


Abstract
Prokaryotes use CRISPR–Cas systems for adaptive immunity, but the reasons for the frequent existence of multiple CRISPRs and cas clusters remain poorly understood. Here, we analysed the joint distribution of CRISPR and cas genes in a large set of fully sequenced bacterial genomes and their mobile genetic elements. Our analysis suggests few negative and many positive epistatic interactions between Cas subtypes. The latter often result in complex genetic organizations, where a locus has a single adaptation module and diverse interference mechanisms that might provide more effective immunity. We typed CRISPRs that could not be unambiguously associated with a cas cluster and found that such complex loci tend to have unique type I repeats in multiple CRISPRs. Many chromosomal CRISPRs lack a neighboring Cas system and they often have repeats compatible with the Cas systems encoded in trans. Phages and 25 000 prophages were almost devoid of CRISPR–Cas systems, whereas 3% of plasmids had CRISPR–Cas systems or isolated CRISPRs. The latter were often compatible with the chromosomal cas clusters, suggesting that plasmids can co-opt the latter. These results highlight the importance of interactions between CRISPRs and cas present in multiple copies and in distinct genomic locations in the function and evolution of bacterial immunity.
INTRODUCTION
CRISPR–Cas systems are an adaptive immune system that protects bacterial and archaeal cells from exogenous mobile genetic elements, such as phages (1–4). They are composed of a CRISPR and a cluster of cas genes. CRISPRs comprise two types of sequences: repeats and spacers. Repeats are short sequences (typically 20–40 bp) identical within a CRISPR. They are interspaced by short and diverse spacer sequences (typically 20–40 bp), which often match sequences from mobile genetic elements. The number of repeats in a CRISPR is an indicator of its activity, because arrays with many spacers can target a larger number of mobile genetic elements (5). The cluster of cas genes encodes the proteins involved in the three stages of CRISPR–Cas immunity: expression, interference, and adaptation (6). During expression, the CRISPR is transcribed and then processed into smaller RNAs called crRNA (CRISPR RNA), each carrying sequences from a repeat and a single spacer. Each of these crRNA serves as a guide for a complex of Cas proteins. If the sequence of a guide is complementary to another DNA sequence in the cell, for example from an infecting bacteriophage (phage), the complex will activate an immune response. For most types of CRISPR–Cas systems this leads to the cleavage and degradation of the invading DNA. During adaptation, a complex of Cas proteins (including Cas1 and Cas2) generates and then incorporates a new spacer in the CRISPR (6,7).
CRISPR–Cas systems are present in fewer than half of Bacteria and in most Archaea (8). They are extremely diverse and have been classified hierarchically according to the composition of the cluster of cas genes. They are grouped in two classes, six types (I to VI) and more than 20 subtypes (8–10). Novel types have been recently proposed (11,12), but they are rare and will thus not be analysed in this study. The last surveys of CRISPR–Cas systems abundance and diversity among fully sequenced bacterial genomes included 2740 and 2751 genomes (8,13). Makarova and colleagues detected 1949 distinct cas clusters and 4210 CRISPRs from 1302 genomes out of 2740. They could assign a subtype to 93% of the cas clusters. Similar results were found by other authors (13). The distribution of Cas types across Prokaryotes reveals that CRISPR–Cas systems are frequently horizontally transferred (8,14,15). They have been detected on diverse mobile genetic elements (MGE) like plasmids, phages or transposons (16–20), but a quantitative assessment of the frequency of CRISPR–Cas systems in MGEs is only now starting to emerge (21) The presence of CRISPR–Cas systems in MGEs singularly complexifies the role of these systems, because MGEs are targeted by chromosomal systems but MGEs may also bring into the genome novel systems (21). Furthermore, CRISPR–Cas systems in MGEs may not only be co-opted by the host to target other MGEs, but can also be used by the MGEs to co-opt the host systems for their own advantage (21).
The ability of CRISPR–Cas systems to acquire new spacers makes them very versatile, because the immune response can evolve in function of the repertoire of spacers, and because this repertoire can target numerous different MGEs. Nevertheless, some genomes have been found to encode several CRISPR–Cas systems (8). This is intriguing: why should a genome encode more than one adaptive system? The existence of anti-CRISPR that are system specific provides MGEs with tools to counteract the host CRISPR–Cas systems and may explain why some bacteria encode multiple systems (22,23). Functional interaction between systems could also improve immunity. This was recently demonstrated in Marinomonas mediterranea, which carries both a subtype I-F and a subtype III-B system (24). There, it was found that the type III-B system can use crRNAs from the type I-F system, enabling the same guide RNA to target phages with different interference modules. These different Cas interference complexes have distinct molecular requirements, thus limiting the emergence of phages escape mutants (24). Many cas genes are found near CRISPRs, but distant arrays (i.e. CRISPRs without neighboring cas genes) have also been identified (8,25). They can be processed by Cas proteins encoded in other regions of the genome (in trans) (7) or they may represent remnant systems.
Even though Cas proteins and CRISPRs are parts of one system and both elements are required for adaptation and interference, there have been few quantitative studies integrating information on both Cas proteins and CRISPRs. Here, we analyse the joint distribution of CRISPR and cas genes in a large set of fully sequenced bacterial genomes and their MGEs. We focus on genomes and loci encoding several of these elements to understand why they co-occur. Our results reveal preferential associations between certain systems, sometimes in complex genetic loci that constitute one single CRISPR–Cas system with one adaptation and several types of interference modules.
MATERIALS AND METHODS
Data
We analysed 13512 complete genomes retrieved in May 2019 from NCBI RefSeq representing 4010 and 220 species of Bacteria and Archaea (http://ftp.ncbi.nih.gov/genomes/refseq/bacteria/). These genomes contained 11805 plasmids that were used in this work. Because plasmids and chromosomes are associated to individual genomes, we know which of these elements co-occur. We retrieved 2498 complete phages genomes from NCBI RefSeq in May 2019. The lifestyle of these phages was predicted using PHACTS v.0.3 (26). Predictions were considered as confident if the average probability score of the predicted lifestyle was at least two standard deviations (SD) away from the average probability score of the other lifestyle, as recommended by the authors (who claim a precision rate of 99% with this parameter). Using these criteria, we classified 54% of the phages into 571 virulent and 779 temperate phages.
Detection of prophages
Putative prophages were detected using VirSorter v.1.0.3 (27) with the RefSeqABVir database in all the complete genomes. The least confident predictions, i.e., categories 3 and 6, which may be prophage remnants or erroneous assignments, were excluded from the analyses. We found 26 987 putative prophages, with a size ranging from 5 to 250 kb, and an average around 42 kb, which is consistent with our previous analyses (28).
CRISPRs and cas clusters detection
CRISPR arrays and cas genes clusters were detected with CRISPR–CasFinder v.4.2.19 (29) (including MacSyFinder v.1.0.2 (30) and CasFinder v.2.0.2 (30)). The program is available at https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index. Only CRISPR arrays with 3 spacers or more were used in our analyses. All results are reported in Supplementary Table S1 and S2.
Linking CRISPRs and cas clusters
In order to be able to associate CRISPRs with cognate cas clusters, we calculated the minimal circular distance between an array and a cluster. When CRISPR and cas clusters were at distances lower than 20 kb, they were put together in one single CRISPR–Cas locus. The clustering was done by transitivity, if other elements, CRISPRs or cas clusters, were less than 20 kb away from the locus, they were also assigned to the locus. Hence, a CRISPR–Cas locus is a region in the chromosome containing at least one cas cluster and one CRISPR, where the elements are never more than 20 kb from the closest element. A specific case occurs when one or several CRISPR are close to one single cas cluster in a CRISPR–Cas locus. In this case, we assigned a subtype to the CRISPR array (the one of the single cas cluster). Subtypes could not be assigned with this method to CRISPRs outside CRISPR–Cas loci neither to CRISPRs in loci containing more than one subtype of cas clusters.
