


Abstract
DNA methyltransferases (DNMTs) are enzymes responsible for establishing and maintaining DNA methylation in cells. DNMT inhibition is actively pursued in cancer treatment, dominantly through the formation of irreversible covalent complexes between small molecular compounds and DNMTs that suffers from low efficacy and high cytotoxicity, as well as no selectivity towards different DNMTs. Herein, we discover aptamers against the maintenance DNA methyltransferase, DNMT1, by coupling Asymmetrical Flow Field-Flow Fractionation (AF4) with Systematic Evolution of Ligands by EXponential enrichment (SELEX). One of the identified aptamers, Apt. #9, contains a stem-loop structure, and can displace the hemi-methylated DNA duplex, the native substrate of DNMT1, off the protein on sub-micromolar scale, leading for effective enzymatic inhibition. Apt. #9 shows no inhibition nor binding activity towards two de novo DNMTs, DNMT3A and DNMT3B. Intriguingly, it can enter cancer cells with over-expression of DNMT1, colocalize with DNMT1 inside the nuclei, and inhibit the activity of DNMT1 in cells. This study opens the possibility of exploring the aptameric DNMT inhibitors being a new cancer therapeutic approach, by modulating DNMT activity selectively through reversible interaction. The aptamers could also be valuable tools for study of the functions of DNMTs and the related epigenetic mechanisms.
INTRODUCTION
DNA methylation constitutes a key epigenetic mechanism that is essential for transcriptional silencing of retrotransposons, genomic imprinting, X-chromosome inactivation and cell lineage commitment (1–3). DNA methylation in mammals, which mainly occurs at the C-5 position of cytosine within the symmetric CpG dinucleotides (4), is established by the de novo DNA methyltransferases (DNMT) 3A/3B during gametogenesis and early embryonic development, and maintained by DNMT1 in a replication-dependent manner (5). Dysregulation of DNMT activities or expressions results in aberrant DNA methylation patterns in the CpG islands of tumor suppressor genes as observed in many cancers (6,7). Therefore, effective DNMT inhibition is being actively pursued in cancer treatment (8,9).
Several small molecular inhibitors, like the cytosine nucleoside analogs, 5-azacytidine and 5-aza-2′-deoxycytidine, have been developed, tested and approved by FDA to treat myelodysplastic syndrome (MDS). Once entering the cells, these compounds could form irreversible covalent complexes with DNMTs (10), triggering DNMT degradation (11). However, complete knock-out of DNMTs by irreversible covalent modification could be lethal, since low levels of DNMTs could induce genomic instability (12). Besides no selectivity towards different DNMT proteins and high toxicity, these molecules also suffer from low stability in aqueous solutions and need metabolic activation. Low-dose, prolonged exposure of these inhibitors when tested in MDS clinical trials in a solid tumor setting turns out to be challenging, limited by the lack of DNA incorporation at low drug concentrations and cytotoxicity at high concentrations (12). Novel DNMT inhibitors are needed for solving all the issues of bioavailability, bioactivation and cytotoxicity (12). It is also highly desirable to develop selective compounds for individual DNMTs to target diseases driven by a particular DNMT enzyme with minimal off-target effects.
Non-covalent and selective inhibition of DNMTs may be achieved by aptamers. Aptamers can be discovered for binding to a specific protein, like one of the DNMTs, with high affinity and selectivity (13). As a single-stranded DNA or RNA (ssDNA or ssRNA), an aptamer is the same type of molecule as the substrates for DNMTs, providing the possibility of competitive binding. Thus, an aptamer that can bind to a specific DNMT stronger than its native substrates, could inhibit its activity selectively via preventing the enzyme-substrate binding. Compared with the protein-based therapeutics, aptameric drugs are easier to be stored, handled, and modified to fit for medical applications; and can penetrate through biological barriers more easily, as well as possess much-reduced immunogenicity with low toxicity (14,15), presenting good promises for disease treatment.
‘Systematic Evolution of Ligands by EXponential enrichment’ (SELEX) has helped discover many aptamers against diverse targets (16–18). But it is challenging to directly apply SELEX for aptamer discovery against DNMTs, which requires target immobilization to facilitate aptamer clean-up (16,17,19–21). DNMTs have a flexible conformation (22–25); and their native folding can be strongly affected by environmental factors like salt content, temperature, and the presence of bio-thiols, making them not sustainable to any immobilization procedures. Filtration using membranes could remove the small ssDNAs from the large protein which retains the bound aptamers on the filter, but non-specific adsorption to the membrane surface could prevent complete removal of the unbound strands. Separation techniques like capillary electrophoresis or flow cytometry requiring no immobilization have been employed to help collection of the target-binding sequences with high purity (26–28), but they are either incompatible with the high-salt buffers required to maintain the proper folding of DNMTs; or not applicable to protein targets.
Asymmetrical flow-field flow fractionation (AF4) is a gentle separation tool that employs two perpendicular liquid flows and a ribbon-like channel in a trapezoidal shape to separate analytes by their hydrodynamic diameters: the channel flow moves the analytes to the outlet, while the cross flow presses them down towards the wall of the channel formed by a sheet of porous membrane, counter-acting the diffusion motion of the analytes (29–31). At the balanced actions of the cross flow and molecular diffusion, the mass center of the analytes would occupy at different heights within the parabolic profile of the channel flow and exit the channel at different times: a smaller molecule would diffuse faster and get closer to the center of the channel and thus move out faster than a larger one (29–31). This technique is well suited for separating free ssDNAs from the protein-bound ones to obtain the bound ssDNAs with high purity in SELEX, because it has no packing material in its separation channel, imposing negligible damage to both the non-covalent aptamer–target binding and the folding of DNMT1; and it is compatible with diverse physiological buffers as its separation buffer, matching the need of SELEX to use the appropriate buffer for keeping the protein and complex structures intact during separation (32,33).
In this study, we employ AF4 to facilitate collection of the protein-bound aptamers without protein immobilization. With the aid of AF4-SELEX, we discovered an aptamer that can bind to the catalytic domain of DNMT1 (Figure 1). The affinity of the aptamer and its inhibition function were fully characterized. Our work also proved the uptake of the aptamer to cells overly expressing DNMT1 and its colocalization with the expressed DNMT1 protein in the nucleus. More importantly, prominent reduction of the DNA methylation level in cells induced by treatment of this aptamer was observed. All these results support that the aptamer could be a new, non-covalent inhibitor for DNMT1, opening a window for development of new therapeutics for disease treatment targeting DNMT proteins.
Figure 1.

(A) Scheme of AF4-SELEX. (B) Ribbon representation of mouse DNMT1 bound to CpG DNA containing a hemi methylated CpG site, in which the target cytosine was replaced by a 5-fluorocytosine (5fC) (PDB 4DA4). The DNA, BAH1, BAH2 and MTase domains are colored in wheat, grey, limon and aquamarine, respectively. The flipped-out 5fC in the catalytic site of DNMT1 is shown in the expanded view. The hydrogen bonds are shown as dashed lines. The Zinc ions and the methyl group on the 5mC are shown in purple and green spheres. The bound-cofactor analog, S-Adenosyl-L-homocysteine (SAH), is shown in sphere representation.
