


Abstract
Inference of active regulatory mechanisms underlying specific molecular and environmental perturbations is essential for understanding cellular response. The success of inference algorithms relies on the quality and coverage of the underlying network of regulator–gene interactions. Several commercial platforms provide large and manually curated regulatory networks and functionality to perform inference on these networks. Adaptation of such platforms for open-source academic applications has been hindered by the lack of availability of accurate, high-coverage networks of regulatory interactions and integration of efficient causal inference algorithms. In this work, we present CIE, an integrated platform for causal inference of active regulatory mechanisms form differential gene expression data. Using a regularized Gaussian Graphical Model, we construct a transcriptional regulatory network by integrating publicly available ChIP-seq experiments with gene-expression data from tissue-specific RNA-seq experiments. Our GGM approach identifies high confidence transcription factor (TF)–gene interactions and annotates the interactions with information on mode of regulation (activation vs. repression). Benchmarks against manually curated databases of TF–gene interactions show that our method can accurately detect mode of regulation. We demonstrate the ability of our platform to identify active transcriptional regulators by using controlled in vitro overexpression and stem-cell differentiation studies and utilize our method to investigate transcriptional mechanisms of fibroblast phenotypic plasticity.
INTRODUCTION
Technological advancements in high-throughput sequencing have made it possible to measure expression of genes at a relatively low cost. However, the direct measurement of regulatory mechanisms, such as transcription factor (TF) activity, in a high-throughput manner is still not readily available. Consequently, there is a need for computational approaches that can identify active regulatory mechanisms from observable gene expression data. The scientific community has developed a multitude of algorithms and biophysical models to study the impact of TF activity on gene expression. Some of these algorithms attempt to infer TF activity and dynamics directly from gene expression data (1,2). Others rely on biophysical approaches to model expression of genes based on known TF–gene interactions (3). Another class of algorithms, which are the main focus of this work, use prior biological knowledge on biomolecular interactions to link a differential gene expression (DGE) profile to upstream regulators (e.g. TFs) (4–7). The essential ingredients of these algorithms are (i) a DGE profile, (ii) a network of biomolecular interactions and (iii) an inference algorithm to query the network.
The DGE profile as obtained from RNA-seq or microarray studies is the observable input and quantifies the difference in transcript abundance between two conditions (e.g. healthy versus disease, stimulated versus not stimulated, etc.). The network of biomolecular interactions encapsulates the prior biological knowledge. The accuracy and ability of inference algorithms to identify upstream molecular drivers of observed DGE profiles rely to a large degree on the quality and coverage of the network and availability of auxiliary information on interactions within the network. There are several sources of publicly available protein–protein interactions (PPIs) and signaling pathways (e.g. STRINGdb (8), Pathway Commons (9), Kyto Encyclopedia of Genes and Genomes (KEGG) Pathways (10), etc.). In case of regulatory networks, Ingenuity (www.ingenuity.com) provides a high-coverage manually curated network of regulatory interactions in an integrated platform, Ingenuity Pathway Analysis tool (4). Among other things, this platform provides pathway inference, enrichment analysis functionality and network visualization tools. However, the Ingenuity tools are inaccessible for the majority of academic applications and there is a need for freely available alternatives for academic purposes.
Several approaches for reconstruction of regulatory networks form gene expression data have been proposed by the scientific community. These approaches can be broadly categorized as (i) co-expression based approaches (11,12), (ii) non-steady state approaches based on time-series analysis (13) and (iii) text-mining approaches (14–16). In co-expression based approaches, the objective is to identify gene–regulator interactions by analyzing correlation patterns using a variety of methods, including direct correlation, partial correlation (e.g. using GGMs) (17), mutual information based methods (18) and Bayesian network reconstruction methods (19,20). A limitation of these approaches is that the recovered interactions are typically associative (undirected). Methods based on non-steady state approaches typically attempt to inference the dynamics of gene–regulator interactions and can yield more accurate results (21). However, they require time-course gene expression data across multiple conditions, which may not be readily available. Text-mining based methods attempt to extract interactions from biomedical literature. There is vast body of literature on these approaches and several data-bases include interactions obtained from text-mining methods (14). These approaches, however, typically yield low coverage and assessing false positives can be very challenging. It should be noted that several of the aforementioned methods utilize other sources of information as prior knowledge to increase the accuracy of the recovered network. In particular methods based on interventional data (e.g. single gene KO) are very promising (22,23). However, these approaches are tailored for a specific condition or particular pathways and generalization to multiple conditions is currently infeasible.
Consequently, several public sources of gene regulatory interactions are derived from aforementioned computational and experimental approaches. These sources include the Transcriptional Regulatory Element Database (TRED) (24), the Transcription Regulatory Regions Database (TRRD) (25) and Transcriptional Regulatory Relationships Unraveled by Sentence-based Text Mining (TRRUST) (14). These databases provide valuable information on gene regulatory mechanisms, but drawbacks exist. The scope of the databases containing experimentally validated interactions are very small, and cover only a fraction of TF–gene interactions. On the other hand, databases of computationally predicted and expression-driven interactions are typically very noisy. Importantly, the majority of the databases do not report the direction of regulation (activation versus repression)—which is crucial to understanding the functional behavior of the cell.
