


Abstract
Alternative splicing is emerging as an oncogenic mechanism. In prostate cancer, generation of constitutively active forms of androgen receptor (AR) variants including AR-V7 plays an important role in progression of castration-resistant prostate cancer (CRPC). AR-V7 is generated by alternative splicing that results in inclusion of cryptic exon CE3 and translation of truncated AR protein that lacks the ligand binding domain. Whether AR-V7 can be a driver for CRPC remains controversial as the oncogenic mechanism of AR-V7 activation remains elusive. Here, we found that KDM4B promotes AR-V7 and identified a novel regulatory mechanism. KDM4B is phosphorylated by protein kinase A under conditions that promote castration-resistance, eliciting its binding to the splicing factor SF3B3. KDM4B binds RNA specifically near the 5′-CE3, upregulates the chromatin accessibility, and couples the spliceosome to the chromatin. Our data suggest that KDM4B can function as a signal responsive trans-acting splicing factor and scaffold that recruits and stabilizes the spliceosome near the alternative exon, thus promoting its inclusion. Genome-wide profiling of KDM4B-regulated genes also identified additional alternative splicing events implicated in tumorigenesis. Our study defines KDM4B-regulated alternative splicing as a pivotal mechanism for generating AR-V7 and a contributing factor for CRPC, providing insight for mechanistic targeting of CRPC.
INTRODUCTION
Alternative splicing is a process during which specific exons are selectively included or excluded during pre-mRNA splicing and is a mechanism for the human genome to generate sufficient proteomic diversity for the functional requirements of complex tissues. More than 90% of human genes are alternatively spliced (1,2). Aberrant RNA splicing due to changes in the RNA splicing machinery is emerging as an important determinant of oncogenesis, response to treatment, and drug resistance, thus representing an important vulnerability with potential to be exploited for therapeutic purposes (3). In prostate cancer (PCa) alternative splicing of androgen receptor (AR) can generate constitutively active forms of AR (AR-Vs) that lack ligand-binding domain and play important roles in castration resistance. Castration-resistant prostate cancer (CRPC) occurs when tumor becomes resistant to hormonal therapies including standard androgen deprivation therapy (ADT). Resistance has also been observed for two new FDA-approved drugs: enzalutamide, which inhibits androgen binding to AR, and abiraterone acetate, which inhibits androgen synthesis (4,5). Among AR-Vs, AR-V7 is the most commonly expressed in human tissues and the best characterized (6,7). AR-V7 is generated by alternative splicing using an alternative 3′-splice site (ss) next to the cryptic exon (CE3) rather than the 3′-ss next to the canonical exon C4, resulting in inclusion of CE3 and translation of a C-terminally truncated form of the AR protein (8). AR-V7 may also be generated due to gene rearrangement of the AR locus in 22Rv1 cells (9,10). It was shown previously that AR-V7 RNA splicing is a dynamic and reversible process and is closely associated with AR gene transcription. Under ADT conditions, recruitment of the spliceosome to the 3′-ss of CE3 is increased in CRPC cells (8), leading to increased AR-V7 production. However, the mechanism of enhanced recruitment of spliceosome at the alternatively spliced exons remains elusive.
KDM4B is a member of the Jumonji C (JmjC) KDM4 histone lysine demethylase (KDM) family (11). KDM4B demethylates H3K9me3 (12), a heterochromatin mark associated with a closed chromatin structure and inhibition of gene transcription. KDM4s are overexpressed in a variety of human cancers including PCa and are emerging as drug targets for cancer therapy (13–15). We found that KDM4B is overexpressed in metastatic CRPC and identified several novel demethylase inhibitors of KDM4 proteins that inhibited the growth of a variety of PCa cell lines (16). KDM4B acts as a co-activator of the transcription factor BMYB, promoting expression of genes involved in the cell cycle (16). Several JmjC histone lysine demethylases such as KDM4D and JMJD1A/KDM3A were known to bind RNA (17) and to promote AR-V7 expression (18), respectively. However, whether and how these KDMs regulates alternative splicing remain elusive. Here, we investigated the role of KDM4B in alternative splicing of AR. We show that KDM4B promotes AR-V7. Mechanistically, KDM4B is phosphorylated by protein kinase A (PKA) in response to conditions that promote castration-resistance, leading to its binding to the splicing factor SF3B3. KDM4B binds specific RNA-sequence near 5′-CE3, recruiting the spliceosome near the alternative exon and promoting the inclusion of nearby exon. Moreover, we found that KDM4B can upregulate the chromatin accessibility near CE3 by removing heterochromatin mark H3K9me3 and couples the spliceosome to the chromatin. Genome-wide analysis of KDM4B-mediated alternative splicing events indicates that KDM4B could regulate additional alternative splicing of genes involved in tumorigenesis, suggesting that KDM4B could be a cancer-specific alternative-splicing regulator that dictates oncogenic alternative splicing.