MATERIALS AND METHODS
Materials
Reagents
Dithiothreitol (DTT), NaCl, MgCl2, 1× PBS buffer, glacial acetic acid, hydrochloric acid, DMSO, titanium (IV) isopropoxide, guanidine HCl (Gu-HCl) and potassium chloride (KCl) were obtained from Fisher Scientific. Polyvinylpyrrolidone (PVP) was acquired from Sigma-Aldrich. Tris base, EDTA and ethanol (EtOH) was obtained from Acros Organics, Promega and Decon Labs, respectively. The 5× Taq polymerase and the 25-mM magnesium chloride (MgCl2) solution used for PCR were from New England Biolabs. The ssDNAs, including the random libraries used for SELEX, the anti-IgE aptamer, as well as those found binding to DNMT1 (Supplementary Tables S1 and S3) were attained from Integrated DNA Technologies, Inc. Beta-casein, cytochrome C, ovalbumin, and hemoglobin were purchased from Sigma-Aldrich. IgE was acquired from Athens Research & Technology, Inc.
Protein expression and purification
For production of DNMT1, the DNA sequence encoding residues 728–1600 of human DNMT1 was inserted into an in-house bacterial expression vector, in which a His6-MBP tag was fused to the DNMT1 sequence via a TEV protease cleavage site. The His6-MBP-DNMT1 fusion protein was expressed in Escherichia coli BL21 DE3 (RIL) cell strains and purified by Ni-NTA affinity chromatography, followed by ion exchange chromatography on a Heparin column. The His6-MBP tag was removed through TEV protease cleavage and subsequent Ni2+-NTA chromatography. The DNMT1 sample was finally purified by size exclusion chromatography on a HiLoad16/600 Superdex 200 pg column (GE Healthcare), and concentrated to 0.1 mM in a buffer containing 20 mM Tris–HCl (pH 7.5), 250 mM NaCl and 5 mM DTT. For protein production of DNMT3A and DNMT3B, the gene fragments encoding residues 628–912 of human DNMT3A or residues 562–853 of human DNMT3B were inserted in tandem with residues 178–386 of regulatory protein, human DNMT3L, into a modified pRSFDuet-1 vector (Novagen). Expression and purification of DNMT3A–DNMT3L and DNMT3B–DNMT3L complexes follows the protocol as described previously (34).
Materials and Methods
AF4-SELEX
The AF2000 manufactured by Postnova Analytics was employed in this work. The trapezoidal separation channel of this system has a tip-to-tip length of 275 mm, an inlet triangle width of 20 mm and an outlet width of 5 mm. The injection loop volume was 50 μl so varied injection volumes ranging from 10 to 50 μL could be used. The accumulation wall was made out of the regenerated cellulose ultrafiltration membrane (Postnova Analytics) with the molecular weight cutoff (MWCO) value of 10 kDa. The eluate exiting AF4 passed through a SPD-20A absorbance detector (λ = 280 nm; Shimadzu) and a fluorescence detector (λex495/λem520 nm; Waters 474 Scanning Fluorometer) followed by a fraction collector (Bio-rad Model 2110).
For AF4-SELEX on DNMT1, a channel of 500 μm thick was used. To avoid DNMT1 denaturation, the DTT-containing Tris buffer (pH 7.5, 20 mM Tris, 500 mM NaCl, 5 mM DTT and 1 mM MgCl2) was used as the separation solution; and kept at 4°C during sample injection and focusing. Fifty pmol ssDNA library containing a random sequence of 20-nt and a fluorescent label in a solution of 50 μL was injected into the AF4 channel directly. Afterwards, 30 pmol DNMT1 was injected and focused for 10 min, using a cross flow, tip flow, and focus flow of 3.00, 0.30 and 3.00 ml/min, respectively. After focusing, the cross flow was reduced to keep the detector flow (the flow exiting the channel from the outlet) at 0.70 ml/min, while keeping the cross flow at 3.00 ml/min.
The selection pressure was enhanced starting from the fifth round of SELEX, by mixing 30 pmol DNMT1 with the standard protein mixture (β-casein, cytochrome C, ovalbumin and hemoglobin; 25 pmol of each protein). In the sixth round, 3 pmol of hemimethylated DNA was injected in addition to the protein standard to compete with the ssDNA pool for binding to DNMT1. At the last few selection rounds (7th–11th round) the DNMT1 amount used was reduced to 15 pmol. During each selection cycle, the elution window of the DNMT1-bound ssDNAs were collected by TiO2 microfibers and then amplified by PCR (Supplementary Methods). The resultant amplification products were treated with streptavidin-coated sepharose beads (GE Healthcare) to obtain the ssDNA pool for the next round of SELEX. The ssDNA pools enriched during the last few selection cycles were submitted to Sangon Biotech (Shanghai, China) for sequencing.
Binding assessment by electrophoretic mobility shift assay (EMSA)
The DNA and DNMT1 samples were incubated in 20 mM Tris buffer containing 25% glycerol and 8% glucose, and 4–20% of gradient polyacrylamide gel was prepared and ran at 60 V in 0.25× TBE buffer for 2.5 h to separate the protein and the free ssDNA, followed by SYBR gold staining for visualization of the DNA bands, and by Coomassie blue staining for protein detection. The aptamer affinity for DNMT1 was measured by incubating a fixed concentration (0.4 μM) of Apt. #9 with increasing concentration of DNMT1 up to 2.8 μM, and calculating the bound ratio from the band intensities for the free and protein-bound forms of the aptamer quantified by using ImageJ. The dissociation constant (Kd) was obtained by plotting the bound ratio versus protein concentration and fitting the curve to the Hill equation using GraphPad Prism 7. DNMT1 binding by the native substrate, i.e. hemiDNA, was measured by fixing the DNMT1 concentration at 0.4 μM and increasing the concentration of hemiDNA up to 4 μM, in the presence or absence of 0.4 μM Apt. #9. Displacement of hemiDNA by Apt. #9 was evaluated by forming the DNMT1–hemiDNA complex at a 1:1 ratio with each at 2 pmol (0.4 μM) and disrupting the complex by the addition of 0.5, 2 and 10 pmol Apt. #9.
In vitro DNA methylation inhibition assay
The synthesized hemiDNA and unmodified (CG-DNA) 36-mer DNA duplex (upper strand: 5′-GAC GAC GAC GAC GAC GAC GAC GAC GAC GAC GAC GAC-3′, lower stand: 5′-GTX GTX GTX GTX GTX GTX GTX GTX GTX GTX GTX GTX-3′, X = 5-methylcytosine or cytosine) were used as a reaction substrate for DNMT1 and DNMT3A/DNMT3B, respectively. The aptamer (Apt.) #9 (5′-AGA AGT GGG GGT GAG TCC ACT TCT G-3′) was used for inhibition, where the modified ssDNA #12 (5′-TGC GAG TGA CAT CTC AAC GG-3′), which showed no binding affinity for DNMT1 and thus called non-binding ssDNA #12 in later text, was used as the negative control. In brief, a 20-μL reaction mixture contained Apt. #9 or the non-binding ssDNA #12 at various concentrations (0, 0.1, 0.5, 1, 2, 5, 10 μM), 0.1 μM DNMT1, 0.4 μM hemiDNA, 2.5 μM S-adenosyl-l-[methyl-3H] methionine (specific activity 18 Ci/mmol, PerkinElmer), 1.9 μM AdoMet in 50 mM Tris–HCl, pH 8.0, 0.05% β-mercaptoethanol, 5% glycerol and 200 μg/mL BSA. The enzymatic assays of DNMT3A and DNMT3B followed the same conditions, except that 0.3 μM DNMT3A-DNMT3L/DNMT3B-DNMT3L and 0.75 μM CG-DNA substrate were used. The DNA methylation assays were carried out in triplicate at 37°C for 20 min for DNMT1 and 40 min for DNMT3A-DNMT3L/DNMT3B-DNMT3L. The reaction was quenched by addition of 5 μL of 10 mM AdoMet. For detection, 15 μL of the reaction mixture was spot on DEAE Filtermat paper (PerkinElmer) and dried out. The DEAE paper was then washed 3 times with 5 mL of 0.2 M cold ammonium bicarbonate (pH 8.2), 5 mL of Milli Q water and 5 mL of EtOH. Subsequently, the DEAE paper was air dried and transferred to scintillation vials filled with 5 mL ScintiVerse (Fisher). The radioactivity of tritium was measured with a Beckman LS6500 counter.
