

Abstract
PathDIP was introduced to increase proteome coverage of literature-curated human pathway databases. PathDIP 4 now integrates 24 major databases. To further reduce the number of proteins with no curated pathway annotation, pathDIP integrates pathways with physical protein–protein interactions (PPIs) to predict significant physical associations between proteins and curated pathways. For human, it provides pathway annotations for 5366 pathway orphans. Integrated pathway annotation now includes six model organisms and ten domesticated animals. A total of 6401 core and ortholog pathways have been curated from the literature or by annotating orthologs of human proteins in the literature-curated pathways. Extended pathways are the result of combining these pathways with protein-pathway associations that are predicted using organism-specific PPIs. Extended pathways expand proteome coverage from 81 088 to 120 621 proteins, making pathDIP 4 the largest publicly available pathway database for these organisms and providing a necessary platform for comprehensive pathway-enrichment analysis. PathDIP 4 users can customize their search and analysis by selecting organism, identifier and subset of pathways. Enrichment results and detailed annotations for input list can be obtained in different formats and views. To support automated bioinformatics workflows, Java, R and Python APIs are available for batch pathway annotation and enrichment analysis. PathDIP 4 is publicly available at http://ophid.utoronto.ca/pathDIP.
INTRODUCTION
Pathways are biological network models defining how biomolecules cooperate to accomplish cellular tasks in different contexts. Pathways are assembled from physically-interacting molecules such as proteins and thus, to obtain a comprehensive image of pathways, we need to know all their participating physical components and the physical interactions among them. In addition, we need to know the context, i.e. tissue or disease, where such interactions and pathways are physiologically meaningful. Despite the advances and the amount of important information available in human pathway databases, and their critical role in bioinformatics workflows and systems-biology research, at the present time, they are still far from being complete.
To increase coverage of human pathway databases and improve results of pathway-enrichment analysis, we have developed pathway Data Integration Portal, pathDIP (1). Through integration of data available in 22 widely used human pathway databases and physical protein–protein interaction (PPI) networks obtained from IID (2) we increased coverage of pathway annotations for human proteins, improved consistency in coverage of individual pathway sources for proteins, predicted pathway annotations for thousands of proteins with no annotation in curated pathway databases (i.e. pathway orphans), and provided annotations on physical connections among each protein and protein members of each pathway. While pathways available in the source databases, i.e. core pathways, annotate 65% of the human proteome, combining them with our network-based predictions, i.e. extended pathways, increases this coverage to 92%. Moreover, leave-one-out cross-validation shows that our predictions recover 87% of core pathway members.
Shortcomings of literature-curated human pathway databases affect non-human organisms even more. It is important to address these caveats, since non-human organisms are widely-used during validation experiments and to find answers to diverse questions such as understanding evolution, discovering molecular mechanisms in cells, studying diseases and developing drugs. Therefore, understanding species-specific molecular networks and pathways can directly and indirectly affect these areas.
At the present time, only a handful of sources, such as BioCyc database collection (3)—including but not limited to MouseCyc (4) and YeastCyc (5), in addition to HumanCyc (6), and 54 other organisms (Tier 1 and 2 Pathway/Genome Databases (PGDBs)), ConsensusPathDB (7), KEGG (8), Panther (9), Reactome (10) and WikiPathways (11) provide pathway annotations for non-human organisms. However, most non-human pathway annotations in ConsensusPathDB, KEGG, Panther, Reactome and WikiPathways are predictions using orthologs of human proteins, leaving only limited sets of literature-curated pathways covering a small fraction of non-human proteins. Importantly, identifying literature-curated and ortholog pathways in these sources is often difficult or impossible. Several non-human organisms in pathDIP 4 have two or three source databases, including chicken, cow, fly, mouse, rat, worm and yeast. Mouse and yeast have the largest number of pathways with 495 and 458 pathways, and 4353 and 1196 annotated proteins, respectively (Table 1).
Table 1.
Coverage of different annotation sets (core, ortholog and extended pathways) for unique proteins and the number of pathways per annotation set across different species, as well as the number and ratio of proteins annotated only through network-based predictions

|
In pathDIP 4, we focused on improving annotations and analysis services for human proteins and sixteen non-human organisms (cat, chicken, cow, dog, duck, fly, guinea-pig, horse, mouse, pig, rabbit, rat, sheep, turkey, worm and yeast). We approached caveats due to lack of available resources and annotations, low coverage for species-specific proteins, and missing physical connectivity among pathway proteins in non-human organisms through our well-established network-based algorithm (1) and ortholog-based consensus method.
PathDIP 4 provides pathway annotations for six model and ten domesticated organisms (Table 1). We annotated proteins with 6401 pathways (total across all species) by: (i) core pathways, i.e. pathways from literature-curated databases, (ii) ortholog pathways, i.e. pathway annotations using protein orthologs in human and (iii) extended pathways, i.e. integration of species-specific PPIs with pathways of the first two sets to obtain species-specific network-based predictions and combining them with core and ortholog pathways. Our predictions extend coverage of pathways for proteins from 23 771 to 120 621 across seventeen species, including human.
Human pathway annotations include the most up-to-date versions of human databases, and two new sources (ACSN2 (12) and Panther (9)) and cover 13,088 proteins in core and 18 454 proteins in extended pathways. Furthermore, we have extended functionality of pathDIP 4 in several ways, including services such as pathway source grouping, search pathways, miRNA-target search and JAVA, Python and R APIs.