To assign a type to the CRISPRs outside CRISPR–Cas loci, we searched for similarities between their repeats and those of CRISPRs that could be typed with the method described above. We used the information on the CRISPR subtypes, taken from the CRISPR–Cas loci with a single Cas subtype, to build a databank of 4979 unique repeats (direct and reverse complement sequences) that we could confidently assign to specific Cas subtypes. This was then used to type the other repeats (those in the remaining CRISPRs). For this, we quantified the sequence similarity between every pair of CRISPR repeats using a global alignment with no gap end penalty and equal gap creation and extension penalties (−3) using the module pairwise2 from Biopython (function align.globalxs). As a result of this procedure, we obtained a table where each pair of repeats is associated with a continuous variable indicating the sequence identity between the two repeats and with a binary variable indicating if the two repeats are from the same Cas subtype. We used this data to make a logistic regression between the identity score of the best hit and the categorical variable assigning the subtype prediction. We used a ROC curve to choose a threshold with a high True Positive Rate (83%). At this threshold, if the best hit among the repeats of an unknown type CRISPR matches a repeat of a CRISPR of a given subtype with >74% identity, the first CRISPR is classed as of the same subtype as the second.
Having defined a minimal sequence identity to class a repeat into a Cas subtype, we used it to assign subtypes to the CRISPRs. For each array, we quantified the sequence identity of its repeats with all repeats of the typed CRISPR repeats. We used a global alignment with no gap end penalty and equal gap creation and extension penalties (−3) using the module pairwise2 from Biopython (function align.globalxs). We took the best hit among those scores. If the identity score was higher than 74%, we classed the array of repeats to the subtype of the best hit.
Phylogenetic analyses
We identified the families of orthologous proteins present in >90% of the genomes (when larger than 1 Mb) of two phyla: Firmicutes (3003 genomes), and Proteobacteria (6986 genomes). The genomes were obtained from GenBank's RefSeq dataset as indicated above. The orthologs were identified as reciprocal best hits using an end-gap free global alignment, between the proteome of a pivot and each of the other strain's proteomes (as in (31)). Escherichia coli K12 MG1655 and Bacillus subtilis str.168 were used as a pivot for each clade. Hits with fewer than 37% similarity in amino acid sequence and >20% difference in protein length were discarded. The persistent genome of each clade was defined as the intersection of pairwise lists of orthologs that were present in at least 90% of the genomes representing 435 protein families for Firmicutes and 387 for Proteobacteria.
We inferred phylogenetic trees for each phyla from the concatenate of the multiple alignments of the proteins encoded by persistent genes, which were obtained with MAFFT v.7.313 (with default options) (32). Alignments were purged of poorly informative sites using BMGE v1.12 (with default options) (33). Missing genes were replaced by stretches of ‘-’ in each multiple alignment. Adding a small number of ‘–’ has little impact on phylogeny reconstruction (34). The length of the resulting multiple alignment was 104 850 residues for Firmicutes and 100 401 residues for Proteobacteria. The trees of the phyla were computed with FastTree v.2.1.10 with the model WAG (35,36), which had lower AIC than the alternative WAG model in both cases. We made 100 bootstrap trees using PHYLIP (37) to generate resampled alignments which were given as input to FastTreeMP (options –n –intree1).
We tested the association between types of Cas systems in two steps. First, to lower the amount of computational load, we assumed that bacteria were phylogenetically independent. We build a contingency table for each pair of types of Cas. We selected the pairs for which the hypothesis of independence was rejected (P < 0.05, Fisher exact test). We then tested these correlations in the light of the phylogeny. This allows to control for the phylogenetic dependency of the data, e.g. if many taxa are monophyletic and homogeneous in terms of a trait this is accounted by the statistical procedure. Therefore, the phylogenetic logistic regression of each association was computed with the function phyloglm from the phylolm v.2.6 R package. We used the logistic MPLE model that maximizes the penalized likelihood of the logistic regression and performed 100 independent bootstrap replicates (options method = logistic_MPLE, boot = 100). We then selected the pairs with significant regression coefficient (P < 0.05, Wald test). All results are reported in Supplementary Table S3.
RESULTS
Epistatic interactions between cas clusters
We identified 14041 CRISPRs and 7060 clusters of cas genes in fully sequenced bacterial (13229) and archaeal genomes (283) (Supplementary Table S1 and 2). The number of spacers of CRISPRs varies widely, from a minimum of three repeats (minimal detection threshold) to a maximum of 587 in the Proteobacteria Haliangium ochraceum DSM 14365 (Supplementary Table S5). Most arrays are small, with 24% of them containing between three and five repeats (Figure 1A). CRISPRs were found in 45%, and cas clusters in 39% of the prokaryotic genomes. The distribution of Cas types is very heterogeneous. The Type I Cas systems are by far the most frequent (present in 30% of all genomes) followed by type II (8%) and type III (6%) (Figure 1B). All of them are present in multiple phyla. The types IV, V and VI are extremely rare in the current genome database— they were found in fewer than 70 genomes—and are mainly restricted to a few clades (Proteobacteria, Actinobacteria, Bacteroidetes). Some subtypes are present in many clades—I-B, I-C, II-C, III-A, III-B, III-D—while others are clade specific, e.g. subtype I-D is mostly found in Cyanobacteria and Archaea, II-A in Firmicutes and II-B in Proteobacteria (Figure 1B). The relative abundances of CRISPR and Cas subtypes are close to those reported previously (8,30), although with our larger dataset type II appear more abundant than type III in Bacteria, in contrast with other studies (8,13,38). Overall, the number of systems per genome does not show systematic variations with genome size, except that small (<1 Mb) genomes rarely encode these systems (Supplementary Figure S1A).
Figure 1.

Distribution of CRISPR arrays and clusters of cas genes in the genomes of Prokaryotes. (A) Histogram of the number of repeats in CRISPRs (histogram truncated at 100, because higher values are very rare, maximum is 587). (B) Distribution per clade (on the top panel only clades with >25 genomes are indicated). The cells indicate the number of genomes with systems detected in the clade, and the colour of the cell is proportional to the average frequency per genome (the darker, the more frequent, see scale). The bottom panel shows the total number of genomes with elements detected in the dataset.
We observe that most bacterial genomes lack cas clusters, but 9% have more than one cluster. To understand if epistatic interactions between different systems could explain the co-occurrence of these multiple cas clusters, we analysed the co-occurrence of all pairs of Cas types. We used phyloglm to integrate the information of the phylogenetic structure in the evaluation of these associations (39). Since phylogenetic inference of all the prokaryotes is very inaccurate, we restricted our analysis to Firmicutes and Proteobacteria, the two clades with more genomes in our dataset (78% of the total). We inferred 100 phylogenetic trees for these clades (to account for uncertainty in phylogenetic inference), and used them to test the associations between every pair of systems (Figure 2A and B).
Figure 2.