Steady-state kinetics assay
The hemiDNA 36-mer DNA duplex described above was used as substrate for measuring the steady-state methylation kinetics of DNMT1, where the Apt. #9 was used for inhibition. The 20-μL reaction contains 50 nM DNMT1, 5–400 nM hemiDNA, 0 or 0.1 μM Apt. #9, 0.5 μM S-adenosyl-l-[methyl-3H] methionine (specific activity 18 Ci/mmol, PerkinElmer), 2 μM AdoMet in 50 mM Tris–HCl, pH 8.0, 0.05% β-mercaptoethanol, 5% glycerol and 200 μg/ml BSA. The reactions were carried out in triplicate at 37°C for 0 min or 60 min, which falls into the linear range of the progression curve (Supplementary Figure S13), and quenched by addition of 5 μL of 10 mM AdoMet. For detection, 10 μL of reaction mixture was spot on Amersham Hybond™-XL membrane (GE Healthcare) and dried out. The membrane was then washed 3 times with 5 mL of 0.2 M cold ammonium bicarbonate (pH 8.2), 5 mL of Milli Q water and 5 mL of EtOH. Subsequently, the membrane was air dried, transferred to scintillation vials filled with 5 mL of ScintiVerse (Fisher) and subject to radioactivity measurement by a Beckman LS6500 counter. The data were analyzed by Michaelis–Menten enzymatic kinetics nonlinear regression fitting (Y = Vmax*X/(KM + X)) using GraphPad Prism7 software.
Transient transfection
To confirm that Apt. #9 can target DNMT1 protein in living cells, we used the transient transfection method to increase DNMT1 expression in HEK 293T cell. Briefly, 1 × 105 cells were seeded in confocal dishes (NEST Biotechnology Co. Ltd) and cultured for 24 hrs. Followed, the cells were transfected with the DNMT1 plasmid (pKanCMV_mRuby_DNMT1, full length) using the TransIT®-2020 Transfection Reagent (Mirus Bio Company) according to the protocol provided by the manufacturer. The transfected cells were cultured for another 24 hrs, before incubation with Apt. #9 or ssDNA #12 and then inspection by confocal laser scanning microscopy.
Confocal laser scanning microscopy (CLSM) for evaluation of aptamer cell uptake
HeLa cells (HeLa ATCC® CCL-2™) were cultured in RPMI-1640 (contain 10% FBS, 1% penicillin and 1% streptomycin). Before the confocal imaging experiment, cells were seeded at desired concentrations in confocal dishes (NEST Biotechnology Co. Ltd). After cultured for 24 hrs, they were washed twice with 1× PBS, then incubated with 3 μM Apt. #9 or the non-binding ssDNA #12, both labeled with Alexa Fluor488, for 3 hrs at 37°C. Two washes were applied before the live cells were imaged by the LSM 880 with Airyscan Microscope (Zeiss). The dye labels on the DNA strands were excited at 488-nm by an argon ion laser and the emission was collected at 500–600 nm. Nuclei were stained by Hoechst 33342 which was excited with a 355 nm UV laser and the emission was collected at 400–480 nm. For the transfection sample, mRuby was excited at 561 nm and the emission was collected at 570–650 nm. These images were stacked and reconstructed by ZEN software. For co-localization analysis, the high resolution Airyscan model was employed for imaging; and the images were analyzed using the colocalization analysis package plugin of ImageJ.
Cell proliferation inhibition assay
In order to evaluate the effect of Apt. #9 on cell proliferation, we treated cells with different concentrations of Apt. #9 for 24 h. Briefly, HeLa and HEK293 cells were cultured in DMEM medium (Corning) containing 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Corning), MCF-10A cell was cultured in DMEM/F12 (Gibco) medium with 5% Horse serum and 0.25% insulin (Corning). The stock solutions were prepared by dissolving the freeze-dried oligonucleotides in PBS and stored at −20°C. The concentrations were measured based on their absorbance at 260 nm and their molar extinction coefficients. Before used, all oligonucleotides were heat-denatured at 95°C for 5 min, and then placed on the ice for 10 min, annealed at room temperature for 30 min, then kept at 4°C overnight to form stable secondary structures. Cells were seeded in 96-well plates (Corning) with 100 μL per well at 104 cells/mL density, after 24-hr culturing, the oligonucleotides were added into each well to get a desired final concentration. To ensure the results’ reliability, each concentration was repeated three wells and each experiment performed at least twice independently. After culturing for another 24 h, cell viability was measured by Cell Counting Kit-8 (CCK-8, Dojindo, Japan) assay according to the protocol. Briefly, the old culture medium was removed by pipetting, 10 μL of CCK-8 was then added into each well with 100 μL of basic medium, and incubated with cells for 0.5–4 hrs. The absorbance was measured on a microplate reader at 450 nm.
Quantification of 5-methyl-2′-deoxycytidine (5-mdC) and 2′-deoxyguanosine (dG) in genomic DNA
HeLa cells were cultured in a six-well plate and incubated with 3 μM Apt. #9 for various duration. Subsequently, the genomic DNA was isolated using a high-salt method. Briefly, the cells were washed with 1× PBS twice, then lysed by 0.5 mL of the lysis buffer containing 20 mM Tris (pH 8.0), 20 mM EDTA, 400 mM NaCl, and 1% SDS (w/v). Next, 25 μL of proteinase K (20 mg/mL) was added and the sample was incubated in a water bath at 55°C overnight. Afterwards, 0.5 volume of the saturated NaCl solution was added to the sample which was vortexed for 1 min and then incubated at 55°C for 15 min. A centrifugation step of 30 min at ∼10 000 rpm was applied to remove the cell debris. The supernatant was collected and subjected to another round of centrifugation. The nucleic acids in the supernatant were then precipitated by ethanol; and the resultant nucleic acids were mixed with 2.5 μL of RNase A (10 mg/ml) and 1 μL of RNase T1 (25 units/μL), and incubated at 37°C for 1 hr. The digested products were precipitated from the aqueous layer by ethanol and isolated by a 15-min centrifugation at 7000 rpm. The sample was air-dried at room temperature for overnight until the ethanol was totally sublimated, and then dissolved in TE buffer. The amount of DNA was quantified by using Nanodrop (Thermo technology).