In this paper, we describe extensions of data and services available through pathDIP 4 (http://ophid.utoronto.ca/pathDIP), and provide examples on how these improvements can help in approaching some important research problems.
MATERIALS AND METHODS
Data collection and processing
Core pathways for human
We obtained protein members of 5380 pathways from 22 pathway databases (Supplementary Table S1A): ASCN2 (12), BioCarta (13), EHMN (14), HumanCyc (6), INOH (15), IPAVS (16), KEGG (17), NetPath (18), OntoCancro (19), Panther (9), PharmGKB (20), PID (21), RB-pathways (22), Reactome (10), Signalink2.0 (23), SIGNOR2.0 (24), SMPDB (25), SPIKE (26), STKE (27), System-biology.org (28), UniProt Pathways (https://www.uniprot.org/help/pathway), WikiPathways (11). It is worth mentioning that although KEGG uses computational methods to reconstruct human-specific pathways from KEGG maps, the maps they use are extracted from the experimental knowledge available in the literature.
Core pathways for model and domesticated organisms
We downloaded and pre-processed pathways for seven model and domesticated organisms (chicken, cow, fly, mouse, rat, worm and yeast) from five sources (MouseCyc (4), Panther, Reactome, WikiPathways and YeastCyc (5)) (Supplementary Table S1B). From any source that combines literature-curated and ortholog pathways to human, we removed ortholog pathways. We did not include KEGG in the sources of non-human pathways as it reconstructs species-specific pathways from KEGG maps through computational methods that partly use protein ortholog information, which is also used in pathDIP 4.
Protein IDs, orthologs and interactions
We downloaded 1:1 orthologs from Ensembl (29) release 92. Mappings between UniProt ID, NCBI Gene ID and protein names are based on UniProt ID conversion service (as of May 2019). Species-specific PPI networks of human and sixteen non-human species were obtained from IID (2) (version 2018–11) (Supplementary Table S1C).
Disease proteins
We obtained a list of 691 proteins associated with psoriasis and 304 proteins associated with osteoarthritis from DisGeNET (http://www.disgenet.org, August 2019) (30). For osteoarthritis, human proteins were mapped to their 1:1 orthologs for each of the other 16 species (DisGeNET only includes human genes), while for psoriasis the mapping was done only to mouse as mouse models are the most frequently used animal models in psoriasis studies (31). Human proteins that had no orthologs in any other species were discarded. Each list of proteins was used to query pathDIP 4 in its relevant species. In human only, we had two sets of extended pathways, one through only experimentally detected PPIs and one through all PPIs (i.e. combination of experimentally detected and computationally predicted). For psoriasis, we used these two sets (both at 0.99 confidence level), in addition to the core pathways. For osteoarthritis, we used core and ortholog pathways.
Pathway member prediction
Predictions based on orthology
For each non-human organism, we replaced members of core human pathways with their orthologs and kept only pathways with at least three ortholog members (i.e. we did not consider a single protein or a single interaction (two proteins) as a pathway) (Supplementary Table S1D).
Predictions based on physical network connectivity
For all seventeen species in pathDIP 4, we used species-specific PPI networks (obtained from IID) to predict statistically significant protein-pathway associations as described in (1). For human, we provide predictions using core pathways and two sets of PPIs: (i) experimentally detected PPIs, and (ii) the full set of human PPIs available in IID (i.e. the combination of experimentally detected and computationally predicted with high confidence). For non-human species, we used only one set of PPIs, i.e. the full set of species-specific PPIs, to predict strong physical associations between each protein and each pathway in core (if available) or ortholog pathway sets.
Calculation of recovery rate for human
We divided the number of protein-pathway pairs annotated both as ‘Known’ (experimentally detected and available in the literature) and ‘Pred’ (network-based predictions) by the number of protein-pathway pairs in the core pathways table whose protein is present in human PPI network (experimental only or experimental and predicted).
Procedure to compare recovery between ortholog pathways and network-based predictions
We used three sets of species-specific protein-pathway pairs for these comparisons:
A subset of core pathways whose titles have exact matches in human and protein members that are available in the species-specific PPI network;
Ortholog pathways;
Predictions based on only core pathways (i.e., predictions based on ortholog pathways were not considered) and species-specific PPI networks.
Next, in each organism, we compared the overlap of protein-pathway pairs in set 1 with protein-pathway pairs in sets 2 and 3.
Procedure to compare conservation of DNA replication pathway across different species
We focused on species with available core pathways (chicken, cow, fly, mouse, rat, worm, yeast). For these species, we collected proteins from core, ortholog and extended pathways whose titles include ‘DNA replication’. We then looked at the number of orthologs present from one species to the next closest in evolutionary terms.
Enrichment analysis
We used Fisher's Exact test followed by correction for multiple hypothesis testing by two different methods, Bonferroni and False Discovery Rate to calculate enrichment of input at two levels of pathways and pathway titles as explained in (1).
Word clouds
Term enrichment was performed as above, and word clouds were generated using Wordle (http://www.wordle.net).
Jaccard Index
Jaccard Index is the result of dividing the size of overlap by the size of union of any two sets of pathways and was performed in R 3.6.0.