In this work, we present Causal Inference Engine (CIE), a platform for active regulator inference on biological networks consisting of a web-server and a user-friendly R-package. The platform provides various inference models, including methods based on Fisher’s exact test (enrichment test) as well as directional enrichment models that can utilize information on mode of regulation (6,7). Moreover, we present an approach based on regularized Gaussian Graphical Models (GGM) to construct an accurate and high-coverage annotated networks of TF–gene regulatory interactions. We achieve this by integrating publicly available high-throughput ChIP-seq experiments deposited in ChIP-Atlas (26) with tissue specific gene expression data from GTEx (27). The key differences of our approach from previous approaches include: (i) TF–gene interactions derived from ChIP-seq experiments are utilized to construct a penalty matrix encoding the prior causal graph of TF–gene interactions. (ii) The penalty matrix is utilized to regularize the log-likelihood of a GGM constructed from tissue-specific gene expression data, from which a posterior TF–gene interaction is constructed. The design of the regularization is such that the GGM essentially eliminates the interactions in the prior ChIP-network that are not supported by the expression data, resulting in posterior tissue-specific and causal ChIP-networks. (iii) An additional advantage is determination of the mode of regulation of the posterior interactions, resulting in network of annotated tissue-specific TF–gene interactions. (iv) In addition to tissue specific interactions, we provide cross-tissue interactions, i.e. interactions that appear in multiple tissues and are in a sense ‘Universally applicable’. (v) Subsequent integrated algorithms in the CIE platform for directional enrichment analysis, designed to identify active transcriptional regulators of DGE data provides a comprehensive pipeline for analysis of transcriptional regulators.
We show the consistency and accuracy of our reconstructed network by benchmarking against manually curated interactions in the gold standard databases. We demonstrate the utility of our platform in identifying active regulators using controlled in vitro overexpression studies as well as more complex gene expression data from a stem cell differentiation experiment. Additionally, we show how our platform can assist in identifying novel transcriptional regulatory mechanisms using gene expression data from primary prostate fibroblast cells stimulated with TGFβ and CXCL12. Although our focus in this work is on transcriptional regulatory networks, the R-package provides functionality to perform inference on any type of user provided network. The CIE platform provides higher-order pathway enrichment analysis on identified active regulators using Reactome pathways (28). Figure 1 shows a schematic overview of the CIE platform.
Figure 1.

CIE platform for inference of active regulators. CIE takes a user provided DGE profile as input (left panel). User selects a prior regulatory network from the provided databases. In the server side (middle panel), the input is processed and the Shiny app calls R functions to perform the inference analysis based on the user selected options. Predicted active regulators associated with the DEG profile are displayed with interactive graphics and are downloadable in table format (right panel). CIE also offers pathway enrichment analysis by mapping the inferred regulators to pathways from the Reactome database.
MATERIALS AND METHODS
Regulatory network
The interaction network can be viewed as a graph G = (V, E), consisting of a set of nodes V (biological entities) and a set of edges E = (u,  ). The network is not limited to interactions between TFs and genes and can include other types of interactions (e.g. interactions between compounds and proteins, PPIs, etc.). Some of these edges may be associations (an undirected edge u −
). The network is not limited to interactions between TFs and genes and can include other types of interactions (e.g. interactions between compounds and proteins, PPIs, etc.). Some of these edges may be associations (an undirected edge u −  , indicating change in u is correlated with change in
, indicating change in u is correlated with change in  ), causal (a directed edge u →
), causal (a directed edge u →  , indicating that u regulates
, indicating that u regulates 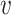 ) or signed causal (with + or −, indicating mode of regulation).
) or signed causal (with + or −, indicating mode of regulation).
Databases
We utilized three sources of TF–gene interaction networks. Our criterion for inclusion was that they either must include high-confidence, manually curated interactions with literature support or must have direct experimental evidence. These sources are:
(i) The TRRUST database: TRRUST (14) is a manually curated database of human transcriptional regulatory network derived from PubMed articles with partial information on mode of regulation. It contains 9396 regulatory interactions between 795 human TFs and 2067 target genes.
(ii) The TRED database: TRED (24) is an integrated repository for both cis- and trans- regulatory elements in mammals with experimental evidence. It includes a total of 6726 interactions between 36 TF and 2910 genes. These interactions are not annotated with mode of regulation.
(iii) ChIP-seq derived TF–gene interactions: we obtained all publicly available ChIP-seq data (>96 000 experiments) that are processed and deposited into ChIP-Atlas (26). A TF–gene interaction network was assembled by merging all experiments and applying various filters for peak signal intensity (0–1000) and distance to the transcription start site (TSS) (1, 5 and 10 kb). These filters are integrated in the CIE platform and can be applied interactively in the web-app. For example, peak intensity score of 500 and distance of 5 kb to TSS results in 185 271 interactions between 642 TFs and 16 148 target genes. Note that ChIP-seq network does not directly provide information on mode of regulation and all TF–gene interactions in this database are unannotated.
(iv) STRINGdb: In addition to TF–gene interactions, we also included Protein-gene interactions from STRINGdb (8). STRINGdb includes PPIs from various sources including curated, experimentally supported and computationally derived interactions. Some of the interactions are causal and annotated, but most interactions in the STRINGdb are undirected. For each undirected PPI u − 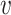 , we constructed two directed interactions u →
, we constructed two directed interactions u → 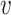 and
and  → u.
→ u.
Differential gene expression profiles
We used several DGE profiles from microarray and RNA-seq experiments to evaluate the utility of our platform. For microarray data, gene expression profiles were normalized and differentially expressed genes were computed using the R limma package (29). We applied a 1.3 absolute value fold change and <0.05 False Discovery Rate (FDR) corrected P-value filter for selecting differentially expressed genes. RNA-seq data was processed using the HISAT2 (30) pipeline and differentially expressed genes were identified for each treatment the edgeR package (31). Similar filters for FDR and fold change were applied to identify differentially expressed genes. The datasets that we utilized for our evaluation are:
(i) Controlled overexpression experiments. We utilized three datasets from (32), in which recombinant antiviruses were used to infect normal human epithelial cell in order to overexpress specific oncogenes. The over expressed genes are E2F3, c-Myc and H-Ras. There are 272, 220 and 268 differentially expressed genes compared to the WT in the experiments respectively.
(ii) Stem cell directed differentiation. We used a time-course invitro differentiation model of pancreatic beta cell development from (33). NEUROG3+ Pancreatic progenitor cells convert to NKX2-2+ endocrine cells, which are able to further differentiate into fully functional insulin producing cells upon implantation into mice (34). A total of 1000 differentially expressed genes were identified from this dataset.