The measurement procedures for 5-mdC and dG in genomic DNA were described previously (34–36). Briefly, 1 μg genomic DNA prepared from cells was enzymatically digested into nucleoside mixtures. Enzymes in the digestion mixture were removed by chloroform extraction, and the resulting aqueous layer was concentrated to 10 μl and subjected directly to LC–MS/MS/MS analysis for quantification of 5-mdC and dG, where an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) coupled with an Agilent 1200 capillary HPLC pump was employed for the quantification. The amounts of 5-mdC and dG (in moles) in the nucleoside mixtures were calculated from area ratios of peaks found in selected-ion chromatograms (SICs) for the analytes over their corresponding isotope-labeled standards, the amounts of the stable isotope-labeled standards added (in moles) and the calibration curves. The final levels of 5-mdC, in terms of percentages of dG, were calculated by comparing the moles of 5-mdC relative to those of dG.
RESULTS
Aptamers against DNMT1 selected by AF4-SELEX
The process of AF4-SELEX is illustrated in Figure 1A. Each SELEX cycle starts by injecting the ssDNA library and target to AF4. The focusing step of AF4 (Supplementary Figure S1) helps concentrate the target and ssDNA and thus formation of the DNA–target complex. The free ssDNAs are separated from the target-bound ones because of their smaller sizes. Because of the high flow rates (typically in 0.3–1 mL/min) used in AF4, the collection volume could be as large as 1 mL, which cannot be directly used for PCR amplification of the obtained ssDNAs. Thus, the electrospun TiO2 microfibers are employed to collect the ssDNAs from the AF4 eluent (Supplementary Figure S2). These fibers have been developed by our group to recover > 90% ssDNAs from solutions utilizing the strong affinity between Ti(VI) and the phosphate backbone of ssDNA (37). The recovered ssDNAs are then eluted in a small volume of Tris-HCl for PCR. The feasibility of AF4-SELEX in aptamer discovery was confirmed using IgE as the protein target and its aptamer (Supplementary Figure S3).
We hypothesized that, using the catalytic domain of DNMT1 as the SELEX target can increase the chance of identifying aptamers with inhibitory function for DNMT1, because the aptamer can compete with the native enzyme substrate for binding to the catalytic domain and displace the substrate from the enzyme. Thus, the active fragment of human DNMT1 (residues 728–1600) spanning a pair of bromo-adjacent-homology (BAH) domains and the methyltransferase domain was used in the present work. The ssDNA library with a random sequence of 20-nt (Supplementary Table S1) and DNMT1 were sequentially injected, so that the impurities in the ssDNA pool were removed through the membrane wall of AF4 during sample focusing, and would not have any impact on the structure of DNMT1. The DTT-containing Tris buffer employed in AF4 separation was adopted from that used in DNMT1 expression and purification, and kept at 4°C during the sample injection and focusing steps. These conditions were found necessary to obtain a decent peak of DNMT1 in AF4 (Supplementary Figure S4).
A DNMT1-bound ssDNA peak was observed starting from the 1st cycle; and continued to increase as SELEX proceeded (Figure 2A). The complex elution window was confirmed by determining the free ssDNA and the DNA dimer peaks via injection of only the enriched ssDNA pool (red trace in Figure 2A). To enhance selection stringency, interfering molecules—a mixture of non-target proteins—were co-injected with DNMT1 starting from the fifth cycle of SELEX; and the amount of DNMT1 injected was reduced by half in the ninth cycle. Moreover, the preferred substrate of DNMT1, hemiDNA (38), was added to the ssDNA library starting from the 6th cycle to induce competitive binding between the aptamer and the native substrate. Close to 50% of the ssDNAs in the pools collected from the 10th and 11th cycles of SELEX were bound to DNMT1. Sequencing these ssDNA pools identified 4 candidates (Apt. #1, #2, #3 and #4) that were gradually enriched in the later SELEX cycles, and present at the highest abundance among all sequences (Supplementary Table S2). In addition, strands with sequences different by only 1–4 nt from these top 4 candidates were also identified at relatively higher frequency in both pools. These evidences strongly support that these four sequences (Apt. #1, #2, #3, and #4) are aptamers with good affinity for DNMT1.
Figure 2.

(A) The elution traces of the FAM-labeled ssDNAs collected during the 11 cycles of AF4-SELEX. The symbols of, ♦ and ▾ label the peaks of the free ssDNAs, the dimers of ssDNAs, and the DNMT1-bound ssDNAs, respectively. (B) Target binding evaluation of identified Aptamer (Apt.) #1, #2, #3 and #4 (sequences listed in Supplementary Table S2) by AF4.
Aptamer characterization and optimization
DNMT1 binding of the four aptamers by AF4-SELEX was first evaluated by AF4. Indeed, when 1 μM individual aptamer was injected with 0.6 μM DNMT1 in the presence of 1 mg/L of the non-DNMT protein mixture and 0.3 μM hemiDNA, most of the aptamers were bound to DNMT1, with the free aptamer peak almost completely disappearing (Figure 2B). Binding to DNMT1 by Apt. #1 and Apt. #2 was also confirmed using the conventional tool of EMSA (Supplementary Figure S5). We then optimized the sequences of the aptamers to identify the most effective regions for DNMT1 binding (Supplementary Table S3). Several of the modified strands showed decent binding towards DNMT1, especially the four truncated sequences (Apt. #8-#11) which exhibited comparable degree of target binding with their full-length sequences (Supplementary Figure S6). Among them, Apt. #9, one of the shortened versions of Apt. #2, displayed the highest DNMT1-bound ratio among all modified strands, and thus its binding features were evaluated in details. Interestingly, although shorter, the affinity of Apt. #9 for DNMT1 was slightly stronger than that of Apt. #2 (Figure 3A), with the dissociation constant (Kd) found to be 0.77 ± 0.11 and 1.8 ± 0.34 μM for Apt. #9 and #2, respectively (39).This result indicates that such a sequence revision has preserved the most effective binding structure.
Figure 3.

(A) Affinity comparison for Apt. # 2 and #9 binding to DNMT1, measured by fixing the aptamer concentration at 0.4 μM and increasing that of DNMT1. (B) Secondary structures of Apt. #9 obtained by Mfold. (C) Affinity of the native substrate of hemiDNA to DNMT1 with or without the presence of 0.4 μM Apt. #9, measured by fixing the DNMT1 concentration at 0.4 μM and increasing the concentration of hemiDNA up to 4 μM. (D) Methylation activity of DNMT1 on hemiDNA measured by mixing 0–10 μM Apt. #9 or the non-binding ssDNA with 0.1 μM DNMT1 and 0.4 μM hemiDNA. No effect was observed from Apt. #9 to the activity of DNMT3A or 3B when mixed with their corresponding substrate, CG-DNA. (E) Methylation rate of DNMT1 on hemiDNA, in the presence or absence of Apt. #9, was plotted as a function of hemiDNA concentration. (F) The steady-state Michaelis–Menten parameters of DNMT1-catalyzed methylation on hemiDNA, derived from (E). Error bars indicate mean±standard deviation (s.d.) (n = 2 for 3A and C; n = 3 for 3D–F).
Four other truncated strands (ssDNA#5–#7 and #12) totally lost the capacity of binding to DNMT1 (Supplementary Figure S6). Since the folded structures of aptamers are key determinants for their target binding capability in general, to better understand the structure-protein binding relationship of the aptamers found in our work, we investigated the potential folding of the ssDNA sequences tested above using the Mflod web application (40). We found that, the four long aptamers identified from AF4-SELEX (Apt. #1–4) all could fold into structures containing multiple stem-loop structures with comparable free energies (Supplementary Figure S7A). More interestingly, the shortened aptamers (Apt. #8–11) also preserve one stem-loop structure from the original sequences, with a stem of 6–8 nt and a loop of 7–8 nt (Supplementary Figure S7B). On the other hand, the non-binding, modified strands either have a bulge of three mismatched bases on their stems (Modified #5 and #12) or a very small loop of 4-nt (Modified #6 and #12) (Supplementary Figure S7B). ssDNA #7 is the weakest binder among all eight aptamers tested. Despite having a stem–loop structure similar to that of Apt. #11, Apt. #7 is shorter than Apt. #11 by 9-nt non-stem–loop sequence, which may contribute to the binding between Apt. #11 and DNMT1 through electrostatic interaction.