PORTAL DESCRIPTION
Data
PathDIP 4 annotates 120 621 protein-coding genes to 6401 pathways across human and 16 non-human organisms (Table 1). Only 23 771 of these genes have core pathway annotations, thus, pathDIP 4 provides novel pathway annotations for 96 850 pathway-orphan genes. Importantly, 36 216 genes are annotated only through network-based pathway association predictions.
Functionalities
We have substantially expanded pathDIP 4 services and functionalities. Novel pathDIP 4 features include categorization of pathway sources, search using miRNA targets, search using pathway terms, and pathDIP APIs. Here we describe all previous and novel features in pathDIP 4.
Search genes tab
Input and settings
One of the novel features in pathDIP 4 is that users can select the species of interest and input pathDIP 4 with a list of proteins or genes specific to that species. Input list can contain UniProt ID, NCBI Entrez Gene ID or protein names. While pathDIP 4 input list is not case-sensitive, it returns case-sensitive protein names which is important in model organisms. Users can also select any subset of pathway source databases, as well as one of the listed annotations sets for each species, i.e. core, ortholog or extended (by predictions at different confidence cut-offs) pathways.
Enrichment analysis
PathDIP 4 performs enrichment analysis at two different levels, pathways and pathway key-terms (words and expressions). To calculate the enrichment score, pathDIP 4 uses the Fisher's Exact Test, followed by correction for multiple hypothesis testing by two different methods, Bonferroni and False Discovery Rate.
- Pathway-enrichment analysis: uses the input list and the selected annotation sets to find the pathways significantly enriched with proteins or genes in the input list.
- Term-enrichment analysis: uses biologically informative terms in enriched pathway titles to find which terms are significantly over-represented in enriched pathway titles. To extract the set of terms, we first defined a set of 393 rules to remove non-informative words and unnecessary characters and define multi-word terms (e.g. cell-cycle). Next, we cleaned this list through manual curation.
This service helps summarize and visualize pathway-enrichment results, and is particularly helpful when result lists from pathway-enrichment analyses contain hundreds of pathways.
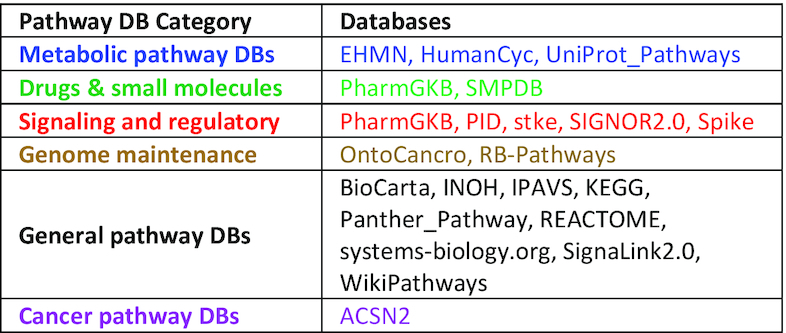
Color-coding of pathway source databases
We have grouped pathway databases according to their content into six categories (32): metabolic, drugs and small molecules, signaling and regulatory, genome maintenance, general and cancer pathway databases (Table 2). It helps users to select subsets of pathway databases suitable for their study.
Table 2.
Classification of pathway source databases according to their context-specificity, and their colour-coding in pathDIP 4. This categorization facilitates selecting pathway databases in the context that is most suitable to each study
|
Search miRNAs tab
PathDIP 4 provides users a pipeline through integration of mirDIP (33), a miRNA–target database, and pathDIP 4, to perform pathway annotation and enrichment analysis of miRNA targets in one step. Instead of querying mirDIP first, taking its output and putting it in pathDIP 4 input format, users can input a list of miRNA IDs and select the level of confidence for miRNA–target pairs (33). Next, they select pathway sources and annotation set. pathDIP 4 first sends the input list of miRNA IDs and confidence settings to mirDIP and receives the list of miRNA–targets. Next, it uses the list of target proteins to perform pathway annotation and analysis using parameters set for pathways. The output contains annotation and enrichment analysis results for the list of miRNA targets. This service is available only for human as mirDIP is a human-specific miRNA–target database.
Search pathways tab
Through this tab, users can perform annotation and enrichment analysis of any subset of pathways (not limited to a particular database). This service, which is new to pathDIP 4, uses the table of terms explained above (see term enrichment analysis) and enables users to select the list of pathways most suitable to their study.
PathDIP 4 provides two approaches to input search keywords. The first approach is to use a list of characters provided in Search Pathway tab. Upon clicking on each character, a list of pathway title keywords starting with that character appears. Users can click on any of the keywords to add it to the list of pathways of their interest. The second option is to use a drop-down search box. Users can type any part of a keyword. If any keyword including the typed character is available in pathDIP 4, a drop-down list containing those keywords appears that allows users to select the full keyword.
After selecting the keywords and databases of interest, users can search and receive a list of pathway titles including those keywords. PathDIP 4 allows users to review and refine this list of pathways. Next, they input a list of protein/gene IDs and select one of the pathways sets for further annotation and enrichment analysis only across their selected pathways. If user selects ‘save as tab delimited’ instead of ‘search’, a file containing input genes annotated with pathways including the selected words will be downloaded.
API
PathDIP 4 provides Java, Python and R APIs to facilitate programmatic use of pathway annotation and enrichment analysis.
