


Abstract
RNA structures that impede ribosome binding or subsequent scanning of the 5′-untranslated region (5′-UTR) for the AUG initiation codon reduce translation efficiency. Yeast DEAD-box RNA helicase Ded1 appears to promote translation by resolving 5′-UTR structures, but whether its paralog, Dbp1, performs similar functions is unknown. Furthermore, direct in vivo evidence was lacking that Ded1 or Dbp1 resolves 5′-UTR structures that impede attachment of the 43S preinitiation complex (PIC) or scanning. Here, profiling of translating 80S ribosomes reveals that the translational efficiencies of many more mRNAs are reduced in a ded1-ts dbp1Δ double mutant versus either single mutant, becoming highly dependent on Dbp1 or Ded1 only when the other helicase is impaired. Such ‘conditionally hyperdependent’ mRNAs contain unusually long 5′-UTRs with heightened propensity for secondary structure and longer transcript lengths. Consistently, overexpressing Dbp1 in ded1 cells improves the translation of many such Ded1-hyperdependent mRNAs. Importantly, Dbp1 mimics Ded1 in conferring greater acceleration of 48S PIC assembly in a purified system on mRNAs harboring structured 5′-UTRs. Profiling 40S initiation complexes in ded1 and dbp1 mutants provides direct evidence that Ded1 and Dbp1 cooperate to stimulate both PIC attachment and scanning on many Ded1/Dbp1-hyperdependent mRNAs in vivo.
INTRODUCTION
Most eukaryotic mRNAs are translated by the scanning mechanism that commences with assembly of a 43S preinitiation complex containing the small (40S) ribosomal subunit, a ternary complex (TC) of eukaryotic initiation factor 2 (eIF2), Met-tRNAiMet and GTP, and factors eIF1, eIF1A, eIF5 and eIF3. The 43S PIC binds to the 5′-end of mRNA, with eIF4F bound to the m7G cap structure (comprised of cap-binding protein eIF4E, scaffolding subunit eIF4G and DEAD-box RNA helicase eIF4A), and scans the 5′-untranslated region (UTR) to identify the start codon. The 48S PIC thus formed joins with the large (60S) subunit to form an 80S initiation complex ready to begin protein synthesis (reviewed in (1,2)). Both 43S PIC attachment to mRNA and subsequent scanning are inhibited by secondary structures in the mRNA 5′-UTR, particularly for certain mammalian mRNAs encoding proteins that regulate growth, proliferation or apoptosis (3), which are thought to be resolved by DEAD-box RNA helicases. The presence of eIF4A within eIF4F is believed to facilitate unwinding of cap-proximal structures; however, it appears that other helicases, including Dhx29 in mammals and Ded1/Ddx3 in budding yeast, are additionally required to resolve cap-distal structures that impede scanning (reviewed in (4)).
eIF4A is required for 48S PIC assembly in vitro for all mRNAs tested regardless of their structural complexity (5–7). Yeast eIF4A is a weak helicase when tested on duplex-containing RNA substrates in vitro (8,9). Recent evidence suggests that mammalian eIF4A remodels the 40S subunit to enhance PIC attachment (10) and facilitates threading of the 5′-end of mRNA into the 40S entry channel (11). Ribosome footprint profiling of a yeast eIF4A temperature-sensitive mutant (tif1-ts) revealed that, despite a strong reduction in bulk polysome assembly, inactivation of eIF4A in cells markedly reduced the relative translational efficiencies of <40 mRNAs (6). These findings suggested that most mRNAs have similar requirements for eIF4A in yeast cells; although mRNAs most dependent on eIF4A show a tendency to contain long, structure-prone 5′-UTRs. These 5′-UTR characteristics are more pronounced, however, for the large cohort of >600 mRNAs found to show a strong dependence on Ded1 in ribosome footprint profiling of a cold-sensitive Ded1 mutant (ded1-cs), suggesting that Ded1 plays a larger role than eIF4A in promoting translation of mRNAs harboring stable 5′-UTR structures (6).
Using an in vitro translation initiation system with purified components, we recently reconstituted the greater stimulation of 48S PIC assembly by Ded1 on four Ded1-hyperdependent mRNAs burdened with 5′-UTR structures compared to two mRNAs containing short, less-structured 5′-UTRs (12). Moreover, Ded1 function in accelerating 48S PIC assembly was enhanced by eIF4F and domains in eIF4G and Ded1 that mediate Ded1–eIF4F association (12), supporting previous results that Ded1 unwinding activity for a model substrate is stimulated by eIF4A and eIF4G (13). Other recent findings provide in vivo evidence that Ded1 acts directly to unwind structures in 5′-UTRs and suggest that impairing this function increases initiation at upstream open-reading-frames (uORFs), which at least for two mRNAs, contributes to diminished translation of downstream coding sequences (14). In that study however, the group of mRNAs with the most prominent increases in 5′-UTR translation in the ded1 mutant was not more likely than other mRNAs to be strongly dependent on Ded1 for translation of their main CDSs, making it unclear whether this uORF-dependent mechanism of Ded1 function applies broadly in the yeast translatome. Hence, it was important to obtain independent, direct evidence that Ded1 stimulation of scanning through 5′-UTRs contributes to its requirement for efficient translation of the large ensemble of mRNAs with heightened dependence on Ded1 in vivo. It was also important to determine whether stimulating 43S PIC attachment at the cap is a widespread function for Ded1 in living cells.
The yeast Dbp1 protein exhibits strong sequence conservation with Ded1, and Dbp1 overexpression can restore growth to an inviable ded1 null mutant (15); although the restoration was not to wild-type growth, being associated with slow-growth and temperature-sensitive phenotypes (16). Moreover, deletion of DBP1 exacerbates the growth defect of a ded1 hypomorph even though the dbp1Δ mutation confers no growth phenotype on its own (15) and, like a ded1 mutation, dbp1Δ selectively impaired expression of a reporter gene containing a long versus short 5′-UTR (17). These findings suggest that Dbp1 is a functional paralog of Ded1, but the role of Dbp1 in translation initiation has not been clearly demonstrated. In particular, it was unknown whether Dbp1 when expressed at native levels acts with Ded1 throughout the translatome or, rather, is restricted to a small set of mRNA targets with essential functions. It was also unclear whether Dbp1 can function as efficiently as Ded1 to stimulate 48S PIC assembly on mRNAs harboring 5′-UTR structures, nor whether it can promote both PIC attachment and scanning to stimulate mRNA translational efficiencies.
By combining conventional 80S ribosome footprint profiling, the newly developed technique TCP-seq for profiling occupancies of 43S and 48S PICs throughout the translatome, and biochemical analysis in the yeast reconstituted system, we show that Dbp1 can resolve 5′-UTR structures to accelerate 48S PIC assembly on Ded1-hyperdependent mRNAs in vitro, and that Dbp1 is polysome-associated and functionally cooperates with Ded1 throughout the translatome, frequently masking the deleterious consequences of impaired Ded1 activity on translation in vivo. Interestingly, Dbp1 also uniquely stimulates translation of a defined cohort of mRNAs with distinct properties that are largely insensitive to Ded1 function. Finally, TCP-seq analysis provides direct evidence that Ded1 and Dbp1 both stimulate scanning of 43S PICs through the 5′-UTRs of many mRNAs found to be hyperdependent on each helicase in vivo by conventional 80S profiling, and further indicates that Ded1 and Dbp1 exert overlapping functions in promoting 43S PIC attachment on many such mRNAs.
MATERIALS AND METHODS
Construction of yeast strains and plasmids
The yeast strains and plasmids used in this study are listed in Supplementary Tables S6 and S7, respectively. Gene deletions were performed by the PCR-mediated gene-replacement strategy using dominant drug-resistance markers (18). All gene disruptions were confirmed by PCR analysis of chromosomal DNA using appropriate primers. The kanMX4 marker of yeast strain F2045 (dbp1Δ::kanMX4) was swapped with the hphMX4 cassette in pAG32 (19) to yield strain NSY14. To generate yeast strain NSY15, strain F2039 (ded1-952::kanMX4) was transformed with a PCR fragment containing dbp1Δ::hphMX4 and ∼200 bp of sequences from both upstream and downstream of DBP1 (amplified from genomic DNA of strain NSY14).
Strains NSY52 and NSY54 were constructed by transforming NSY4 and NSY5, respectively, with empty vector p1376 (2μ HIS3). Strain NSY56 was generated by transforming NSY5 with plasmid pNDS43 (2μ HIS3 DBP1). For generating strain NSY67, NSY10 was transformed with empty vector p1378 (2μ LEU2). Strains NSY71 and NSY73 were generated by transforming strain NSY11 with plasmids p1378 and pNDS44, respectively.
pNDS42 was constructed by amplifying DBP1 with its native upstream (546 bp) and downstream (352 bp) flanking sequences by PCR amplification from genomic DNA of WT Research Genetics strain BY4741 using primers DBP1F-XhoI (CCAAATcTCGAGATTTAATTTTCGAAGGAATAGCGG) and DBP1R-SpeI (GACCAaCTAgTTCAAAAATATTCCGTATTTTTCCCTCTC), digesting the resulting PCR product with XhoI and SpeI, and inserting it between the same restriction sites in pRS416. pNDS42 was confirmed by restriction enzyme digestion and sequencing. To generate plasmids pNDS43 and pNDS44, a fragment containing DBP1 and its flanking sequences were isolated from pNDS42 digested with Xho1 and Spe1 and cloned into same sites in pRS423 and pRS425, respectively.
Luciferase reporter assays
To assay LUC reporters, cells were disrupted with glass beads in 1× PBS containing 2 Complete EDTA-free Protease Inhibitor Cocktail Tablets (Roche)/50 ml, and firefly luciferase activities were measured using the Dual-Luciferase® Reporter Assay System (Promega), according to the supplier’s protocol, and normalized to the total protein levels in the extracts as determined by the Bradford assay.
Polysome profiles, polysome gradient fractionation, RNA isolation and quantitative RT-PCR
For polysome analysis, strains were cultured under the conditions specified in RESULTS to A600 ∼0.8, cycloheximide was added to 50 μg/ml for 5 min prior to harvesting. Whole cell extracts (WCEs) were prepared by vortexing the cell pellet with glass beads in cold breaking buffer (20 mM Tris–HCl [pH 7.5], 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 200 μg/ml heparin, 50 μg/ml cycloheximide and 1 Complete EDTA-free Protease Inhibitor cocktail Tablet [Roche]/50 ml buffer). For analysis of bulk polysome A260 profiles, 20 A260 units of WCEs were separated by velocity sedimentation on a 4.5–45% sucrose gradient by centrifugation at 39 000 rpm for 3.1 h at 4°C in a SW41Ti rotor (Beckman), and gradient fractions were scanned at 254 nm to visualize ribosomal species. To monitor polysome distributions of specific mRNAs, WT (NSY10) and dbp1Δ (NSY79) strains were cultured in SC-His medium at 30°C to log-phase growth, sedimented at room temperature, transferred to flasks containing pre-warmed SC medium and incubated at 37°C for 2 h. About 30 A260 units of WCEs were resolved by sedimentation through 10–50% sucrose gradients by centrifugation at 35 000 rpm for 2 h and 40 min. Gradient fractions were scanned at 254 nm and collected. About 4.8 ng of ‘spike-in RNA’ was added to each fraction to control for differences in RNA recovery, comprised of a mixture of in vitro transcribed capped mRNAs: 160 pg/μl RLUC and 320 pg/μl FLUC. Purification of total RNA from gradient fractions and qRT-PCR analysis of mRNA abundance was conducted as described previously (20).
To examine association of Dbp1 with native polysomes, cultures of DBP1-TAP strain grown in SC-His media were cross-linked with 2% formaldehyde (21)). Cells were washed once with lysis buffer (20 mM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 mM NaF and 2 Complete EDTA-free Protease Inhibitor cocktail Tablet [Roche]/50 ml buffer). WCEs were prepared by vortexing the cell pellet with glass beads in cold lysis buffer supplemented with 1 mM AEBSF, and 10 μg/ml of pepstatin A, aprotinin and leupeptin. For RNase I treatment prior to centrifugation, 30 A260 units of WCE were treated with 450U of RNAse I (Ambion, #AM2295) at 25°C for 1 h on thermomixer at 700 rpm. RNaseI-treated or untreated WCEs were separated by sedimentation through 10–50% sucrose gradients by centrifugation at 35 000 rpm for 2 h and 40 min in a SW41Ti rotor (Beckman), and proteins were precipitated from gradient fractions with two volumes of 100% ethanol. Protein pellets were washed twice with 1 ml 70% ethanol, resuspended and boiled for 3 min in 30 μl Tris-Glycine SDS sample buffer (Invitrogen, #LC2676 with 10% β-mercapatoethanol), which also served to reverse the cross-links. Total protein from each fraction was resolved by SDS-PAGE using 4–20% Criterion TGX pre-cast gels (Biorad) and subjected to western blot analysis using antibodies against TAP-tag (Thermo Fisher Scientific Cat# CAB1001, RRID:AB_10709700) and Rps22. Immune complexes were detected by chemiluminescence using reagents of Immobilon Western kit (Millipore).
In vitro assays of purified Dbp1
Preparation and purification of mRNAs, charged initiator tRNA and translation initiation factors
Plasmids for in vitro run-off mRNA and initiator tRNA transcription are described in (12,22,23). mRNAs and initiator tRNA plasmids were digested with appropriate restriction enzymes to linearize the plasmids, transcribed by run-off transcription using His6 tagged T7 RNA polymerase and gel purified as described previously (22,23). Initiator tRNA was methionylated using purified E. coli methionyl-tRNA synthetase (24). Charged Met-tRNAiMet was then purified by phenol–chloroform extraction followed by a desalting column (5 ml desalting column, GE Healthcare) (7). mRNAs were capped (m7GpppG) in vitro using vaccinia virus D1/D12 capping enzyme with either α-32P radiolabeled GTP (Perkin Elmer) or unlabeled GTP (for pulse-chase) (23). Eukaryotic initiation factors, 40S ribosomal subunits and Ded1, were expressed and purified as described previously (12,22,23,25).
