


Abstract
JASPAR (http://jaspar.genereg.net) is an open-access database of curated, non-redundant transcription factor (TF)-binding profiles stored as position frequency matrices (PFMs) for TFs across multiple species in six taxonomic groups. In this 8th release of JASPAR, the CORE collection has been expanded with 245 new PFMs (169 for vertebrates, 42 for plants, 17 for nematodes, 10 for insects, and 7 for fungi), and 156 PFMs were updated (125 for vertebrates, 28 for plants and 3 for insects). These new profiles represent an 18% expansion compared to the previous release. JASPAR 2020 comes with a novel collection of unvalidated TF-binding profiles for which our curators did not find orthogonal supporting evidence in the literature. This collection has a dedicated web form to engage the community in the curation of unvalidated TF-binding profiles. Moreover, we created a Q&A forum to ease the communication between the user community and JASPAR curators. Finally, we updated the genomic tracks, inference tool, and TF-binding profile similarity clusters. All the data is available through the JASPAR website, its associated RESTful API, and through the JASPAR2020 R/Bioconductor package.
INTRODUCTION
Transcription factors (TFs) are proteins involved in the regulation of gene expression at the transcriptional level (1). They interact with DNA in a sequence-specific manner through their DNA-binding domains (DBDs), which are used to classify TFs into structural families (2). The genomic locations where TFs bind to DNA are known as TF binding sites (TFBSs), which are typically short (6–20 bp) and exhibit sequence variability (3). Genome-wide identification of TFBSs is key to understanding transcriptional regulation. As it is not possible to identify all TFBSs for every cell type and cellular condition experimentally, computational modeling of TF-binding specificities has been instrumental to predict TFBSs in the genome. These computational models aim at representing the complex interplay between nucleotide and/or DNA shape readout at TFBSs (4), and can be used to predict not only the precise location where TFs interact in the genome (5), but also TFs with enriched TFBSs in a set of sequences (6), or the impact of mutations on TF binding (7,8), amongst others.
From the plethora of existing computational models (9), position frequency matrices (PFMs) (10) are one of the simplest and (still) most commonly used, although more complex models, for instance based on hidden Markov models or deep learning (11–13), are becoming more common. A PFM is a TF-binding profile that models the DNA-binding specificity of a TF by summarizing the frequencies of each nucleotide at each position from observed TF-DNA interactions. These interactions are usually derived from in vitro assays (e.g. SELEX (14) or protein binding microarrays (15)), which assess the binding affinity of TFs to DNA sequences, or from ChIP-based experiments (e.g. ChIP-seq (16), ChIP-exo (17), or ChIP-nexus (18)), which capture TF-DNA interactions in vivo, by looking for over-represented DNA sequences in regions bound by the ChIP’ed TF.
With the advent of high-throughput sequencing more than a decade ago, the number of PFMs derived from in vivo and in vitro experiments has increased dramatically, leading to the creation of multiple databases storing PFMs or more complex TF-binding profiles such as JASPAR (19), CIS-BP (20) and HOCOMOCO (21) (see (22) for a comprehensive review). The JASPAR database (http://jaspar.genereg.net/) is one of the most popular databases of TF-binding profiles, and has been maintained for over 15 years (23). As such, many computational tools dedicated to the study of gene regulation incorporate profiles from JASPAR (e.g. TFBSshape (24,25), RSAT (26), MEME (27) or i-cisTarget (6)). At the heart of JASPAR is its CORE collection, which contains TF-binding profiles that are: (i) manually curated (meaning that orthogonal supporting evidence from the literature is required for each profile); (ii) non-redundant (one profile per TF with the exception of TFs with multiple DNA-binding sequence preferences (28)); (iii) associated with TFs from one of six taxa (vertebrates, nematodes, insects, plants, fungi, and urochordata) and (iv) freely available to the community through a user-friendly web interface, a RESTful API (29), and a dedicated R/Bioconductor data package (‘JASPAR2020’).
Here, we present the 8th release of JASPAR, which comes with a major expansion and update of its CORE collection. Moreover, we introduce a new collection of unvalidated profiles, which stores quality-controlled PFMs for which our curators could not find orthogonal support. This collection has a dedicated web interface to engage the community of users in the curation of TF-binding profiles. Finally, we have updated the hierarchical clusters of TF-binding profiles, the genomic tracks of predicted TFBSs (now available for 8 genomes), and the profile inference tool.