MATERIALS AND METHODS
Plasmids, AR-V7 mini-genes, cell culture, transfection, stable cell line and minigene reporter assays
KDM4B plasmids were described previously (16). AR-V7 minigene-A2 was obtained from Dr. Dong (8). AR-V7 minigenes A1 and A3 were constructed by subcloning PCR-amplified human AR genomic sequences into AR-V7 minigene-A2. Detailed information on construction of A1 and A3-minigenes were available upon request. siRNA duplexes were purchased from Sigma (Sigma-Aldrich). HEK293T and human PCa cell line LNCaP, VCaP, and 22Rv1 were purchased from ATCC (Manassas VA, USA). Cells were cultured in RPMI1640 (LNCaP) or DMEM (VCaP and 22Rv1) medium supplemented with FBS or charcoal-stripped FBS (CFBS) (Highclone, Logan, UT, USA) as indicated. Stable cell lines LNCaP-ctl and LNCaP-4B were established by infection of LNCaP cells with lentiviruses expressing vector and Flag-KDM4B, respectively, and subsequent selection with blasticidin. Plasmids and siRNA transfections were performed using lipofectamine 3000 and RNAimax (Invitrogen), respectively. Minigene reporter assays were performed in HEK293T cells following the protocol described in (8).
RNA isolation, quantitative real-time polymerase chain reaction (qRT-PCR), and RNA-seq
Total RNA was isolated using TRIzol reagent (Invitrogen) according to manufacturer and treated with Ambion DNase I before downstream applications. RNA (2 μg) was used to generate cDNA using Superscript III (Invitrogen). qRT-PCR was performed using SYBR Green (BioRad) with gene-specific primers. Data were normalized to an internal standard as indicated (ΔCT method). RNA-seqs of LNCaP-ctl and LNCaP-4B cells were performed in BGI Genomics (Shenzhen, Guangdong, China). Genomic data is deposited to GEO (GSE130620). Primer sequences are listed in Supplementary Table S3.
RNA immunoprecipitation (RIP), chromatin immunoprecipitation (ChIP) and chromatin RNA-immunoprecipitation (ChRIP)
RIP experiments were performed using total cell extracts and antibody against KDM4B or control IgG using RIP assay kit (MBL international, Woburn, MA, USA). RNAs associated with IgG- or KDM4B-immunoprecipitate were extracted using Trizol, treated with DNase I, converted to cDNA, and used for qRT-PCR. For ChIP and ChRIP, chromatin fractions were crosslinked with 1% paraformaldehyde, fragmented to ∼500 bp via sonication, and immunoprecipitated with anti-KDM4B antibody. Chromatin precipitates were split into two parts after reversing the cross-link. One part was treated with RNase and remaining DNA fragments were quantified by SYBR-based qPCR, normalized using the percent input method (Invitrogen) (ChIP-qPCR). The other part was extracted with Trizol for RNA, followed by DNase I treatment, cDNA synthesis and qPCR (ChRIP-qPCR).
Antibodies, immunoblotting, immunoprecipitation, immunofluorescence and proximity ligation assays
Antibodies against following proteins were used in this study: KDM4B (#8639), pCREB(#9198), HA (#3724), and hnRNAPA1 (#8443) (Cell Signaling); H3K9me3 (ab8898, Abcam); AR (PG21, Miliipore); AR-V7 (AG10008, Precision antibody); Flag (F3165, Sigma-Aldrich); AR (441, sc-7305), HA (sc-7392), SF3B3 (sc-398670), Tubulin (sc-5274) and GAPDH (sc-137179) (Santa Cruz). Immunoblotting, immunoprecipitation, and immunofluorescence were performed according to the standard protocol. Proximity ligation assays (19) were performed using Duolink-PLA kit according to manufacturer's protocol (Sigma-Aldrich).