Purification of recombinant Dbp1
To purify Dbp1, the DBP1 coding sequence was cloned in the pET22b vector with an N-terminal His6-tag (Genscript) and the resulting plasmid was introduced into E. coli strain BL21(DE3) RIL CodonPlus cells (Agilent). Cells were grown at 37°C to OD600 of 0.5, cooled to 20°C and induced with 1 mM IPTG for 16 h. Cells were harvested and resuspended in 35 ml lysis buffer (10 mM HEPES-KOH, pH 7.4, 200 mM KCl, 0.1% IGEPAL CA-630, 10 mM imidazole, 10% glycerol, 10 mM 2-mercaptoethanol and cOmplete protease inhibitor cocktail (Roche)) and lysed using a French Press. The cell lysate was treated with DNaseI (1 U/ml) for 20 min and the KCl concentration was adjusted to 500 mM. Dbp1 was affinity-purified using a nickel column (5 ml His-Trap column, GE Healthcare). Fractions containing Dbp1 were collected, the glycerol concentration was adjusted to 30%, and Dbp1 was further purified by phosphocellulose chromatography (P11, Whartman). Purified Dbp1 was dialyzed against dialysis buffer (10 mM HEPES-KOH, pH 7.4, 200 mM KOAc, 50% glycerol, 2 mM DTT) and stored at −80°C.
mRNA recruitment kinetics
mRNA recruitment kinetics was performed using a native gel shift assay as described previously (7,23). This assay determines the apparent rates (kapp) and maximal rates (kmax) of recruitment as well as the concentration of a factor required to achieve half-maximal rate of recruitment (K1/2). Briefly, PICs were assembled with 300 nM eIF2, 0.5 mM GDPNP·Mg2+, 200 nM Met-tRNAiMet, 1 μM eIF1, 1 μM eIF1A, 300 nM eIF5, 300 nM eIF4B, 300 nM eIF3, 75 nM eIF4E·eIF4G, 7 μM eIF4A (15 μM for SFT2 mRNA) and 30 nM 40S subunits in 1× Recon buffer (30 mM HEPES-KOH, pH 7.4, 100 mM KOAc, 3 mM Mg(OAc)2 and 2 mM DTT). The concentration of Dbp1 was varied in the reactions (0–1000 nM). Complexes were incubated at 26°C for 10 min. Reactions were initiated by addition of 15 nM 32P-m7G capped mRNA and 5 mM ATP·Mg2+. Reactions were stopped by addition of 1000–1500 nM non-radiolabeled m7G-mRNA at appropriate times (7,23), and 48S PICs were separated from the free mRNA on a 4% non-denaturing PAGE gel. The fraction of the mRNA recruited to the 48S PIC at each time-point was calculated using ImageQuant software (GE Healthcare). Data were fitted with a single exponential rate equation and hyperbolic equations to calculate kmax and K1/2 of Dbp1 for each mRNA using KaleidaGraph software (Synergy). Bar-graph data representations were prepared using Prism 7 (GraphPad).
ATPase assay
NADH-coupled ATPase assays were performed as described in (7). Briefly, reactions were assembled in 1× Recon buffer with 500 nM Dbp1, 2.5 mM phosphoenolpyruvate, 1 mM NADH, 1/250 dilution of the PK/LDH mix (pyruvate kinase (600–1000 units/ml) and lactate dehydrogenase (900–1400 units/ml)), 2 μM uncapped RPL41A mRNA and equilibrated to 26°C. Reactions were initiated by the addition of varying concentrations of ATP·Mg2+ and absorbance A340 was measured over time on a Tecan Infinite M1000PRO microplate reader. The slope of a linear plot of NADH oxidation over time was used to measure the reaction rates (V0), rates were plotted against the concentration of the ATP and fitted with the Michaelis–Menten equation to calculate kcat and KmATP using KaleidaGraph software (Synergy).
Fluorescence anisotropy assay of RNA binding
Fluorescent anisotropy assays were performed as described previously (26). Briefly, reactions were assembled with fluorescein-labeled single-stranded RNA, no nucleotide or 5 mM ADP·Mg2+ or 5 mM ADPNP·Mg2+ in 1× Recon buffer and incubated at 26°C with different Dbp1 concentrations (0–750 nM). Fluorescent anisotropies were measured with excitation and emission wavelengths of 495 and 520 nM, respectively. The data were fitted with a hyperbolic equation using KaleidaGraph software (Synergy).
Generation, processing and analysis of sequence libraries of 80S ribosome-protected footprints or total mRNA fragments
Ribo-Seq was performed as previously described (27). Briefly, yeast strains growing exponentially in SC medium at 30°C were harvested by centrifugation at room temperature (R.T.) and resuspended in SC medium at 37 or 15°C (depending on which ded1 allele was involved in the experiment). DBP1 deletion strains, NSY14 (dbp1Δ) and ded1-ts dbp1Δ (NSY15), ded1-ts strains carrying empty vector (NSY71) or hcDBP1 plasmid (NSY73) and WT strain with empty vector (NSY67) were grown under conditions similar to those employed previously for isogenic WT (NSY10) and ded1-ts (NSY11) strains (6) including a temperature shift at 37°C for 2 h. Similarly, ded1-cs strains carrying empty vector (NSY54) or hcDBP1 plasmid (NSY56) and WT strain with empty vector (NSY52) were grown under conditions similar to those employed previously for isogenic WT (NSY4) and ded1-cs (NSY5) strains (6) including a temperature shift at 15°C for 10 min. All cultures were treated with 100 μg/ml cycloheximide for 2 min. Cells were harvested by vacuum filtration at room temperature and quick-frozen in liquid nitrogen. Cells were lysed in a freezer mill with lysis buffer (20 mM Tris [pH 8.0], 140 mM KCl, 1.5 mM MgCl2, 1% Triton, 100 μg/ml cycloheximide). For ribosome footprint library preparation, 30 A260 units of extract were treated with 450U of RNAse I (Ambion, #AM2295) at 25°C for 1 h on a thermomixer at 700 rpm, and ribosomes were pelleted by centrifugation on a 1 M sucrose cushion. Ribosome-protected mRNA fragments (RPFs) were purified using a miRNeasy Mini kit (Qiagen) per the manufacturer’s instructions. Following size selection and dephosphorylation, a Universal miRNA cloning linker (New England Biolabs, #S1315S) was ligated to the 3′ ends of RPFs, followed by reverse transcription, circular ligation, rRNA subtraction, PCR amplification of the cDNA library and DNA sequencing with an Illumina HiSeq system. For RNA-seq library preparation, total RNA was purified using miRNeasy Mini kit (Qiagen) from aliquots of the same extracts used for RPF library preparation. Polyadenylated mRNA was isolated from total RNA using the Poly(A)Purist MAG Kit (Ambion, #AM1922), and randomly fragmented at 70°C for 8 min in fragmentation reagent (Ambion, #AM8740). Fragment size selection, library generation and sequencing were carried out as above. Ribo-Seq and RNA-seq libraries were prepared and sequenced from two independent cultures (biological replicates) for each pair of WT and mutant strains under comparison.
Linker sequences were trimmed from Illumina reads and the trimmed fasta sequences were aligned to the Saccharomyces cerevisiae ribosomal database using Bowtie (28). The non-rRNA reads (unaligned reads) were then mapped to the S. cerevisiae genome using TopHat (29). The total number of reads from sequencing libraries of each strain and their biological replicates that mapped to the yeast transcriptome are summarized in Supplementary File S1. Only uniquely mapped reads from the final genomic alignment were used for subsequent analyses. Statistical analysis of differences in RPF or mRNA read counts, or TE values, between WT and mutant samples using data from their biological replicates was conducted in DESeq2 (30), a statistical package well-suited to genome-wide analyses with only two or three biological replicates for each condition under comparison. This program (as well as EdgeR employed below to analyze the SSU profiling data) operates by pooling information about variances of read counts across the thousands of genes analyzed to model count variances for genes of similar expression levels. The modeled variances are used in the framework of a generalized linear model (GLM) to identify expression changes and place confidence intervals on the magnitude of changes, and also to exclude genes showing aberrantly high variability. Transcriptional and translational changes are analyzed together in a GLM by including library type (mRNA-Seq or Ribo-Seq) as one of the variables, in addition to genotype, in a multi-factor design. The translational efficiency (TE) emerges as the effect of the Ribo-seq library type against the mRNA-Seq baseline, and significant interactions of TE with the genotype indicates translational control by the gene product under consideration (31). Genes with <128 total mRNA reads in the four samples combined (two replicates of both WT and mutant strains under comparison) were excluded, as were RPF reads mapping to the first 15 codons of all CDS, in the calculation of TE values using DESeq2.
Hierarchical cluster analysis of TE changes in mutants was conducted with the R heatmap.2 function from the R ‘gplots’ library, using the default hclust hierarchical clustering algorithm. For all notched box-plots, constructed using a web-based tool at http://shiny.chemgrid.org/boxplotr/, the upper and lower boxes contain the second and third quartiles and the band gives the median. If the notches in two plots do not overlap, there is roughly 95% confidence that their medians are different. Statistical significance of the difference between the medians of two distributions was computed using the Mann–Whitney U test.
Generation of sequencing libraries for small subunit (SSU) protected footprints or total RNA fragments
The same yeast strains used for conventional 80S profiling were also employed for SSU profiling, and were cultured under conditions identical to those used for the 80S profiling experiments. SSU profiling was carried out as described previously (32), with minor modifications. Briefly, 500 ml cell cultures were poured into bottles containing 125 g ice and 50 ml of freshly depolymerized 30% w/v paraformaldehyde. Samples were mixed by inversion and left on ice for 20 min with intermittent mixing. Cross-linking was quenched by addition of 25 ml of 2.5 M glycine to the samples. The fixed cells were pelleted by centrifugation at 7000 rpm for 5 min at 4°C and resuspended in 40 ml of buffer A (25 mM HEPES-KOH, pH 7.5, 100 mM KCl and 2 mM MgCl2). The cells were washed twice by sequential centrifugation and resuspension in 40 ml of buffer A and resuspended finally in 2 ml buffer A supplemented with 0.5 mM DTT, and 1× Complete EDTA-free Mini Protease Inhibitor (Roche) per 10 ml of buffer A. The cell suspension was dropped into liquid nitrogen to make frozen beads, which were ground in a freezer mill to produce WCEs. For SSU footprint library preparation, 50 A260 units of WCE were treated with 750 U of RNAse I (Ambion, #AM2295) at 23°C for 45 min on a thermomixer at 700 rpm. Samples were sedimented through 7.5–30% sucrose gradients (prepared in buffer B; 50 mM Tris-HCl, pH 7.0 at 25°C, 50 mM NH4Cl, 4 mM MgCl2, 1 mM DTT) for 4 h at 38k rpm and 350 μl gradient fractions were collected. SSU gradient fractions were supplemented with 50 μl stop buffer [final concentration: 1% w/v SDS, 10 mM EDTA, 10 mM Tris-HCl (pH 7.4 C), 10 mM glycine]. Acid phenol–chloroform solution (5:1, pH 4.5; Ambion, # AM9720) was then added, and the mixture was shaken at 1300 rpm in a thermomixer for 45 min at 65°C to reverse the cross-links. Aqueous phase was recovered by centrifugation in swinging bucket rotor in Beckman JHC centrifuge for 10 min at 4200 rpm at 4°C. Aqueous phase was re-extracted once with 1× volume of acid phenol–chloroform that involves vortexing samples for 5 min at room temperature followed by 5 min spin at 132 000 rpm at 4°C. Subsequently, aqueous phase was reextracted once with 1× volume of chloroform–IAA (24:1). RNA was precipitated with 1/10 volume of 3M NaOAc pH 5.5 and 1× volume of isopropanol. RNA pellets were washed once with 1 ml 75% ethanol and were air dried. Each RNA pellet was resuspended in 30 μl 10 mM Tris, pH 8. Sequencing libraries from SSU protected fragments and total RNAs were prepared as described above for 80S profiling (including rRNA depletion) from biological replicates of each pair of WT and mutant strain under comparison.
Analysis of SSU profiling sequence data
Read trimming and filtering of SSU-protected fragments and total RNA
Fastq files from sequencing were quality-trimmed and adapter-trimmed at the 3′ ends using Trimmomatic (33) as outlined in Step 147 of (34), except that when trimming and selecting for reads having 3′ adapter sequences, the Cutadapt parameters were adjusted for the adapter used in the library preparation protocol (CTGTAGGCACCATCAATAGATCGGAAGA) instead of a poly-A sequence. Reads were then adapter-trimmed at the 5′ end and reads mapping to rRNA, tRNA and other structural RNAs were removed by mapping against custom indices of these sequences using STAR (35), as outlined in Steps 148–150 of (34). Total number of reads that mapped to the yeast transcriptome from sequencing libraries of each strain and their biological replicates are summarized in Supplementary File S1.