Significant associations between Cas subtypes in the same genome in Proteobacteria (A) and Firmicutes (B). Each circle corresponds to the association between two subtypes. Associations are represented in grey (not significant), blue (negative), and orange (positive). Only sub-types with systems present in >1% of the genomes in the clade are represented (the others never have significant statistics). Statistical significance was assessed at two stages. First, we assumed that distributions are independent of phylogeny and made 2 × 2 contingency tables where independence was tested using a Fisher exact test (P < 0.05). Second, for the tests revealing significant effects, we made a phylogenetic logistic regression to control for the effect of phylogeny and selected only the significant associations (P < 0.05, Wald test). Coloured circles surrounding grey disks correspond to statistically significant interactions for Fisher exact test that were found not significant after the phylogenetic logistic regression.
We found lower co-occurrence of Cas subtypes than expected in both phyla. In Proteobacteria, subtype I-E is negatively associated to both II-C (observed three times, expected 46) and I-F (observed 71 times, expected 135). In Firmicutes, only subtypes II-A and I-B co-occur less than expected (observed 13, expected 45). Under-represented co-occurrences between Cas subtypes could be explained by counter-selection of the presence of the two types of Cas systems in the genome (negative epistasis), or by functional redundancy leading to the loss of one of them by genetic drift. The study of specific interactions provides some clues on these points. For example, Types I-E and I-F are very similar and co-occur in several genomes, suggesting that their joint presence in certain genetic backgrounds is not deleterious. Hence, their rare co-occurrence is likely to result from functional redundancy leading to a system loss by genetic drift. In contrast, type I-B and I-E are never observed together, even if the expected number of co-occurrences is low (7). Subtypes II-A and I-B are from the two different classes of Cas systems and are not expected to be functionally redundant. In both cases, the lower than expected co-occurrence of these pairs of systems suggests that negative epistasis, e.g. because of incompatibility between their machineries, leads to counter-selection of individuals expressing both systems.
Higher than expected co-occurrences of Cas subtypes were very frequent. This may indicate selection for certain combinations of adaptive immunity mechanisms. In Proteobacteria, we observed positive associations between subtype I-U with I-C (but only 55 occurrences are observed). The deviations from the expected values are higher for the co-occurrence between subtype I-F and III-A/B systems (87 observed, 20 expected). This is in agreement with experimental work showing that in Marinomonas mediterranea (a Proteobacteria), there is synergy between the action of type I-F and type III-B systems (24). In Firmicutes, systems I-B co-occur more than expected with all type III systems (87 observed, 20 expected), subtype I-E co-occurs more than expected with subtype II-A (40 observed, 14 expected), and subtype III-D is more often present in genomes encoding any of the other type III systems than expected (16 observed, 3 expected). Overall, there is a clear excess of positive co-occurrences of type I and type III systems, relative to other combinations.
Many CRISPR–Cas loci are complex
We observed that many genomes with a cas cluster had more than one cluster (23%), and most genomes with a CRISPR had more than one array (61%). This challenges the canonical view of the organization of a CRISPR–Cas locus as the association of one CRISPR with one cas cluster (40). Accordingly, genomes encode more CRISPRs than cas clusters and, in genomes encoding several of the latter, the number of CRISPR arrays grows faster than the number of cas clusters (Figure 3A). This suggests that increasing the number of CRISPRs per cas cluster is beneficial in bacteria were CRISPR–Cas immunity plays an important role.
Figure 3.

Organization of CRISPR–Cas loci. (A) Number of CRISPR arrays in function of the number of cas clusters in bacterial genomes (mean, CI 95%). The straight line indicates the identity (number of CRISPRs equal to the number of cas clusters) (B) Frequency of CRISPRs and cas clusters in terms of their genetic context. Loci were classed as complete CRISPR–Cas loci when they included at least one CRISPR and one cas cluster, ‘distant’ when the element (CRISPR or cas cluster) is >20 kb from the closest cognate element, and ‘orphan’ when the cognate element is absent from the genome. (C) Quantification of the different organizations of CRISPR–Cas loci.
To shed some light on the multiplicity of CRISPRs and cas clusters, we must first solve the problem of how to associate them in CRISPR–Cas loci. This problem is trivial when there is one single cas cluster and a contiguous single CRISPR, but not when there are multiple cas clusters or CRISPRs. The distributions of the distances between cas clusters and the closest CRISPR and between a CRISPR and the closest cas cluster (they are not identical because there are more CRISPR than cas clusters), reveal three groups: very close elements (<700 bp), intermediate (between 700 and 20 kb apart) and further apart (>20 kb of distance) (Supplementary Figure S2A and B). The careful analysis of the ‘intermediate’ group showed that the sequences intervening between the CRISPR and the cas cluster were often either other CRISPRs or genes that might be associated with the cas clusters. The latter were not annotated by our pipeline because we focus on the most conserved genes (41). The probability of finding pairs of elements <20 kb apart by chance is much lower than that observed in genomes. Based on these arguments, we defined a CRISPR–Cas locus as a region in the genome containing at least one cas cluster and one CRISPR, and eventually other such elements when two consecutive elements are spaced by less than 20 kb (clustered by transitivity, Supplementary Figure S2C). Hence, multiple cas clusters and CRISPRs can be part of the same locus. The elements not included in CRISPR–Cas loci were classed in two categories. Distant elements are CRISPRs or cas clusters >20 kb away from the closest cognate element (or present in another replicon). Orphan elements are those present in genomes lacking the cognate element (i.e. CRISPRs in genomes without cas clusters and vice-versa). Using this classification, the vast majority of cas clusters (88%), and a small majority of CRISPRs (65%) are part of CRISPR–Cas loci. Around 23% of the CRISPRs are distant and 12% are orphans (Figure 3B). Hence, there is an asymmetry in the genetic organization of the components of these systems: cas clusters are much more often co-localized with CRISPRs than the latter are co-localized with cas clusters.
We classified CRISPR–Cas loci in function of the number of CRISPRs and cas clusters (Figure 3C). The canonical CRISPR–Cas system—a locus with one CRISPR and one cas cluster—represents slightly more than half of all loci (58%). Almost a third (31%) of all loci have one cas cluster and two CRISPRs. Many other combinations are observed, even if they are rarer. Among these, we observed that 3% of the loci encode more than one cas cluster. This shows that the organization of the loci can be much more complex than the prototypical one cas to one CRISPR textbook example.
Distant CRISPR systems could function in trans
The above classification allows to investigate more closely the association between CRISPRs and cas clusters in CRISPR–Cas loci. Within CRISPR–Cas loci with a single cas cluster, the number of spacers in a CRISPR depends on the subtype of the cas cluster (Supplementary Table S5, ANOVA, P < 0.001), as described previously (38). Type IV, VI and subtype II-A tend to have short CRISPRs (<17 repeats on average). On the other hand, subtype I-A, I-B, I-D have the longest CRISPRs with >35 repeats on average (with a maximum for type I-A of 52 repeats on average). CRISPRs outside CRISPR–Cas loci are different. Orphan CRISPR arrays are smaller (eight repeats on average) than distant arrays (13), which are smaller than arrays within CRISPR–Cas loci (24, Figure 4A). The latter result, is consistent with previous findings (42). In consequence, the presence, proximity and subtype of cas clusters impact the number of repeats in CRISPRs.
Figure 4.

Characterization of CRISPRs according to their association with cas clusters. (A) Number of repeats in CRISPRs in function of their distance to cas clusters (Tukey HSD, all pairs, P < 0.001). (B) ROC curve (orange) of the results of the study using logistic regression to predict the subtype of Cas systems for the best hit of the set of repeats of a CRISPR. In grey, the threshold chosen to assign subtype to unknown arrays (74% identity). (C) Percentage of orphan and distant arrays with subtype assignment.
