell sorting.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences [R01GM109110 to D.M.F.]; National Human Genome Research Institute [RM1HG010461]; American Cancer Society Fellowship [PF-15-221-01 to K.A.M.]; National Cancer Institute Interdisciplinary Training Grant in Cancer [T32CA080416 to K.A.M.]; CIFAR Azrieli Global Scholarship (to D.M.F.); National Science Foundation Graduate Research Fellowship (to M.A.C.); National Cancer Institute[F30CA236335 to N.H.]. Funding for open access charge: National Institute of General Medical Sciences [R01GM109110 to D.M.F].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gasperini M., Starita L., Shendure J.. The power of multiplexed functional analysis of genetic variants. Nat. Protoc. 2016; 11:1782–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forsyth C.M., Juan V., Akamatsu Y., DuBridge R.B., Doan M., Ivanov A.V., Ma Z., Polakoff D., Razo J., Wilson K. et al.. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs. 2013; 5:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heredia J.D., Park J., Brubaker R.J., Szymanski S.K., Gill K.S., Procko E.. Mapping interaction sites on human chemokine receptors by deep mutational scanning. J. Immunol. 2018; 200:3825–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Majithia A.R., Tsuda B., Agostini M., Gnanapradeepan K., Rice R., Peloso G., Patel K.A., Zhang X., Broekema M.F., Patterson N. et al.. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016; 48:1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giacomelli A.O., Yang X., Lintner R.E., McFarland J.M., Duby M., Kim J., Howard T.P., Takeda D.Y., Ly S.H., Kim E. et al.. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018; 50:1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sack L.M., Davoli T., Xu Q., Li M.Z., Elledge S.J.. Sources of error in mammalian genetic screens. G3. 2016; 6:2781–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hill A.J., McFaline-Figueroa J.L., Starita L.M., Gasperini M.J., Matreyek K.A., Packer J., Jackson D., Shendure J., Trapnell C.. On the design of CRISPR-based single-cell molecular screens. Nat. Methods. 2018; 15:271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Findlay G.M., Daza R.M., Martin B., Zhang M.D., Leith A.P., Gasperini M., Janizek J.D., Huang X., Starita L.M., Shendure J.. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018; 562:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hiatt J.B., Patwardhan R.P., Turner E.H., Lee C., Shendure J.. Parallel, tag-directed assembly of locally derived short sequence reads. Nat. Methods. 2010; 7:119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu F., Gamboa M., Farruggio A.P., Hippenmeyer S., Tasic B., Schüle B., Chen-Tsai Y., Calos M.P.. DICE, an efficient system for iterative genomic editing in human pluripotent stem cells. Nucleic Acids Res. 2014; 42:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duportet X., Wroblewska L., Guye P., Li Y., Eyquem J., Rieders J., Rimchala T., Batt G., Weiss R.. A platform for rapid prototyping of synthetic gene networks in mammalian cells. Nucleic Acids Res. 2014; 42:13440–13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaidukov L., Wroblewska L., Teague B., Nelson T., Zhang X., Liu Y., Jagtap K., Mamo S., Tseng W.A., Lowe A. et al.. A multi-landing pad DNA integration platform for mammalian cell engineering. Nucleic Acids Res. 2018; 46:4072–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matreyek K.A., Stephany J.J., Fowler D.M.. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017; 45:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matreyek K.A., Starita L.M., Stephany J.J., Martin B., Chiasson M.A., Gray V.E., Kircher M., Khechaduri A., Dines J.N., Hause R.J. et al.. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 2018; 50:874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheung R., Insigne K.D., Yao D., Burghard C.P., Wang J., Hsiao Y.-H.E., Jones E.M., Goodman D.B., Xiao X., Kosuri S.. A multiplexed assay for exon recognition reveals that an unappreciated fraction of rare genetic variants cause large-effect splicing disruptions. Mol. Cell. 2018; 73:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 17. Rubin A.F., Gelman H., Lucas N., Bajjalieh S.M., Papenfuss A.T., Speed T.P., Fowler D.M.. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 2017; 18:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen W.Y., Bailey E.C., McCune S.L., Dong J.Y., Townes T.M.. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc Natl Acad Sci U S A. 1997; 94:5798–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma P., Yan F., Doronina V.A., Escuin-Ordinas H., Ryan M.D., Brown J.D.. 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res. 2012; 40:3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gritz L., Davies J.. Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene. 1983; 25:179–188. [DOI] [PubMed] [Google Scholar]
- 21. Kimura M., Kamakura T., Tao Q.Z., Kaneko I., Yamaguchi I.. Cloning of the blasticidin S deaminase gene (BSD) from Aspergillus terreus and its use as a selectable marker for Schizosaccharomyces pombe and Pyricularia oryzae. MGG. 1994; 242:121–129. [DOI] [PubMed] [Google Scholar]
- 22. Lacalle R.A., Pulido D., Vara J., Zalacaín M., Jiménez A.. Molecular analysis of the pac gene encoding a puromycin N-acetyl transferase from Streptomyces alboniger. Gene. 1989; 79:375–380. [DOI] [PubMed] [Google Scholar]
- 23. Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. et al.. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009; 27:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Straathof K.C., Pulè M.A., Yotnda P., Dotti G., Vanin E.F., Brenner M.K., Heslop H.E., Spencer D.M., Rooney C.M.. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005; 105:4247–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang H.W., Chung M., Kudo T., Meyer T.. Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature. 2017; 549:404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong A.S.L., Choi G.C.G., Cheng A.A., Purcell O., Lu T.K.. Massively parallel high-order combinatorial genetics in human cells. Nat. Biotechnol. 2015; 33:952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roe J.-S., Kim H., Lee S.-M., Kim S.-T., Cho E.-J., Youn H.-D.. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell. 2006; 22:395–405. [DOI] [PubMed] [Google Scholar]
- 28. Tiainen M., Ylikorkala A., Mäkelä T.P.. Growth suppression by Lkb1 is mediated by a G1 cell cycle arrest. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9248–9251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karuman P., Gozani O., Odze R.D., Zhou X.C., Zhu H., Shaw R., Brien T.P., Bozzuto C.D., Ooi D., Cantley L.C. et al.. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol. Cell. 2001; 7:1307–1319. [DOI] [PubMed] [Google Scholar]
- 30. Jusiak B., Jagtap K., Gaidukov L., Duportet X., Bandara K., Chu J., Zhang L., Weiss R., Lu T.K.. Comparison of integrases identifies Bxb1-GA mutant as the most efficient site-specific integrase system in mammalian cells. ACS Synth. Biol. 2019; 8:16–24. [DOI] [PubMed] [Google Scholar]
- 31. Kotler E., Shani O., Goldfeld G., Lotan-Pompan M., Tarcic O., Gershoni A., Hopf T.A., Marks D.S., Oren M., Segal E.. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell. 2018; 71:178–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An R markdown script recreating the analysis from the datafiles can be found at the Fowler lab GitHub repository (https://github.com/FowlerLab/LentiLandingPad).
The Illumina raw sequencing files can be accessed at the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE135717.