These results are consistent with the previous finding that RNAs with stem–loop structures may serve as strong binders of DNMT1 (41). To further inspect the importance of the stem-loop structure in DNMT1 binding, we derived a few DNA sequences from Apt. 9, which contains a stem of 8-bp and a loop of 8-nt (Figure 3B), by varying the size of its stem and loop region (Supplementary Figure S8 and Table S4). Interestingly, increasing the stem size to 10 bp, or introduction of two mismatched base-pairs to the 8-bp stem of Apt. 9 (through one G·C→C·C and one A·T→T·T mutation) did not affect the aptamer–DNMT1 interaction. In contrast, shortening the stem to 6 or 4 bp, or reducing the loop size to 5 or 3 nt drastically decreased the binding affinity of the aptamer; while the decrease was not as noticeable for a 12-nt loop or a stem with one mismatched base-pair. We also did a simple comparison for DNMT1 binding by the RNA sequence that was used in the previous study (R2) and our Apt. #9 using EMSA and found that, incubation of equal molar quantity of Apt. #9 with DNMT1 yielded 4 times more binding than with R2 (Supplementary Figure S9), for which a Kd of 0.045 μM was reported when binding to full-length DNMT1. More detailed investigation needs to be conducted in the future to affirmatively reveal the impacts from aptamer secondary structures to DNMT1 binding, and to compare the binding affinity of the aptamers discovered here with other reported nucleic acid structures in the context of full-length DNMT1. Still, these results indicate the proper sizes of the stem and loop region, as well as the overall length of the ssDNA are important determinants for high affinity to DNMT1.
Inhibition of DNMT1 activity by aptamer
Since we included the hemiDNA as the competing molecule in our SELEX, we reasoned this native substrate of the enzyme would have little influence on the binding between the aptamers and DNMT1. By EMSA assay, we found that Apt. #9 binds to DNMT1 about four times stronger than the hemiDNA (Figure 3A and C). Consistently, the presence of equimolar Apt. #9 significantly decreased the affinity of the hemiDNA for DNMT1 (Figure 3C and Supplementary Figures S10 and S11). To further illustrate the competitive binding between Apt. #9 and hemiDNA, we incubated various concentrations (0.1, 0.4 and 2 μM) of Apt. #9 with the complex formed between 0.4 μM DNMT1 and hemiDNA. We found that 0.1 μM Apt. #9 was sufficient to decrease the amount of the hemiDNA–DNMT1 complex by 50%, and higher Apt. #9 concentration caused further reduction of the hemiDNA-DNMT1 complex (Supplementary Figure S12). On the other hand, increasing amount of the Apt. #9–DNMT1 complex was formed with increasing concentration of Apt. #9 (Supplementary Figure S12). These results support that Apt. #9 can compete with the hemiDNA for binding to DNMT1 and displace it from DNMT1.
The capability of outcompeting the substrate in binding to the enzyme indicates the potential of Apt. #9 in inhibiting the methylation activity of DNMT1. We thus performed DNA methylation assays for DNMT1 in the presence of various amounts of Apt. #9 or ssDNA #12, which is a truncated form of Apt. #1 but showed no DNMT1-binding activity. As shown in Figure 3D, the activity of DNMT1 was gradually decreased with increasing amount of Apt. #9, with an IC50 value of 0.20 μM, indicating that Apt. #9 is an effective inhibitor of DNMT1. In fact, detailed steady-state kinetics analysis (Figure 3D) indicated that the presence of two-fold molar excess of Apt. #9 increased the KM value of DNMT1-mediated methylation from 27.8 to 212 μM, but did not lead to any appreciable change for the kcat, which further supports the role of Apt. #9 as the substrate competitive inhibitor (Figure 3E and F). More interestingly, Apt. #9 could not inhibit DNMT3A and DNMT3B-mediated de novo methylation (Figure 3D), nor did we detect any binding between Apt. #9 and these two enzymes (Supplementary Figure S13A), proving that it is a specific inhibitor for DNMT1. By contrast, the non-binding ssDNA #12 failed to affect the DNMT1-mediated methylation appreciably (Figure 3D); and no methylation activity was observed on Apt. #9 with any of the DNMT enzyme (Supplementary Figure S13B).
Cell entry and inhibition of cellular DNMT1 activity
In order to achieve in vivo inhibition, the aptamer should be able to bind to intracellular targets. Previous works have found that aptamers with stem–loop structures can enter cells by simple aptamer-cell incubation (42–44). In this regard, the half-life of the non-binding ssDNA #12 and Apt. #9 were around 8 and 11 hrs, respectively, in the cell culture media (Supplementary Figure S14). We therefore incubated them with HEK 293T cells transfected with the DNMT1 plasmid (pKanCMV_mRuby_DNMT1, full length) for 3 h, which led to significant cellular uptake and co-localization with both the expressed DNMT1 and Hoechst 33342 stain (for nuclei location) for Apt. #9, but not for the non-binding ssDNA#12 (Supplementary Figure S15A). To further confirm the co-localization of the aptamer and DNMT1, we treated the cells with 25 μM 2′-deoxycytidine to synchronize them in the S phase. Inspection of the nucleus region of the cell clearly indicates that the majority of the green Alexa 488-labeled Apt. #9 overlapped with the mRuby-labeled DNMT1 (red spots), and all of the Apt. #9 and DNMT1 molecules located inside the nucleus (Figure 4A). There remained a few red spots that did not overlap with the green ones, which may be attributed to the unsaturated binding of DNMT1 under this over-expression condition and the dynamics of nucleus entry of Apt. # 9. We further quantified the degree of colocalization by obtaining the Pearson's correlation coefficient (PCC) (45) and the overlap coefficient (OC) using the colocalization analysis package plugin of ImageJ (Supplementary Figure S15B). The PCC and OC were found to be 0.7932 and 0.9307 in Figure 4A, respectively, indicating a high level of co-localization of these two molecules.
Figure 4.

(A) Alexa Fluor 488 (green)-labeled Apt. #9 (bottom row) bound to the mRuby (red)-tagged DNMT1 in the nucleus of the transfected HEK293T cell detected by confocal laser scanning microscope in the high-resolution Airyscan mode; while no nuclear enrichment was detected for the Alexa Fluor 488-labeled non-binding ssDNA #12 (upper row). Blue – Hoechst 33342 stain for cell nuclei. Scale bar – 5 μm. (B) Cell viability measured after 24-h incubated with Apt. #9 or ssDNA #12 in various cell lines (n = 3 biological repeats; **P < 0.01; ***P < 0.005). (C) Significant reduction of DNA methylation level in HeLa cells by incubation with 3 μM Apt. #9 for up to 12 h (n = 3 biological repeats; *P < 0.05).