(iii) fibroblast phenotypic plasticity. We utilized data from RNA-seq experiments performed on prostatic stromal fibroblasts stimulated with Vehicle, TGFβ and CXCL12 (35). A total of 10 032 differentially expressed genes were identified in fibroblasts treated with TGFβ or CXCL12. The DGE profile of TGFβ and CXCL12 were 75% similar (7502 transcripts). A total of 1012 (10%) were induced by TGFβ treatment only and 1357 (13%) by CXCL12 treatment only. A total of 161 (2%) were differentially regulated in opposite directions by CXCL12 and TGFβ.
Construction of transcriptional regulatory networks
The network constructed from ChIP-data is noisy as the experiments are performed under various conditions in different cell lines. Moreover, the interactions are not annotated with mode of regulation (activation versus repression). To reduce the noise and annotate the interactions, we utilized a regularized Gaussian Graphical Model (glasso) (36) to integrate the ChIP-derived network with tissue specific RNA-seq data obtained form the Genotype-Tissue Expression project (The GTEx Consortium) (27). Processed normalized gene expression values were obtained from (37), where GTEx RNA sequencing reads from 15 tissues from 2585 paired-end RNA-seq samples were re-processed, uniformly realigned and normalized to remove batch effects and tissues with low number of samples. Every ChIP-derived interaction was taken account without any filters (≈4 × 106 interactions). To construct the tissue-specific annotated regulatory networks, we estimated a sparse covariance matrix using each tissue-expression data separately, while softly enforcing ChIP-derived interaction using an ℓ1 penalty matrix. Gene expression was log transformed prior to analysis. Only protein-coding genes were utilized and genes with no one-to-one map between Ensemble ID and HGNC symbol were excluded.
The process of constructing a posterior network form gene expression data and ChIP-network is as follows. Let S denote the empirical covariance matrix estimated from the RNA-seq expression data for a given tissue, Σ be the (unknown) covariance matrix and Θ = Σ−1 be the precision matrix. Glasso directly estimates the precision matrix Θ by maximizing the ℓ1 penalized log likelihood
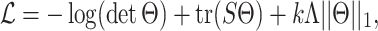 |
(1) |
on the space of positive semi-definite matrices. Here Λ is a shrinkage parameter matrix and k is a scaler tuning parameter. The prior ChIP-network structure was incorporated in the penalty as follows. First, we constructed an adjacency matrix A from the ChIP-network. The rows and columns of A were arranged by TFs first and then by genes. If there is a connection between TF i and gene j the corresponding entry in the adjacency matrix is set to 1 (i.e. Aij = 1), and otherwise it is set to 0. The penalty matrix Λ has the same size as the adjacency matrix. The entries of this matrix are constant values and are set to differentially penalize the connections based on the information in the adjacency matrix as follows:
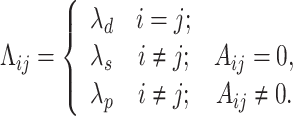 |
(2) |
In our implementation, diagonal elements were not penalized (i.e. λd = 0). Interacting TFs and genes (i.e. Aij = 1) were penalized by a small nonzero value (λs = 0.05) and non-interacting pairs were penalized by a relatively large value (λp = 0.5). The matrix was then scaled by a constant value k. We utilized a path of values ranging from 1 to 6 with step size 0.1 for k. For each scaling value, we fitted the model by maximizing the log likelihood and calculated the corresponding precision matrix Θ, from which a posterior regulatory network was constructed based on the conditional independence property of GGMs (38). More precisely there is a connection between TF i and gene j if and only if Θij ≥ ε. The threshold value ε was selected 1e − 4 empirically as a small value. For each posterior network, we calculated the scale-free property using the R-squared (R2) value between log (p(d)) and log (d) , where p(d) represents the proportion of nodes in the network with d interactions (39,40). We chose a value of k for each tissue that generated the highest R2 value. Figure 2 illustrates the approach. Once the final posterior network was constructed, the signs of the interactions were determined using the partial correlation matrix:
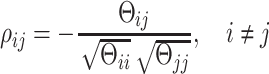 |
(3) |
Figure 2.

Assembly of tissue-specific regulatory networks. The empirical covariance matrix was estimated from GTEx tissue-specific gene expression data. ChIP-Atlas prior network was converted into an adjacency matrix of prior TF–gene interactions and softly enforced into graphical lasso using a penalty matrix Λ with various degrees of stringency. The sparse precision matrix Θ was estimated by optimizing the penalized log likelihood, from which the posterior network was constructed. The precision matrix encodes the direct interaction between entities in the network. A non-zero value Θij ≥ ε indicates that there is an interaction between ith TF and jth target gene. The sign of interaction (activation versus repression) is calculated from the partial correlation matrix.
Note that the connections in each posterior network are supported by both ChIP-seq data as well as by partial correlation of gene expression values. A total of 15 tissue-specific posterior network were constructed using this process. Additionally, we examined the overlap between these networks to identify the connections that appear across multiple tissues. Such connections can be viewed as consistent universal interactions. We refer to these network as merged networks. In our implementation, we constructed merged network using interactions that are consistent in at least 2, 3, 4 or 5 tissues.
Inferring active regulators
CIE provided several (directional and un-directional) enrichment tests to query the networks and identify transcriptional regulators from a user provided DGE profile. The starting point of the inference is selection of one of the causal networks of interactions that are provided by CIE. The type of inference depends on the availability of information on the mode of regulation in the causal graph. The first method is the Fisher’s exact test or the enrichment scoring (ES) statistic, which is the standard for gene set enrichment analysis (41). This method does not take information on mode regulation into account. The next enrichment method is Ternary scoring (TS) statistic proposed by Chindelevitch et al. (7). This method is suitable for fully annotated networks. For networks with a mixture of annotated and unannotated edges, we utilized the Quaternary scoring (QS) statistic proposed by Fakhry et al. (6).