Summarizing and classifying SSU and total RNA reads by position within transcripts
The remaining SSU reads were aligned against an index consisting of spliced mRNA sequences with 1 kb flanking sequence added at the 5′ and 3′ ends of the ORFs. Certain genes exhibited artefactual hotspots of alignments with identical 5′ and 3′ ends but with very low read density elsewhere; and alignments were filtered out from 11 such hotspots identified manually in the SSU fractions (Supplementary Table S8). Alignments shorter than 14 nucleotides or with 3 or more internal mismatches were removed. Alignments from the SSU footprint fractions were classified into different position-classes, as either ‘5′-UTR’, ‘start codon’, ‘ORF’, ‘stop codon’, ‘3′-UTR’ or ‘uncertain’, as outlined in Supplementary Figure S12F. Alignments from the total RNA fractions were initially similarly classified. However, the longer length of total RNA fragments relative to the SSU footprint libraries causes a bias against 5′-UTR classification, as long fragments are less likely to fit entirely within the short yeast 5′-UTRs. Therefore, a second classification scheme was employed for the total RNA fractions for the purpose of gene-wise statistical testing: a binary ‘5′-UTR’ or ‘ORF’ classification, with the boundary between the two adjusted for fragment length and 5′ position within each individual biological sample such that, if the length of the 5′-UTR changes in different mutants, the resultant change in 5′-UTR:ORF occupancy ratio will be similar in both SSU and total RNA fractions from that sample (for details of algorithm see R script pcntl_norm_posclass.R in the TCPseq git repository (34).
Testing for gene-specific differences between samples
Alignment counts were aggregated by gene and by position (i.e. 5′-UTR, start, ORF, stop or 3′-UTR) for each sample. The position-classes were then simplified to ‘5′-UTR’ and ‘control’ for the purpose of gene-wise statistical testing for effects of mutant on scanning, (‘control’ region being start-codon associated FPs for the SSU fraction and ORF-aligned fragments for the total RNA fraction), and all other position-classes were ignored. Genes for which 5′-UTR splice junctions have been detected (36) were removed from the testable gene set. For genes having no annotated 5′-UTR (37), 200 nt upstream region or the distance to the nearest ORF, whichever is less, was treated as the presumptive 5′-UTR. For each pair-wise comparison between mutants, genes were filtered out if they did not have at least 10 reads in at least 1 replicate from both total-RNA and SSU footprint fractions from both relevant mutants. EdgeR (38) was then applied to detect the effect of the mutation on the ratio between different position-classes for each gene (i.e. the interaction between position-class and mutant). For viewing SSU or RNA reads in the IGV browser, normalized bigwig files were used. The bigwig files are raw coverage (a number of aligned reads covering each basepair) that were normalized by the number of millions of aligned reads in the original bed file (rpm).
RESULTS
Ribosome footprint profiling reveals a cohort of Dbp1-hyperdependent mRNAs
To determine the role of Dbp1 in regulating translation of yeast mRNAs, we deleted DBP1 in isogenic WT and temperature-sensitive ded1-952 mutant (ded1-ts) strains. Consistent with previous findings (15), loss of DBP1 in WT cells does not impair growth (Supplementary Figure S1A); however, dbp1Δ exacerbates the growth defect of ded1-ts cells (Supplementary Figure S1B). As expected (15), the growth defects of the ded1-ts dbp1Δ double mutant were fully suppressed by a high-copy (hc) DBP1 plasmid (Supplementary Figure S1B). Examination of bulk polysomes revealed no differences in the polysome:monosome (P/M) ratios between dbp1Δ and WT cells; whereas dbp1Δ exacerbated the depletion of polysomes in ded1-ts cells (Supplementary Figure S1C–F). Thus, the synthetic-sick phenotype of ded1-ts dbp1Δ cells can be attributed to the additive effect of these mutations in reducing the rate of translation initiation.
To identify Dbp1-regulated mRNAs, ribosome footprint profiling (Ribo-Seq) and RNA-Seq were conducted for biological replicates of the dbp1Δ and ded1-ts dbp1Δ strains following a shift from 30 to 37°C for 2 h (as in Supplementary Figure S1D and F), and comparing the results to those we obtained under identical conditions for the isogenic WT and ded1-ts strains (GSE66411) (6). The ratio of sequencing reads of ribosome-protected fragments (RPFs) to mRNA reads for each gene provides a measure of translational efficiency (TE). The RPFs and mRNA reads for each gene were normalized to the total RPF and mRNA reads for that strain, masking the overall reductions in translation in the mutants but yielding the changes in TE relative to all other mRNAs (for brevity, normalized RPF reads, normalized mRNA reads and relative TE values will be referred to simply as RPF reads, mRNA reads and TEs). Ribo-Seq and RNA-Seq results were highly reproducible between biological replicates (Pearson’s r ≈ 0.99) (Supplementary Figure S2A–D). To assess the quality of our Ribo-Seq data, we examined the frequency distribution of RPF lengths and reading frame, as well as the triplet periodicity of RPFs in coding regions. A typical analysis is shown in Supplementary Figure S3 for one of the biological replicates of the dbp1Δ strain, showing the expected major peak of RPFs at 28 nt (Supplementary Figure S3A), of which the majority are in the 0-frame (Supplementary Figure S3B). A metagene analysis of the distribution of 5′ends of 28 nt RPFs reveals the expected start codon peak at ∼12 nt upstream, the preponderance of RPFs mapping to CDS versus 5′-UTRs, and the 3 nt 0-frame periodicity of RPFs (Supplementary Figure S3C).
Comparing RPF reads between dbp1Δ and WT cells revealed a set of ∼226 mRNAs (of ∼5300 expressed mRNAs) with substantially altered translation in the mutant (Supplementary Figure S2E; red dots, >2-fold changes), whereas RNA-Seq data identified ∼350 mRNAs with >2-fold changes in mRNA abundance (Supplementary Figure S2F). Integrating the RPF and mRNA data revealed 87 mRNAs showing ≥2-fold reductions in TE in dbp1Δ versus WT cells (ΔTEdbp1Δ = TEdbp1Δ/TEWT < 0.5) at a high stringency for statistically significant changes (FDR < 0.01), thus indicating a heightened dependence on Dbp1 (Figure 1A, red dots below diagonal). Only 12 mRNAs display less than average Dbp1-dependence and exhibit ≥2-fold higher TE in the mutant (ΔTEdbp1Δ = TEdbp1Δ/TEWT > 2.0; FDR < 0.01) (Figure 1A, red dots above diagonal). Hereafter, we refer to the 87 mRNAs with ΔTEdbp1Δ < 0.5 as Dbp1-hyperdependent, and the 12 mRNAs with ΔTEdbp1Δ > 2 as hypodependent on Dbp1 (mRNAs listed in Supplementary File S1). Including smaller fold-changes of >1.4-fold and relaxing the stringency to FDR < 0.05 reveals 394 and 107 mRNAs showing ΔTEdbp1Δ values of <0.7 or >1.4-fold (Figure 1A, red plus blue dots; Supplementary Table S1). We conclude that Dbp1 contributes significantly to the TEs of a distinct fraction of yeast mRNAs. As shown below, Dbp1 additionally functions at many other mRNAs where its contribution is masked by Ded1.
Figure 1.

Dbp1 regulates translation of a substantial fraction of mRNAs. (A andB) Scatter-plots of translational efficiencies (TEs) in mutants versus WT for (A) dbp1Δ or (B) ded1-ts dbp1Δ mutant cells. Those mRNAs exhibiting ≥2-fold, or between 1.4- and 2-fold, changes in dbp1Δ or ded1-ts dbp1Δ cells versus WT at FDR < 0.01 (as determined by DESeq2 using biological replicates of each strain) are highlighted in red and blue, respectively. (C) Venn diagram of overlap between mRNAs exhibiting ≥2-fold reductions in TE in dbp1Δ, ded1-ts or ded1-ts dbp1Δ versus WT cells; N, number of mRNAs. (D) Notched box-plot analysis of 226 Ded1-hyperdependent mRNAs exhibiting ≥2-fold TE reductions in ded1-ts versus WT cells, comparing the distributions of TE changes observed in dbp1Δ, ded1-ts or ded1-ts dbp1Δ mutant versus WT cells. Each box depicts the interquartile range containing 50% of the data, intersected by the median; the notch indicates a 95% confidence interval (CI) around the median. Median TE changes are shown below the x-axis. (E) As in Figure 1D, but for 523 mRNAs exhibiting ≥2-fold reductions in TE in ded1-ts dbp1Δ versus WT cells. (F) As in Figure 1D, but for 87 Dbp1-hyperdependent mRNAs. (D and F) P-values computed from Mann–Whitney U-test are indicated (*, P< 10−6; **, P< 10−16).
Dbp1 functionally cooperates with Ded1 in regulating a substantial fraction of the yeast translatome
Analyzing our previous Ribo-Seq data on the ded1-ts mutant (6) and applying the same criteria for hyperdependence used above, we identified a cohort of 226 mRNAs hyperdependent on Ded1 in the presence of WT DBP1 (ΔTEded1-ts < 0.5, FDR < 0.01, Supplementary Figure S4A, red dots), considerably more than the 87 Dbp1-hyperdependent mRNAs noted above. Remarkably, Ribo-Seq analysis of the ded1-ts dbp1Δ double mutant revealed a much larger set of 523 mRNAs exhibiting a comparable >2-fold reduction in TE compared to WT cells (ΔTEded1-ts dbp1Δ = TEded1-ts dbp1Δ/TEWT < 0.5, FDR < 0.01) (Figure 1B, red dots; mRNAs listed in Supplementary File S1), consistent with the relatively greater polysome depletion seen in the double mutant (Supplementary Figure S1E–F).
Most (∼94%) of the 226 Ded1-hyperdependent mRNAs down-regulated in the ded1-ts mutant were also down-regulated in the ded1-ts dbp1Δ double mutant (Figure 1C) and, importantly, the median reduction in TE for the group of 226 mRNAs was considerably greater in the double mutant versus the single ded1-ts mutant (Figure 1D; for this and all other notched box plots, the absence of overlap between the notches of two adjacent plots indicates that their medians differ with >95% confidence). Similarly, the reduction in TE for the group of 523 hyperdependent mRNAs identified in the double mutant is much greater in this strain versus the ded1-ts single mutant (Figure 1E). Thus, in cells lacking DBP1, ded1-ts reduces the TEs of many more mRNAs, and to a greater extent, than when DBP1 is present. These findings indicate that Dbp1 masks the involvement of Ded1 at a large proportion of the 523 mRNAs found to be down-regulated in the double mutant, presumably by carrying out a redundant function. Both groups of 226 and 523 hyperdependent mRNAs exhibit relatively small, but significant, ∼20% reductions in median TE in the dbp1Δ single mutant compared to WT (Figure 1D and E; median log2ΔTE values < 0); however, as shown below, the function of Dbp1 is masked by Ded1 at many of these mRNAs.
To identify more directly mRNAs for which Ded1 function is masked by Dbp1, we looked for mRNAs where the ded1-ts mutation produced a strong reduction in TE only in cells lacking DBP1. To this end, we identified 417 mRNAs exhibiting a ≥2-fold TE reduction in ded1-ts dbp1Δ versus dbp1Δ cells (defined as ΔTEded1-ts(dbp1Δ)= TEded1-ts dbp1Δ /TEdbp1Δ < 0.5) at FDR < 0.01 (Supplementary Figure S4B and Supplementary File S1). Since 198 of these 417 mRNAs already belong to the group of 226 mRNAs that are hyperdependent on Ded1 in the presence of WT DBP1 (Figure 2A), this analysis increased the number of Ded1-hyperdependent mRNAs by 219, for a total of 445 mRNAs (Figure 2A, 226 + 219 non-overlapping mRNAs). This group of 219 mRNAs, dubbed ‘conditionally Ded1-hyperdependent’ to reflect their marked response to ded1-ts only in dbp1Δ cells, displays a greater reduction in median TE in the ded1-ts dbp1Δ double mutant relative to either WT or dbp1Δ cells compared to that seen for the ded1-ts single mutant versus WT (Figure 2B, cols. 3–4 versus 2). Note that the median TE of these 219 mRNAs is reduced in the ded1-ts single mutant, but by less than the 2-fold change that defines Ded1-hyperdependence; and the median TE reduction exceeds the 2-fold cut-off only in the ded1-ts dbp1Δ double mutant. These findings support our conclusion that Dbp1 masks the contribution of Ded1 in stimulating translation of a large group of mRNAs (219), comparable in size to the group for which impairing Ded1 alone evoked a strong TE reduction (226).
Figure 2.

Dbp1 functionally cooperates with Ded1 in regulating the yeast translatome. (A) Venn diagram of overlap between mRNAs exhibiting ≥2-fold TE reductions in response to ded1-ts in otherwise WT or dbp1Δ strains. P values for significance of overlap are based on the hypergeometric distribution. (B) Notched box-plot analysis of 219 ‘conditionally Ded1-hyperdependent’ mRNAs, defined in ‘Results’ section, comparing TE changes observed in the four indicated comparisons. (C) Venn diagram of overlap between mRNAs exhibiting ≥2-fold reductions in TE on DBP1 deletion in a WT or ded1-ts strain. (D) Similar to (B), but for 209 ‘conditionally Dbp1-hyperdependent’ mRNAs, defined in ‘Results’ section. In (B) and (D), P-values from Mann–Whitney U-test are indicated (*, P< 10−11; **, P< 10−16; n.s., not significant). (E) Venn diagram of overlap between 445 mRNAs exhibiting ≥2-fold reductions in TE conferred by ded1-ts in either WT or dbp1Δ cells and a set of 296 mRNAs exhibiting ≥2-fold reductions in TE conferred by dbp1Δ in either WT or ded1-ts cells. (F) Hierarchical clustering analysis of ΔTE values conferred by ded1-ts, dbp1Δ or ded1-ts dbp1Δ versus WT for 1957 mRNAs with significant TE changes in ded1-ts dbp1Δ versus WT cells (FDR < 0.05), presented as a heatmap. Ten mRNAs with >64-fold changes in TE were excluded because they would diminish color differences among the remaining mRNAs. Bracket includes mRNAs with TEs reduced much more by dbp1Δ versus ded1-ts (col. 3 versus 2).