Several natural and synthetic systems revealed that CRISPR arrays can be used in trans by cas genes (7,43). We therefore tested whether CRISPRs distant from CRISPR–Cas loci could function with the latter for immune defence. If true, one would expect that the repeats of these CRISPRs should be similar to those of the cas clusters in trans. To test this hypothesis, we typed the CRISPRs outside CRISPR–Cas loci and checked if they matched the subtype of CRISPR–Cas systems in the genome. We typed these CRISPRs using the information on the best hit of the repeats of the CRISPR to a databank of 4978 unique repeats that could be unambiguously assigned to a Cas subtype (because they were taken from CRISPRs of CRISPR–Cas loci with one single cas cluster, Supplementary Table S2). We used a logistic regression to set the identity score threshold above which a best hit could be reliably associated with a Cas subtype. We chose a threshold that corresponds to an identity score of 74% (Figure 4B, Supplementary Figure S3A). The analysis of the original dataset shows 3961 correct and 843 incorrect assignments (accuracy of 82%). This method could be useful in metagenomic studies, where most of the detected elements are effectively orphan because most contigs are very small. We show in supplementary material a proof of concept for the classification of CRISPRs from metagenomics data (Supplementary Text1, Figure S3C and Table S6).
The method allowed the assignment of subtypes to 54% of the orphan arrays and 81% of the distant arrays (Figure 4C). The different levels of success in typing the two classes of CRISPR may be explained by the presence of spurious CRISPR arrays in the orphan dataset (44), i.e. sequences that were erroneously annotated as CRISPRs. Accordingly, untyped orphan CRISPRs are shorter on average (five repeats) than the typed ones (10 repeats, Supplementary Figure S3B, Mann Whitney, P < 0.001). This suggests that many of the untyped CRISPRs might be either false positives or elements ongoing genetic degradation (which is presumably facilitated by the lack of a cognate cas cluster in the genome rendering the CRISPR silent). The analysis of distant CRISPRs revealed that 75% of them had repeats of the same subtype as the cas cluster present in the genome in trans. This trend did not change when varying the True Positive Rate (TPR) chosen to assign a subtype (72% and 78% respectively for TPR of 50% and 90%). The relatively high number (25%) of non-matching repeats changed only slightly (22%) when the analysis was restricted to arrays with a number of repeats higher than 5 (Supplementary Table S4). Hence, the majority of CRISPRs distant from cas clusters have repeats matching the subtype of the latter. This suggests that such CRISPRs could be used by the latter for immune defence.
Complex loci have unique adaptation and multiple interference mechanisms
The analysis of co-occurrence of cas clusters and the ability to type CRISPRs using the sequence of their repeats paves the way to study in detail the complex CRISPR–Cas loci, especially those with more than one Cas subtype. Analysis of these loci shows that different cas clusters are often clearly separated by other elements, such as CRISPRs (Figure 5A). Other genes that were not annotated by our procedure are also found between cas clusters or between pairs of CRISPRs. Some of them have previously been proposed to be associated with CRISPR–Cas systems (41). Some Cas subtypes are more likely to co-occur in a locus than others (Figure 5B). For example, type II systems rarely co-occur with other systems in the same locus. This is also the case of most type I systems, with notable exception of type I-B and at a lesser extent I-A. Type III elements are much more likely to be in complex CRISPR–Cas loci. In particular, subtypes I-B and III-B were very often found together (20% of all complex loci). This fits the observation described above of positive genome-wide associations between subtype I-B and type III systems in Firmicutes (Figure 2B, Supplementary Figure S4). Interestingly, although few cas clusters in genomes could not be typed (fewer than 4%), they are often found in complex CRISPR–Cas loci. This could be explained by functional interaction between cas clusters leading to the loss of certain cas genes that render the specific system hard to type.
Figure 5.

Association between Cas clusters. (A) Examples of complex CRISPR–Cas loci found in Bacteria. Arrows represent cas genes and cas clusters are coloured by subtypes. Genes that were not identified as cas genes were omitted and replaced by a slash (/). CRISPRs colours match the Cas subtype to which their repeats were assigned. Grey indicates that no subtype could be assigned. (B) Number of loci with a given Cas subtype found in simple or complex loci.
Complex CRISPR–Cas loci often have several CRISPRs and cas clusters. We wondered if there were preferential associations between the two. The CRISPRs in complex loci with multiple cas clusters have identical repeats in 56% of the cases, and repeats are >80% identical in 34% of the remaining cases. Hence, there is less heterogeneity among CRISPRs than expected given that these loci combine proteins from different Cas subtypes. This is in line with our observation that these loci have one single pair of cas1–cas2 genes, the key genes involved in adaptation, in the majority of cases (86% have only one cas1). To further test the hypothesis that loci with multiple cas clusters often share one single adaptation module, we searched to identify the Cas subtype associated with the CRISPRs. In142 out of 177 loci, all the CRISPRs that could be typed in a locus were from the same subtype. In particular, clusters with Cas of types I-B and types III had repeats classed as I-B 60% of the times (38 out of 63 cases) showing that these loci tend to have the adaptation module of type I-B systems. These results suggest that complex loci use multiple mechanisms for interference and a single, often type I-like, mechanism for adaptation.
CRISPRs in mobile elements match chromosomal Cas systems
It has been shown that bacterial lysogens are more likely to encode CRISPR–Cas systems than bacteria devoid of prophages (28). Recently, the ICP1 phage of Vibrio cholerae was shown to carry a CRISPR–Cas system to subvert its host immune defences (19). Following on these studies, we wished to assess the frequency with which CRISPR–Cas systems occur in phages and if there are significant associations between these systems and those of the host. We quantified the occurrence of CRISPR–Cas systems in the 2498 phages of RefSeq. Using PHACTS, we could characterize the lifestyle of more than half of the phages, among which 42% are virulent and 58% are temperate. We found only one complete CRISPR–Cas system and 21 CRISPRs in these genomes (Figure 6). We then analysed the sequences of 26987 prophages to search if temperate phages that successfully infected a host were more likely to encode CRISPR–Cas systems than the other phages. We only detected 27 complete CRISPR–Cas systems, seven cas clusters and 212 CRISPRs. These values are very similar to those identified in the phage dataset, once the different number of elements is accounted for. We conclude that, within the limits given by the size and diversity of current genome databases, CRISPR–Cas systems are extremely rare in phages. Their frequency in prophages is not significantly different from that of the average temperate phage, suggesting that prophages carrying these traits do not significantly increase in frequency by natural selection on the host.
Figure 6.
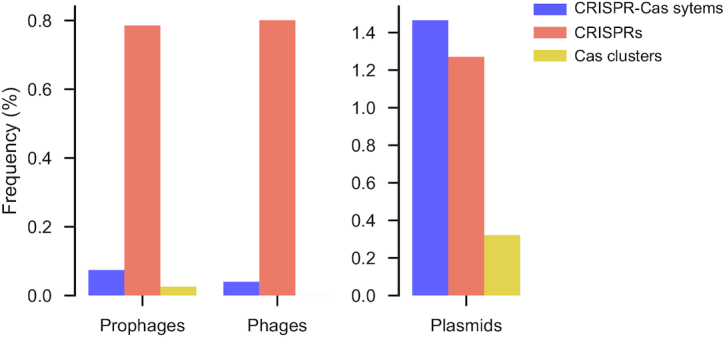
Frequency of CRISPR–Cas systems in prophages, phages and plasmids. Frequency notes the percentage of such mobile elements encoding at least one CRISPR–Cas system, a cas cluster or a CRISPR. Note that the scales of the axes are different, since CRISPR–Cas are much more abundant in plasmids.