The fact that Apt. #9 demonstrates a great potential in binding to DNMT1 in vitro prompted us to further evaluate the impact of Apt. #9 on the DNA methylation level in cells with endogenous DNMT1. We first tested cellular uptake of these two strands in non-transfected cell lines, including the non-transfected HEK293T cells, HeLa cells, and MCF-10A cells. Overexpression of DNMT1 in HeLa (46) and HEK293T cells (47), but low expression in MCF-10A cells (48) has been reported. Indeed, aptamer uptake and colocalization with DAPI in the nucleus was only observed in the HeLa or HEK293T cells; but not in MCF-10A cells (Supplementary Figure S16). Interestingly, in agreement with these cell uptake results, we observed about 10–15% reduction in the cell survival rate at the higher Apt. #9 concentrations of 2 and 3 μM in HeLa and HEK293 cells. Compared to the non-tumor cells (i.e. MCF-10A) and the treatment with the non-binding ssDNA #12, the reduction is significant with a P value < 0.01 for HeLa cells, and < 0.001 for HEK293 cells with 3 μM Apt. #9 (Figure 4B). In contrast, the non-binding ssDNA #12 did not induce any cell toxicity.
With significant cellular uptake of Apt. #9 by HeLa cells observed, we then extracted the genomic DNA from the cells upon aptamer treatment, and digested with nuclease P1 and alkaline phosphatase. The levels of 5-methyl-2′-deoxycytidine and 2′-deoxy-guanosine in the resulting nucleoside mixture were quantified by LC–MS/MS/MS (34,36). Strikingly, incubation with 3 μM Apt. #9 for 12 hrs led to a significant decrease in the methylation level by >40% in HeLa cells (105 cells/mL) (Figure 4C). The reduction should not be due to cell death, because >90% of the cells remained viable under this treatment condition (Figure 4B). Since the 12 h-duration corresponds to less than one cell division, such a high reduction in the DNA methylation level suggests a nearly complete inhibition of DNMT1. To further examine this result, we conducted the restriction enzyme digestion experiment on the DNA extracted from HeLa cells using the methylation-sensitive enzyme HpaII and the methylation-insensitive enzyme MspI. The growth of HeLa cells was synchronized with 10 μM Lovastatin, and the cells were treated with Apt. #9 for 12 and 24 h (the aptamer was replenished after 12 h to avoid reduction of aptamer concentration due to degradation). The DNA digest was analyzed by gel electrophoresis. Our result indicates that, the restriction enzyme HpaII barely digested the genomic DNA extracted from the cells treated for 12 h, but 24-h treatment with Apt. #9 induced significant cleavage of the extracted DNA by HpaII (Supplementary Figure S17), indicating a decrease in the methylation level. Furthermore, we performed fluorescence-based measurement of the intracellular concentration of the aptamer after 3-hr incubation, and found it to be close to 5.5 μM (Supplementary Figure S18). With the Kd value of 0.77 μM reported in Figure 3A, we calculated the bound ratio for DNMT1 in each cell based on the 1:1 binding stoichiometry and the binding equilibrium, which was about 87%. These results corroborate our LC–MS/MS data and suggest an almost complete inhibition of DNMT1. In contrast, incubation with the non-binding ssDNA #12 did not yield any increase in fluorescence when the cells were treated and measured under the conditions.
Mapping of Apt. #9-binding site on DNMT1
To better understand the inhibitory activity of Apt. #9 on DNMT1, we explored the binding site of Apt. #9 on DNMT1 with limited proteolysis, in which the kinetics of proteolytic cleavage of the protein is measured to reveal the potential binding interface of protein-interacting molecules. We found that proteolysis of the peptide region of a.a. #1100–1240 of DNMT1 was significantly reduced upon aptamer binding (Supplementary Figure S19A). The peak areas of the corresponding peptides detected by LC–MS/MS decreased by more than 8-fold in the presence of Apt. #9 (Supplementary Figure S19B), indicating potential blockage of protease access upon binding by Apt. #9. Structural analysis of DNMT1 in complex with hemimethylated DNA (24) indicated that this segment is located in the DNA-contacting surface of the catalytic domain, permitting us to generate a structural model in which the Apt. #9 is docked near to the catalytic center of DNMT1 (Figure 5), thereby explaining the inhibitory effect on DNMT1. Crystal structure of the aptamer-DNMT1 complex needs to be acquired in the future to reveal unambiguously the binding sites and the possible inhibitory mechanism.
Figure 5.
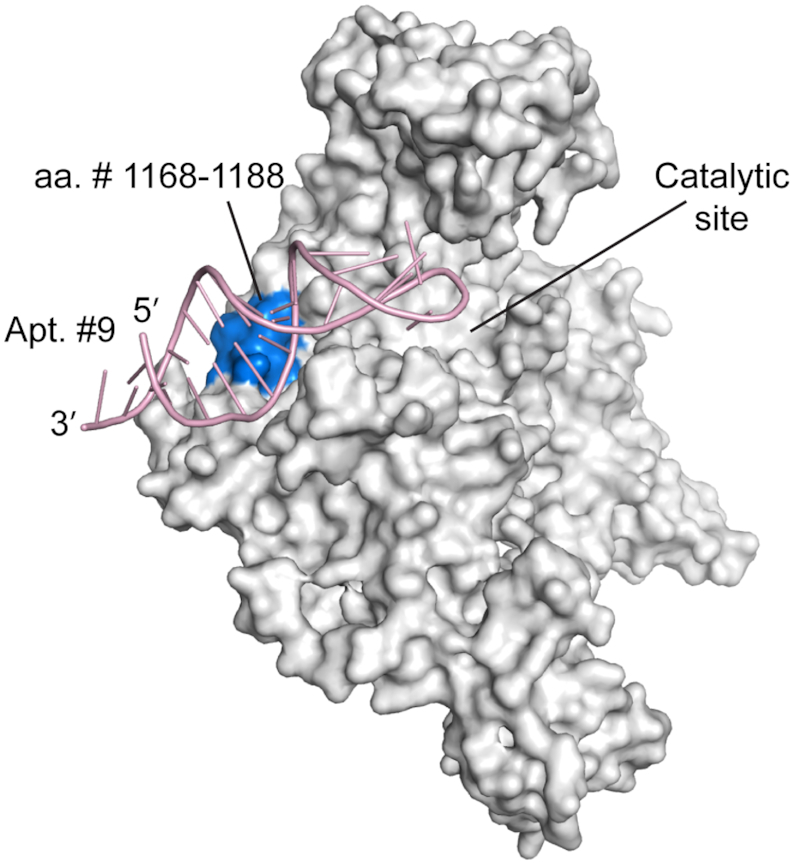
The structural model of human DNMT1 (728-1600)-Apt. #9 complex proposed based on the proteolysis result. The protein crystal structure was adapted from that of human DNMT1 (351-1600) (PDB: 4WXX).