RNA gel-shift assay
RNA oligonucleotide probes were synthesized by Sigma and end-labeled with T4 polynucleotide kinase and γ-32P-ATP (NEB). Binding of RNA probe with bacterially expressed recombinant KDM4B (1–350) were carried out in 30 μl volumes binding buffer (10 mM Trip pH 7.4, 1 mM MgCl, 20 mM KCl, 1 mM DTT, 5% glycerol) at 37°C for 30 min in the presence or absence of various concentration of KDM4 inhibitor B3. Samples were separated on 5% native polyacrylamide gels in 0.5xTBE at RT.
Immunoprecipitation and mass spectrometry (IP-MASS)
LNCaP-4B cell lysates were immunoprecipitated with anti-flag or IgG antibodies, treated with or without RNAse, washed, and eluted with 3xFlag peptide (Sigma-Aldrich). Eluted proteins were run on SDS-PAGE gel until they reached the separating gel. Protein gel pieces were reduced and alkylated with DTT (20 mM) and iodoacetamide (27.5 mM) and digested with a 0.1 μg/μl solution of trypsin in 50 mM triethylammonium bicarbonate (TEAB) overnight (Pierce). Following solid-phase extraction, the resulting peptides were reconstituted in 10 ul of 2% (v/v) acetonitrile (ACN) and 0.1% trifluoroacetic acid in water. 5 ul of this were injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography systems (Dionex). Samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo), and eluted with a gradient from 1–28% buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol and 0.1% formic acid in water. Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification and oxidation of Met was set as a variable modification. Phosphorylation of Ser, Thr and Tyr was also set as a variable modification for phosphorylation samples and the ptmRS node was used for phosphorylation site location. The false-discovery rate (FDR) cutoff was 1% for all peptides.
In vitro kinase assay and phosphor-mass spectrometry
Kinase assay was performed using 1 μg recombinant KDM4B protein (Active Motif) and 1 μl of active PKA (NEB, #P6000S) following manufacturer's protocol (NEB). Phosphorylated proteins were separated by Phos-Tag gel (Fujifilm Wako Chemicals). For mass spectrometry to determine the phosphorylation site, 4 μg recombinant KDM4B protein and 2 μl PKA were used. pKDM4B protein was cut from the SDS-PAGE gel and subjected for mass-spectrometry.
Targeted-deletion of KDM4B with CRISPR/CAS9
22Rv1 cells were electroporated with U6 target gRNA expression vector containing KDM4B-targeted gRNA (GCTGCAGCCATGGGGTCTCAGG) or lentiCRISP-GFP-v2 containing non-targeted control gRNA (CGCTTCCGCGGCCCGTTCA) along with plasmid expressing CAS9. Cells were FACS sorted in single cell to 96-well plates. Clonal cells were genotyped. Deletion or knockdown of KDM4B was confirmed by qPR-PCR and WB analysis. Multiple clonal controls were selected and showed no difference in growth rate.
Cell fractionation
Cells (2 × 107) were collected by centrifugation at 600g 5 min. Chromatin associated RNA and RNA in soluble nuclear fraction and cytoplasmic fraction were collected and extracted respectively according to the protocol (20).
Analysis of the SU2C dataset
All RNA-Seq data from the PRJNA283923 study (21) was downloaded as raw paired-end fastq data using dbGAP. Quality assessment of the raw sequencing reads was performed using NGS-QC-Toolkit (22). Sequencing reads with quality score under phred score <20 were discarded. Quantification of transcript-level expression of the samples was performed using the SALMON pipeline (23) against HG38 transcriptome reference downloaded from the Ensemble database.
Statistical analysis
All data are shown as mean ± SD or SEM as stated. Student's t test (two-tailed) was used to compare the difference between two groups. P < 0.05 was considered statistically significant.