We then turned our attention to plasmids. We observed that genomes encoding CRISPR–Cas systems are slightly more likely to have plasmids (37%) than the others (34%, χ2P < 0.0001). We searched for CRISPR–Cas systems among 11 805 plasmids that were sequenced in the whole-genome projects analysed in this study. This means that we know the chromosome of the bacterial host of every plasmid in the database. We found 173 complete systems and 150 CRISPRs in plasmids devoid of cas clusters (Figure 6). Plasmid CRISPRs have on average 16 repeats, significantly fewer than chromosomal arrays (20 repeats) (Supplementary Figure S5A Mann–Whitney, P < 0.001). The relative abundance of subtypes is also significantly different on plasmids and chromosomes (Supplementary Figure S5B). In particular, no plasmids encode type II-A CRISPR–Cas systems while type IV systems are encoded almost exclusively on plasmids, as described previously (45). Plasmids with CRISPR arrays and encoding cas clusters were larger (1.5 times when only encoding a CRISPR and 2.5 times when encoding a cas) than the other plasmids (Supplementary Figure S5C). These results show that plasmids are much more likely to encode CRISPR and especially CRISPR–Cas loci than phages, even if this concerns fewer than 5% of all plasmids. The differences observed between the distributions of Cas subtypes in plasmids and chromosomes suggests that plasmid CRISPR–Cas systems are not just a random sample of chromosome systems. Instead, they may reflect selection for systems influencing the interactions between the plasmid and its host. Our analysis shows that plasmids over-represent systems I-A to I-D, which have mostly positive interactions with other systems, and under-represent systems I-E which have mostly negative interactions with other systems. This suggests that type IV systems, extremely over-represented in plasmids, could also have positive interactions with other systems.
The large number of plasmids carrying CRISPRs but lacking cas clusters suggests the existence of interactions between plasmid-associated CRISPRs and the chromosomal-encoded Cas proteins. More than half (54%) of the genomes with CRISPRs in plasmids (but no cas clusters) have chromosomal cas clusters (Supplementary Figure S6A). We typed these CRISPRs and then tested if they matched the subtype of cas clusters found on the chromosome. We assigned a subtype to 55 of these plasmid CRISPR arrays, and in 38 cases these matched the type of the chromosomal cas clusters (Supplementary Figure S6B). We tested the significance of this result by simulating the expected number of matches between plasmid and chromosomal subtypes. In 1000 simulations, the average number of matches was 19.53 and the highest number was 27, which is well below the 38 observed cases (Supplementary Figure S6C). While the number of observations is low, these results suggests that when plasmids with CRISPRs, but no cas clusters, are in genomes with cas clusters, the array is more likely to be classed in that precise Cas subtype than expected by chance.
DISCUSSION
We detected and analysed thousands of CRISPRs and cas clusters in fully sequenced bacterial and archaeal genomes. We used the entire RefSeq complete genome database, and therefore there was no bias in our analysis of the data, apart from the bias of the database itself, which is known to over-represent cultivable Bacteria over other Prokaryotes. We have not used draft genomes because they would have greatly increased the sampling biases, since the drafts database is overwhelmingly dominated by a dozen species of bacterial pathogens, and because one cannot reliably analyse chromosomal distances between genetic elements in drafts nor to discriminate accurately plasmids from chromosomes. It is possible that extremely rare systems in our dataset—types IV, V, and VI—are more frequent in poorly sampled clades. However, it should be noted that in our study these three systems tend to be overrepresented in phyla that are well sampled in the database (Proteobacteria, Actinobacteria and Bacteroidetes). Further work and much broader samples will be needed to understand if these systems are rare and why this is so. In this study, their low frequency resulted in no significant association with other systems. Our analysis also revealed that 18% of CRISPRs have fewer than five repeats. One third of these small CRISPRs were in CRISPR–Cas loci, but part of the other small CRISPRs might be false positives. To control for their impact on our results—when necessary—we made replicates of the analyses using only CRISPRs with five or more repeats. These analyses resulted in qualitatively similar conclusions (Table S3). We used CasFinder to identify and type cas clusters, which was previously shown to provide accurate classifications (29). As our detection takes into account the architectures and signature proteins of cas clusters, it provides a robust subtype assignment compared to a previous study where subtypes were only inferred from Cas1 (13). To associate the CRISPRs outside CRISPR–Cas loci to Cas types we used the sequence similarity to a database of known repeats. This part of our method might gain from the diversification of the genome database, since rare CRISPR–Cas systems are under-represented. A larger reference database might also allow to define subtype-specific sequence identity thresholds. Such developments could pave the way to understand if our inability to type half of the orphan CRISPRs (because they lack high sequence similarity to known repeats) is due to the lack of a sufficiently comprehensive repeat database or to the rapid evolution by drift of orphan arrays.
Consistent with previous analyses (8,13), we observe that most bacteria lack CRISPR–Cas systems. This is puzzling, because most species have mobile genetic elements against which these systems might presumably provide protection. For example, half of the genomes in the database are lysogens (28), and many have plasmids (this work). If CRISPR–Cas are universally efficient immune systems, why is it that most bacteria lack them? And why is that many of the remaining bacteria have small CRISPRs, which presumably provide protection against few MGEs? This cannot be caused by phylogenetic inertia, i.e. the fact that certain lineages have not developed such systems, since CRISPR–Cas systems are frequently transferred across phyla (14,15,46,47). Instead, it has been proposed that the deleterious effects of CRISPR–Cas systems on the host, either by spacers targeting the chromosome (48), or by interfering with DNA repair functions (49) could explain the relative paucity of CRISPR–Cas systems across Bacteria. These processes may also explain why so many CRISPRs contain so few spacers: they could result from decaying inactive CRISPR–Cas systems. The number of spacers in CRISPRs may also be affected by the balance of the rates of acquisition and loss of the spacers. Experimental observations on primed adaptation (acquisition of spacers from mobile elements already targeted by a spacer in the CRISPR) (50) and some mathematical models predict selective sweeps of lineages with CRISPR–Cas systems effective in providing immunity against phages present in the community (51). CRISPRs could thus acquire several spacers within a short time-frame, rapidly increasing in size, whereas the loss of old spacers could be more gradual (52,53). As a result, short CRISPRs could result from the gradual shrinkage of CRISPR arrays that have not undergone recent acquisition—selection events. The small CRISPRs found in this and previous studies suggest that CRISPRs target a relatively small number of mobile genetic elements in most individual genomes.
Orphan or distant CRISPRs represent 40% of all the CRISPRs raising the question of how they arise in the first place. Long gaps in the activity of CRISPR–Cas systems might explain the abundance of CRISPRs without a neighboring cas cluster because when the system is not being expressed, and therefore is not adaptive, cas clusters may be lost in a neutral manner. Similar neutral processes may take place when CRISPR–Cas systems are acquired by a host that is incapable of expressing the cas genes. The deletion of cas genes should be much accelerated, relative to the neutral cases, when the function of the system decreases the fitness of the host. Cas systems lacking CRISPRs might be more frequently counter-selected than CRISPRs lacking cognate Cas systems, because the expression of these genes can be costly either because of protein synthesis or because cas genes may interact deleteriously with the host genetic background (49,54). In addition, MGEs have many more CRISPRs lacking cas genes than the converse, their acquisition by horizontal transfer will then bring such CRISPRs into the genome. Multiple CRISPRs may also arise by the split of chromosomal CRISPRs by genome rearrangements, even if these are rarely fixed in bacterial populations (55), or DNA insertions. The split of cas loci is less likely. Splitting a CRISPR will often only inactivate one spacer, whereas splitting a cas locus may inactivate a gene and therefore the whole system.