DISCUSSION
Aberrant DNA methylation serves as a hallmark of cancer, with a prominent feature of global hypomethylation but regional hypermethylation of gene promoters associated with tumor suppressor genes (44). Therefore, development of DNMT-specific inhibitors has become an important strategy for cancer therapy. Our previous studies provided structural basis for the recognition between DNMT1 and the double-stranded DNAs (22,24). However, increasing evidence has indicated that single-stranded DNA or RNA molecules may serve as strong DNMT1 binders in vitro and in cells (41,49). For instance, as mentioned above, a regulatory RNA from the CEBPA locus, with a stem-loop structure, has been shown to interact with DNMT1 to block its DNA methylation activity around the CEBPA promoter (49). Through an independent SELEX approach, this study identified DNA aptamers as potent binders and DNA-competitive inhibitors of DNMT1. Intriguingly, the stem-loop structural feature of these aptamers (Figure 3B; Supplementary Figures S7 and S8) appears to be important for the DNMT1-inhibitor interaction, implying a similar mode of inhibition as that of cellular RNAs. Affinity measurement (Figure 3A), substrate binding analysis (Figure 3C; Supplementary Figures S10–S12), and enzymatic inhibition study (Figure 3D–F) conducted in the present work strongly support that, the identified aptamer inhibits DNMT1 activity by competing with the native DNA substrate for DNMT1 binding. Importantly, the structural complexity of the DNA aptamer permits its discrimination of DNMT1 from the de novo DNA methyltransferases DNMT3A and DNMT3B (Supplementary Figure S13), thereby providing a DNMT1-specific inhibition mechanism, a trait that was not attainable with currently clinical DNMT1 inhibitors. Moreover, since aptameric inhibitors exert their inhibition function through non-covalent but selective binding to DNMT1, this approach provides high target specificity and reversibility to reduce DNMT1 degradation and cytotoxicity.
Our work also proves that, the aptamer not only performs well in in vitro inhibition of DNMT1 activity, but also can enter the cell nuclei readily and co-localize with DNMT1 (Figure 4A), probably owing to its stem-loop structure that enhances membrane penetration as well as its high affinity to DNMT1 that prevents it from diffusing out of the nuclei. The aptamer exhibits excellent inhibitory function for DNMT1 in cells, as demonstrated in our restriction enzyme digestion experiment (Supplementary Figure S17) and LC–MS/MS/MS analysis of the global methylation level in cellular DNA (Figure 4C). Some cytotoxicity was observed in cells with higher DNMT1 expression and thus higher aptamer uptake (Figure 4B). Altogether, the cellular experiments support the aptamers discovered in this study could be effective tools for functional interrogation of DNMT1. They have good biocompatibility and high aqueous solubility (50–52). Additionally, the structures of aptamers can be chemically modified to improve stability in physiological environment and binding affinity (53,54), providing promises in being used in low-dose to minimize side-effects. Moreover, under proper conditions, aptamers could penetrate biological barriers more easily than proteins because of their smaller physical sizes; and are lack of immunogenicity due to the absence of T cell-dependent immunity (14,15). These properties are favorable if aptamers are used as therapeutics. Thus, future studies could explore the potential of the DNMT-inhibiting aptamers being anti-tumor drugs.
The capability of our aptamers in effectively competing with the native DNA substrate for DNMT1 binding and thus inhibiting its activity is the result of our strategic design of the SELEX procedure, which used the catalytic domain of DNMT1 as the target for aptamer selection and included the native substrate as the competing molecule during SELEX. Both were enabled by the approach of AF4-SELEX that offers the advantage of separating the protein-bound ssDNAs from the free ones using only liquid flows and an unpacked channel (29,30,55). Its open separation channel and high compatibility with physiological buffers well maintain the native structure of DNMTs to ensure good in vivo activity of the discovered aptamers; and exert negligible detrimental force to the aptamer-target complex, improving collection of the high-affinity strands during the early selection cycles (32,33,56,57). AF4-SELEX is suitable for DNMT1-like protein targets whose protein stability is strongly affected by the environmental conditions, representing a valuable tool for aptamer discovery. We expect with the aid of this tool, aptamers selective for other DNMT proteins, like DNMT3A and 3B, and other domains of DNMT1 can be discovered for modulation of DNA methylation activities in cells.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are also thankful for Dr Tao Bing for advice on SELEX.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01CA188991 to W.Z., R35GM119721 to J.S., R01CA210072 to Y.W.]; Research Training Grant in Environmental Toxicology from the National Institute of Environmental Health Sciences [T32ES018827 to G. B. A.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Li E., Beard C., Jaenisch R.. Role for DNA methylation in genomic imprinting. Nature. 1993; 366:362–365. [DOI] [PubMed] [Google Scholar]
- 2. Razin A., Riggs A.D.. DNA methylation and gene function. Science. 1980; 210:604–610. [DOI] [PubMed] [Google Scholar]
- 3. Walsh C.P., Chaillet J.R., Bestor T.H.. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998; 20:116–117. [DOI] [PubMed] [Google Scholar]
- 4. Ehrlich M., Gama-Sosa M.A., Huang L.H., Midgett R.M., Kuo K.C., McCune R.A., Gehrke C.. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982; 10:2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goll M.G., Bestor T.H.. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005; 74:481–514. [DOI] [PubMed] [Google Scholar]
- 6. Baylin S., Bestor T.H.. Altered methylation patterns in cancer cell genomes: cause or consequence. Cancer Cell. 2002; 1:299–305. [DOI] [PubMed] [Google Scholar]
- 7. Feinberg A.P., Tycko B.. The history of cancer epigenetics. Nat. Rev. Cancer. 2004; 4:143–153. [DOI] [PubMed] [Google Scholar]
- 8. Gjerstorff M.F., Burns J., Ditzel H.J.. Cancer-germline antigen vaccines and epigenetic enhancers: future strategies for cancer treatment. Expert Opin. Biol. Ther. 2010; 10:1061–1075. [DOI] [PubMed] [Google Scholar]
- 9. Subramaniam D., Thombre R., Dhar A., Anant S.. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014; 4:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santi D.V., Norment A., Garrett C.E.. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:6993–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghoshal K., Datta J., Majumder S., Bai S., Kutay H., Motiwala T., Jacob S.T.. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005; 25:4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Foulks J.M., Parnell K.M., Nix R.N., Chau S., Swierczek K., Saunders M., Wright K., Hendrickson T.F., Ho K.K., McCullar M.V. et al.. Epigenetic drug discovery: targeting DNA methyltransferases. J. Biomol. Screen. 2012; 17:2–17. [DOI] [PubMed] [Google Scholar]
- 13. Liu J., Cao Z., Lu Y.. Functional nucleic acid sensors. Chem. Rev. 2009; 109:1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang X., Tan W.. Aptamers generated from cell-SELEX for molecular medicine: a chemical biology approach. Acc. Chem. Res. 2010; 43:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keefe A.D., Pai S., Ellington A.. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010; 9:537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 17. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 18. Berezovski M.V., Lechmann M., Musheev M.U., Mak T.W., Krylov S.N.. Aptamer-facilitated biomarker discovery (AptaBiD). J. Am. Chem. Soc. 2008; 130:9137–9143. [DOI] [PubMed] [Google Scholar]