RESULTS
KDM4B promotes AR-V7 expression
All four KDM4 family members (KDM4A-D) are reported to be overexpressed in PCa cells (13,14,16) and KDM4A has been shown to play a role in PCa initiation (16,24). We analyzed their expression in the TCGA and SU2C (25,26) databases derived from mainly primary tumors and mCRPC patients, respectively. Both KDM4A and KDM4B are upregulated in prostate tumors compared to non-cancerous tissues in a pairwise comparison in TGCA dataset (Supplementary Figure S1A). However, only KDM4B showed significantly increased expression in tumor specimens compared to normal tissues when all samples were used in comparison (Supplementary Figure S1B). The expressions of KDM4C and KDM4D remain similar in both tumor and normal tissues (Supplementary Figure S1A and S1B). Among the four KDM4 family members, only KDM4B expression was significantly correlated with that of AR in the SU2C dataset (Supplementary Figure S1C). Since AR hyperactivation remains the central mechanism associated with CRPC progression, these data suggest that KDM4B is the major KDM4 isoform involved in CRPC progression.
Since AR-V7 plays important role in CRPC (6,27), we tested whether KDM4B can promote AR-V7 expression. The LNCaP and VCaP cells are androgen-sensitive. AR-V7 is only detectable at mRNA level in LNCaP cells but is expressed at high protein level in VCaP cells. 22Rv1 cells are castration-resistant and express high levels of AR-V7. KDM4B expression is relatively low in LNCaP cells compared to that in VCaP and 22Rv2 cells (Supplementary Figure S1D). Overexpression (OE) of KDM4B in LNCaP cells preferentially promoted AR-V7 compared to that of AR (Figure 1A). KDM4B OE in C4-2 cells, an androgen-independent cell line derived from LNCaP, also preferentially promoted AR-V7 as well (Supplementary Figure S1E). Since vector-transfected control LNCaP cells express no/low levels of AR-V7 and high levels of AR, the relative increase of AR-V7 compared to that of AR in KDM4B OE LNCaP cells may be overestimated. To test the generality of our observation, we also overexpressed KDM4B in VCaP and 22Rv1 cells that have comparable AR-V7 and AR expression. KDM4B OE in both VCaP and 22rv1 cells also preferentially upregulated AR-V7 expression (Figure 1A). The demethylase activity of KDM4B is required as the demethylase-dead mutant KDM4BH188A(KDM4Bm) failed to activate AR-V7 (Figure 1A). The overall transcription of AR as measured by qRT-PCR with a primer pair encompassing exons 1 and 2 is also upregulated by KDM4B in LNCaP cells (Supplementary Figure S1F).
Figure 1.

KDM4B promotes AR-V7 expression. (A) Relative fold change of mRNAs of AR-V7 and AR in PCa cells transfected with KDM4B (4B) or mutant KDM4BH188A (4Bm) expression plasmid. mRNA was normalized against internal Calnexin and expressed relative to that of vector transfected cells (n = 3, mean ± SEM). (B) WB of indicated proteins in LNCaP-ctl, LNCaP-4B and VCaP cells. Antibody against N-terminal AR (SP107) was used to detect both AR and AR-V7. (C) WB of AR-V7 and AR in LNCaP-4B cells cultured in CFBS in the absence or presence of bicalutamide (bic, 10 μM) or enzalutamide (enz, 5 μM). (D) The mRNA and (E) protein levels of AR-V7 and AR in VCaP and 22Rv1 cells transfected with control (ctl) or KDM4B siRNAs. mRNA was normalized against internal Calnexin and expressed relative to that of control siRNA-transfected cells (n = 3, mean ± SEM). (F) WB of KDM4B, AR-V7 and AR in clonal 22Rv1 cells transfected with control (ctl) or KDM4B-targeted gRNA (cl4 and cl7). GAPDH was used as loading control. (G) WB of AR-V7 and AR in 22Rv1 cells treated with vehicle (veh) or KDM4 inhibitor B3. (H) Spearman's correlation coefficient analysis between mRNAs of KDM4B and AR-V7 from the SU2C database.
We established a stable LNCaP cell line with ectopically expressed Flag-KDM4B (LNCaP-4B). AR-V7 is un-detectable by Western blot (WB) in vector-transfected LNCaP-ctl cells but can be detected in LNCaP-4B cells (Figure 1B). AR-V7 is upregulated in PCa patients following ADT and further upregulated after abiraterone acetate or enzalutamide treatment (7). LNCaP-4B cells showed similar characteristics; treatment of LNCaP-4B cells with bicalutamide (bic) or enzalutamide (enz) resulted in the upregulation of AR-V7 (Figure 1C).