We used a similar approach to identify mRNAs whose dependence on Dbp1 is masked by the presence of Ded1, identifying 234 mRNAs exhibiting a ≥2-fold reduction in TE in ded1-ts dbp1Δ versus ded1-ts cells (ΔTEdbp1Δ(ded1-ts)= TEded1-ts dbp1Δ /TEded1-ts < 0.5) at FDR < 0.01 (Supplementary Figure S4C and Supplementary File S1). Because only 25 of these 234 mRNAs belong to the group of 87 Dbp1-hyperdependent mRNAs defined above (Figure 2C), the remaining 209 mRNAs exhibit ‘conditional Dbp1-hyperdependence’. Indeed, they display a much larger reduction in median TE in the ded1-ts dbp1Δ double mutant compared to either WT or ded1-ts cells than observed in the dbp1Δ mutant versus WT (Figure 2D, cols. 3–4 versus 1). Thus, although deleting DBP1 alone reduces the TEs of most of these 209 mRNAs to a small degree, the reduction is considerably greater and exceeds the 2-fold threshold for TE changes, only in ded1-ts dbp1Δ cells. Clearly, Ded1 masks the role of Dbp1 at a substantial group of mRNAs. The total number of 296 mRNAs showing heightened dependence on Dbp1 (Figure 2C, 87 + 209 non-overlapping mRNAs) is only marginally smaller than the group of 445 Ded1-stimulated mRNAs shown in Figure 2A, indicating comparably widespread functions for Dbp1 and Ded1. Importantly, there is a much greater overlap between these expanded groups of mRNAs showing heightened dependence on Ded1 or Dbp1 (Figure 2E), compared to that shown in Figure 1C for the 226 and 87 mRNAs judged to hyperdependent on Ded1 or Dbp1, respectively, indicating that extensive functional cooperation between Ded1 and Dbp1 was unveiled by analysis of the ded1-ts dbp1Δ double mutant.
As noted above, the three groups of Ded1-hyperdependent, conditionally Ded1-dependent, and conditionally Dbp1-dependent mRNAs exhibit a greater TE reduction in the double mutant than in either single mutant (Figures 1D, 2B and D, respectively). Surprisingly, the 87 mRNAs judged to be Dbp1-hyperdependent in the dbp1Δ versus WT comparison exhibit a TE reduction in the double mutant that is actually smaller than that seen in the dbp1Δ single mutant (Figure 1F, col. 3 versus 1). This observation explains why more than half (55) of the 87 Dbp1-hyperdendent mRNAs are not hyperdependent in the ded1-ts dbp1Δ strain (Figure 1C). Examining the data for each of these 87 mRNAs individually revealed that 65 show a TE reduction in the dbp1Δ single mutant equal to or greater than that seen in the ded1-ts dbp1Δ double mutant (data not shown), indicating no involvement of Ded1 at all. Thus, the majority (∼75%) of the 87 Dbp1-hyperdependent mRNAs appear to rely almost entirely on Dbp1 rather than Ded1 for WT translation. By contrast, individually examining all 226 Ded1-hyperdependent mRNAs (i.e. showing TE reductions in ded1-ts versus WT cells) revealed that only ∼12% (26/226) appear to rely solely on Ded1 in showing TE reductions in ded1-ts versus WT cells equal to or greater than those seen in the double mutant (data not shown). Thus, the majority of Ded1-dependent mRNAs also utilize Dbp1 to achieve WT translation. Interestingly, 18 mRNAs showed substantial TE reductions only in the double mutant (exhibiting <20% TE reduction in one single mutant and none in the other single mutant) indicating nearly complete functional redundancy between Ded1 and Dbp1 for this small group of mRNAs.
The foregoing conclusions, based on analysis of mRNAs exhibiting TE changes exceeding the 2-fold threshold, were corroborated by examining TE changes of any magnitude for a group of ∼1950 mRNAs showing significant TE changes in the ded1-ts dbp1Δ mutant versus WT at FDR < 0.05. Hierarchical clustering of TE changes reveals that the ded1-ts mutation reduces the TEs of more mRNAs, and to a greater extent, than does dbp1Δ in otherwise WT cells (Figure 2F, more/darker red colors in lower half of heatmap in col. 2 versus 3). It also confirms that Ded1 and Dbp1 cooperate on many mRNAs, as the magnitude of TE reductions are generally greater in the double mutant versus either single mutant (Figure 2F col. 1 versus cols. 2–3). A more extensive cluster analysis in Supplementary Figure S4D reveals the sizable group of conditionally Ded1-dependent mRNAs for which the effect of ded1-ts is exacerbated by the absence of DBP1 (Supplementary Figure S4D, darker shades of red in col. 2 versus col. 3), and also the numerous conditionally Dbp1-dependent mRNAs for which the effect of dbp1Δ is exacerbated by the presence ded1-ts (Supplementary Figure S4D, darker shades of red in col. 4 versus col. 5). Overall, these findings indicate extensive functional cooperation between Ded1 and Dbp1, varying in degree with the mRNA, for many mRNAs that depend on one or the other helicase for robust translation in WT cells.
Overexpression of Dbp1 ameliorates translation defects in ded1 mutants
Results above indicate that deletion of DBP1 exacerbates the translational defects conferred by the ded1-ts mutation, indicating extensive functional overlap between Ded1 and Dbp1. To further support this conclusion, we asked if overexpressing Dbp1 from a high-copy plasmid (hcDBP1) would rescue the translation defects of specific mRNAs in ded1 mutants. We first examined the effects of Dbp1 overexpression on translation of two luciferase reporter mRNAs, one with an unstructured 5′-UTR consisting of mostly (CAA)n repeats and the other also containing a cap-distal stem–loop (SL) structure. As observed previously (6), expression of the reporter lacking a SL was reduced only marginally in the ded1-cs strain at a semi-permissive temperature of 23°C compared to that seen in WT DED1 cells or the ded1-cs mutant complemented by a hc DED1 plasmid (Figure 3A, -SL, red versus green/blue). The presence of the SL reduced LUC expression considerably (by ∼80%) in WT cells (Figure 3A, blue, +SL versus -SL), and nearly abolished reporter expression in ded1-cs cells (Figure 3A, red, +SL versus -SL). Interestingly, the hcDBP1 plasmid rescued expression of the +SL reporter in ded1-cs cells by >10-fold while having little effect on the -SL reporter (Figure 3A, purple versus red, +SL versus -SL). These findings suggest that overexpressed Dbp1 can compensate for the inability of the ded1-cs product to promote scanning through a structured 5′-UTR.
Figure 3.

Overexpression of Dbp1 substantially rescues translation defects in ded1 mutants. (A) LUC reporters containing the RPL41A promoter, modified 5′-UTR of 91 nt comprised of CAA repeats alone (-SL) or a SL of -3.7 kcal/mol predicted stability (+SL), and first 4 codons of RPL41A fused to LUC CDS, plus a truncated RPL41A 3′-UTR, were assayed in WT cells (strain NSY4) containing an empty vector or ded1-cs cells (strain NSY5) containing empty vector (p1376), hc DED1 (pNDS47) or hcDBP1(pNDS43) plasmids. Strains were cultured in SC-Leu-His-Ura medium at 30°C, diluted to OD600 of ∼0.1 and grown for ∼3 doublings at 23°C (∼18 h for ded1-cs, ∼10h for WT, ded1-cs (hcDED1) and ∼15 h for ded1-cs (hcDBP1) cells)). Luciferase activities were assayed in cell extracts, normalized to total protein, and reported in relative light units (RLUs) per mg of protein, as the means (±S.E.M.) determined from six transformants. P-values of Student’s t-test for statistical significance of the difference between means in comparing results from ded1-cs(vector) transformants with each of the other three transformant types are shown (*, P< 0.03; **, P< 0.002). (B) Hierarchical clustering analysis of TE changes in ded1-cs or ded1-cs(hcDBP1) versus WT, or ded1-cs(hcDBP1) versus ded1-cs cells for 3077 mRNAs with significant ΔTE changes of any magnitude in ded1-cs versus WT cells at FDR < 0.01 (as determined by DESeq2 using biological replicates of each strain) presented as in Figure 2E. Six mRNAs with >64-fold changes in TE were excluded. (C) Notched box-plot analysis of 1101 mRNAs exhibiting ≥2-fold reductions in TE in ded1-cs versus WT cells (FDR < 0.01), comparing TE changes in ded1-cs versus WT (col. 1) to those in ded1-cs(hcDBP1) versus WT cells. (D) Overlap between 1101 Ded1-hyperdependent mRNAs and 614 mRNAs exhibiting ≥2-fold reductions in TE in ded1-cs(hcDBP1) versus WT cells (FDR < 0.01). (E) As in panel (C), but for 603 mRNAs exhibiting ≥2-fold reductions in TE in ded1-cs(hcDBP1) versus WT cells (FDR < 0.01). In (C) and (D), P-values from Mann–Whitney U-test are indicated (**, P< 10−16).
We next investigated if Dbp1 overexpression can rescue translation defects in ded1 mutants genome-wide. Ribo-Seq analysis of ded1-cs and ded1-ts mutants containing hcDBP1 or empty vector, and the corresponding WT strains, was conducted under the same non-permissive conditions described both here and previously (6). The results from the empty vector transformants of the ded1-cs and ded1-ts mutants were very similar to our previous findings (6), whether considering all expressed mRNAs (Supplementary Figure S5A and C), groups of 3412 and 743 mRNAs showing statistically significant TE changes in the new analyses for ded1-cs and ded1-ts mutants, respectively (Supplementary Figure S5E), or groups of mRNAs judged to be Ded1-hyperdependent (≥2-fold reductions in TE at FDR < 0.01) (Supplementary Figure S5B and D). We presume that a >2.5-fold greater sequencing depth in the new ded1-cs analysis allowed us to identify a greater number of Ded1-hyperdendent mRNAs than previously observed (1101 versus 617 mRNAs) (Supplementary Figure S5D and Supplementary Table S1).
Remarkably, Ribo-Seq analysis revealed a broad reduction in the magnitude of TE changes in both ded1 mutants on overexpression of DBP1 (the RPFs for DBP1 itself indicated a ∼25-fold increase in DBP1 expression conferred by hcDBP1). Cluster analysis of 3077 mRNAs exhibiting significant TE changes of any magnitude in ded1-cs versus WT cells (FDR < 0.01) showed that hcDBP1 conferred a broad dampening of TE reductions evoked by ded1-cs (Figure 3B, generally lighter shades of red in col. 2 versus 1), producing widespread TE increases compared to the ded1-cs mutant containing vector alone (Figure 3B, note color switch between cols. 2–3). Consistently, introducing hcDBP1 into ded1-cs cells (i) diminished the TE reductions compared to WT cells for a large proportion of the ∼1100 Ded1-hyperdependent mRNAs (Figure 3C), (ii) reduced by nearly one-half (from 1101 to 603) the number of such hyperdependent mRNAs (Figure 3D) and (iii) diminished the TE reductions compared to WT for the ∼600 mRNAs that still exceeded the 2-fold threshold of TE reductions in the ded1-cs/hcDBP1 strain (Figure 3E). A highly similar rescue of translation by hcDBP1 was observed in the ded1-ts strain (Supplementary Figure S6A–D). Thus, independent of temperature effects, Dbp1 overexpression enhances translation of Ded1-hyperdependent mRNAs in both cold- and temperature-sensitive ded1 mutants. We conclude that overexpressed Dbp1 can partially compensate for loss of Ded1 function throughout the yeast translatome.
Dbp1 cooperates with Ded1 on a group of long, structured mRNAs with weak closed-loop forming potential
Our findings thus far suggest that Dbp1 acts similarly to Ded1 in overcoming the inhibitory effects of long, structure-prone 5′-UTRs on translation of many mRNAs. To test this interpretation, we analyzed the 5′-UTR features of both conditional Dbp1- and conditional Ded1-hyperdependent mRNAs using a compilation of 5′-UTR lengths and propensities for secondary structure in the yeast transcriptome (39). Each nucleotide in 3000 different yeast transcripts was assigned a ‘parallel analysis of RNA structure’ (PARS) score based on its susceptibility to digestion with single- or double-stranded specific nucleases in yeast mRNA reannealed in vitro; with a higher PARS score denoting a higher probability of double-stranded conformation. For each transcript, we calculated various cumulative PARS scores for 5′-UTRs (Figure 4A), including the sum of scores for (i) all 5′-UTR nucleotides (Total PARS); (ii) the first 30 nt (First30 PARS); (iii) 30 nt surrounding the start codon (Start30 PARS; for mRNAs with a 5′-UTR ≥ 15 nt); and (iv) the highest cumulative score in any 30 nt window (Max30 PARS). We also summed PARS scores for successive intervals downstream of the AUG, including +1 to +30 (Plus15), +16 to +45 (Plus30), +31 to +60 (Plus45), +46 to +75 (Plus60) and +61 to +90 (Plus75); as well as the average scores over the 5′-UTR (Avg 5′-UTR PARS) and CDS (Avg CDS PARS), which are independent of 5′-UTR or CDS lengths, respectively. To obtain groups of hyper- and hypodependent mRNAs large enough for statistical analysis, we adjusted the thresholds of TE changes to >1.4 and <0.7, while maintaining FDR at <0.01 (39).
Figure 4.