- 19. Meng H.M., Liu H., Kuai H., Peng R., Mo L., Zhang X.B.. Aptamer-integrated DNA nanostructures for biosensing, bioimaging and cancer therapy. Chem. Soc. Rev. 2016; 45:2583–2602. [DOI] [PubMed] [Google Scholar]
- 20. Gotrik M.R., Feagin T.A., Csordas A.T., Nakamoto M.A., Soh H.T.. Advancements in Aptamer Discovery Technologies. Acc. Chem. Res. 2016; 49:1903–1910. [DOI] [PubMed] [Google Scholar]
- 21. Liu X., Li H., Jia W., Chen Z., Xu D.. Selection of aptamers based on a protein microarray integrated with a microfluidic chip. Lab Chip. 2016; 17:178–185. [DOI] [PubMed] [Google Scholar]
- 22. Song J., Rechkoblit O., Bestor T.H., Patel D.J.. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011; 331:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeshita K., Suetake I., Yamashita E., Suga M., Narita H., Nakagawa A., Tajima S.. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9055–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song J., Teplova M., Ishibe-Murakami S., Patel D.J.. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012; 335:709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Z.M., Liu S., Lin K., Luo Y., Perry J.J., Wang Y., Song J.. Crystal Structure of Human DNA Methyltransferase 1. J. Mol. Biol. 2015; 427:2520–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis K.A., Abrams B., Lin Y., Jayasena S.D.. Use of a high affinity DNA ligand in flow cytometry. Nucleic Acids Res. 1996; 24:702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendonsa S.D., Bowser M.T.. In vitro evolution of functional DNA using capillary electrophoresis. J. Am. Chem. Soc. 2004; 126:20–21. [DOI] [PubMed] [Google Scholar]
- 28. Mendonsa S.D., Bowser M.T.. In vitro selection of aptamers with affinity for neuropeptide Y using capillary electrophoresis. J. Am. Chem. Soc. 2005; 127:9382–9383. [DOI] [PubMed] [Google Scholar]
- 29. Wahlund K.G. Flow field-flow fractionation: critical overview. J. Chromatogr. A. 2013; 1287:97–112. [DOI] [PubMed] [Google Scholar]
- 30. Wagner M., Holzschuh S., Traeger A., Fahr A., Schubert U.S.. Asymmetric flow field-flow fractionation in the field of nanomedicine. Anal. Chem. 2014; 86:5201–5210. [DOI] [PubMed] [Google Scholar]
- 31. Ashby J., Schachermeyer S., Duan Y., Jimenez L.A., Zhong W.. Probing and quantifying DNA-protein interactions with asymmetrical flow field-flow fractionation. J. Chromatogr. A. 2014; 1358:217–224. [DOI] [PubMed] [Google Scholar]
- 32. Mayer G. The chemical biology of aptamers. Angew. Chem. Int. Ed. Engl. 2009; 48:2672–2689. [DOI] [PubMed] [Google Scholar]
- 33. Ruff K.M., Snyder T.M., Liu D.R.. Enhanced functional potential of nucleic acid aptamer libraries patterned to increase secondary structure. J. Am. Chem. Soc. 2010; 132:9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Z.M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S.B., Wang Y. et al.. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018; 554:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volz D.C., Leet J.K., Chen A., Stapleton H.M., Katiyar N., Kaundal R., Yu Y., Wang Y.. Tris(1,3-dichloro-2-propyl)phosphate Induces Genome-Wide Hypomethylation within Early Zebrafish Embryos. Environ. Sci. Technol. 2016; 50:10255–10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu Y., Guerrero C.R., Liu S., Amato N.J., Sharma Y., Gupta S., Wang Y.. Comprehensive assessment of oxidatively induced modifications of DNA in a rat model of human Wilson's disease. Mol. Cell Proteomics. 2016; 15:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jimenez L.A., Gionet-Gonzales M.A., Sedano S., Carballo J.G., Mendez Y., Zhong W.. Extraction of microRNAs from biological matrices with titanium dioxide nanofibers. Anal. Bioanal. Chem. 2018; 410:1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hermann A., Goyal R., Jeltsch A.. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004; 279:48350–48359. [DOI] [PubMed] [Google Scholar]
- 39. Pagano J.M., Clingman C.C., Ryder S.P.. Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA. 2011; 17:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Di Ruscio A., Ebralidze A.K., Benoukraf T., Amabile G., Goff L.A., Terragni J., Figueroa M.E., De Figueiredo Pontes L.L., Alberich-Jorda M., Zhang P. et al.. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013; 503:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan Y., Shi Y.S., Wu X.D., Liang H.Y., Gao Y.B., Li S.J., Zhang X.M., Wang F., Gao T.M.. DNA aptamers that target human glioblastoma multiforme cells overexpressing epidermal growth factor receptor variant III in vitro. Acta Pharmacol. Sin. 2013; 34:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trinh T.L., Zhu G., Xiao X., Puszyk W., Sefah K., Wu Q., Tan W., Liu C.. A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLoS One. 2015; 10:e0136673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiang Q., Tan G., Jiang X., Wu K., Tan W., Tan Y.. Suppression of FOXM1 transcriptional activities via a single-stranded DNA aptamer generated by SELEX. Sci. Rep. 2017; 7:45377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gomes de Castro M.A., Hobartner C., Opazo F.. Aptamers provide superior stainings of cellular receptors studied under super-resolution microscopy. PLoS One. 2017; 12:e0173050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. You J.S., Kang J.K., Lee E.K., Lee J.C., Lee S.H., Jeon Y.J., Koh D.H., Ahn S.H., Seo D.W., Lee H.Y. et al.. Histone deacetylase inhibitor apicidin downregulates DNA methyltransferase 1 expression and induces repressive histone modifications via recruitment of corepressor complex to promoter region in human cervix cancer cells. Oncogene. 2008; 27:1376–1386. [DOI] [PubMed] [Google Scholar]
- 47. Cheng J., Yang H., Fang J., Ma L., Gong R., Wang P., Li Z., Xu Y.. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 2015; 6:7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elangovan S., Pathania R., Ramachandran S., Ananth S., Padia R.N., Srinivas S.R., Babu E., Hawthorn L., Schoenlein P.V., Boettger T. et al.. Molecular mechanism of SLC5A8 inactivation in breast cancer. Mol. Cell Biol. 2013; 33:3920–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mao S.Q., Ghanbarian A.T., Spiegel J., Martinez Cuesta S., Di Antonio M., Marsico G., Hansel-Hertsch R., Tannahill D., Balasubramanian S.. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018; 25:951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu G., Chen X.. Aptamer-based targeted therapy. Adv. Drug. Deliv. Rev. 2018; 134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou J., Rossi J.. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017; 16:181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou G., Latchoumanin O., Bagdesar M., Hebbard L., Duan W., Liddle C., George J., Qiao L.. Aptamer-based therapeutic approaches to target cancer stem cells. Theranostics. 2017; 7:3948–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gong P., Shi B.H., Zheng M.B., Wang B., Zhang P.F., Hu D.H., Gao D.Y., Sheng Z.H., Zheng C.F., Ma Y.F. et al.. PEI protected aptamer molecular probes for contrast-enhanced in vivo cancer imaging. Biomaterials. 2012; 33:7810–7817. [DOI] [PubMed] [Google Scholar]
- 54. Wang C.H., Kang S.T., Lee Y.H., Luo Y.L., Huang Y.F., Yeh C.K.. Aptamer-conjugated and drug-loaded acoustic droplets for ultrasound theranosis. Biomaterials. 2012; 33:1939–1947. [DOI] [PubMed] [Google Scholar]
- 55. Ashby J., Schachermeyer S., Duan Y., Jimenez L., Zhong W.. Probing and quantifying DNA-protein interactions with asymmetrical flow field-flow fractionation. J. Chromatogr., A. 2014; 1358:217–224. [DOI] [PubMed] [Google Scholar]
- 56. Mayer G. The chemical biology of aptamers. Angew. Chem. Int. Ed. 2009; 48:2672–2689. [DOI] [PubMed] [Google Scholar]
- 57. Ruff K.M., Snyder T.M., Liu D.R.. Enhanced functional potential of nucleic acid aptamer libraries patterned to increase secondary structure. J. Am. Chem. Soc. 2010; 132:9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.