We knocked down (KD) KDM4B in high AR-V7 expressing 22Rv1 and VCaP cells using KDM4B specific siRNAs. KDM4B KD resulted in a significantly reduced level of AR-V7, but not AR, at both the mRNA and protein levels (Figure 1D-E) whereas KDM4A and KDM4C KD had no effect on AR-V7 expression (Supplementary Figure S1G). We also generated clonal 22Rv1 cells with KDM4B KD using CRISPR/CAS9 (22Rv1-cl4 and 22Rv1-cl7), which also downregulated AR-V7 without much effect on AR (Figure 1F). KDM4B specific shRNA also downregulated AR-V7 (Supplementary Figure S1H). Inhibition of KDM4B with KDM4 inhibitor B3 also downregulated AR-V7 expression significantly and to a less extend, AR expression as well (Figure 1G). The effect of KDM4B KD on the expression of AR-V7 is more prominent under hormone-deprived CFBS culture condition than FBS condition (Supplementary Figure S1I). KDM4B expression also significantly correlated with that of AR-V7 in both SU2C (Figure 1H) and TGCA datasets (Supplementary Figure S1J). JMJD1A/KDM3A was recently shown to promote AR-V7 expression (18). We analyzed its correlation with AR-V7 and AR in both TGCA and SU2C database. JMJD1A expression significantly correlated with that of AR (Supplementary Figure S1K) but minimally with that of AR-V7 in TGCA database (Supplementary Figure S1L). JMJD1A expression in SU2C database also failed to correlate with that of AR and AR-V7 (Supplementary Figure S1M and N), suggesting that KDM4B and JMJD1A likely regulate AR-V7 expression via different mechanisms.
KDM4B promotes PCa cell growth via AR-V7 under androgen-deprivation conditions
We next tested whether KDM4B can promote PCa cell growth via AR-V7. Overexpression of KDM4B in LNCaP cells promoted cell growth under both FBS and CFBS conditions (Figure 2A). To test whether upregulation of AR-V7 underlies the growth promoting effect of KDM4B on LNCaP cells, we knocked down AR-V7 (Supplementary Figure S2A) in KDM4B transfected LNCaP cells. AR-V7 KD did not influence the KDM4B-elicited LNCaP cell growth under FBS culture condition but exhibited a significant impact when cells were cultured in CFBS (Figure 2A), suggesting the growth-promoting effect of KDM4B under androgen-free condition is mediated through AR-V7 whereas in the presence of androgen KDM4B may promote PCa cell growth via the BMYB-dependent mechanism as shown previously (16). Conversely, KDM4B KD in 22Rv1 cells reduced cell growth under both CFBS and FBS culture conditions compared to non-targeting control-transfected cells (ctl) (Figure 2B). Re-expression of V5-tagged AR-V7 in KDM4B KD cells (Supplementary Figure S2B) rescued cell growth in CFBS but had only a mild effect under FBS condition (Figure 2B).
Figure 2.

KDM4B promotes PCa cell growth and drug-resistance. (A) Relative growth of LNCaP cells transfected with vector or KDM4B in the presence of control or AR-V7 siRNA. Equal number of cells from each experimental group was seeded and cells were counted 6 days after transfection, Cell growth was expressed relative to vec/siRNA transected cells (n = 6, mean ± SEM, *, P < 0.05). (B) Relative growth of ctl and KDM4B KD cells (cl4) transfected with AR-V7 siRNA or AR-V7 expression plasmid in FBS or CFBS (n = 5, mean ± SEM). *, P < 0.05. (C) Relative growth of LNCaP cells transfected with vector or KDM4B in the presence of control or AR-V7 siRNA (siAR-V7-1, siAR-V7-2), and various concentrations of enzalutamide (n = 6, mean ± SEM). (D) Relative cell growth of 22Rv1- ctl or KDM4B KD 22Rv1-cl4 cells with or without transfected AR-V7 plasmid in medium containing various concentrations of enzalutamide (n = 6, mean ± SEM).