EXPANSION AND UPDATE OF THE JASPAR CORE COLLECTION
For this 8th release of JASPAR, we added to the CORE collection 245 new TF-binding profiles for TFs in the following taxa: vertebrates (169 profiles, corresponding to an expansion of 29% for this taxon), plants (42 profiles, 9% expansion), nematodes (17 profiles, 65% expansion), insects (10 profiles, 8% expansion) and fungi (7 profiles, 4% expansion). We updated 156 profiles (Table 1). The new PFMs were derived from HT-SELEX (30), PBMs (20), ChIP-seq and DAP-seq experiments (data sourced from CistromeDB (31), ReMap (32,33), GTRD (34), ChIP-atlas (35) and ModERN (36), see Supplementary Text for method details). As previously described, the newly introduced profiles were manually curated to be supported by an orthogonal reference from the literature, which is provided in the metadata of the profiles. Moreover, the TF DBD class and family (following the TFClass classification (2)), the TF UniProt ID (37), and links to the TFBSshape (24,25), ReMap (32,33) and UniBind (38) databases are provided in the profiles metadata (whenever possible). Finally, the profiles previously associated with ID2, ID4 and TRB2 were removed from the CORE collection as these proteins are not TFs (1).
Table 1.
Overview of the growth of the number of PFMs in the JASPAR 2020 CORE and unvalidated collections compared to the JASPAR 2018 CORE collection
| Taxonomic Group | Non-redundant PFMs in JASPAR 2018 | New non-redundant PFMs in JASPAR 2020 | Removed profiles | Updated PFMs in JASPAR 2020 | Total PFMs (non-redundant) in JASPAR 2020 | Total PFMs (all versions) in JASPAR 2020 |
|---|---|---|---|---|---|---|
| Vertebrates | 579 | 169 | 2 | 125 | 746 | 1011 |
| Plants | 489 | 42 | 1 | 28 | 530 | 572 |
| Insects | 133 | 10 | 0 | 3 | 143 | 153 |
| Nematodes | 26 | 17 | 0 | 0 | 43 | 43 |
| Fungi | 176 | 7 | 0 | 0 | 183 | 184 |
| Urochordata | 1 | 0 | 0 | 0 | 1 | 1 |
| Total CORE | 1404 | 245 | 3 | 156 | 1646 | 1964 |
| unvalidated | 337 | 337 |
Overall, the JASPAR 2020 CORE collection includes 1646 non-redundant PFMs (746 for vertebrates, 530 for plants, 183 for fungi, 143 for insects, 43 for nematodes and 1 for urochordates) (Table 1; Figure 1). Moreover, we continued with the incorporation of novel transcription factor flexible models (TFFMs), which are hidden Markov-based models capturing dinucleotide dependencies in TF–DNA interactions (11). We introduced new TFFMs for 217 TFs (136 for vertebrates, 38 for plants, 21 for insects, 17 for nematodes, and 5 for fungi) and updated TFFMs for 20 vertebrates TFs, which represents a 50% increase in the number of TFFMs available. All data is available on the JASPAR website, its associated RESTful API, and through the JASPAR2020 R/Bioconductor package.
Figure 1.
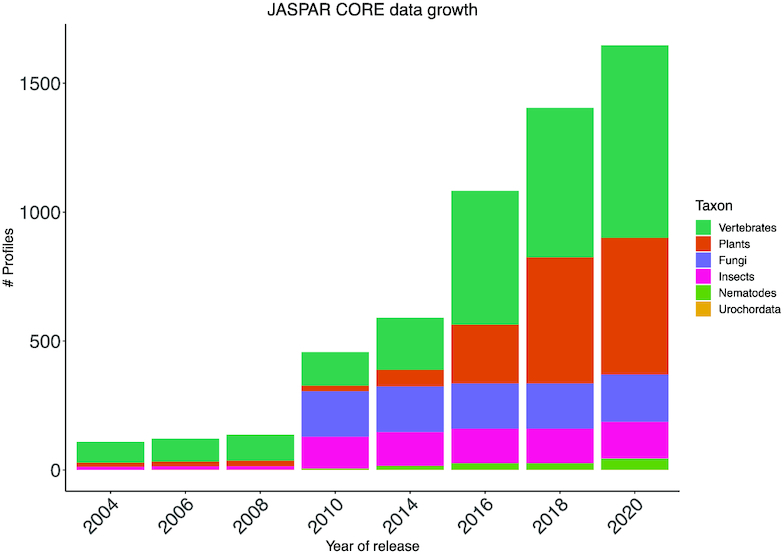
JASPAR CORE growth. The number of profiles in each taxon and overall (see legend) through all JASPAR releases.