All these methods are build on a common core, calculating the goodness of the fit of the score, which measure the agreement between predictions made by regulators in the graph and the observed DEG profile. For each regulator in the selected network, a contingency table is constructed that tabulates the agreement between the predictions made by the regulator according to the graph and the observations based on the DGE profile. The rows of the table represent the prediction made by the regulator and the columns represent observed differential expression profile. In case of the ES statistic, a 2 × 2 contingency is constructed, with rows representing genes that are predicted to be regulated or unregulated by the regulator according to the network, and the columns representing genes that are observed to be regulated, or unregulated according to the observed DGE profile. In case of TS statistics a 3 × 3 contingency table is constructed, with rows representing genes that are predicted to be upregulated, downregulated, or unregulated by the regulator and the columns represent genes that are observed to be upregulated, downregulated or unregulated according to the DGE profile. At last, in case of QS statistic a 4 × 3 contingency table is constructed in a similar manner with an additional row representing regulated or unregulated corresponding to unsigned edges in the network. The statistical significance of the score is calculated using a permutation (generalized hyper-geometric) test and the P-values are reported (see Supplementary Figure S6 for the details of inference algorithm).
The CIE web server
The CIE web server was implemented using R Shiny, Docker, ShinyProxy and a NGINX web server. The applet RCytoscapeJs was used for network visualization. The core of functionality of the web server is driven by the CIE R package. The open-source version of Shiny is a single-threaded web server for web applications implemented with R. If it is used to run a web application that takes a few seconds at most to load, this will not cause any noticeable impairment. However, CIE takes up to several minutes to load and the open-source Shiny web server cannot allow another user to connect during this time as it can only do one task at a time. To overcome this limitation, we used ShinyProxy and Docker container to allow creation of multiple instances. ShinyProxy detects a new user’s request and starts a Docker container of CIE application specifically for the user. NGINX takes the request from the user and forwards it through reverse proxy to ShinyProxy. The CIE web server is located at ‘https://umbibio.math.umb.edu/cie/app’, and is accessible by all major browsers. The inference result table consists of the regulator’s symbol, total number of target genes, number of the significant target genes (i.e. differentially expressed genes), and the corresponding P-values. The result table can be downloaded in a text format. Users can also run higher-order pathway enrichment analysis by mapping the inferred regulators to Reactome pathways (28).
The CIE R-package
We also provide CIE R-package for offline and local usage available to download at ‘https://github.com/umbibio/CIE-R-Package’, under the GNU public license. The package is capable of producing the same plots and results as the web server with a simple function call and allows for more fine-tune control, automation and customized input networks. The CIE R package is parallelized to provide efficient and fast inference. It utilizes the multidplyr and dopar packages to implement this parallel computation. Multidplyr allows the table of statistics from which enrichment is calculated to be produced quickly, and dopar calculates the P-values in parallel by wrapping a function call to their calculator. Our package is documented and includes a comprehensive manual and instructional vignettes.
RESULTS
We performed our GGM approach to integrate ChIP-derived network from ChIP-Atlas (26) with tissue-specific gene expression data from GTEx (27) (Supplementary Table S1). For each tissue, a grid of regularization parameters was applied, and the best network based on the highest R-squared value was selected (Supplementary Table S2). We also identified the optimal networks using a cross validation strategy and compared the results with the R-squared metric (Supplementary Table S4).
Summary statistics
We examined the overlap between recovered interactions in each tissue. Figure 3A shows the number of recovered regulatory interactions shared between tissues. Interactions that appear across several tissues are called consistent and reflect non-tissue specific, universal TF–gene interactions. As expected, we observed a significant drop in number of consistent interactions as number of tissues increase. There are a total number of 95 745 interactions between 739 TFs and 14 660 genes that appear across at least five tissues. We further examined the consistency of recovered signs (mode of regulation) across tissues. This analysis was carried out on interactions that appear in at least 5 tissues. For each interaction, the proportion of times that the interaction was annotated as positive (or negative) across the tissues in which the interaction appeared was calculated (see Supplementary Figure S5 for the consistency changes across tissues). Completely consistent interactions will have either positive proportion of 1 (i.e. recovered always as positive) or 0 (i.e. recovered always as negative). Figure 3B shows the frequency of the positive proportions. As can be observed the distribution is bimodal, with most interactions recovered consistently as either positive or negative, indicating that TF–gene interactions tend to be activation or repression independent of the tissue. For cross-tissue merged networks, a majority voting scheme was used to annotate inconsistent interactions.
Figure 3.

(A) Number of interactions shared across tissues. (B) Proportion of positive interactions shared in at least five tissues. The bimodal distribution shows that interactions are consistently annotated as positive (1.0) or negative (0).
Furthermore, we investigated the optimal networks selected by the cross-validation strategy, and similar pattern can be observed between recovered interactions among tissues (Supplementary Figure S3). A cross tissue analysis of fitted log-likelihoods indicates that the recovered interactions have high tissue specificity (Supplementary Figure S4).
Benchmark results
We compared our inferred mode of regulation with the signs reported in the TRRUST and STRING databases including high-quality and manually curated sources of human TF–gene interactions, which can be considered as the gold standard for our purpose. For this benchmark, we merged all annotated interactions across all tissues. Figure 4A shows the number of posterior interactions and their associated sign distribution recovered by the GGM approach. Of the interactions in the ChIP-network, 47% were supported by a tissue-specific gene expression data and annotated by our GGM approach (Figure 4A, top). The final sign of recovered interactions were decided according to a majority voting scheme resulting in 55% positive and 45% negative interactions (Figure 4A, top). We compared the sign of annotated interactions with the signs reported in the TRRUST and STRING databases as a gold standard. The overlap between the ChIP-network and the gold standard is 5701 TF–gene interactions (Figure 4A, bottom), of which 2619 are annotated in both gold standard and the tissue corrected ChIP-network. Further restricting the interactions in the ChIP-network to consistent interactions that appear across at least three tissues, results in 390 overlaps with gold standard interactions (see also Supplementary Figures S1 and 2). Figure 4B shows classification performance of these 2619 and 390 overlapping interactions between three-tissue merged (corrected) ChIP-networks with the gold standard. As can be seen the agreement is high in both cases (F1-scores 0.74 and 0.92), demonstrating that our approach is highly accurate in identifying signs of regulation (see Supplementary Equation S1 for F1-score calculation details).