AR-V7 has been shown to promote the resistance of PCa cells to anti-androgen drugs such as enzalutamide (6). Likewise, KDM4B OE also downregulated the sensitivity of LNCaP cells to enzalutamide inhibition (ca. 9-fold increase on IC50, Figure 2C) that can be partially reversed by AR-V7 KD. Conversely, KDM4B KD reduced enzalutamide-resistance of 22Rv1 that can be partially reversed by re-expression of AR-V7 (Figure 2D). AR-V9 is another AR-V often detected in CRPC and is also increased in KDM4B OE LNCaP cells (Supplementary Figure S2C) and decreased in KDM4B KD 22Rv1 cells (Supplementary Figure S2D), suggesting KDM4B-activated AR-V9 may contribute to drug-resistance as well.
KDM4B regulates AR-V7 alternative splicing
To understand the mechanism by which KDM4B promotes AR-V7, we performed RNA-immunoprecipitation assays (RIP) with anti-KDM4B antibody to identify potential KDM4B-binding regions. A strong KDM4B-binding peak was observed in the intron near 5′-CE3 (Figure 3A, peak I3f). We constructed a series of AR-V7 minigene reporters that contain CE3, constitutive exons 3 (C3) and 4 (C4), and various lengths of 5′ intron sequences near CE3 (Figure 3B). KDM4B preferentially activated AR-V7 over AR in the A1-minigene reporter in a dose-dependent manner (Figure 3C, lanes 1–3). Upregulation of AR-V7 relative to AR was downregulated in the A2-minigene reporter that lacks the I3f region (Figure 3C, lanes 4–6, 10-fold vs 3.5-fold), and was abolished in the A3-minigene reporter (Figure 3C, lanes 7–9). A2-minigene is also upregulated under baseline condition compared to that of A1-minigene (Figure 3C, lane 4 vs 1), suggesting that the intro in the A1-minigene may contain a negative cis-element. The mRNAs of reporter A1, A2 and A3 at baseline without KDM4B are similar as assayed by exon3-CE3 and exon3-exon4 primer pairs (Supplementary Figure S3A), suggesting that the deletion of the intronic sequences alone in A2 and A3 minigenes has no effect on the splicing activity of minigenes. We observed increased transcription of shorter minigene reporter (A3 > A2 > A1) (Supplementary Figure S3A). This is most likely due to more shorter-minigene is transfected on the molar-base since equal amounts of reporters on weight-base are transfected. This also implicates that the downregulation of AR-V7/AR in shorter minigene-transfected cells cannot be due to downregulation of its transcription. Based on this control experiment, we believe that the differential amounts of AR-V7/AR in various reporter-transfected cells along with KDM4B are due to KDM4B and not transcriptional level of the reporter.
Figure 3.

KDM4B promotes alternative splicing of AR-V7 as a trans-acting splicing factor. (A) Relative occupancy of KDM4B over the AR pre-mRNA. IgG was used as the negative control. n = 3, mean ± SD. (B) Schematics of AR-V7 minigenes with various intronic sequences 5′-upstream of CE3. (C) Minigene reporters (A1, A2 or A3) (1.5 μg) with indicated expression vectors (400 ng, 1×) were transfected into HEK293T cells. RNAs were extracted 48 h post-transfection. Expression levels of AR and AR-V7 were determined by qRT-PCR using primer pair p1/p3 and p1/p2, respectively. Ratios of AR-V7 over AR were expressed relative to the A1-minigene alone (n = 4, mean ± SD). (D) RNA-gel shift assay with KDM4B (1–350) and 32P-labeled RNA probes (WT-4BRBS, mut-4BRBS) in the absence or presence of B3. (E) Coomassie-blue stained gels with biotin–4BRBS bound proteins. Biotin-4BRBS was incubated with recombinant GST, KDM4B (1–350) or KDM4Bm (1–350) proteins. The RNA-protein complex was pulled down with streptavidin-beads, washed and separated by SDS-PAGE.