A NEW COLLECTION OF UNVALIDATED PROFILES FOR COMMUNITY ENGAGEMENT
We introduced a novel ‘unvalidated’ collection to store high-quality (i.e. passing multiple quality controls, see Supplementary Text) TF-binding profiles for which no independent support was found in the literature by our curators. This collection contains 337 PFMs. As these profiles are not yet supported by an orthogonal evidence, we recommend users to use this collection with caution. We encourage the community to engage in the curation of these profiles by providing the JASPAR curators with supporting complementary evidence (from their own work or others) whenever possible. This is facilitated by the availability of an individual submission form for each profile in the ‘unvalidated’ collection (Figure 2).
Figure 2.

Unvalidated TF-binding profile collection. Example with the ZNF793 profile. This high-quality PFM was derived from a ChIP-seq experiment and was built from thousands of potential TFBSs. Further, the TFBSs are enriched around the ChIP-seq peak summits. However, no orthogonal evidence supporting this profile was found by our curators. Users can upload relevant information about the profile in the unvalidated collection through the ‘Community curation’ box.
Further, we started a Q&A forum (https://groups.google.com/forum/#!forum/jaspar) to ease the communication between JASPAR curators and the community; we welcome the community to send us their questions and suggestions, or to report errors in JASPAR.
CLUSTERED PROFILES, GENOMIC TRACKS AND PROFILE INFERENCE TOOL
In the previous releases, we introduced novel features such as hierarchical clustering of TF-binding profiles in the CORE collection to visualize profile similarities, genomic tracks of predicted TFBSs, and an inference tool to predict TF-binding profiles likely recognized by TFs not available in the JASPAR CORE. We improved the profile inference tool using our own implementation of a recently described similarity regression method (20). We updated the generation of genomic tracks that are publicly available through the UCSC Genome Browser data hub (39) for 7 organisms: human (hg19, hg38), mouse (mm10), zebrafish (danRer11), Drosophila melanogaster (dm6), Caenorhabditis elegans (ce10), Arabidopsis thaliana (araTha1) and baker's yeast (sacCer3). For more details on the updated genomic tracks and inference tool, refer to the Supplementary Text. Finally, we generated the hierarchical clusters of available TF-binding profiles for each taxon with RSAT matrix-clustering (40). Users can explore the CORE/unvalidated collection through the trees and access directly the corresponding profiles by clicking on the TF name.
CONCLUSIONS AND PERSPECTIVES
Similar to previous releases, we substantially expanded the CORE collection of the JASPAR database. For this 8th release, we processed more than 18,000 ChIP-seq datasets. As a large number of the obtained high-quality TF-binding profiles were not supported with orthogonal supporting evidence, it motivated us to create the novel ‘unvalidated’ collection of profiles. We expect that upcoming experiments and publications will provide additional supporting evidence to some profiles to be incorporated into the JASPAR CORE collection. Meanwhile, we would like to extend our invitation to the research community to 1) help us curate these unvalidated profiles (e.g. by pointing us to supporting literature), and 2) send us their own novel profiles (e.g. determined experimentally) for incorporation in the next release of JASPAR.
The JASPAR CORE vertebrates collection now contains 746 profiles, 637 of which are associated with human TFs with known DNA-binding profiles (1), which corresponds to a 58% of the 1,107 reported by Lambert et al. (1). While this is an impressive collective achievement by the field (the original JASPAR database only contained 81 profiles, a ∼7% coverage for human TFs), it suggests that targeted experimental efforts to find the binding preferences for remaining TFs will be important. Although computational approaches can be used to infer missing TF-binding profiles (20,41), especially for non-model organisms, the JASPAR approach is conservative, including profiles supported by at least two experiments in the literature. This is very important as we stand by the reliability of our data. Since its initial publication in 2004 (23), the JASPAR database has been committed to provide the research community with high-quality, manually curated, non-redundant TF-binding profiles.