RESULTS AND DISCUSSION
PathDIP 4 content
PathDIP 4 annotates 120 621 proteins with 6401 pathways across seventeen organisms, in three pathway tables.
Core pathways
Core pathways integrate literature-curated pathways available in 24 pathway databases and annotate 23 771 proteins in eight species (human: 13 088, chicken: 390, cow: 857, fly:935, mouse: 4353, rat: 1597, worm: 1355, yeast: 1196) (Table 1). However, while twenty-two of these resources focus on human, we found only five sources with literature-curated pathways for non-human organisms and the number of these pathways remains limited (Supplementary Table S1B).
Ortholog pathways
Ortholog pathways are predictions based on direct mapping of human core pathway members to their ortholog proteins in other organisms (Supplementary Table S1D). In PathDIP 4, 68 035 proteins across 16 species are annotated to ortholog pathways. Although this approach improves the coverage of pathway annotation for non-human organisms, the annotations are bound to the limitations of core human pathways, i.e. high rate of pathway orphans and low overlap across different source databases. In addition, only fractions of the proteins in these organisms have orthologs available in human.
Figure 1A shows the distribution of conservation of pathways across organisms. The least conserved pathways (conserved in no or one organism) are enriched in terms ‘defective’ and ‘biosynthesis’ while the pathways conserved in all organisms are enriched in words ‘signaling’, ‘degradation’, ‘RNA’, ‘transcription’, ‘response’, ‘wnt’, ‘APC-C’, ‘mitotic’, ‘Cell-cycle’, ‘apoptosis’, etc. The term ‘metabolism’ was over-represented in both groups (Supplementary Table S2A).
Figure 1.

(A) Distribution of the number of non-human organisms covered by each ortholog pathway. Pathways with count organism ‘16’ are human pathways for which we provide ortholog pathways in all non-human organisms, while pathways with count organism ‘0’ are human pathways with no ortholog pathways in pathDIP 4. (B) Recovery rate for core protein-pathway pairs through ortholog and network-based predictions. In chicken and cow, where almost all available PPIs used for network-based predictions are orthologous ones, networks do not improve recovery, whereas in fly, yeast and worm, in which experimental PPIs are more prevalent, networks improve recovery drastically. (C) Comparing coverage of the three largest pathway databases pathDIP 4 (core, ortho, extended), full Reactome (i.e. combination of core and ortholog pathways) and full WikiPathways for proteins across different species shows that extended pathways in pathDIP 4 annotate the largest number of proteins compared with the other two databases. The only two exceptions are chicken and pig. Among all 24 source databases, Reactome and WikiPathways provide the two largest number of core pathways in human (separately) and in non-human organisms (combined) (details in Supplementary Tables S1A and B). Plots generated using R package ggplot2 (version 3.2.1).
Extended pathways
Although predicting pathway annotations for non-human proteins through their human orthologs significantly improves coverage of pathways for non-human organisms, it is not sufficient due to caveats in human pathways and limitations of ortholog mapping.
Extended pathways combine core and ortholog pathways, with predictions based on significance of physical connectivity of each protein with members of each core or ortholog pathway. Association-prediction based on network properties of proteins and genes has been applied to different research questions in molecular and systems biology (34–38). Compared to other network-based measures, connectivity-significance has been shown to be one of the best predictors of association (for example, to a group of disease genes or pathways) (39). PathDIP 4 annotates 120 621 proteins in human and sixteen non-human organisms. Furthermore, despite its relatively conservative approach (compared with other available algorithms such as (39)), extended pathways annotate 36 216 pathway orphan proteins in sixteen species, i.e., proteins with no pathway annotations available in core or ortholog pathways.
In human, extended pathways increase coverage of pathway annotations for proteins from 65% (13 088 proteins) to 92% (18 454 proteins) and cross-validation determined recovery rate of up-to 87% for core pathways (Table 3).
Table 3.
Coverage and recovery rate of core pathways, extended pathways using only experimental PPIs, and extended pathways using full (combination of experimental and predicted) PPIs for human proteins (top row) and their pathway memberships (bottom row) in pathDIP 4.
| Human pathways | Curated | Extended using experimental PPIs (0.95) | Extended using experimental and predicted PPIs (0.95) |
|---|---|---|---|
| Coverage for protein-coding genes | 65% | 89% | 92% |
| Recovery for known pathway members | - | 59% | 87% |
Extended pathways are based on 95% confidence cut-off.
Comparison of ortholog versus extended pathways
Coverage for proteins
Comparing coverage of ortholog versus network-based predicted pathways for proteins shows that in all seventeen species combined, almost 30% of extended pathways are pathway orphans in core and ortholog pathways and have obtained their annotations through only network-based predictions (Table 1). Across non-human species, network-based-only predictions cover between ∼0.2% (guinea-pig) and 70% (yeast) of extended pathways with median 27%. This highlights the importance of network-based predictions, even though in many organisms all network-based predictions are based on connectivity of proteins with only ortholog pathways and through only orthologous PPIs (due to lack of available experimental data).
Recovery for core pathways
To compare the recovery rate of ortholog-based versus network-based predictions, we focused on the seven organisms for which both core and ortholog pathways are available, i.e. chicken, cow, fly, mouse, rat, worm and yeast. Details are provided in ‘methods’ section and (Supplementary Table S2B).