We tested whether KDM4B can bind RNA directly using electrophoretic mobility shift. A 40 bp RNA-oligomer (4BRBS) corresponding to the sequences around I3f region in A1 minigene was identified that binds KDM4B (1–350) (Figure 3D, lane 2). Mutation of nucleotides highly conserved between human and mouse (italicized) abolished binding (Figure 3D, lane 3). KDM4B binding to the 4BRBS was also inhibited by KDM4B inhibitor B3 (Figure 3D, lanes 4 and 5). Full-length KDM4B is required to activate AR-V7 as KDM4B (1–710) or KDM4B (710-C) failed to activate AR-V7 (Figure 3C, lanes 11 and 12). KDM4B also failed to activate a mutant A2-minigene where the splicing factor U2AF2-binding site ISE is mutated (Figure 3C, lane 10). As U2AF2 is required for spliceosome assembly (8), these data suggest that activation of AR-V7 by KDM4B requires the spliceosome. Consistently, pull-down assays with a biotin-labeled 4BRBS and 22Rv1 cell lysates revealed that KDM4B is in a complex with the core components of spliceosome SF3B1 and SF3B3 (Supplementary Figure S3B). Pull-down assays with biotin-labeled 4BRBS and recombinant KDM4B (1–350) and KDM4Bm (1–350) proteins also indicated that inactivation of demethylase activity of KDM4B also downregulated its ability to bind RNA 4BRBS (Figure 3E).
KDM4B binds to splicing factors in response to androgen deprivation
We performed immunoprecipitation with anti-Flag antibody in LNCaP-4B cells followed by mass spectrometry to identify potential binding partners of KDM4B. Approximately 125 proteins were identified that are >2-fold enriched in Flag-KDM4B-immunoprecipitates (Figure 4A, Supplementary Table S1). Gene ontology (GO) analysis indicated that >70% of them are involved in RNA binding and/or splicing (Figure 4B). RNase treatment of immunoprecipitates prior to mass spectrometry resulted in loss of association of KDM4B with some splicing factors such as SRSF1 and SFPQ, association of KDM4B with other splicing factor/RNA binding proteins (RBP), i.e. EWSR1, TRIM28, SF3B3 and hnRNPA1, remained (Figure 4A, Supplementary Table S2), and was confirmed by co-immunoprecipitation (co-IP) assays (Figure 4C). These data suggest that KDM4B may associate with splicing factors via binding to RNA and/or direct protein-protein interaction. Size-exclusion chromatography of 22Rv1 cell lysates revealed that KDM4B co-elutes with general splicing factors, including SF3B3 and hnRNPA1, in protein complexes with molecular weight ranging from >600 kD to ca. 130 kD (Supplementary Figure S4A). Further characterization of interactions between KDM4B and its binding partners indicated KDM4B can bind these proteins via different domains (Figure 4D). Co-IP assays also indicated that amino acid residues 340–710 of KDM4B and 525–1077 of SF3B3 are sufficient to interact with each other (Figure 4E) and demethylase activity is not required for KDM4B binding to SF3B3 (Figure 4F). Interaction between SF3B3 and KDM4B is specific as no binding of SF3B3 and KDM4A is observed in co-IP assays (Supplementary Figure S4B). SF3B3 is required for AR-V7 activation as SF3B3 KD downregulated AR-V7 expression (Figure 4G, Supplementary Figure S4C).
Figure 4.

KDM4B is associated with splicing factors. (A) Representative KDM4B-associated proteins in LNCaP-4B cells identified by Flag co-immunoprecipitation (IP) and mass spectrometry. Components of the spliceosome are highlighted in red, chromatin modifiers in green, and proteins involved in transcription in blue. (B) GO analysis of the KDM4B-interactive proteins. (C) WB of indicated proteins from IgG or Flag-immunoprecipitates of LNCaP-4B cells. (D) HEK293T cells were transfected with HA-KDM4B plasmid expressing different regions of KDM4B. Cell lysates were immunoprecipitated with anti-HA (rat) antibody and probed with antibodies indicated. (E) Co-IP assays of HEK293T cells transfected with various fragment of myc-SF3B3 and HA-KDM4B (340–710). (F) Co-IP assays of HEK293T cells transfected with HA-KDM4B (4B) or HA-KDM4Bm (4Bm) in the presence or absence of myc-SF3B3 (525–1077). (G) WB of indicated proteins from 22Rv1 cells transfected with control (ctl) or SF3B3 siRNAs.