Lastly, although PFMs have dominated the field of gene regulation for decades, new profile representations have emerged. For example, profiles with expanded alphabets to represent methylated bases (42,43), modelling binding energy (44) or derived from deep learning importance scores (45). Depending on how the field evolves and how popular these profiles become, we will consider them for inclusion in JASPAR in the future.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the user community for useful input and the scientific community for performing experimental assays of TF–DNA interactions and for publicly releasing the data. We thank Giovanna Ambrosini for her help with PWMScan, the UCSC Genome Browser Project Team for their assistance with the genome tracks, WestGrid (https://www.westgrid.ca), Compute Canada (https://www.computecanada.ca), Georgios Magklaras and Georgios Marselis for their IT support, Jacques van Helden and Adam Handel for contacting us to add and validate TF binding profiles, and Dora Pak and Ingrid Kjelsvik for administrative support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Norwegian Research Council [187615]; Helse Sør-Øst; University of Oslo through the Centre for Molecular Medicine Norway (NCMM) (to A.M., J.A.C.-M., A.K., M.G.); Norwegian Research Council [288404 to J.A.C.-M. and Mathelier group]; The Norwegian Cancer Society [197884 to Mathelier group]; O.F., X.Z., P.A.R., S.C. and W.W.W. were supported by grants from the Canadian Institutes of Health Research [BOP-149430 and PJT-162120]; Genome Canada and Genome British Columbia [255ONT and 275SIL]; Michael Smith Foundation for Health Research [17746]; Natural Sciences and Engineering Research Council of Canada Discovery Grant [RGPIN-2017-06824]; CREATE programs; Weston Brain Institute [20R74681]; BC Children's Hospital Foundation and Research Institute; Netherlands Organization for Scientific Research [Rubicon fellowship to R.v.d.L., 452172015]; Genome British Columbia [SIP007 to B.P.M.]; A.S. was supported by grants from the Lundbeck Foundation, the Danish Cancer Foundation, the Danish Innovation Fund and the Danish Council for Independent Research. F.P. was supported by the French National Agency for Research [FloPiNet ANR-16-CE92-0023-01; GRAL, ANR-10-LABX-49-01]; D.B. is a recipient of a Rutherford Fund Fellowship.
Conflict of interest statement. None declared.
This paper is linked to: https://doi.org/10.1093/nar/gkz945.
REFERENCES
- 1. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 2. Wingender E., Schoeps T., Haubrock M., Krull M., Dönitz J.. TFClass: expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018; 46:D343–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reid J.E., Evans K.J., Dyer N., Wernisch L., Ott S.. Variable structure motifs for transcription factor binding sites. BMC Genomics. 2010; 11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R.. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014; 39:381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wasserman W.W., Sandelin A.. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004; 5:276–287. [DOI] [PubMed] [Google Scholar]
- 6. Imrichová H., Hulselmans G., Atak Z.K., Potier D., Aerts S.. i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 2015; 43:W57–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fornes O., Gheorghe M., Richmond P.A., Arenillas D.J., Wasserman W.W., Mathelier A.. MANTA2, update of the Mongo database for the analysis of transcription factor binding site alterations. Sci. Data. 2018; 5:180141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu Y., Liu Z., Lou S., Bedford J., Mu X.J., Yip K.Y., Khurana E., Gerstein M.. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014; 15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weirauch M.T., Cote A., Norel R., Annala M., Zhao Y., Riley T.R., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S. et al.. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013; 31:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000; 16:16–23. [DOI] [PubMed] [Google Scholar]
- 11. Mathelier A., Wasserman W.W.. The next generation of transcription factor binding site prediction. PLoS Comput. Biol. 2013; 9:e1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kiesel A., Roth C., Ge W., Wess M., Meier M., Söding J.. The BaMM web server for de-novo motif discovery and regulatory sequence analysis. Nucleic Acids Res. 2018; 46:W215–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 14. Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J. et al.. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010; 20:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W. 3rd, Bulyk M.L.. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006; 24:1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson D.S., Mortazavi A., Myers R.M., Wold B.. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007; 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 17. Pugh B.F., Franklin Pugh B.. Ultra-high resolution mapping of protein-genome interactions using ChIP-exo. BMC Proc. 2012; 6:O27. [Google Scholar]