Distant CRISPRs often have repeats compatible with the cas locus in trans. This may be explained by several mechanisms. Multiple CRISPRs with similar repeats may arise from the fission of an ancestral single CRISPR. They may also result from multiple acquisitions of a similar CRISPR–Cas systems that leads to loss of one of Cas locus and maintenance of the cognate CRISPR because it is functionally compatible with the existing complete CRISPR–Cas locus. Finally, recent data suggests that non-specific integration of spacers at non-CRISPR locus could lead to the creation of CRISPRs with repeats similar to the existing CRISPR–Cas system (56). The compatibility between distant CRISPR and CRISPR–Cas systems may also result from natural selection. If MGEs bringing into the genome isolated CRISPRs use them to co-opt the CRISPR–Cas system of the host, then the repeats of their CRISPRs must be compatible those of the host for co-option to work. What functions provided by distant CRISPRs might be different from those of the CRISPRs encoded next to the Cas locus? The fewer spacers in the CRISPRs distant from the cas genes cluster, relative to those that are contiguous to it, could result from reduced efficiency at incorporating spacers when arrays are distant from cas clusters or less efficient selection for the spacers of the distant CRISPR if expression of these arrays is weaker than those located next to cas clusters (57). Interestingly, if these clusters have different acquisition or deletion rates, they may allow to explore different trade-offs between long-term memory of past infections and rapid acquisition of novel spacers (58). Recombination between CRISPRs (59) could switch spacers between arrays with different turnover rates (57). Finally, a recent study has shown that CRISPRs spacers can facilitate horizontal transfer of neighbouring bacterial genes by a process resembling specialized transduction (60). One is tempted to speculate that the presence of multiple CRISPRs in the chromosome could be selected to increase the local rates of genetic exchanges.
Little was known about the frequency of CRISPR–Cas in phages and plasmids, in spite of the previous studies describing their existence and relevance (16–19). While this paper was in review, an analysis was published showing that most of the phage-encoded CRISPR–Cas loci lack the genes required for adaptation, suggesting that, like we observed for plasmids, phages tend to encode either the interference mechanisms or just the CRISPR (21). Interestingly, this study showed that most spacers of phage-encoded CRISPRs have good matches in the database, usually matching other phages or prophages (45). It must however be considered that phages rarely encode such systems. A recent preprint identified CRISPR–Cas systems in phages with very large genomes (61), but even in this case their frequency was very low. Here, we show that CRISPR–Cas are almost never carried by the phages available in the genome databases. This does not invalidate the previous results showing that CRISPR–Cas carried by phages may provide the latter with a mechanism to escape host innate immunity (19). Yet, if the current phage database is representative of the natural diversity of these elements, such mechanisms are rarely used by phages and may be specific of a few families. Interestingly, the much higher frequency of plasmids carrying CRISPR–Cas and especially CRISPRs compatible with the chromosomal cas clusters opens the possibility for plasmids to manipulate the host immunity by using the host Cas proteins and their own CRISPRs. The differences in terms of the distributions of Cas subtypes in plasmids and chromosomes reinforces this hypothesis, because it suggests that plasmid systems are not just random samples of chromosomal systems. This is particularly striking in the case of the type IV systems, previously reported in plasmids (8), that we show are almost exclusively encoded on these mobile elements. As these systems do not encode Cas1 and Cas2, the main proteins for adaptation (8), it is not known how they acquire new spacers. It is tempting to speculate that type IV CRISPR are able to use the adaptation machinery of the host's CRISPR–Cas systems, in a way resembling the CRISPRs in plasmids matching chromosomal Cas systems, and that of type III systems in complex loci that share the adaptation machineries of type I systems. Interestingly, a recent reprint finds some bioinformatic evidence that type I and type IV systems interact (62). If true, and given the vast over-representation of type IV systems in plasmids, it suggests that these systems may have evolved as specialists in subverting chromosomal systems. This is consistent with the observation that these systems frequently lack not only the adaptation machinery but also the enzymes necessary for target cleavage (8), and in some cases for the processing of crRNAs (63).
Our work revealed several negative associations between Cas subtypes. These may have selective or neutral causes. Some systems may be functionally very similar. In this case, the presence of the two systems in the genome is redundant and one of them is expected to be lost by genetic drift. Some genomes have several clusters of the same Cas subtype (272 out of 5281 genomes with cas clusters), of which most (186) are type I. Systems of the same subtype among Type II-A and B systems rarely co-occur (0 and 1 case) which shows that these systems may be compatible, and their low co-occurrence might be explained by loss by drift. Alternatively, two systems may not work well together, e.g. because they compete for a substrate or because their mechanisms interfere in a deleterious manner. It should be emphasized that many of the negative interactions observed in our study correspond to co-occurrences that are observed in a few genomes, and some have been shown to be functional in the lab (54). Negative interactions should be understood as combinations that are generally unfavorable in natural populations: if a system is acquired by a genome and is incompatible with another system already functioning in the cell, natural selection will lead to the loss of one of them. This mechanism should result in rapid deletion of cas clusters, because they prevent CRISPRs from functioning, and result in a CRISPR distant from functional cas clusters. Such negative interactions could be behind the peculiar system preventing the horizontal transfer of type I-F system in E. coli that encode the type I-E system (64). In this case, CRISPRs of type I-F contain spacers matching sequences of the cognate (absent) type I-F cas genes (64). Upon entry in the cell of a type I-F Cas system by horizontal transfer, spacers guide the incoming I-F CRISPR–Cas system to degrade the cas genes, and thus preventing the acquisition of the system (65). The existence of such a mechanism suggests selection for preventing simultaneous presence of these two systems in the same genome.
The interactions observed between Cas systems in Proteobacteria differ from those observed in Firmicutes. Although there is no incompatibility between them, i.e. no inversion of sign in the significant associations, many interactions that are significant in one clade aren’t in the other. This may result from different effects. The power of the statistical tests is higher when the two systems are abundant. Since, as shown in Figure 1, the frequency of the sub-types is very different in the two phyla, the associations that can be detected given the current datasets are different. This may explain why many interactions concern the system IB in Firmicutes (where it's abundant) and not in Proteobacteria (where it's rare). Furthermore, the negative interaction between system I-B and II-A could hardly have been observed in Proteobacteria, because II-A is almost absent in the phyla. Hence, it's not clear at this stage if such interactions are phyla-specific or just reflect the underlying distribution of sub-types. Phylum-specific interactions could be caused by interactions of the systems with the genetic background, e.g. with DNA repair and recombination systems that differ markedly across the clades (49). For example, the relevance of CRISPR in providing resistance to phages, relative to mutations in surface phage receptors, depends on the mutation rate, modulated by the presence of mutS (66) a gene absent in certain species and in multiple copies in others (67).
The influx of novel CRISPR–Cas systems in genomes by mobile elements leads to co-existence of different systems in the same genome. Since horizontal gene transfer tends to accumulate in a small number of regions of the bacterial chromosome (68), this leads to the accumulation of certain defence systems in these hotspots (69,70). When such associations increase the immune competence of the cells, they may evolve to become integrated functional systems. This may explain why we found many more positive than negative associations between Cas subtypes. Different systems may provide different degrees of protection, e.g. by targeting DNAs or RNAs in a specific or non-specific manner (71,72). The existence of different systems may also allow bacteria to counteract the action of anti-CRISPRs in MGEs. Indeed, recent data revealed the presence of numerous systems with the ability to counteract the action of CRISPR–Cas systems (22,73–75). These systems tend to be Cas-type specific (76), and the diversification of the repertoires of Cas systems provides a way to increase the likelihood that the MGE does not escape immune response.