mRNAs hyperdependent on Ded1 or Dbp1 exhibit heightened propensities for RNA structures in somewhat different regions. (A) Schematic showing a subset of the 5′-UTR and CDS intervals assigned for calculating cumulative PARS scores for each mRNA. (B–E) Notched box-plots showing distribution of 5′-UTR and CDS features 5′-UTR length (B), Avg 5′-UTR PARS score (C), Max30 PARS (D) or CDS length (E), for all mRNAs for which PARS data is compiled in (39), comparing all mRNAs (col. 1) to mRNAs exhibiting >1.4-fold reduction in TE in the indicated mutants versus WT (cols. 2–4) or mRNAs conditionally dependent on Dbp1 or Ded1 as defined in ‘Results’ section (cols. 5–6). Numbers of mRNAs in each dataset exceed 150. P-values from Mann–Whitney U-test are compiled in Table 1 and Supplementary Tables S1–5, and also indicated with asterisks for significant differences with the ‘All mRNAs’ group (*, P< 0.002; **, P< 10−8). (F) Spearman coefficients from correlations between the indicated mRNA features in 5′-UTRs (left) or CDS (right) and ΔTE values conferred by the indicated single mutations versus WT (blue, cyan, light cyan) or the ded1-ts dbp1Δ double mutation versus ded1-ts (pink) or dbp1Δ (violet) single mutations, for all 2603 curated mRNAs (*, P< 10−16). Number of mRNAs in CDS length dataset, N = 5296.
Interestingly, the 190 conditionally Dbp1-hyperdependent mRNAs (strongly impaired by dbp1Δ in ded1-ts, but not in otherwise WT cells) for which PARS scores exist exhibit a median 5′-UTR length of ∼100 nt, substantially greater than the median length of ∼50 nt for all 2603 expressed mRNAs compiled in the PARS database (Figure 4B, pink versus gray), indicating that longer 5′-UTRs are associated with increased conditional dependence on Dbp1 for efficient translation in ded1-ts cells. Remarkably, the median PARS scores for all 5′-UTR features, but only Plus 15 for the CDS, were also significantly greater for this group of mRNAs versus all mRNAs (Figure 4C and Supplementary Figure S7A, B and D, pink versus gray; Table 1, cols. 3–4 versus col. 2). Moreover, the average PARS scores for 5′-UTRs, but not for CDS, was higher for the conditionally Dbp1-hyperdependent mRNAs (Figure 4D versus Supplementary Figure S7C, pink versus gray). Since the average 5′-UTR PARS scores were significantly higher for the conditionally Dbp1-hyperdependent mRNAs (Supplementary Figure S7A), it appears that their 5′-UTRs are both more structurogenic and longer than average. For the 173 conditionally Dbp1-hypodependent mRNAs, by contrast, the median 5′-UTR Total PARS score was ∼2-fold lower, the median CDS Avg PARS score was slightly higher, and no other PARS features were significantly different compared to all mRNAs (Table 1, cols. 5–6 versus col. 2). Thus, as concluded previously for Ded1-hyperdependent mRNAs (summarized in Supplementary Table S2), both 5′-UTR length and propensity for secondary structure in 5′-UTRs are associated with Dbp1 enhancement of translation for those mRNAs where Dbp1 function was unmasked by impairing Ded1 in ded1-ts cells. Not surprisingly, similar conclusions were reached from analysis of the mRNA features of conditionally Ded1 hyperdependent mRNAs (impaired by ded1-ts in dbp1Δ, but not in otherwise WT cells) (Supplementary Table S3); and also for the mRNAs showing >1.4-fold TE reductions between ded1-ts dbp1Δ versus WT cells (Supplementary Table S4).
Table 1.
Comparison of 5′-UTR lengths and PARS features for conditionally Dbp1-dependent mRNAs (exhibiting >1.4-fold TE changes at FDR < 0.01 in ded1-ts dbp1Δ versus ded1-ts cells but no significant changes in dbp1Δ versus WT cells)
| All mRNAs | ΔTEconditional Dbp1 < 0.7 | ΔTEconditional Dbp1 >1.4 | |||
|---|---|---|---|---|---|
| Feature | (N = 2603) | (n = 190) | P-value | (n = 158) | P-value |
| 5′-UTR | |||||
| Total Length | 51 | 96 | <10−13 | 37 | <10−6 |
| Total PARS | 3.79 | 14.62 | <10−8 | 1.73 | <0.02 |
| Avg. PARS | 0.06 | 0.14 | <0.0003 | 0.06 | 0.35 |
| First30 PARS | 2.14 | 4.27 | <0.005 | 1.66 | 0.61 |
| Start30 PARS | 4.23 | 6.81 | <0.003 | 3.6 | 0.63 |
| Max30 PARS | 13.07 | 17.21 | <10−5 | 11.28 | <0.05 |
| CDS | |||||
| CDS length | 1068 | 1406 | 0.0008 | 1397 | <10−8 |
| Avg PARS | 0.27 | 0.28 | 0.78 | 0.34 | <10−4 |
| Plus15 PARS | 7.16 | 9.52 | <0.04 | 7.54 | 0.32 |
| Plus30 PARS | 7.23 | 8.24 | 0.77 | 8.27 | 0.23 |
| Plus45 PARS | 7.19 | 8.16 | 0.71 | 7 | 0.45 |
| Plus60 PARS | 7.29 | 7.38 | 0.76 | 7.621 | 0.63 |
| Plus75 PARS | 7.37 | 7.91 | 0.58 | 8.438 | 0.49 |
Median 5′-UTR length or indicated PARS feature was calculated from the database compiled in (39) for all 2603 mRNAs, or the sets of 190 or 158 conditionally Dbp1 hyper- or hypo-dependent mRNAs, respectively. P-values are from Mann–Whitney U-test of statistical significance of the difference between the medians of two distributions.
We also examined the properties of the two groups of non-overlapping mRNAs shown in Figure 2E, representing the largest groups of mRNAs we identified that are strongly dependent on only Ded1 (298 mRNAs in the top group) or only Dbp1 (149 mRNAs in the bottom group). The subset of 164 mRNAs in the top group compiled in the PARS dataset exhibits the same features of longer 5′-UTRs, greater 5′-UTR PARS scores for the Max30, Average and Start 30 features, and longer CDSs compared to all mRNAs that were shown by the 226 Ded1-hyperdependent mRNAs identified in the ded1-ts single mutant (Figure 4B–E and Supplementary Figure S7B). Similarly, the subset of 47 mRNAs in the bottom group with available PARS data show the longer 5′-UTR lengths, greater 5′-UTR PARS scores for the Max30, Average and First30 features, and greater Plus15 and Average CDS PARS scores observed for the 87 Dbp1-hyperdependent mRNAs identified in the dbp1Δ single mutant (Figure 4B–E and Supplementary Figure S7B). Thus, the mRNAs that are most exclusively dependent on Ded1 or Dbp1 have very similar features as those observed for the groups of Ded1- or Dbp1-hyperdependent mRNAs, respectively, that strongly depend on that helicase even when the other one is present.
Spearman correlation analysis of TE changes for all expressed mRNAs allowed us to extend the foregoing conclusions to the entire translatome, as global TE changes conferred by deleting DBP1 in ded1-ts cells (ΔTEdbp1Δ(ded1-ts) = TEded1-ts dbp1Δ /TEded1-ts) showed significant negative correlations with 5′-UTR length, Total PARS and Max30 PARS scores (Figure 4F, left, light pink bars marked with asterisks), whereas no correlations were observed for any CDS PARS features (Figure 4F, right, light pink bars). Similar results were obtained for the global TE changes conferred by ded1-ts in dbp1Δ cells (ΔTEded1-ts(dbp1Δ)= TEded1-ts dbp1Δ /TEdbp1Δ), and by the ded1-ts dbp1Δ double mutation compared to WT cells, with negative correlations for 5′-UTR features comparable or higher than those observed in the ded1-ts single mutant versus WT (Figure 4F, left, violet and blue versus cyan). It should be noted that the correlations with 5′-UTR length and CDS length are generally stronger than for any of the significant correlations observed for PARS features.
As the Max30 PARS score yielded one of the strongest negative correlations with TE changes in the various helicase mutants (Figure 4F), we asked if a particular location of the Max30 PARS window in the 5′-UTR is associated with Ded1-hyperdependence. To this end, we calculated the Max30 position with respect to mRNA 5′-end (dubbed 5′-end Max30 Pos, Supplementary Figure S8A) for all mRNAs with 5′-UTR lengths >30 nt. Interestingly, for the Ded1 hyperdependent mRNAs identified in ded1-cs and ded1-ts versus WT cells (Figure 3C and Supplementary Figure S6B), the median 5′-end Max30 Pos is substantially larger than the median for all mRNAs (Supplementary Figure S8B and C, col. 2 versus col. 1), but for the groups of Ded1 hypodependent mRNAs is substantially smaller, or not significantly different, than the median for all mRNAs (Supplementary Figure S8B and C, col. 3 versus col. 1). These findings suggest that Max30 windows located at relatively greater distances from the 5′-end (cap-distal structures) show a greater association with Ded1 hyperdendence versus cap-proximal structures. The same conclusion emerged on binning the ∼2000 mRNAs on the basis of 5′-end Max30 Pos values and plotting their TE changes in ded1-cs cells. The mRNAs in the bins with the largest distances of Max30 windows from the 5′-end, i.e. those with the most cap-distal structures, tend to show the greatest TE reductions in ded1-cs cells (Supplementary Figure S8D, for bins 8–10, the upper notches are well below the red line indicating no change in TE, i.e. log2ΔTEded1-cs= 0). These findings are consistent with our previous conclusion, based on analysis of luciferase reporter mRNAs with stem–loop structures at different locations, that a cap-distal structure confers a greater dependence on Ded1 versus a cap-proximal structure (6).
The length of CDSs, reflecting overall transcript length (40), is another mRNA feature associated with TE reductions in ded1 single mutants, comparable in importance to 5′-UTR length (41) (Figure 4E, cyan versus gray). Here as well, we observed that the groups of mRNAs exhibiting the strongest TE reductions in the ded1-ts dbp1Δ double mutant in comparison to each single mutant, or to WT, also have longer than average CDS lengths (Figure 4E, blue, pink and violet versus gray). In addition, longer CDS lengths are negatively correlated with global TE reductions in the same strain comparisons (Figure 4F, right, pink, violet, blue and cyan).
There is evidence that longer mRNAs are less efficient in forming the closed-loop intermediate mediated by interactions of eIF4E/eIF4G and PABP bound to the cap and poly(A) tail, respectively (42), consistent with the fact that longer mRNAs tend to have relatively low TEs in WT yeast (43,44). Based on occupancies of eIF4E/eIF4G and PABP and the inhibitory eIF4E-binding proteins Caf20 and Eap1 (capable of disrupting closed loop assembly), Costello et al. assigned yeast mRNAs to four groups (I–IV), of which the 374 mRNAs with relatively high eIF4E/eIF4G/PABP and low Caf20/Eap1 occupancies (group III) were judged to have strong closed-loop (SCL) potential (45). Previously, we noted that these SCL mRNAs tend to be Ded1-hypodependent, exhibiting increased relative TE in the ded1-ts mutant (Figure 5A, col. 4 (Gp III) versus col. 1 (All mRNAs). Similar results were observed here for the TE changes conferred in ded1-ts dbp1Δ versus WT cells (Figure 5B, Gp III versus all mRNAs). These findings suggest that functional cooperation between Ded1 and Dbp1 is particularly important for longer mRNAs with relatively weak closed-loop formation.
Figure 5.

A subset of highly translated mRNAs with elevated closed-loop potential or PABP occupancies exhibit marked Dbp1-dependence. (A–C) Notched box-plots showing ΔTEs conferred by the indicated mutations versus WT for four groups of mRNAs (numbers in parenthesis) described in (45) that vary in occupancies of proteins that promote or oppose closed-loop formation (see text). (D) Notched box-plots showing TE distributions in WT cells for all mRNAs (cols. 1 and 3, N ≅ 5300) or subsets of mRNAs exhibiting ≥2-fold TE reductions (indicated by #) in dbp1Δ versus WT (col. 2, N = 87) or ded1-ts versus WT cells (col. 4, N = 226). In panels (A–D), results from Mann–Whitney U-test are shown only for comparisons with significant P-values (*, P< 10−10; **, P< 10−16). (E–G) qRT-PCR analysis of polysome size distributions of Dbp1-hyperdependent mRNAs in dbp1Δ versus WT cells. WT (NSY10) and dbp1Δ (NSY79) strains were cultured in SC-His medium at 30°C to log-phase growth, sedimented at room temperature, transferred to flasks containing pre-warmed SC medium, and incubated at 37°C for 2 h. Following treatment with cycloheximide, cell extracts were resolved by sedimentation through sucrose density gradients. Gradient fractions were collected, spiked with equal amounts of control luciferase RNA, and total RNA was extracted and subjected to qRT-PCR analysis to quantify abundance of the indicated native mRNAs relative to spike-in control RNA. Three technical replicates for each fraction were analyzed and mean values are plotted as percentages of total mRNA in the gradient in each fraction. Positions of polysomes and 80S monosomes were determined by monitoring A254 during fractionation. Similar results were obtained in an independent experiment using biological replicate cultures (Supplementary Figure S10E–G). (H) Dbp1 associates with translationally active polysomes. WT cells expressing Dbp1-TAP were grown in SC-His at 30°C to OD600 of ∼0.8 and cross-linked with 2% formaldehyde prior to harvesting. Cell extracts digested with ribonuclease I (+RNase I) or left untreated (-RNase I) were separated by sedimentation through sucrose gradients, and proteins from each gradient fraction were precipitated by ethanol. Dbp1 was detected by western blotting with anti-Tap antibody and 40S subunits were detected using anti-Rps22 antibody.