Figure 4.

(A) Summary statistics of annotated interactions in the ChIP-network and overlap with gold standard. (B) Classification performance of annotated interactions in ChIP-network compared with gold standard.
We further compared our result with the CV-based approach and benchmarked our method against a closely related prior-based integrative framework for regulatory network inference (MerlinP) (11). Our merged inferred network shows significant agreements of 91% and 86% with CV and MerlinP approaches respectively in predicting mode of regulation (Supplementary Tables S5 and 7).
Recovering known perturbations in controlled overexpression experiments
To test the performance of our tissue-corrected ChIP-network and inference algorithms, we used the CIE platform to identify drivers of differential expressed genes in controlled overexpression studies. For this analysis, we utilized three differential expression profiles, all of which were obtained by over expressing an oncogene. The genes are E2F3, c-Myc and H-Ras. The number of differentially expressed genes in experiment are 272, 220 and 268, respectively. Table 1 outlines the top 10 regulators predicted by CIE on each experiment sorted by the FDR corrected P-values of the enrichment statistics (ES). The FDR corrected P-values of the Ternary score (TS) along with the predicted direction of regulation by the Ternary method are also presented in the table.
Table 1.
Top 10 predicted regulators by CIE on tissue corrected ChIP-network
| E2F3 | c-Myc | H-Ras | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | ES | TS | Reg. | Name | ES | TS | Reg | Name | ES | TS | Reg. |
| E2F1 | 3.9e-8 | 4.9e-11 | up | TP53 | 8.8e-18 | 2.5e-20 | up | FOSL1 | 2.6e-28 | 2.0e-42 | up |
| PSIP1 | 1.7e-6 | 1.6e-6 | up | SSRP1 | 3.5e-14 | 2.2e-22 | up | TP63 | 4.5e-13 | 9.6e-2 | up |
| FOXM1 | 3.7e-6 | 1.5e-8 | up | WDR5 | 1.9e-12 | 5.6e-20 | up | KDM6B | 2.4e-12 | 6.2e-19 | up |
| EZH2 | 3.7e-6 | 2.2e-4 | up | SMARCA4 | 1.0e-11 | 2.7e-12 | up | ELF3 | 2.1e-11 | 1.6e-12 | up |
| BRCA1 | 1.5e-5 | 3.1e-3 | up | E2F4 | 2.1e-9 | 3.3e-17 | up | EGR1 | 3.1e-11 | 3.0e-6 | up |
| E2F2 | 4.3e-5 | 1.1e-5 | up | MYC | 2.1e-8 | 1.9e-5 | up | MAFF | 3.2e-10 | 3.3e-15 | up |
| MYBL2 | 5.7e-5 | 5.6e-4 | up | TRIM28 | 1.7e-7 | 9.6e-14 | up | FOS | 1.7e-9 | 1.0e-5 | up |
| E2F7 | 6.8e-5 | 5.4e-6 | up | TEAD4 | 1.9e-7 | 3.7e-12 | up | JUNB | 2.9e-9 | 1.9e-12 | up |
| ETS1 | 1.2e-4 | 6.6e-3 | up | NFKB1 | 6.2e-7 | 1.7e-6 | up | BCL3 | 6.1e-8 | 2.7e-8 | up |
| CEBPD | 1.0e-3 | 1.8e-2 | down | ILF3 | 8.4e-7 | 3.4e-12 | up | SMAD3 | 6.7e-8 | 7.7e-9 | up |
Columns: Predicted regulator (Name), FDR corrected P-value (Enrichment Score: ES and Ternary Score: TS), predicted direction of regulation (Reg.).
In the case of E2F3 experiment, E2F1 is returned as the top putative regulator along with E2F2 as another top regulator. E2F1, E2F2 and E2F3 are close related family of TFs with very similar roles that function to control the cell cycle and are implicated in cancer (42). The direction of regulation for these factors are correctly predicted as upregulated by Ternary method. Another predicted regulator is EZH2, which is a downstream of the pRB-E2F pathway and is essential for proliferation and amplified in several primary tumors (43). Interestingly, the Ternary method predicts the direction of regulation of CEBPD as down. It is documented that CEBPD reverses E2F1-mediated gene repression and increased level of CEBPD attenuates E2F1-induced cancer cell proliferation (44). In the c-Myc experiment, the algorithm recovered MYC as one of the top putative regulators and it is predicted to be upregulated. CIE also predicted WDR5, a required interactor of MYC that associates with the same target genes in vivo and is implicated in driving tumorigenesis (45). Finally, in the H-Ras experiment, EGR1 is the top putative regulator returned by the algorithm with a significant P-value and predicted to be upregulated. EGR1 is a key regulator of oncogenic processes and is downstream of H-RAS (46). In all cases the biology behind the recovered regulators is sufficiently evident, demonstrating the ability of the CIE platform and the tissue corrected ChIP-network in recovering correct regulators of DGE.
Note that Quaternary score is equal to Ternary score when we have annotated interactions only. Quaternary score is useful for mixed signed and unsigned graphs. For completely unsigned graphs, Quaternary is equivalent to Fisher’s exact test. Ternary (and Quaternary) scores are generally more stringent than the enrichment score. Unlike the enrichment score, they also match the direction of regulation between the network and the DEG profile. Additionally, the Ternary and Quaternary are able to make inference on the direction of perturbation of the predicted active regulators.