Complex CRISPR–Cas loci may produce more efficient immune response when they reflect functional associations between CRISPR–Cas systems of various types. This is suggested by the presence of a single adaptation module and by similar CRISPR repeats across many of the loci. Type III-B and type I-F are positively associated in Proteobacteria, and this could be explained by the experimentally demonstrated ability of type III-B to process and use guide RNAs expressed from a type I-F CRISPR array (24). The immunity provided by type III systems involves the production of an intracellular signal which activates a non-specific RNAse, Csm6. This mechanism can lead to cell death or dormancy when high levels of target mRNA are detected or when the target is mutated. As such, type III systems have been proposed to form a second line of defence able to block phage infection when type I systems fail (24). It should be noted that this experimentally verified interaction between systems is based on two cas clusters that are not in a single locus. Hence, interactions between systems may start before they merge in a single complex locus. Together, these results suggest that associations between type I and type III systems combine the adaptation and interference functions of the former with a diversity of mechanisms associated with interference and abortive mechanisms of the latter. This suggests that the integration of multiple CRISPRs on complex loci including multiple Cas systems can improve the immune defence of Prokaryotes against infection by mobile elements. Altogether, our results highlight the importance of interactions between CRISPRs and cas present in multiple copies and in distinct genomic locations in the function and evolution of bacterial immunity.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Pasteur Institute and the CNRS; European Research Council (ERC) under the Europe Union's Horizon 2020 research and innovation program [677823]; French Government's Investissement d’Avenir program and by Laboratoire d’Excellence ‘Integrative Biology of Emerging Infectious Diseases’ [ANR-10-LABX-62-IBEID]; INCEPTION project [PIA/ANR-16-CONV-0005]; This work used the computational and storage services (TARS cluster) provided by the IT department at Institut Pasteur, Paris. Funding for open access charge: Pasteur Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garneau J.E., Dupuis M.-È., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S.. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010; 468:67–71. [DOI] [PubMed] [Google Scholar]
- 5. Gophna U., Kristensen D.M., Wolf Y.I., Popa O., Drevet C., Koonin E. V. No evidence of inhibition of horizontal gene transfer by CRISPR Cas on evolutionary timescales. ISME J. 2015; 9:2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marraffini L.A. CRISPR–Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 7. Hille F., Richter H., Wong S.P., Bratovič M., Ressel S., Charpentier E.. The biology of CRISPR–Cas: backward and forward. Cell. 2018; 172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 8. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H. et al.. An updated evolutionary classification of CRISPR Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koonin E. V, Makarova K.S., Zhang F.. Diversity, classification and evolution of CRISPR–Cas systems. Curr. Opin. Microbiol. 2017; 37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.V., Van der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR–Cas systems. Science. 2016; 353:aad5147. [DOI] [PubMed] [Google Scholar]
- 11. Harrington L.B., Burstein D., Chen J.S., Paez-espino D., Ma E., Witte I.P., Cofsky J.C., Kyrpides N.C., Banfield J.F., Doudna J.A.. Programmed DNA destruction by miniature CRISPR–Cas14 enzymes. Science. 2018; 842:839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan W.X., Yan W.X., Hunnewell P., Alfonse L.E., Carte J.M., Keston-smith E., Sothiselvam S., Garrity A.J., Chong S., Makarova K.S. et al.. Functionally diverse type V CRISPR–Cas systems. Science. 2018; 7271:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burstein D., Sun L., Brown C., Sharon I., Anantharaman K., Probst A., Thomas B., Banfield J.. Major bacterial lineages are essentially devoid of CRISPR–Cas viral defense systems. Nat. Commun. 2016; 7:10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Godde J.S., Bickerton A.. The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol. 2006; 62:718–729. [DOI] [PubMed] [Google Scholar]
- 15. Chakraborty S., Snijders A.P., Chakravorty R., Ahmed M., Tarek A.M., Hossain M.A.. Comparative network clustering of direct repeats (DRs) and cas genes confirms the possibility of the horizontal transfer of CRISPR locus among bacteria. Mol. Phylogenet. Evol. 2010; 56:878–887. [DOI] [PubMed] [Google Scholar]
- 16. Scholz I., Lange S.J., Hein S., Hess W.R., Backofen R.. CRISPR-cas systems in the cyanobacterium synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One. 2013; 8:e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Millen A.M., Horvath P., Boyaval P., Romero D. A.. Mobile CRISPR/Cas-mediated bacteriophage resistance in lactococcus lactis. PLoS One. 2012; 7:e51663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo P., Cheng Q., Xie P., Fan Y., Jiang W., Qin Z.. Characterization of the multiple CRISPR loci on Streptomyces linear plasmid pSHK1. Acta Biochim. Biophys. Sin. (Shanghai). 2011; 43:630–639. [DOI] [PubMed] [Google Scholar]
- 19. Seed K.D., Lazinski D.W., Calderwood S.B., Camilli A.. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013; 494:489–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters J.E., Makarova K.S., Shmakov S., Koonin E.V.. Recruitment of CRISPR–Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E7358–E7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faure G., Shmakov S.A., Yan W.X., Cheng D.R., Scott D.A., Peters J.E., Makarova K.S., Koonin E.V.. CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol. 2019; 17:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pawluk A., Davidson A.R., Maxwell K.L.. Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 2017; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 23. Borges A.L., Davidson A.R., Bondy-denomy J.. The discovery, mechanisms, and evolutionary impact of anti-CRISPRs. Annu. Rev. Virol. 2017; 4:37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silas S., Lucas-Elio P., Jackson S.A., Aroca-Crevillén A., Hansen L.L., Fineran P.C., Fire A.Z., Sánchez-Amat A.. Type III CRISPR–Cas systems can provide redundancy to counteract viral escape from type I systems. Elife. 2017; 6:e27601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shmakov S., Smargon A., Scott D., Cox D., Pyzocha N., Yan W., Abudayyeh O.O., Gootenberg J.S., Makarova K.S., Wolf Y.I. et al.. Diversity and evolution of class 2 CRISPR Cas systems. Nat. Rev. Microbiol. 2017; 15:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mcnair K., Bailey B.A., Edwards R.A.. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics. 2012; 28:614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roux S., Enault F., Hurwitz B.L., Sullivan M.B.. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 2015:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Touchon M., Bernheim A., Rocha E.P.. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016; 10:2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., Rocha E.P.C., Vergnaud G., Gautheret D., Pourcel C.. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018; 46:W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abby S.S., Néron B., Ménager H., Touchon M., Rocha E.P.C.. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR–Cas systems. PLoS One. 2014; 9:e110726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rendueles O., de Sousa J.A.M., Bernheim A., Touchon M.. Genetic exchanges are more frequent in bacteria encoding capsules. PLoS Genet. 2018; 14:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katoh K., Misawa K., Kuma K., Miyata T.. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Criscuolo A., Gribaldo S.. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010; 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Filipski A., Murillo O., Freydenzon A., Tamura K., Kumar S.. Prospects for building large timetrees using molecular data with incomplete gene coverage among species. Mol. Biol. Evol. 2014; 31:2542–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Price M.N., Dehal P.S., Arkin A.P.. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009; 26:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Price M.N., Dehal P.S., Arkin A.P.. FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. 2001; Washington: Dep. Genome Sci. Univ. [Google Scholar]