Figure 1B shows recovery rate of ortholog pathways versus network-based pathways. Importantly, network-based predictions improve recovery of ortholog pathways for core protein-pathway pairs differently across different species. While in chicken and cow, PPIs improve recovery rate only slightly (1.03, 1.06), in fly, worm and yeast, networks improve the recovery by a factor of 2.20, 1.41, 2.37. In mouse and rat, the improvements are 1.11 and 1.10, respectively. Our further investigation showed that in chicken and cow, <0.5% of PPIs are experimentally detected; thus, the major source of PPIs are orthologous PPIs whose proteins are a subset of proteins we used for ortholog pathway predictions. Similarly, in mouse and rat, experimentally detected PPIs constitute 10 and 2.8% of the full network. However, in worm, fly and yeast, experimentally detected PPIs make ∼22%, ∼57% and 80% of the PPI network, respectively. A similar trend is observed for the number of proteins in experimental versus orthologous PPI networks (Supplementary Figure S1A and Supplementary Tables S2C and D). These numbers can explain the differences in recovery rates of ortholog versus network-based protein-pathway predictions across different species, and reinforce the importance of integration of data with different types of sources to extract relevant information. In fact, experimental PPIs, whose detection method is independent of ortholog proteins, improve recovery-rate markedly better than using only orthologous PPIs.
Comparison of pathDIP 4 with other databases
Comparison of core, ortholog and extended pathways in pathDIP 4 with the combination of literature-curated and ortholog pathways in Reactome and WikiPathways, the two largest publicly available databases for ortholog pathways in organisms, shows that coverage of pathDIP 4 for proteins in most of the organisms is higher than coverage of the other two databases (Figure 1C). For several organisms, the protein coverage of ortholog pathways in PathDIP 4 is lower than the coverage of ortholog pathways in Reactome (release 68). However, when the extended pathways are selected, the coverage of PathDIP 4 becomes higher than that of Reactome. Two exceptions are chicken and pig. Lower coverage of ortholog pathways in pathDIP 4 for proteins is due to using a more reliable set of orthologs (inclusion of only 1:1 orthologs) compared with Reactome. Supplementary Figure S1B compares the number of pathways available in different pathway sets in pathDIP 4, full Reactome and full WikiPathways per species.
While network-based predictions are specific to PathDIP 4, to the best of our knowledge, it is the only available integrated resource that provides direct distinction between core and ortholog pathways. In addition, direct miRNA–target pathway analysis, pathway source classification, and term-enrichment analysis are additional useful features specific to pathDIP 4.
Example applications
Conservation of DNA replication pathway across different species
To show the quality of our predictions across species, we looked at a pathway that is highly conserved: DNA replication (40). Figure 2A shows that proteins annotated only with core pathways are conserved among mammals or among lower organisms (fly, worm and yeast), while inclusion of orthologs and predicted pathway annotations extends the overlap from human to yeast. Interestingly, 14 genes present in the ortholog pathway set in cow have orthologs in the core pathways in fly (PRIM2, POLE, RPA1, RFC3, RFC5, MCM7, CDC45, ORC5, ORC2, CDC6, CDT1, MCM10, ORC4, ORC1), providing experimental support for the ortholog approach. When considering core and predicted pathway annotations for human, the overlap of DNA replication proteins between human and other seven species increases. 40 proteins (listed in Supplementary Table S3A) are present in all the species from human to yeast. Of these, 9 are present in yeast core pathways, 11 in yeast ortholog pathways and the remaining 20 in predicted pathways. Interestingly, all 20 are present only in predicted pathways in all the 7 species considered, even though among them there are proteins that have been described being involved in DNA replication (i.e. MRE11A (41), XPO1 (42), SMC2 (43)). Of note, three proteins annotated with predicted DNA replication pathways in human have orthologs in other species that are annotated with core pathways (HIST1H3J, HIST2H3D and TOP1MT between human and mouse; HIST2H3D and TOP1MT between human and rat).
Figure 2.

(A) Conservation of DNA-replication pathways across multiple species. For DNA replication pathways in each species, orthologs to human were considered, and overlap of orthologs per consecutive pairs of species are shown. Each bar represents an organism and each color represents genes present in ortholog (bottom), expanded (middle) and core (top) DNA replication pathways. Flow among bars depicts the number of proteins with orthologs from the starting organism to the landing one. Flow color grows darker with the number of organisms in which a set of proteins is conserved. (B) Overlap among core pathways in different species after enrichment analysis using osteoarthritis genes or their orthologs. (C) Heatmap of Jaccard indices of pathways across different species. Human pathways were obtained using core pathways while for every other species only ortholog pathways were used. Plots generated based on R package alluvial (version 6.3.0) modified by authors, UpSetR (version 1.3.3), and ggplot2 (version 3.1.0).
Ranking the non-human organisms suitable for disease-related functional genomics studies
Osteoarthritis-associated genes were obtained from DisGeNET and their orthologs in other species were used to query pathDIP 4 using core pathways to observe disease-specific pathway conservation. Overlap of core pathways among the species with available core pathways is quite poor (Figure 2B), while the overlap notably improves among all species when we consider ortholog pathways (Figure 2C and Supplementary Table S3B). The three invertebrate species, fly, worm and yeast did not have any enriched pathway. Duck had only one enriched pathway, which was also present in all remaining twelve species and in human: Endochondral Ossification, a process essential for osteoarthritis development (44). A total of 76 pathways were present in 11 out of 14 vertebrates, and these included processes linked to osteoarthritis such as TGF-beta signaling (45), GPCRs (46) and inflammation (47), as visible in (Supplementary Figures S2A and B).