We compared the performance of the inference algorithms on the original ChIP-Atlas, the CV-based networks, MerlinP and three-tissue corrected ChIP-network. The analysis was performed using Fisher’s exact and the Ternary algorithms (Supplementary Tables S3, 6 and 8). Furthermore, we extracted all identified regulators with enrichment P-value < 0.05 to investigate unrelated prediction and false positive rate (Supplementary File S1.xlsx). The results show that the tissue-corrected ChIP-network encapsulates the TF–gene interactions more accurately is able to successfully recover correct regulator.
Drivers of stem cell directed differentiation
To test the utility of the CIE platform and the higher-order pathway enrichment on inferred regulators in generating biological insight, we utilized a more complex data set of stem cell directed differentiation of pancreatic beta cells (33) and the mixed signed STRINGdb network with QS method. Figures 5A and B show the CIE causal regulatory inference results. Among the top predictors, Gastrin (GAST), IL6 and NEUROG3 are predicted to be upregulated by CIE, all of which are involved in the development of the pancreatic endocrine cell lineage (47,48). Users can interactively select top regulators predicted by CIE and perform pathway enrichment analysis using the Reactome (28) pathways. Figure 5C shows the enriched Reactome pathways returned by CIE using the top five predicted regulators. Several significant pathways were acquired through this analysis such as regulation of gene expression in late stage (branching morphogenesis) pancreatic bud precursor cells, which is essentially pointing to the endocrine differentiation of the epithelial cells (49). Furthermore, Regulation of beta-cell development is also identified, which provides a direct link to the transient cellular stages leading to the generation of all pancreatic endocrine cells including insulin-producing beta cells (49). At last, transcriptional regulation of pluripotent stem cells is identified, which encodes regulatory networks underlying embryonic stem cells differentiation into any cell type or tissue type in body (50).
Figure 5.

CIE output on differentiation of pancreatic beta cell. (A) Inferred regulatory network corresponding to top five predicted regulators and their target genes. (B) Table of top five predicted active regulators. (C) Reactome pathways corresponding to the top five predicted regulators by CIE.
Signaling mechanisms underlying fibroblast-to-myofibroblast phenoconversion
In this section we show how the CIE platform can be utilized to test a specific biological hypothesis regarding transcriptional regulators. We demonstrate this use case in the context of fibroblast phenotypic plasticity. Fibroblasts are an abundant cell type within the human connective tissue and play a primary role in secretion of the components of the extracellular matrix (51). Fibroblasts have striking similarities with mesenchymal stem cells (MSCs) and share some functions with MSCs, including phenotypic plasticity governed at the genetic level (52). It is known that Fibroblast can phenotypically convert to myofibroblasts in response to pro-fibrotic proteins such as TGFβ and CXC-type chemokines, secreted by the aging and/or inflammatory cells (35,53,54). Phenotypic conversion can occur through two independent cellular signaling mechanisms: one that depends upon TGFβ/TGFβR axis activation and Smad singling, and another that depends upon CXCL12/CXCR4-axis activation, EGFR transactivation and downstream signaling through MEK/ERK and PI3K/Akt pathways—all of which converge in the nucleus to promote the expression of multiple collagen genes (53,54).
We previously reported that TGFβ and CXCL12 induced or repressed a transcriptional molecular signature that was 75% similar and 25% dissimilar (35). There is evidence that both TGFβ and CXCL12 may be acting upon the same set of bHLH (basic Helix-Loop-Helix) E-box and Egr-1/Egr-2 TFs that bind to consensus sequences in the promoters of the COL1A1 and COL1A2 and other genes (55,56). To test this hypothesis we utilized the DGE profiles from the RNA-seq experiments for both TGFβ and CXCL12 treatment (35) and performed a CIE analysis using TS statistic and merged tissue corrected ChIP-network.
The algorithm predicts many active regulators. Notably, AHR, BHLHE40, TCF4, TCF12, ARNT, ARNTL, MYC and NEUROG2 bHLH TFs, and Egr-1, Egr-2 TFs are predicted by the algorithm as top putative regulators. We also examined the promoter of COL1A1 and COL1A2 and identified multiple binding sites for several of these TFs, including AHR, Egr-1, BHLHE40, ARNT, TCF4. In particular, Egr-1 has multiple binding sites in the promoters of these genes. Taken together, these results support the hypothesis that Egr- bHHL-family TFs can drive the expression of collagen genes in response to TGFβ and CXCL12.
CONCLUSION AND DISCUSSION
In this work, we present CIE, an integrated platform for identification and interpretation of active regulators of transcriptional response. The platform offers visualization tools and pathway enrichment analysis to map predicted regulators to Reactome pathways. We provide a parallelized R-package for fast and flexible directional enrichment analysis that can run the inference on any user provided custom regulatory network. Multiple inference algorithms are provided within the CIE platform along with regulatory networks from curated sources TRRUST and TRED as well as a causal protein–gene interactions derived from the STRINGdb. Importantly we provide a high-confidence annotated causal transcriptional regulatory network by combining publicly available ChIP-seq data with tissue-specific gene expression data. Using a novel regularized gaussian graphical model, we softly enforce the TF–gene interaction identified by ChIP-seq experiments in estimating the precision and partial correlation matrices form tissue gene-expression data, from which we drive tissue-specific annotated transcriptional regulatory networks. Further by merging the networks, we obtained a set of consistent TF–gene interactions that are universally applicable independent of the context. Benchmarks against the gold standard TRRUST database demonstrate that our approach is well able to recover mode of regulation with high accuracy. We demonstrated the utility of our approach in discovering known and novel biology using controlled in vitro over-expression studies as well as stem cell differentiation. Moreover, we demonstrated how our platform can be utilized to investigate specific biological hypotheses of transcriptional regulatory mechanisms in the context of fibroblast phenotypic plasticity in response to signaling events. Our approach and platform can be adopted for other settings, such as identifying candidate co-activators of specific TFs and reconstructing regulatory networks from single cell gene expression data. We hope that this platform provides the scientific community an open source alternative tool to interpret DGE and to generate new biological insights. In the future, we plan to integrate additional networks and pathway inference methods in our platform. We also plan to pursue biological validation of our results on fibroblast phenotypic plasticity using ChIP-seq methodologies.