- 38. Crawley A.B., Henriksen J.R., Barrangou R.. CRISPRdisco: an automated pipeline for the discovery and analysis of CRISPR–Cas systems. Cris. J. 2018; 1:crispr.2017.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pagel M., Meade A.. Bayesian analysis of correlated evolution of discrete characters by reversible jump Markov chain Monte Carlo. Am. Nat. 2013; 167:808–825. [DOI] [PubMed] [Google Scholar]
- 40. Lander E.S. The heroes of CRISPR. Cell. 2015; 164:18–28. [DOI] [PubMed] [Google Scholar]
- 41. Shmakov S.A., Koonin E.V., Severinov K. V., Wolf Y.I., Makarova K.S.. Systematic prediction of genes functionally linked to CRISPR–Cas systems by gene neighborhood analysis. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E5307–E5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Toms A., Barrangou R.. On the global CRISPR array behavior in class I systems. Biol. Direct. 2017; 12:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Q., Ye Y.. Not all predicted CRISPR–Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics. 2017; 18:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faure G., Shmakov S.A., Yan W.X., Cheng D.R., Scott D.A., Peters J.E., Makarova K.S., Koonin E.V.. CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol. 2019; 17:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anupama S., Mp A.R., Gurusaran M., Radha P., Ks D.K., Chitra R., Am H.V., Sekar K.. Evolutionary analysis of CRISPRs in archaea: an evidence for horizontalGene Transfer. J. Proteomics Bioinform. 2014; S9:doi:10.4172/jpb.S9-005. [Google Scholar]
- 47. Makarova K.S. A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res. 2002; 30:482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stern A., Keren L., Wurtzel O., Amitai G., Sorek R.. Self-targeting by CRISPR: gene regulation or autoimmunity. Trends Genet. 2010; 26:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bernheim A., Bikard D., Touchon M., Rocha E.P.C.. A matter of background: DNA repair pathways as a possible cause for the sparse distribution of CRISPR–Cas systems in bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2019; 374:20180088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Staals R.H.J., Jackson S.A., Biswas A., Brouns S.J.J., Brown C.M., Fineran P.C., Nishihama S., Yoshizuka K., Li X., Kawano T.. Interference dominates and amplifies spacer acquisition in a native CRISPR–Cas system. Nat. Commun. 2016; 23:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weinberger A.D., Sun C.L., Pluciński M.M., Denef V.J., Thomas B.C., Horvath P., Barrangou R., Gilmore M.S., Getz W.M., Banfield J.F.. Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol. 2012; 8:e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mick E., Stern A., Sorek R.. Holding a grudge. RNA Biol. 2013; 10:900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun C.L., Thomas B.C., Barrangou R., Banfield J.F.. Metagenomic reconstructions of bacterial CRISPR loci constrain population histories. ISME J. 2015; 10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bernheim A., Calvo Villamanan A., Basier C., Rocha E.P.C., Bikard D.. Inhibition of NHEJ repair by type II-A CRISPR–Cas systems. Nat. Commun. 2017; 8:170647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Touchon M., Bobay L.-M., Rocha E.P.. The chromosomal accommodation and domestication of mobile genetic elements. Curr. Opin. Microbiol. 2014; 22C:22–29. [DOI] [PubMed] [Google Scholar]
- 56. Grainy J., Garrett S., Graveley B.R., P. Terns M.. CRISPR repeat sequences and relative spacing specify DNA integration by Pyrococcus furiosus Cas1 and Cas2. Nucleic Acids Res. 2019; 47:7518–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weissman J.L., Fagan W.F., Johnson P.L.F.. Selective maintenance of multiple CRISPR arrays across prokaryotes. Cris. J. 2018; 1:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Martynov A., Severinov K., Ispolatov I.. Optimal number of spacers in CRISPR arrays. PLoS Comput. Biol. 2017; 13:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kupczok A., Landan G., Dagan T.. The contribution of genetic recombination to CRISPR array evolution. Genome Biol. Evol. 2015; 7:1925–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Varble A., Meaden S., Barrangou R., Westra E.R., Marraffini L.A. Recombination between phages and CRISPR−cas loci facilitates horizontal gene transfer in staphylococci. Nat. Microbiol. 2019; 4:956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Al-Shayeb B., Sachdeva R., Chen L.X., Ward F., Munk P., Castelle C.J., Olm M.R., Gregson K.B., Amano Y., Méheust R. et al.. Clades of huge phage from across Earth's ecosystems. 2019; bioRxiv doi:11 March 2019, preprint: not peerreviewed 10.1101/572362. [DOI] [PMC free article] [PubMed]
- 62. Pinilla-redondo R., Mayo-muñoz D., Russel J., Garrett R.A.. Type IV CRISPR–Cas systems are highly diverse and involved in competition between plasmids. 2019; bioRxiv doi:24 September 2018, preprint: not peerreviewed 10.1101/780106. [DOI] [PMC free article] [PubMed]
- 63. Özcan A., Pausch P., Linden A., Wulf A., Schühle K., Heider J., Urlaub H., Heimerl T., Bange G., Randau L.. Type IV CRISPR RNA processing and effector complex formation in Aromatoleum aromaticum. Nat. Microbiol. 2019; 4:89–96. [DOI] [PubMed] [Google Scholar]
- 64. Touchon M., Rocha E.P.C.. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One. 2010; 5:e11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Almendros C., Guzmán N.M., García-Martínez J., Mojica F.J.M.. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR–Cas I-F systems. Nat. Microbiol. 2016; 1:16081. [DOI] [PubMed] [Google Scholar]
- 66. Chevallereau A., Meaden S., Van Houte S., Westra E.R., Rollie C.. The effect of bacterial mutation rate on the evolution of CRISPR–Cas adaptive immunity. Philos. Trans. R. Soc. B Biol. Sci. 2019; 374:20180094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burby P.E., Simmons L.A.. MutS2 promotes homologous recombination in Bacillus subtilis Peter. J. Bacteriol. 2017; 199:e00682-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Oliveira P.H., Touchon M., Cury J., Rocha E.P.C.. The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun. 2017; 8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Makarova K.S., Wolf Y.I., Snir S., Koonin E.V.. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011; 193:6039–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Doron S., Melamed S., Ofir G., Leavitt A., Lopatina A., Keren M., Amitai G., Sorek R.. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018; 359:eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Meeske A.J., Nakandakari-Higa S., Marraffini L.A.. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature. 2019; 570:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hille F., Charpentier E.. CRISPR–Cas: biology, mechanisms and relevance. Philos. Trans. R Soc. Lond. B Biol. Sci. 2016; 371:20150496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Marino N.D., Zhang J.Y., Borges A.L., Sousa A.A., Leon L.M., Rauch B.J., Walton R.T., Berry J.D., Joung J.K., Kleinstiver B.P. et al.. Discovery of widespread type I and type V CRISPR–Cas inhibitors. Science. 2018; 362:240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Knott G.J., Thornton B.W., Lobba M.J., Liu J.-J., Al-Shayeb B., Watters K.E., Doudna J.A.. Broad-spectrum enzymatic inhibition of CRISPR–Cas12a. Nat. Struct. Mol. Biol. 2019; 26:315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dong L., Guan X., Li N., Zhang F., Zhu Y., Ren K., Yu L., Zhou F., Han Z., Gao N. et al.. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 2019; 26:308–314. [DOI] [PubMed] [Google Scholar]
- 76. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H. et al.. A unified resource for tracking anti-CRISPR names. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.