Interestingly, model organisms used to study osteoarthritis include mouse (Jaccard Index (JI) with human is 0.88), rat, guinea pig (JI for both: 0.63), dog (JI: 0.57), rabbit (JI: 0.53), horse (JI: 0.42) and sheep (JI: 0.23) (48). Our data suggest not studying molecular mechanisms of osteoarthritis parallel to human in sheep, whereas cow, as the second highly overlapping organism (after mouse, JI: 0.68), is a good candidate (Supplementary Table S3C). Cow has naturally occurring osteoarthritis (49) and has been previously proposed as a model organism for knee osteoarthritis due to the similarity of its knee to the human's (50). Cat, turkey and duck have very little overlap (JI: 0.3, 0.008 and 0.001, respectively) while pig and chicken have an intermediate overlap (JI: 0.47 and 0.46, respectively). Commercial pigs have been used as model organisms for naturally occurring osteoarthritis (51), even if their knee structure is less similar to the human one than cow (50), while chicken has been proposed in the past but never really used (52), to the best of our knowledge.
This analysis highlights the application of pathDIP 4 data in selecting the right model organism (even beyond the most commonly used ones if needed) for the type of molecular mechanism to be studied, especially when the aim is to translate any finding to human diseases and treatments.
Psoriasis: network-based predictions extend the set of disease-related pathways
We used the genes associated to psoriasis to obtain pathway-enrichment using human core and extended sets as described in ‘Materials and Methods’ section. We obtained 1148 pathways in core pathways, 1737 in extended using experimental PPIs and 2531 in extended using all PPIs (experimental and predicted) (Supplementary Tables S3D and E). A total of 972 such pathways were common among the three sets, and included pathways linked to MAPK and NFKB signaling and to immune processes (Supplementary Figures S3A and B). A total of 719 pathways were exclusively annotated as extended using experimental and predicted PPIs and included signaling and metabolism (Supplementary Figure S3C).
A total of 25 and 69 pathways were specific to the core and extended pathways respectively. Two psoriasis experts reviewed the two sets of pathways, aiming to verify whether they are linked to psoriasis. The two experts strongly disagreed on nine pathways (three from literature and six from extended), for a discordance rate of 9.5% (Supplementary Tables S3F and G). Sixteen pathways from the literature set were voted as related to psoriasis, five were ‘maybe related’ and one was considered unrelated. Of the extended set, 37 were labeled as related, 16 as ‘maybe’ or ‘don’t know’ and 10 as non-related to psoriasis. Addition of the 37 psoriasis related pathways to the set of enriched pathways only through predicted pathways shows the importance of our predictions in performing comprehensive pathway analysis. Moreover, having only 10 non-related out of 69 extended pathways, along with 37 related and 16 gray zone (‘maybe’ or ‘don’t know’) shows that the extended pathways mostly include pathways that are known to be or that can be meaningful to the proteins being analysed rather than only random pathways.
Psoriasis is a disease restricted to humans and it has been reported sporadically in few other animals, making it difficult to identify a proper model organism and translate findings from the model organism to human. At the present time, mouse models mimicking the disease are the most frequently used (31), so we queried mouse orthologs of human psoriasis related genes in mouse pathDIP 4, using core, ortholog and extended pathways. Twenty-seven pathways were enriched in core, 1129 in ortholog and 2534 in extended pathways. Thirteen pathways were present in all three sets, while two and 1075 were present in the overlap of extended pathways with either core or ortholog pathways (respectively) (Supplementary Table S3H). Term-enrichment performed on the union of these 1090 mouse pathways shows very similar terms when compared to 972 overlapping human pathways (Supplementary Figure S4A and B). This suggests conservation of molecular mechanisms of psoriasis in mouse and explains why mouse models have been successful even though mice do not spontaneously develop psoriasis.
SUMMARY AND CONCLUSION
PathDIP 4, compared with its previous versions, is substantially expanded and improved in several ways. In addition to human, it extends pathway annotations for sixteen non-human species through integrating different data types from several sources, provides new services such as pathway database classification and search pathways, as well as Java, Python and R APIs, and offers features specific to pathDIP 4 such as direct miRNA-target search for human and term enrichment analysis.
Limited coverage of core pathways for protein-coding genes of human and non-human organisms (Table 1), along with the slow growth rate of core pathway annotations (57% in 2016 versus 65% in 2019 in human) demonstrates the importance of devising computational methods that use data integration to predict physically and biologically relevant protein-pathway associations. PathDIP 4 integrates core pathway data from 24 pathway databases (nineteen only for human, three for both human and organisms and two only for organisms). Limited coverage of literature-curated source databases affects pathDIP 4 too. Thus, we used two well-established prediction methods to improve coverage of pathway annotations for proteins, and improve consistency across data from different pathway sources. In pathDIP 4, extended pathways in human increase coverage for protein coding genes to 92%, while they extend protein coverage to 9.56 times for model organisms (Table 1).