DATA AVAILABILITY
CIE is an open source collaborative initiative available in the GitHub repository: ‘https://github.com/umbibio/CIE-R-Package’ All of the used databases can be downloaded through CIE web server via Download menu: ‘https://umbibio.math.umb.edu/cie/app’.
Supplementary Material
ACKNOWLEDGEMENTS
Authors would like to acknowledge Yasaman Rezvani for help with R-code.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
UMB Joseph P. Healey Research Grant [JHG-18-15 to K.Z.]; NIH/NIDDK U54 Grant [U54DK104310 to J.A.M.]; NIH-funded Initiative for Maximizing Student Development (IMSD) at University of Massachusetts Boston (to C.O.). Funding for open access charge: UMB.
Conflict of interest statement. None declared.
REFERENCES
- 1. Asif H.M.S., Sanguinetti G.. Large-scale learning of combinatorial transcriptional dynamics from gene expression. Bioinformatics. 2011; 27:1277–1283. [DOI] [PubMed] [Google Scholar]
- 2. Bulashevska S., Eils R.. Inferring genetic regulatory logic from expression data. Bioinformatics. 2005; 21:2706–2713. [DOI] [PubMed] [Google Scholar]
- 3. Djordjevic M., Sengupta A.M., Shraiman B.I.. A biophysical approach to transcription factor binding site discovery. Genome Res. 2003; 13:2381–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krämer A., Green J., Pollard J., Tugendreich S.. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014; 30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zarringhalam K., Enayetallah A., Gutteridge A., Sidders B., Ziemek D.. Molecular causes of transcriptional response: a Bayesian prior knowledge approach. Bioinformatics. 2013; 29:3167–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fakhry C.T., Choudhary P., Gutteridge A., Sidders B., Chen P., Ziemek D., Zarringhalam K.. Interpreting transcriptional changes using causal graphs: new methods and their practical utility on public networks. BMC Bioinformatics. 2016; 17:318–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chindelevitch L., Ziemek D., Enayetallah A., Randhawa R., Sidders B., Brockel C., Huang E.S.. Causal reasoning on biological networks: interpreting transcriptional changes. Bioinformatics. 2012; 28:1114–1121. [DOI] [PubMed] [Google Scholar]
- 8. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. et al.. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur Ö., Anwar N., Schultz N., Bader G.D., Sander C.. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011; 39:D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siahpirani A.F., Roy S.. A prior-based integrative framework for functional transcriptional regulatory network inference. Nucleic Acids Res. 2016; 45:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fakhry C.T., Zarringhalam K., Chen P.. Biomedical relation extraction using stochastic difference equations. 2015 IEEE High Performance Extreme Computing Conference (HPEC). 2015; Waltham, MA: IEEE,Jeremy Kepner; 1–6. [Google Scholar]
- 13. Cardner M., Meyer-Schaller N., Christofori G., Beerenwinkel N.. Inferring signalling dynamics by integrating interventional with observational data. Bioinformatics. 2019; 35:i577–i585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han H., Shim H., Shin D., Shim J.E., Ko Y., Shin J., Kim H., Cho A., Kim E., Lee T. et al.. TRRUST: a reference database of human transcriptional regulatory interactions. Sci. Rep. 2015; 5:11432–11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerner M., Sarafraz F., Bergman C.M., Nenadic G.. BioContext: an integrated text mining system for large-scale extraction and contextualization of biomolecular events. Bioinformatics. 2012; 28:2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farahmand S., Riley T., Zarringhalam K.. ModEx: A text mining system for extracting mode of regulation of transcription factor-gene regulatory interaction. 2019; bioRxiv doi:15 June 2019, preprint: not peer reviewed 10.1101/672725. [DOI] [PubMed]
- 17. Schafer J., Strimmer K.. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005; 21:754–764. [DOI] [PubMed] [Google Scholar]
- 18. Olsen C., Meyer P.E., Bontempi G.. On the impact of entropy estimation on transcriptional regulatory network inference based on mutual information. EURASIP J. Bioinform. Syst. Biol. 2009; 2009:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Werhli A.V., Husmeier D.. Reconstructing gene regulatory networks with bayesian networks by combining expression data with multiple sources of prior knowledge. Stat. Appl. Genet. Mol. Biol. 2007; 6:Article15. [DOI] [PubMed] [Google Scholar]
- 20. Margolin A.A., Nemenman I., Basso K., Wiggins C., Stolovitzky G., Dalla Favera R., Califano A.. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006; 7:S7–S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greenfield A., Hafemeister C., Bonneau R.. Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics. 2013; 29:1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yeang C.-H., Ideker T., Jaakkola T.. Physical network models. J. Comput. Biol. 2004; 11:243–262. [DOI] [PubMed] [Google Scholar]
- 23. Markowetz F., Bloch J., Spang R.. Non-transcriptional pathway features reconstructed from secondary effects of RNA interference. Bioinformatics. 2005; 21:4026–4032. [DOI] [PubMed] [Google Scholar]
- 24. Jiang C., Xuan Z., Zhao F., Zhang M.Q.. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007; 35:D137–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolchanov N.A., Podkolodnaya O.A., Ananko E.A., Ignatieva E.V., Stepanenko I.L., Kel-Margoulis O.V., Kel A.E., Merkulova T.I., Goryachkovskaya T.N., Busygina T.V. et al.. Transcription Regulatory Regions Database (TRRD): its status in 2000. Nucleic Acids Res. 2000; 28:298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C.. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO reports. 2018; 19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC)-Analysis Working Group, Statistical Methods groups-Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site-NDRI, Biospecimen Collection Source Site-RPCI, Biospecimen Core Resource-VARI, Brain Bank Repository-University of Miami Brain Endowment Bank, Leidos Biomedical-Project Management, ELSI Study, Genome Browser Data Integration & Visualization-EBI, Genome Browser Data Integration & Visualization-UCSC Genomics Institute, University of California Santa Cruz, analysts: L., Laboratory, Data Analysis & Coordinating Center (LDACC):, program management: N., collection: B., Pathology: manuscript∼working group: E. Battle A., Brown C.D., Engelhardt B.E., Montgomery S.B.. Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213.29022597 [Google Scholar]