Dbp1 acts uniquely on a subset of highly translated, structured mRNAs in WT cells
Analysis of the dbp1Δ single mutant described above identified a group of 87 mRNAs that are hyperdependent on Dbp1 in otherwise WT cells; >60% of which were relatively unaffected by the ded1-ts single mutation (Figure 1C and F). Such mRNAs whose TEs are reduced by dbp1Δ but nearly unaffected by ded1-ts, even in cells lacking DBP1, were also revealed by the cluster analysis in Figure 2E (bottom clusters marked with bracket, col. 3. versus 2). To determine whether mRNAs that are uniquely hyperdependent on Dbp1 have features distinct from those of Ded1-hyperdependent mRNAs, we analyzed the 5′-UTR features of 154 mRNAs with ΔTEdbp1Δ < 0.7 for which PARS data are available. The median 5′-UTR length, Max30, Start30 and Avg 5′-UTR PARS scores, as well as the CDS length of these Dbp1-hyperdependent mRNAs are all significantly larger than the genome-median values (Figure 4B–E, Supplementary Figure S7B; Table S5, cf. cols. 2–3), as noted above for Ded1-hyperdependent mRNAs. Unlike the latter, however, all CDS PARS scores are significantly higher for the Dbp1-hyperdependent group versus all mRNAs (Supplementary Table S5, col. 2–3 versus col.1).
The same overall trends emerged from considering TE changes for all expressed mRNAs in the dbp1Δ single mutant versus WT, as the ΔTEdbp1Δ values showed negative correlations with all CDS PARS features, especially the average PARS score for the entire CDS (Figure 4F, right, light cyan), in addition to the 5′-UTR length, Max30 and Total 5′-UTR PARS scores (Figure 4F, left, light cyan). Whereas the strongest negative correlation with ΔTEdbp1Δ values occurred with the average CDS PARS scores, TE changes conferred by ded1-ts (ΔTEded1-ts) were by contrast negatively correlated most strongly with 5′-UTR length, Total and Max30 5′-UTR PARS scores and CDS length (Figure 4F, right and left, cyan versus light cyan).
The weaker negative correlation between TE changes and CDS length for dbp1Δ versus ded1-ts mutants was further revealed by sorting all mRNAs into 14 bins of different CDS lengths. Whereas ΔTEdbp1Δ values are lower than average only for the last three bins containing the longest CDSs (Supplementary Figure S9A, the upper notches for bins 12–14 are below the red line at log2ΔTEdbp1Δ = 0), ΔTEded1-ts values progressively decline with increasing CDS lengths (Supplementary Figure S9B). In addition, the TEs of the shortest mRNAs (in the first bin) are decreased in dbp1Δ cells but increased in ded1-ts cells (Supplementary Figure S9A versus B, bin 1), indicating a unique requirement for Dbp1 for a subset of the shortest mRNAs. Consistent with greater than average TE reductions in dbp1Δ versus WT cells for a subset of the shortest mRNAs, SCL mRNAs show a modest decrease in TE in dbp1Δ cells, as opposed to the increased TE they exhibit in ded1-ts cells (Group III, Figure 5C versus 5A). Thus, a fraction of shorter mRNAs with strong closed-loop potential displays a heightened dependence on Dbp1, but not Ded1, in WT cells.
In accordance with their strong closed-loop potential, SCL (Group III) mRNAs comprise one of the two most efficiently translated groups of mRNAs in WT yeast, whereas the second group (designated Group I), was enriched only for PABP (45). The SCL/Group III mRNAs exhibit increased median TE in ded1-ts cells; however, group I mRNAs are virtually unchanged compared to all mRNAs (Figure 5A, col. 2 versus col. 4). By contrast, both Group I and SCL/Group III mRNAs exhibit decreased TE in dbp1Δ cells (Figure 5C). Consistent with this, Dbp1-hyperdependent mRNAs tend to be highly translated in WT cells in comparison to Ded1-hyperdependent (Figure 5D). Interestingly, although TE is directly correlated with mRNA abundance in WT cells (44), the Dbp1-hyperdependent mRNAs have less than average abundance compared to all mRNAs (Supplementary Figure S9C). Together, these findings suggest that the group of mRNAs uniquely dependent on Dbp1 in WT cells is distinct from Ded1-hyperdependent mRNAs in being highly translated but not unusually abundant in WT cells, in showing a tendency for high closed-loop forming potential or high PABP occupancy, and in displaying greater than average structurogenic potential within the CDS.
To determine whether Dbp1 enhances translation of mRNAs at the initiation stage, we examined the distribution of several Dbp1-hyperdependent mRNAs in polysomes, 80S monosomes, and free mRNPs in cell extracts resolved by sucrose density gradient centrifugation. Relative mRNA abundance in each fraction was quantified by normalization to spike-in control firefly luciferase (FLUC) mRNA. Five of six Dbp1-hyperdependent mRNAs tested showed shifts in their distributions from heavier to lighter polysomes in dbp1Δ versus WT cells (results for three of the five are shown in Figure 5E–G; results for biological replicates for these three mRNAs are in Supplementary Figure S10E–F; replicate analyses of the other two mRNAs, FLO11 and FIG2, are shown in Supplementary Figure S10A–D). These findings are consistent with a reduced initiation rate for these mRNAs in the dbp1Δ mutant. Considering the important role played by Dbp1 in regulating translation of many mRNAs, we asked whether Dbp1 associates with actively translating ribosomes in WT cells by monitoring sedimentation of Dbp1 protein across ribosomal fractions in sucrose density gradients. Cells of a strain expressing Tap-tagged Dbp1 from a chromosomal DBP1-TAP allele were treated with formaldehyde to cross-link Dbp1-polysome complexes and the resulting cell extracts were treated with RNase I or left untreated prior to ultracentrifugation. A proportion of Dbp1-TAP in untreated extracts sedimented in polysome-containing fractions, which was greatly diminished, along with heavy polysomes, by RNAse I digestion—behaving similar to 40S subunit protein Rps22 (Figure 5H, left versus right panels). The association of Dbp1-TAP with actively translating ribosomes in polysomes is consistent with a role for Dbp1 in translation initiation, if we stipulate that Dbp1 interacts with scanning 43S PICs bound to the 5′-UTRs of mRNAs in polysomes.
Dbp1 stimulates 48S PIC assembly in the yeast reconstituted system
We recently reconstituted Ded1 function in stimulating assembly of 48S PICs on both native and synthetic mRNAs using a purified yeast translation initiation system (12), and we sought to accomplish the same here for Dbp1. We first examined the RNA-binding and RNA-dependent ATPase activities of Dbp1. Similar to Ded1 (12), purified Dbp1 (Supplementary Figure S11A) displayed higher affinity for single-stranded RNA in the presence of nonhydrolyzable ADPNP versus ADP or no nucleotide (Supplementary Figure S11B and C). As expected, Dbp1 ATPase activity was stimulated by RNA, and the kcat and Km for ATP in the presence of saturating RNA are similar to those obtained for purified Ded1 (Supplementary Figure S11D) (12).
Next, we sought to recapitulate Dbp1’s stimulation of 48S PIC assembly in vitro. 43S PICs (comprised of 40S subunits and canonical initiation factors eIFs 1, 1A, eIF2·GDPNP·Met-tRNAiMet, eIF3, eIF5, eIF4B, eIF4E·eIF4G and eIF4A) were pre-assembled and then incubated with radiolabeled capped mRNA and ATP for increasing times to allow 48S PIC assembly. Reaction aliquots were quenched with excess unlabeled capped mRNA and 48S PICs and resolved from unbound mRNAs by native gel electrophoresis. This assay was performed with five mRNAs (Figure 6A) previously shown to exhibit different degrees of stimulation by Ded1 in vitro (12). These include native RPL41A mRNA (henceforth, simply RPL41A), containing a short 5′-UTR of 24 nt and low degree of predicted secondary structure (23), which was classified as Ded1-hypodependent in our Ribo-Seq experiments (6). We also examined a reporter mRNA containing the 92nt 5′-UTR and the first 60 nt of CDS from SFT2 mRNA—a Ded1-hyperdependent mRNA (6) harboring a well-defined SL located 47 nt from the 5′ end (46)—as well as three model mRNAs containing synthetic 5′-UTRs appended to the CDS and 3′-UTR of RPL41A. The latter include a 67 nt 5′-UTR primarily comprised of CAA repeats (No SL) and derivatives of No SL mRNA containing a SL insertion (of predicted ΔGo = –8.1 kcal/mol) in a cap-proximal position 5 nt from the cap (CP-SL) or a cap-distal location 45 nt downstream from the cap (CD-SL).
Figure 6.

Dbp1 stimulates 48S PIC assembly on native and synthetic mRNAs in the yeast reconstituted system. (A) Schematics of mRNAs for in vitro analysis, including full-length native RPL41A and the 5′-UTR and first 60 nt of SFT2 ORF. No SL, CD-SL and CP-SL are synthetic mRNAs with unstructured 5′-UTRs (CAA repeats) and the CDS and 3′-UTR of RPL41A. Hairpins indicate stem–loops, and numbers give lengths in nucleotides. (B) Apparent rates of recruitment of different mRNAs measured as a function of Dbp1 concentration. Data are fitted with a hyperbolic equation. Error bars represent one standard deviation (n = 3). (C) Top: Maximal rate (kmax) of mRNA recruitment observed without Dbp1 (blue bars) and with saturating Dbp1 (orange bars) from data in (B). Bottom: Fold-change in maximal rate with and without Dbp1 from data in top panel. (D) Concentrations of Dbp1 required to achieve half-maximal rates of recruitment (K1/2) from data in (B). (C and D) Bars represent mean values and dots indicate values from individual experiments. P-values from Student’s t-test are indicated (*, P< 0.004; **, P< 0.0008).
As observed previously (12), in the absence of Dbp1 or Ded1, the rates of 48S assembly at saturating concentrations of all other components are substantially greater for the two mRNAs lacking SLs (No SL and RPL41A) versus the three containing SLs (SFT2, CP-SL and CD-SL) (Figure 6B, 0 nM Dbp1; 6C, blue), reflecting the inhibitory effects of SLs on PIC recruitment or scanning in the reconstituted system. Adding Dbp1 accelerated 48S assembly on all five mRNAs (Figure 6B), with the highest kmax achieved for No SL mRNA, and the lowest kmax values observed for the two SL-containing synthetic mRNAs (Figure 6C, orange versus blue). Notably, the fold-increases in kmax values achieved with Dbp1 were considerably greater for CD-SL and SFT2, both containing SLs, compared to the No SL and RPL41A mRNAs lacking SLs (Figure 6C, gray). Moreover, the Dbp1 concentrations required to achieve half-maximal rates (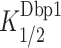 ) were also relatively higher for all three SL-containing mRNAs CP-SL, CD-SL and SFT2 (Figure 6D). These findings indicate that Dbp1 can increase the rate of 48S PIC assembly even for mRNAs with little 5′-UTR structure (No SL and RPL41A), but confers greater acceleration for mRNAs whose recruitment rate is diminished by cap-distal SL structures (CD-SL and SFT2). The increases in kmax conferred by Dbp1 are remarkably similar to those observed previously for Ded1 for these same mRNAs (Supplementary Figure S11E, cf. cols. 3–4); however, the
) were also relatively higher for all three SL-containing mRNAs CP-SL, CD-SL and SFT2 (Figure 6D). These findings indicate that Dbp1 can increase the rate of 48S PIC assembly even for mRNAs with little 5′-UTR structure (No SL and RPL41A), but confers greater acceleration for mRNAs whose recruitment rate is diminished by cap-distal SL structures (CD-SL and SFT2). The increases in kmax conferred by Dbp1 are remarkably similar to those observed previously for Ded1 for these same mRNAs (Supplementary Figure S11E, cf. cols. 3–4); however, the 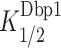 values are ∼2- to ∼4.5-fold lower than the corresponding
values are ∼2- to ∼4.5-fold lower than the corresponding 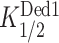 values, except for No SL mRNA, with the largest differences seen for the three SL-containing mRNAs (Supplementary Figure S11E, last two cols., rows 2,3,5). The fact that lower concentrations of Dbp1 versus Ded1 are required to achieve the same degree of rate stimulation suggests that Dbp1 is relatively more efficient in accelerating 48S PIC assembly, particularly with mRNAs with structured 5′-UTRs. Our finding that higher Dbp1 concentrations are required to achieve the maximum rate stimulation for mRNAs containing cap-distal SLs versus SL-lacking mRNAs was observed previously for Ded1, and might be explained by proposing that accelerating the rate of scanning through SL structures requires higher Dbp1/Ded1 concentrations than the step(s) of 48S PIC assembly enhanced by these helicases, regardless of 5′-UTR structures. Thus, Dbp1 mimics Ded1 function in stimulating 48S PIC assembly on a range of structured and unstructured mRNAs in vitro, consistent with our conclusion that these helicases cooperate on many mRNAs in vivo.
values, except for No SL mRNA, with the largest differences seen for the three SL-containing mRNAs (Supplementary Figure S11E, last two cols., rows 2,3,5). The fact that lower concentrations of Dbp1 versus Ded1 are required to achieve the same degree of rate stimulation suggests that Dbp1 is relatively more efficient in accelerating 48S PIC assembly, particularly with mRNAs with structured 5′-UTRs. Our finding that higher Dbp1 concentrations are required to achieve the maximum rate stimulation for mRNAs containing cap-distal SLs versus SL-lacking mRNAs was observed previously for Ded1, and might be explained by proposing that accelerating the rate of scanning through SL structures requires higher Dbp1/Ded1 concentrations than the step(s) of 48S PIC assembly enhanced by these helicases, regardless of 5′-UTR structures. Thus, Dbp1 mimics Ded1 function in stimulating 48S PIC assembly on a range of structured and unstructured mRNAs in vitro, consistent with our conclusion that these helicases cooperate on many mRNAs in vivo.