- 18. He Q., Johnston J., Zeitlinger J.. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol. 2015; 33:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., van der Lee R., Bessy A., Chèneby J., Kulkarni S.R., Tan G. et al.. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018; 46:D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lambert S.A., Yang A.W.H., Sasse A., Cowley G., Albu M., Caddick M.X., Morris Q.D., Weirauch M.T., Hughes T.R.. Similarity regression predicts evolution of transcription factor sequence specificity. Nat. Genet. 2019; 51:981–989. [DOI] [PubMed] [Google Scholar]
- 21. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A. et al.. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stormo G.D. DNA motif databases and their uses. Curr. Protoc. Bioinformatics. 2015; 51:doi:10.1002/0471250953.bi0215s51. [DOI] [PubMed] [Google Scholar]
- 23. Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B.. JASPAR: an open‐access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004; 32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiu T.P., Xin B., Markarian N., Wang Y., Rohs R.. TFBSshape v2.0: an expanded motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2019; doi:10.1093/nar/gkz970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L., Zhou T., Dror I., Mathelier A., Wasserman W.W., Gordân R., Rohs R.. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014; 42:D148–D155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen N.T.T., Contreras-Moreira B., Castro-Mondragon J.A., Santana-Garcia W., Ossio R., Robles-Espinoza C.D., Bahin M., Collombet S., Vincens P., Thieffry D. et al.. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018; 46:W209–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME suite. Nucleic Acids Res. 2015; 43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X. et al.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khan A., Mathelier A.. JASPAR RESTful API: accessing JASPAR data from any programming language. Bioinformatics. 2018; 34:1612–1614. [DOI] [PubMed] [Google Scholar]
- 30. Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F. et al.. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017; 356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng R., Wan C., Mei S., Qin Q., Wu Q., Sun H., Chen C.-H., Brown M., Zhang X., Meyer C.A. et al.. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019; 47:D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chèneby J., Gheorghe M., Artufel M., Mathelier A., Ballester B.. ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2017; 46:D267–D275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chèneby J., Ménétrier J., Mestdagh M., Rosnet T., Douida A., Rhalloussi W., Bergon A., Lopez F., Ballester B.. ReMap 2020: A database of regulatory regions from an integrative analysis of Human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 2019; doi:10.1093/nar/gkz945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yevshin I., Sharipov R., Kolmykov S., Kondrakhin Y., Kolpakov F.. GTRD: a database on gene transcription regulation—2019 update. Nucleic Acids Res. 2019; 47:D100–D105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C.. ChIP‐Atlas: a data‐mining suite powered by full integration of public ChIP‐seq data. EMBO Rep. 2018; 19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kudron M.M., Victorsen A., Gevirtzman L., Hillier L.W., Fisher W.W., Vafeados D., Kirkey M., Hammonds A.S., Gersch J., Ammouri H. et al.. The ModERN Resource: Genome-Wide binding profiles for hundreds of drosophila and caenorhabditis elegans transcription factors. Genetics. 2018; 208:937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. The UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gheorghe M., Sandve G.K., Khan A., Chèneby J., Ballester B., Mathelier A.. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 2019; 47:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Wang T., Nguyen N., Paten B., Zweig A.S., Karolchik D. et al.. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014; 30:1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castro-Mondragon J.A., Jaeger S., Thieffry D., Thomas-Chollier M., van Helden J.. RSAT matrix-clustering: dynamic exploration and redundancy reduction of transcription factor binding motif collections. Nucleic Acids Res. 2017; 45:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathelier A., Fornes O., Arenillas D.J., Chen C.-Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al.. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Viner C., Johnson J., Walker N., Shi H., Sjöberg M., Adams D.J., Ferguson-Smith A.C., Bailey T.L., Hoffman M.M.. Modeling methyl-sensitive transcription factor motifs with an expanded epigenetic alphabet. bioRxiv doi:15 March 2016, preprint: not peer reviewed 10.1101/043794. [DOI] [PMC free article] [PubMed]
- 43. Chang Y.K., Granas D., Stormo G.D.. Measuring quantitative effects of methylation on transcription factor–DNA binding affinity. Science. 2017; 3:eaao1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruan S., Swamidass S.J., Stormo G.D.. BEESEM: estimation of binding energy models using HT-SELEX data. Bioinformatics. 2017; 33:2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shrikumar A., Tian K., Shcherbina A., Avsec Ž., Banerjee A., Sharmin M., Nair S., Kundaje A.. TF-MoDISco v0.4.2.2-alpha: Technical Note. 2019; arXiv doi:31 October 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1811.00416.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.