It is worth mentioning that despite all the improvements, pathDIP 4 annotations and analysis results are not free of errors and limitations in their sources. For example, in addition to their incompleteness and inconsistency, different data sources have been created for different purposes, cover different contexts and include different levels of detail. However, with the improvement of our source databases, coverage and quality of data in pathDIP 4 will improve, too. For example, while pathDIP 2.5 cross-validation in 2016 showed recovery rate for core pathways in human to be 71% (1), in pathDIP 4 recovery rate has increased to 87% (Table 3).
Combined, our annotations cover 120 621 unique proteins in human, six model organisms, and ten domesticated animals, from which only 23 771 proteins have pathway annotations in the literature (for details see Table 1). While in most of the organisms, network-based annotations overlap with high fraction of ortholog pathways (See ‘comparison of ortholog versus extended pathways’ section), 36 216 proteins are annotated only through network-based predictions, supporting the importance of data integration to extract present, but hidden information in different data sources and types.
Our three use-cases are only a few examples to highlight how improved coverage for proteins in multiple species provides a unique resource that enables comprehensive enrichment analysis, hypotheses generation, in silico validation and explanations in basic, translational and clinical research.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ontario Research Fund [#34876 to I.J.]; Natural Sciences Research Council [NSERC #203475 to I.J.]; Canada Foundation for Innovation [#225404, #30865 to I.J.]; Ian Lawson Van Toch Memorial Fund; IBM; Atlantic Canada Opportunities Agency [# 781-19095-209843 to P.R.]. Funding for open access charge: Ontario Research Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rahmati S., Abovsky M., Pastrello C., Jurisica I.. PathDIP: an annotated resource for known and predicted human gene-pathway associations and pathway enrichment analysis. Nucleic Acids Res. 2017; 45:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kotlyar M., Pastrello C., Malik Z., Jurisica I.. IID 2018 update: context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019; 47:D581–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Mueller L.A. et al.. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016; 44:D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Evsikov A. V, Dolan M.E., Genrich M.P., Patek E., Bult C.J.. MouseCyc: a curated biochemical pathways database for the laboratory mouse. Genome Biol. 2009; 10:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caspi R., Foerster H., Fulcher C.A., Kaipa P., Krummenacker M., Latendresse M., Paley S., Rhee S.Y., Shearer A.G., Tissier C. et al.. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008; 36:D623–D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trupp M., Altman T., Fulcher C.A., Caspi R., Krummenacker M., Paley S., Karp P.D.. Beyond the genome (BTG) is a (PGDB) pathway genome database: HumanCyc. Genome Biol. 2010; 11:O12. [Google Scholar]
- 7. Herwig R., Hardt C., Lienhard M., Kamburov A.. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016; 11:1889–1907. [DOI] [PubMed] [Google Scholar]
- 8. Kanehisa M., Sato Y.. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2019; doi:10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X., Thomas P.D.. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019; 14:703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B. et al.. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018; 46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Slenter D.N., Kutmon M., Hanspers K., Riutta A., Windsor J., Nunes N., Mélius J., Cirillo E., Coort S.L., DIgles D. et al.. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018; 46:D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuperstein I., Bonnet E., Nguyen H.A., Cohen D., Viara E., Grieco L., Fourquet S., Calzone L., Russo C., Kondratova M. et al.. Atlas of Cancer Signalling Network: a systems biology resource for integrative analysis of cancer data with Google Maps. Oncogenesis. 2015; 4:e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishimura D. BioCarta. Biotech Softw. Internet Rep. 2001; 2:117–120. [Google Scholar]
- 14. Ma H., Sorokin A., Mazein A., Selkov A., Selkov E., Demin O., Goryanin I.. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 2007; 3:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamamoto S., Sakai N., Nakamura H., Fukagawa H., Fukuda K., Takagi T.. INOH: Ontology-based highly structured database of signal transduction pathways. Database. 2011; 2011:doi:10.1093/database/bar052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sreenivasaiah P.K., Rani S., Cayetano J., Arul N., Kim D.H.. IPAVS: Integrated Pathway Resources, Analysis and Visualization System. Nucleic Acids Res. 2012; 40:D803–D808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M.. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kandasamy K., Mohan S.S.S., Raju R., Keerthikumar S., Kumar G.S.S., Venugopal A.K., Telikicherla D., Navarro D.J., Mathivanan S., Pecquet C. et al.. NetPath: a Public Resource of Curated Signal Transduction Pathways. Genome Biol. 2010; 11:R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simão É.M., Cabral H.B., Castro M.A.A., Sinigaglia M., Mombach J.C.M., Librelotto G.R.. Modeling the Human Genome Maintenance network. Phys. A Stat. Mech. its Appl. 2010; 389:4188–4194. [Google Scholar]