Small subunit (SSU) profiling reveals scanning defects on Ded1/Dbp1-hyperdependent mRNAs in vivo
Our conclusions that Ded1 and Dbp1 function broadly to stimulate scanning through 5′-UTRs with greater than average structurogenic potential was based largely on correlations between TE changes in the helicase mutants and 5′-UTR PARS scores (Figure 4C, D and F). Because PARS scores are derived from purified mRNAs folded in vitro, and thus might only approximate mRNA structures formed in vivo, we sought to obtain direct evidence for widespread scanning defects in ded1 and dbp1Δ cells. To this end, we employed Translation Complex Profiling (TCP-seq) (32), which employs formaldehyde cross-linking to stabilize small (40S) ribosomal subunits (SSUs) on mRNA, followed by RNAse I treatment, purification of SSUs, reversal of cross-links, mRNA purification, and deep-sequencing of DNA libraries prepared from the purified mRNA. TCP-seq was conducted for ded1-cs, ded1-ts, dbp1Δ and ded1-ts dbp1Δ mutants versus WT cells using the same culture conditions and growth temperatures employed both here and previously (6) for conventional 80S profiling of the same strains.
We reasoned that a reduced rate of scanning in ded1 mutants would lead to accumulation of SSUs on 5′-UTRs relative to SSUs at AUG codons (ΔSSU_5′-UTR/ ΔSSU_AUG ratio; with SSU reads defined as in Supplementary Figure S12F). This outcome was widespread for the Ded1 hyperdependent mRNAs we defined previously by conventional 80S profiling of ded1 mutants, as shown for three specific mRNAs in Figure 7A; Supplementary Figure S12A and B, and for the group of mRNAs as a whole by the increased median ΔSSU_5′-UTR/ ΔSSU_AUG ratios in ded1-cs and ded1-ts versus WT cells (Figure 7B and Supplementary Figure S12D, col. 2 versus col. 1). By contrast, the ΔSSU_5′-UTR/ ΔSSU_AUG ratios are reduced for the groups of Ded1 hypodependent mRNAs (Figure 7B and Supplementary Figure S12D, col. 3 versus 1). Similar trends were observed in the dbp1Δ and ded1-ts dbp1Δ mutants, except that the median ΔSSU_5′-UTR/ ΔSSU_AUG ratios for the hyper- and hypodependent mRNAs identified in these strains do not differ at a 95% confidence level (Figure 7C and Supplementary Figure S12E, col. 2 versus 3).
Figure 7.

SSU profiling reveals functional cooperation between Ded1 and Dbp1 in stimulating PIC attachment and scanning in vivo. (A) SSU and mRNA reads on the Ded1-hyperdependent mRNA YBR221C in two biological replicates each (_a, _b) of WT or ded1-cs cells in units of rpm (reads per million mapped reads), with the position of the CDS shown (gray arrow) below the tracks, revealing accumulation of SSUs in the 5′-UTR in ded1-cs versus WT cells. Depicted using Integrated Genomics Viewer (Broad Institute). (B andC) Notched box-plots showing distributions of the log2 ratios ΔSSU_5′-UTR/ΔSSU_AUG (cols. 1–3) or ΔSSU_5′-UTR/ΔRNA (cols. 4–6) for all mRNAs (white) and those exhibiting TE decreases (hyper, red) or TE increases (Hypo, blue) exceeding 2-fold in ded1-cs versus WT cells (B), or 1.4-fold in ded1-ts dbp1Δ versus WT cells (C), both at FDR < 0.05. Median ratios for each group are below the x-axis, and numbers of mRNAs in each group are in parenthesis. Results from Mann–Whitney U-test are shown only for comparisons with significant P-values (*, P< 10−10). (D) Spearman coefficients from correlations between the ratios ΔSSU_5′-UTR/ΔSSU_AUG (top 4 bars) or ΔSSU_5′-UTR/ΔRNA (bottom 4 bars) versus ΔTE values (measured by 80S profiling) conferred by the indicated mutation versus WT, for all mRNAs yielding data in both analyses, ranging from ∼900–1300 mRNAs. (*, P< 10−3; **, P< 10−16). (E) Plot showing shift in SSU densities from start codon into 5′-UTRs in ded1-cs cells (right) versus WT cells (left). All mRNAs annotated with a 5′-UTR (37) were ranked according to 5′-UTR length (vertical axis) with longest 5′-UTRs at the top. SSU footprint 5′-end positions are plotted (horizontal axis), and were scaled to the 5′-UTR length such that the 5′-end and start codon of all transcripts are vertically aligned (solid and dotted red lines, respectively). (F) As in (A) but for Ded1-hyperdependent mRNA YDR178W, exhibiting reducd SSUs in the 5′-UTR in ded1-cs versus. WT cells. (G) Notched box-plots showing distributions of the ΔSSU_5′-UTR/ΔSSU_AUG ratios (light gray boxes) or ΔSSU_5′-UTR/ΔRNA ratios (dark gray boxes) across all mRNAs sorted into 13 bins (100 genes each) on the basis of increasing ΔTEded1-cs from 80S profiling, with the 300 most Ded1-hyperdependent mRNAs falling in bins 1–3.
We reached similar conclusions by analyzing all mRNAs for which both SSU and 80S profiling data were available (rather than just the helicase hyper- or hypo-dependent subsets of mRNAs), as ΔSSU_5′-UTR/ ΔSSU_AUG ratios in ded1 mutants yielded significant negative correlations with TE changes determined by 80S profiling in ded1-cs, ded1-ts, and ded1-ts dbp1Δ mutants (Figure 7D, upper 4 rows, blue, royal blue, and light blue bars; asterisks indicate significant correlations). Consistent with a widespread scanning defect in ded1-cs cells, we observed an upstream shift in SSU abundance from AUGs into 5′-UTRs for many mRNAs (Figure 7E, right versus left).
We reasoned that defects in PIC attachment could also be detected by TCP-seq, as a reduced abundance of SSUs in the 5′-UTR relative to mRNA abundance. Indeed, the ΔSSU_5′-UTR/ ΔRNA ratio was reduced on certain Ded1-dependent mRNAs in ded1-cs versus WT cells, including YDR178W mRNA (Figure 7F). While the ΔSSU_5′-UTR/ ΔRNA ratio was not reduced for the group of Ded1-hyperdependent mRNAs as a whole (Figure 7B), binning the ∼1300 mRNAs yielding SSU data by the TE changes in ded1-cs cells from 80S profiling (Figure 7G) revealed that mRNAs with the greatest TE reductions tend to show reduced ΔSSU_5′-UTR/ ΔRNA ratios (dark gray boxplot, bins 1–3, upper notches below red line) in addition to elevated ΔSSU_5′-UTR/ ΔSSU_AUG ratios (light grey boxplot, bins 1–3, lower notches above red line). These data indicate that dual defects in PIC attachment and scanning are associated with TE reductions in ded1-cs cells for the mRNAs with strongest Ded1-dependence. Moreover, the median ΔSSU_5′-UTR/ ΔRNA ratio was significantly reduced in the ded1-ts dbp1Δ mutant for the group of mRNAs whose TEs were reduced in that strain versus WT (Figure 7C, col. 5 versus col. 4), indicating dual PIC attachment and scanning defects on many of the mRNAs impaired in the double mutant. That the median ΔSSU_5′-UTR/ ΔRNA ratio was not as strongly reduced in the ded1-ts or dbp1Δ single mutant for the groups of Ded1 or Dbp1 hyperdependent mRNAs identified in these mutants (Supplementary Figure S12D–E, col. 5 versus col. 4; <95% confidence), suggests that functional cooperation between Ded1 and Dbp1 is particularly important at the PIC attachment step on many mRNAs in which these helicases cooperate. Supporting observations were made when considering all mRNAs, as the ΔSSU_5′-UTR/ ΔRNA ratio showed a significant positive correlation with ΔTEded1-ts dbp1Δ (i.e. reduced SSU abundance in 5′-UTRs associated with reduced TE), whereas no significant correlations were observed for the ded1-ts or dbp1Δ single mutant (Figure 7D, lower bars; royal blue with asterisks). Thus, it appears that Ded1 and Dbp1 functionally cooperate in stimulating both PIC attachment and scanning on a substantial fraction of mRNAs in vivo.
As Ded1 hyperdependent mRNAs exhibit higher propensities for secondary structures in 5′-UTRs, we examined whether secondary structures in 5′-UTRs is associated with stronger scanning defects as judged by SSU profiling. Supporting this possibility, sorting the 343 Ded1-hyperdependent mRNAs identified by 80S profiling of ded1-cs cells according to 5′-UTR total PARS scores revealed that the two bins containing mRNAs with the highest scores, i.e. most structure-prone 5′-UTRs, had higher median ΔSSU_5′-UTR/ΔSSU_AUG ratios indicating scanning defects (Figure S12C, light gray boxes, bins 1–2, lower notches above red line), while the two bins of mRNAs with lowest scores had lower median ratios compared to all other mRNAs (Supplementary Figure S12C, light gray boxes, bins 6–7, upper notchs below red line). By contrast, PIC attachment defects, as indicated by reduced ΔSSU_5′-UTR/ΔRNA ratios in ded1-cs versus WT cells were not specifically associated with elevated 5′-UTR total PARS scores (Supplementary Figure S12C, dark gray boxes), possibly because only structures near the 5′-end interfere with PIC attachment. In any event, it appears that greater potential for secondary structure across 5′-UTRs is associated with scanning defects, leading to PIC accumulation in 5′-UTRs, when Ded1 activity is impaired.
DISCUSSION
In this study, we explored whether Ded1 and its paralog Dbp1 functionally cooperate to stimulate translation of yeast mRNAs in vivo. Consistent with this hypothesis, we found that DBP1 overexpression partially rescues the translation of a reporter mRNA harboring a strong SL structure in ded1-ts cells; and by ribosome footprint profiling, we showed that hcDBP1 mitigates the TE reductions conferred by ded1-ts throughout the translatome. We further demonstrated that dbp1Δ exacerbates the dissociation of bulk polysomes in ded1-ts cells; and showed that TE reductions conferred by ded1-ts were exacerbated by dbp1Δ for hundreds of mRNAs. These findings suggest that Dbp1 functions on these mRNAs in WT cells and that it masks the severity of Ded1 defects conferred by ded1-ts. Also supporting this view, the majority of ∼500 mRNAs whose TEs are strongly reduced in the ded1-ts dbp1Δ double mutant also show TE reductions of lesser magnitude in the dbp1Δ single mutant (Figure 1E).
For a group of >200 mRNAs for which strong (≥2-fold) TE reductions were conferred by ded1-ts only in the absence of Dbp1 in the ded1-ts dbp1Δ double mutant, dubbed ‘conditionally’ Ded1-dependent mRNAs, the median 5′-UTR length, propensity for 5′-UTR structure (based on PARS scores), and CDS length were all found to be greater than average, as observed previously for mRNAs strongly impaired by ded1-ts in DBP1+ cells. Thus, it appears that Dbp1 contributes with Ded1 to overcome these inhibitory features on these mRNAs. We came to similar conclusions for ‘conditionally’ Dbp1-dependent mRNAs, for which deleting DBP1 strongly reduced translation only when Ded1 was also impaired in the double mutant, as expected if Ded1 masks defects conferred by eliminating Dbp1. Again, because the median TE of these mRNAs is reduced in the dbp1Δ single mutant (Figure 2D), we conclude that Dbp1 and Ded1 cooperate on many of these mRNAs to diminish the inhibitory effects of long, structured 5′-UTRs and long transcript lengths, and that the activity of each helicase can mitigate the deleterious effects of impairing or removing the other paralog.
In vitro support for our conclusion that Ded1 and Dbp1 cooperate on many yeast mRNAs came from the reconstitution of Dbp1 function with purified components using an assay that measures the kinetics of 48S PIC assembly, including reactions from PIC attachment through scanning to the start codon. With this assay, we recently demonstrated that Ded1 confers a larger increase in maximum rate constant (kmax) of 48S assembly for mRNAs harboring the structured 5′-UTRs of Ded1-hyperdependent mRNAs compared to the less structured leaders of Ded1-hypodependent mRNAs. This difference in fold-stimulation by Ded1 was recapitulated for synthetic mRNAs containing or lacking SL insertions in an otherwise unstructured 5′-UTR (12). Here we demonstrated analogous results using purified Dbp1 for the Ded1 hyper- and hypodependent mRNAs SFT2 and RPL41A, respectively, and for the synthetic mRNAs containing or lacking a cap-distal SL insertion. Thus, Dbp1 largely mimics Ded1 function in stimulating 48S PIC assembly on a range of structured and unstructured mRNAs in vitro, consistent with our conclusion that these helicases functionally cooperate on many mRNAs in vivo.
Despite our extensive evidence for analogous functions of Ded1 and Dbp1, we identified a group of mRNAs that are highly dependent on Dbp1 in WT cells and minimally impaired by ded1-ts, even in cells lacking DBP1. This group of ‘uniquely Dbp1-dependent’ mRNAs differed from Ded1-hyperdependent mRNAs in showing greater than average structure within CDS and a less pronounced tendency for longer, structured 5′-UTRs and CDS lengths. It also included mRNAs with elevated eIF4E·eIF4G/PABP occupancies, thought to be optimized for closed-loop assembly, whereas such ‘SCL’ mRNAs are generally hypodependent on Ded1. The uniquely Dbp1-hyperdependent group also includes mRNAs enriched only for PABP association—a group that resembles SCL mRNAs in being highly translated in WT cells (45). Interestingly, these two groups of mRNAs are reported to have the fastest predicted initiation speeds (47), and consistent with this, the Dbp1-hyperdependent mRNAs have a greater than average median TE in WT cells.
To summarize, the uniquely Dbp1-dependent mRNAs tend to be largely independent of Ded1, highly translated with higher than average closed-loop potential or PABP occupancy, harbor moderately long, structured 5′-UTRs, and have greater than average secondary structures within CDS. By contrast, Ded1-hyperdependent mRNAs, including those in which Ded1 and Dbp1 cooperate, have relatively longer and more highly structured 5′-UTRs, longer CDS, lack enrichment for eIF4E·eIF4G and PABP, and do not contain unusually structured CDS.