- 28. Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B. et al.. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018; 46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smyth G.K. limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor. 2005; NY: Springer; 397–420. [Google Scholar]
- 30. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat.Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bild A.H., Yao G., Chang J.T., Wang Q., Potti A., Chasse D., Joshi M.-B., Harpole D., Lancaster J.M., Berchuck A. et al.. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006; 439:353–357. [DOI] [PubMed] [Google Scholar]
- 33. Gutteridge A., Rukstalis J.M., Ziemek D., Tié M., Ji L., Ramos-Zayas R., Nardone N.A., Norquay L.D., Brenner M.B., Tang K. et al.. Novel pancreatic endocrine maturation pathways identified by genomic profiling and causal reasoning. PLoS One. 2013; 8:e56024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kroon E., Martinson L.A., Kadoya K., Bang A.G., Kelly O.G., Eliazer S., Young H., Richardson M., Smart N.G., Cunningham J. et al.. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008; 26:443–452. [DOI] [PubMed] [Google Scholar]
- 35. Patalano S., Rodríguez-Nieves J., Colaneri C., Cotellessa J., Almanza D., Zhilin-Roth A., Riley T., Macoska J.. CXCL12/CXCR4-mediated procollagen secretion is coupled to cullin-RING Ubiquitin Ligase Activation. Sci. Rep. 2018; 8:3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Friedman J., Hastie T., Tibshirani R.. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008; 9:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Q., Armenia J., Zhang C., Penson A.V., Reznik E., Zhang L., Minet T., Ochoa A., Gross B.E., Iacobuzio-Donahue C.A. et al.. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data. 2018; 5:180061–180069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uhler C. Gaussian Graphical Models: an algebraic and geometric perspective. 2017; arXiv doi:13 July 2017,preprint: not peer reviewedhttps://arxiv.org/abs/1707.04345.
- 39. Saha A., Kim Y., Gewirtz A.D., Jo B., Gao C., McDowell I.C., Engelhardt B.E., Battle A.. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017; 27:1843–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang B., Horvath S.. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005; 4:Article17. [DOI] [PubMed] [Google Scholar]
- 41. Maciejewski H. Gene set analysis methods: statistical models and methodological differences. Brief. Bioinform. 2013; 15:504–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen H.-Z., Tsai S.-Y., Leone G.. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat. Rev. Cancer. 2009; 9:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K.. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003; 22:5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pan Y.C., Li C.F., Ko C.Y., Pan M.H., Chen P.J., Tseng J.T., Wu W.C., Chang W.C., Huang A.M., Sterneck E. et al.. CEBPD reverses RB/E2F1-mediated gene repression and participates in HMDB-induced apoptosis of cancer cells. Clin. Cancer Res. 2010; 16:5770–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomas L.R., Wang Q., Grieb B.C., Phan J., Foshage A.M., Sun Q., Olejniczak E.T., Clark T., Dey S., Lorey S. et al.. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell. 2015; 58:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nandan M.O., Yoon H.S., Zhao W., Ouko L.A., Chanchevalap S., Yang V.W.. Krüppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004; 23:3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gradwohl G., Dierich A., LeMeur M., Guillemot F.. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krause M.d.S., Bittencourt A., de Bittencourt P.I.H., McClenaghan N.H., Flatt P.R., Murphy C., Newsholme P.. Physiological concentrations of interleukin-6 directly promote insulin secretion, signal transduction, nitric oxide release, and redox status in a clonal pancreatic β-cell line and mouse islets. J. Endocrinol. 2012; 214:301–311. [DOI] [PubMed] [Google Scholar]
- 49. Servitja J.M., Ferrer J.. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004; 47:597–613. [DOI] [PubMed] [Google Scholar]
- 50. Guenther M.G. Transcriptional control of embryonic and induced pluripotent stem cells. Epigenomics. 2011; 3:323–343. [DOI] [PubMed] [Google Scholar]
- 51. Sriram G., Bigliardi P.L., Bigliardi-Qi M.. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015; 94:483–512. [DOI] [PubMed] [Google Scholar]
- 52. Denu R.A., Nemcek S., Bloom D.D., Goodrich A.D., Kim J., Mosher D.F., Hematti P.. Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematol. 2016; 136:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gharaee-Kermani M., Kasina S., Moore B.B., Thomas D., Mehra R., Macoska J.A.. CXC-type chemokines promote myofibroblast phenoconversion and prostatic fibrosis. PLoS One. 2012; 7:e49278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rodríguez-Nieves J.A., Patalano S.C., Almanza D., Gharaee-Kermani M., Macoska J.A.. CXCL12/CXCR4 axis activation mediates prostate myofibroblast phenoconversion through non-canonical EGFR/MEK/ERK signaling. PLoS One. 2016; 11:e0159490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fang F., Ooka K., Bhattachyya S., Wei J., Wu M., Du P., Lin S., Del Galdo F., Feghali-Bostwick C.A., Varga J.. The early growth response gene Egr2 (alias Krox20) is a novel transcriptional target of transforming growth factor-β that is up-regulated in systemic sclerosis and mediates profibrotic responses. Am. J. Pathol. 2011; 178:2077–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jung F., Johnson A.D., Kumar M.S., Wei B., Hautmann M., Owens G.K., McNamara C.. Characterization of an E-box-dependent cis element in the smooth muscle α-actin promoter. Arterioscler. Thromb. Vasc. Biol. 1999; 19:2591–2599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CIE is an open source collaborative initiative available in the GitHub repository: ‘https://github.com/umbibio/CIE-R-Package’ All of the used databases can be downloaded through CIE web server via Download menu: ‘https://umbibio.math.umb.edu/cie/app’.