- 20. Whirl-Carrillo M., McDonagh E.M., Hebert J.M., Gong L., Sangkuhl K., Thorn C.F., Altman R.B., Klein T.E.. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012; 92:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H.. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009; 37:D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calzone L., Gelay A., Zinovyev A., Radvanyi F., Barillot E.. A comprehensive modular map of molecular interactions in RB/E2F pathway. Mol. Syst. Biol. 2008; 4:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fazekas D., Koltai M., Türei D., Módos D., Pálfy M., Dúl Z., Zsákai L., Szalay-Bekő M., Lenti K., Farkas I.J. et al.. SignaLink 2 – a signaling pathway resource with multi-layered regulatory networks. BMC Syst. Biol. 2013; 7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perfetto L., Briganti L., Calderone A., Perpetuini A.C., Iannuccelli M., Langone F., Licata L., Marinkovic M., Mattioni A., Pavlidou T. et al.. SIGNOR: A database of causal relationships between biological entities. Nucleic Acids Res. 2016; 44:D548–D554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jewison T., Su Y., Disfany F.M., Liang Y., Knox C., MacIejewski A., Poelzer J., Huynh J., Zhou Y., Arndt D. et al.. SMPDB 2.0: Big improvements to the small molecule pathway database. Nucleic Acids Res. 2014; 42:D478–D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paz A., Brownstein Z., Ber Y., Bialik S., David E., Sagir D., Ulitsky I., Elkon R., Kimchi A., Avraham K.B. et al.. SPIKE: a database of highly curated human signaling pathways. Nucleic Acids Res. 2011; 39:D793–D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gough N.R. Science's signal transduction knowledge environment. Ann. N. Y. Acad. Sci. 2002; 971:585–587. [DOI] [PubMed] [Google Scholar]
- 28. Kitano H., Funahashi A., Matsuoka Y., Oda K.. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005; 23:961–966. [DOI] [PubMed] [Google Scholar]
- 29. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piñero J., Bravo Á., Queralt-Rosinach N., Gutiérrez-Sacristán A., Deu-Pons J., Centeno E., García-García J., Sanz F., Furlong L.I.. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017; 45:D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bochénska K., Smolińska E., Moskot M., Jakóbkiewicz-Banecka J., Gabig-Cimińska M.. Models in the research process of psoriasis. Int. J. Mol. Sci. 2017; 18:2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rahmati S., Pastrello C., Rossos A.E.M., Jurisica I.. Two Decades of Biological Pathway Databases: results and challenges. Encycl. Bioinform. Comput. Biol. 2019; 1071–1084. [Google Scholar]
- 33. Tokar T., Pastrello C., Rossos A.E.M., Abovsky M., Hauschild A.C., Tsay M., Lu R., Jurisica I.. MirDIP 4.1—integrative database of human microRNA target predictions. Nucleic Acids Res. 2018; 46:D360–D370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hishigaki H., Nakai K., Ono T., Tanigami A., Takagi T.. Assessment of prediction accuracy of protein function from protein-protein interaction data. Yeast. 2001; 18:523–531. [DOI] [PubMed] [Google Scholar]
- 35. Guo J., Wu X., Zhang D.-Y., Lin K.. Genome-wide inference of protein interaction sites: lessons from the yeast high-quality negative protein-protein interaction dataset. Nucleic Acids Res. 2008; 36:2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bass J.I.F., Diallo A., Nelson J., Soto J.M., Myers C.L., Walhout A.J.M.. Using networks to measure similarity between genes: association index selection. Nat. Methods. 2013; 10:1169–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Piovesan D., Giollo M., Ferrari C., Tosatto S.C.E.. Protein function prediction using guilty by association from interaction networks. Amino Acids. 2015; 47:2583–2592. [DOI] [PubMed] [Google Scholar]
- 38. Wang S., Huang E., Cairns J., Peng J., Wang L., Sinha S.. Identification of pathways associated with chemosensitivity through network embedding. PLoS Comput. Biol. 2019; 15:e1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghiassian S.D., Menche J., Barabási A.-L.. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 2015; 11:e1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gaboriaud J., Wu P.Y.J.. Insights into the link between the organization of DNA replication and the mutational landscape. Genes (Basel). 2019; 10:E252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maser R.S., Mirzoeva O.K., Wells J., Olivares H., Williams B.R., Zinkel R.A., Farnham P.J., Petrini J.H.. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol. Cell Biol. 2001; 21:6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knauer S.K., Bier C., Habtemichael N., Stauber R.H.. The Survivin–Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep. 2006; 7:1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thadani R., Kamenz J., Heeger S., Muñoz S., Uhlmann F.. Cell-cycle regulation of dynamic chromosome association of the condensin complex. Cell Rep. 2018; 23:2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hosaka Y., Saito T., Sugita S., Hikata T., Kobayashi H., Fukai A., Taniguchi Y., Hirata M., Akiyama H., Chung U. et al.. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shen J., Li S., Chen D.. TGF-β signaling and the development of osteoarthritis. Bone Res. 2014; 2:14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo J., Sun P., Siwko S., Liu M., Xiao J.. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019; 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sokolove J., Lepus C.M.. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013; 5:77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuyinu E.L., Narayanan G., Nair L.S., Laurencin C.T.. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016; 11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vaughan L.C. Osteoarthritis in cattle. Aust. Vet. J. 1961; 37:329–329. [Google Scholar]
- 50. Proffen B.L., McElfresh M., Fleming B.C., Murray M.M.. A comparative anatomical study of the human knee and six animal species. Knee. 2012; 19:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Macfadyen M.A., Daniel Z., Kelly S., Parr T., Brameld J.M., Murton A.J., Jones S.W.. The commercial pig as a model of spontaneously-occurring osteoarthritis. BMC Musculoskelet. Disord. 2019; 20:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Anderson-Mackenzie J., Hulmes D.J., Thorp B.. Degenerative joint disease in poultry — differences in composition and morphology of articular cartilage are associated with strain susceptibility. Res. Vet. Sci. 1997; 63:29–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.