The properties of the Ded1-hyperdependent mRNAs can be rationalized by proposing that Ded1 acts to resolve secondary structures in 5′-UTRs to facilitate PIC attachment or scanning, and that the requirement for Ded1 is intensified for long mRNAs with low propensity for closed-loop formation owing to their relative inability to maintain high occupancies of eIF4E·eIF4G needed to recruit and activate helicase eIF4A. As Ded1 is stimulated by eIF4A and eIF4G (13), the relatively low eIF4E·eIF4G occupancies of these mRNAs would also be expected to diminish activation of Ded1, and thus render these mRNAs more sensitive to reductions in Ded1 function conferred by the ded1-ts mutation. The uniquely Dbp1-hyperdependent mRNAs, by contrast, appear to more readily form the closed loop with attendant efficient recruitment of eIF4A and activation of Ded1, which together with their only moderately long, structured 5′-UTRs, confers a lower requirement for Ded1 to facilitate PIC attachment and scanning. While this might account for the modest effects of the ded1-ts mutation on the TEs of uniquely Dbp1-hyperdependent mRNAs, it would not explain their considerably stronger impairment by dbp1Δ. One possibility is that these mRNAs exhibit higher occupancies of Dbp1 versus Ded1. Alternatively, their greater propensity for structured CDS might require unwinding of structures formed between sequences in the 5′-UTR or 5′ end of the CDS and sequences further downstream in the CDS, for which Dbp1 might be better suited than Ded1 to unwind. The latter might reflect the more efficient helicase activity of Dbp1, or possibly its greater access to these structures within the folded mRNP. Given our finding that several Dbp1-hyperdependent mRNAs shifted to smaller polysomes in dbp1Δ versus WT cells, indicating reduced initiation, we do not favor the possibility that Dbp1 enhances translation elongation on its target mRNAs by resolving structures within CDS.
The broad role of Dbp1 in collaborating with Ded1 on long mRNAs with long, structured 5′-UTRs, and acting as the principle paralog on the Dbp1-hyperdependent mRNAs is surprising given that Dbp1 is expressed at a level ∼50-fold lower than Ded1 in WT cells. As enzymes however, they need not function as stoichiometric components of the PIC. Recent evidence suggests that Ded1 is effectively a stoichiometric subunit of eIF4F in yeast, functions as the principle catalytic component of the eIF4F/Ded1 complex, and is activated by both eIF4A and eIF4G in this complex (13). Perhaps Dbp1 is less dependent on association with eIF4F for helicase activity or access to secondary structures, and thus functions at much lower, substoichiometric levels compared to Ded1. Our biochemical analysis also revealed that Dbp1 and Ded1 achieve similar maximum rates of 48S PIC assembly on structured 5′-UTRs but at concentrations (K1/2 values) ∼2.5- to ∼5-fold lower for Dbp1 versus Ded1, indicating that Dbp1 is more efficient than Ded1. Finally, there is evidence that Ded1 also functions in mRNA export, mRNA splicing, and ribosome biogenesis (48), and might also function in mRNA granule formation during conditions of low translational activity (49). Thus, perhaps only the fraction of cytoplasmic Ded1 that interacts stoichiometrically with eIF4G actually functions in stimulating translation initiation, which (based on eIF4G and Dbp1 abundance) is only ∼10-fold in excess of Dbp1. Relative abundance of eIF4G and Dbp1 in cells was estimated from their RPF reads in WT strain (NSY10) at 37°C.
Applying the newly developed TCP-seq methodology for SSU profiling, which provides a snapshot of the occupancies of 43S PICs in 5′-UTRs and 48S PICs at start codons, we obtained direct evidence implicating Ded1/Dbp1 in scanning through 5′-UTRs, as mRNAs found to be hyperdependent on Ded1, Dbp1, or both, by 80S profiling tend to accumulate SSUs in 5′-UTRs relative to SSUs at the AUG codons of the same mRNAs, as expected for delayed progression of scanning PICs to the start codon. There is also a tendency for Ded1/Dbp1-hyperdependent mRNAs to exhibit reduced SSU occupancies in 5′-UTRs relative to mRNA levels, consistent with PIC attachment defects. On many Ded1/Dbp1-dependent mRNAs, the TCP-Seq data suggest dual defects in scanning and impaired PIC attachment (or reduced processivity) on the same mRNAs in helicase mutants. These findings provide strong evidence that the reduced translation of Ded1/Dbp1-hyperdependent mRNAs observed in mutants lacking these helicases results directly from defects in PIC attachment or scanning in a manner exacerbated by secondary structures in 5′-UTRs. It has been proposed that a delay in scanning conferred by reduced Ded1 or Dbp1 activity provokes increased translation of uORFs, which in turn attenuates scanning to the downstream CDS, as was demonstrated for a few Ded1-dependent mRNAs (14). However, in our view, impaired scanning through secondary structures may be adequate on its own to reduce translation of downstream CDS, without a coupled increase in translation of a nearby uORF, at many Ded1 hyperdependent genes in ded1 cells. Indeed, we have reconstituted here and elsewhere (12) the strong stimulatory effects of Ded1 and Dbp1 in accelerating 48S PIC assembly on Ded1-hyperdependent mRNAs in a purified system lacking 60S subunits where initiation at upstream uORFs cannot occur.
In conclusion, in this report we have shown that the Ded1-paralog Dbp1 functionally interacts with Ded1 throughout the yeast translatome in stimulating translation of mRNAs with long, structure-prone 5′-UTRs. For many such mRNAs, Dbp1 masks the involvement of Ded1, and Dbp1 overexpression can mitigate loss of Ded1 function, in stimulating translation. Interestingly, a sizable group of mRNAs with distinctive properties was found to be uniquely dependent on Dbp1 versus Ded1. Dbp1 function in accelerating preinitiation complex (PIC) assembly on mRNAs harboring 5′-UTR stem–loop structures was reconstituted in a purified system. Finally, genome-wide profiling of small (40S) ribosomal subunits provided direct evidence implicating Ded1 and Dbp1 in both PIC attachment and scanning to stimulate translation of many mRNAs in vivo.
DATA AVAILABILITY
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE 111255 and GSE 124863.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of our laboratories and those of Tom Dever and Jon Lorsch for many helpful suggestions and Sumit Sen for help with bioinformatics. We also acknowledge assistance from the Monash Bioinformatics Platform.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health; Australian Research Council Discovery Project grant [DP180100111 to T.P.]; National Health and Medical Research Council of Australia Senior Research Fellowship [APP1135928]. Funding for open access charge: Intramural Research Program of the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 2. Shirokikh N.E., Preiss T.. Translation initiation by cap-dependent ribosome recruitment: Recent insights and open questions. Wiley Interdiscip. Rev. RNA. 2018; 9:e1473. [DOI] [PubMed] [Google Scholar]
- 3. Sonenberg N., Hinnebusch A.G.. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011; 75:434–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pestova T.V., Kolupaeva V.G.. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002; 16:2906–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sen N.D., Zhou F., Ingolia N.T., Hinnebusch A.G.. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res. 2015; 25:1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yourik P., Aitken C.E., Zhou F., Gupta N., Hinnebusch A.G., Lorsch J.R.. Yeast eIF4A enhances recruitment of mRNAs regardless of their structural complexity. Elife. 2017; 6:e31476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajagopal V., Park E.H., Hinnebusch A.G., Lorsch J.R.. Specific domains in yeast translation initiation factor eIF4G strongly bias RNA unwinding activity of the eIF4F complex toward duplexes with 5′-overhangs. J. Biol. Chem. 2012; 287:20301–20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogers G.W. Jr, Richter N.J., Merrick W.C.. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem. 1999; 274:12236–12244. [DOI] [PubMed] [Google Scholar]
- 10. Sokabe M., Fraser C.S.. A helicase-independent activity of eIF4A in promoting mRNA recruitment to the human ribosome. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:6304–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar P., Hellen C.U., Pestova T.V.. Toward the mechanism of eIF4F-mediated ribosomal attachment to mammalian capped mRNAs. Genes Dev. 2016; 30:1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta N., Lorsch J.R., Hinnebusch A.G.. Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. Elife. 2018; 7:e38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao Z., Putnam A.A., Bowers H.A., Guenther U.P., Ye X., Kindsfather A., Hilliker A.K., Jankowsky E.. Coupling between the DEAD-box RNA helicases Ded1p and eIF4A. Elife. 2016; 5:e16408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guenther U.P., Weinberg D.E., Zubradt M.M., Tedeschi F.A., Stawicki B.N., Zagore L.L., Brar G.A., Licatalosi D.D., Bartel D.P., Weissman J.S et al.. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature. 2018; 559:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jamieson D.J., Beggs J.D.. A suppressor of yeast spp81/ded1 mutations encodes a very similar putative ATP-dependent RNA helicase. Mol. Microbiol. 1991; 5:805–812. [DOI] [PubMed] [Google Scholar]
- 16. Banroques J., Cordin O., Doere M., Linder P., Tanner N.K.. Analyses of the functional regions of DEAD-box RNA “helicases” with deletion and chimera constructs tested in vivo and in vitro. J. Mol. Biol. 2011; 413:451–472. [DOI] [PubMed] [Google Scholar]
- 17. Berthelot K., Muldoon M., Rajkowitsch L., Hughes J., McCarthy J.E.. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 2004; 51:987–1001. [DOI] [PubMed] [Google Scholar]
- 18. Longtine M.S., McKenzie A. III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R.. Additonal modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998; 14:953–961. [DOI] [PubMed] [Google Scholar]
- 19. Goldstein A.L., McCusker J.H.. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999; 15:1541–1553. [DOI] [PubMed] [Google Scholar]
- 20. Chiu W.L., Wagner S., Herrmannova A., Burela L., Zhang F., Saini A.K., Valasek L., Hinnebusch A.G.. The C-terminal region of eukaryotic translation initiation factor 3a (eIF3a) promotes mRNA recruitment, scanning, and, together with eIF3j and the eIF3b RNA recognition motif, selection of AUG start codons. Mol. Cell Biol. 2010; 30:4415–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valasek L., Szamecz B., Hinnebusch A.G., Nielsen K.H.. In vivo stabilization of preinitiation complexes by formaldehyde cross-linking. Methods Enzymol. 2007; 429:163–183. [DOI] [PubMed] [Google Scholar]
- 22. Acker M.G., Kolitz S.E., Mitchell S.F., Nanda J.S., Lorsch J.R.. Reconstitution of yeast translation initiation. Methods Enzymol. 2007; 430:111–145. [DOI] [PubMed] [Google Scholar]
- 23. Mitchell S.F., Walker S.E., Algire M.A., Park E.H., Hinnebusch A.G., Lorsch J.R.. the 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and block an alternative pathway. Mol. Cell. 2010; 39:950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker S.E., Fredrick K.. Preparation and evaluation of acylated tRNAs. Methods. 2008; 44:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Munoz A.M., Yourik P., Rajagopal V., Nanda J.S., Lorsch J.R., Walker S.E.. Active yeast ribosome preparation using monolithic anion exchange chromatography. RNA Biol. 2017; 14:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walker S.E., Zhou F., Mitchell S.F., Larson V.S., Valasek L., Hinnebusch A.G., Lorsch J.R.. Yeast eIF4B binds to the head of the 40S ribosomal subunit and promotes mRNA recruitment through its N-terminal and internal repeat domains. RNA. 2013; 19:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ingolia N.T. Ribosome footprint profiling of translation throughout the genome. Cell. 2016; 165:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T.. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016; 535:570–574. [DOI] [PubMed] [Google Scholar]
- 33. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shirokikh N.E., Archer S.K., Beilharz T.H., Powell D., Preiss T.. Translation complex profile sequencing to study the in vivo dynamics of mRNA-ribosome interactions during translation initiation, elongation and termination. Nat. Protoc. 2017; 12:697–731. [DOI] [PubMed] [Google Scholar]
- 35. Dobin A., Gingeras T.R.. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015; 51:doi:10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Z., Hesselberth J.R., Fields S.. Genome-wide identification of spliced introns using a tiling microarray. Genome Res. 2007; 17:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M.. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008; 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E.. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010; 467:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thompson M.K., Rojas-Duran M.F., Gangaramani P., Gilbert W.V.. The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs. Elife. 2016; 5:e11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sen N.D., Zhou F., Harris M.S., Ingolia N.T., Hinnebusch A.G.. eIF4B stimulates translation of long mRNAs with structured 5′ UTRs and low closed-loop potential but weak dependence on eIF4G. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amrani N., Ghosh S., Mangus D.A., Jacobson A.. Translation factors promote the formation of two states of the closed-loop mRNP. Nature. 2008; 453:1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D.. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weinberg D.E., Shah P., Eichhorn S.W., Hussmann J.A., Plotkin J.B., Bartel D.P.. Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep. 2016; 14:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Costello J., Castelli L.M., Rowe W., Kershaw C.J., Talavera D., Mohammad-Qureshi S.S., Sims P.F., Grant C.M., Pavitt G.D., Hubbard S.J et al.. Global mRNA selection mechanisms for translation initiation. Genome Biol. 2015; 16:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rouskin S., Zubradt M., Washietl S., Kellis M., Weissman J.S.. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014; 505:701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Costello J.L., Kershaw C.J., Castelli L.M., Talavera D., Rowe W., Sims P.F.G., Ashe M.P., Grant C.M., Hubbard S.J., Pavitt G.D.. Dynamic changes in eIF4F-mRNA interactions revealed by global analyses of environmental stress responses. Genome Biol. 2017; 18:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharma D., Jankowsky E.. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 2014; 49:343–360. [DOI] [PubMed] [Google Scholar]
- 49. Hilliker A., Gao Z., Jankowsky E., Parker R.. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell. 2011; 43:962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE 111255 and GSE 124863.