We confirmed the endogenous interactions between KDM4B and SF3B3 in VCaP cells by proximity ligation assays (PLA) and found that binding of KDM4B to SF3B3 is dramatically increased under ADT conditions (CFBS + enzalutamide) (Figure 5A). ADT is known to activate PKA that promotes androgen-independent growth and neuroendocrine differentiation of PCa cells (28–33). PKA is among the KDM4B-interactive proteins identified by mass-spectrometry (Supplementary Table S2). Binding of KDM4B to SF3B3 is regulated by PKA as it is abolished in the presence of PKA specific inhibitor H89 (Figure 5B). H89 treatment also downregulated AR-V7 expression in cells cultured under ADT conditions (Figure 5C). We confirmed the interaction between PKA and KDM4B in Co-IP (Figure 5D) and kinase assays (Figure 5E), and identified several PKA phosphorylation sites including Ser666 on KDM4B by mass spectrometry (Supplementary Figure S5). Mutation of Ser666 to Ala resulted in inability of KDM4B to promote AR-V7 transcription (Figure 5F and G) and to bind SF3B3 (Figure 5H and I).
Figure 5.

KDM4B binds SF3B3 in response to androgen-deprivation. (A) In situ PLAs were performed using antibodies against KDM4B and SF3B3 in VCaP cells cultured in FBS or CFBS plus enzalutamide (enz. 10 μM). PLA signals (red), representing interactions between KDM4B and SF3B3, were significantly upregulated in androgen-deprivation conditions. Nuclei were stained with Dapi. (B) Cell lysates from VCaP cells cultured under FBS, CFBS, CFBS + H89 were immunoprecipitated with anti-KDM4B antibody and western blotted with antibodies indicated. H89 abolished the interaction between KDM4B and SF3B3. (C) WB of indicated proteins in VCaP cells cultured under the conditions indicated. H89 downregulated AR-V7 expression that was upregulated by androgen-deprivation. (D) HEK293T cells were transfected without or with HA-PKA. Cell lysates were immunoprecipitated with anti-HA antibody and western blotted with anti-HA and anti-KDM4B antibodies. (E) Recombinant KDM4B protein was incubated without or with catalytically active PKA and analyzed by Phos-Tag gel followed by WB with antibody indicated. (F) AR-V7 minigene reporter assay with cell transfected with KDM4B, KDM4Bm, or KDM4B(S666A) (n = 4, mean ± SD), *P < 0.05. (G) Relative mRNA of AR-V7 and AR in VCaP cells transfected with KDM4B or KDM4B(S666A). (H-I) HEK293T cells were transfected with myc-SF3B3 (525–1077) together with HA-KDM4B (H) or HA-KDM4B WT or S666A (I), treated with or without PKA inhibitor H89 (H). Lysates were immunoprecipitated with anti-HA antibody and western blotted with indicated antibodies.
KDM4B regulates AR-V7 pre-mRNA and alternative splicing at the chromatin
Co-IP and mass spectrometry also identified TRIM28 as a KDM4B-interactive protein (Figure 4A and C). TRIM28 is closely associated with heterochromatin rich in methylated H3K9 in the genome (34). TRIM28 can bind heterochromatin protein 1 (HP1), regulating RNApol II pausing. Given that KDM4B demethylase activity is required for promoting AR-V7 expression, we speculated that KDM4B might regulate AR-V7 alternative splicing at the chromatin. Assay for transposase-accessible chromatin (ATAC) indicated that chromatins in the I3f and CE3 region in KDM4B-overexpressed LNCaP cells are more open than those in KDM4Bm-transfected LNCaP cells (Figure 6A). KDM4B binds chromatins near 5′-CE3 that is associated with downregulation of H3K9me3 (Figure 5B, left and middle panels, respectively). Overexpression of KDM4B in LNCaP cells also significantly upregulated the occupancy of RNApolII near the CE3 locus (Figure 6B, right panel) whereas KDM4B KD downregulated the occupancy of RNApolII near the CE3 locus (Supplementary Figure S6A). Knockdown of KDM4B also significantly downregulated the occupancy of SF3B3 at the CE3 locus (Supplementary Figure S6B), suggesting the recruitment of spliceosome to the CE3 locus requires KDM4B.
Figure 6.
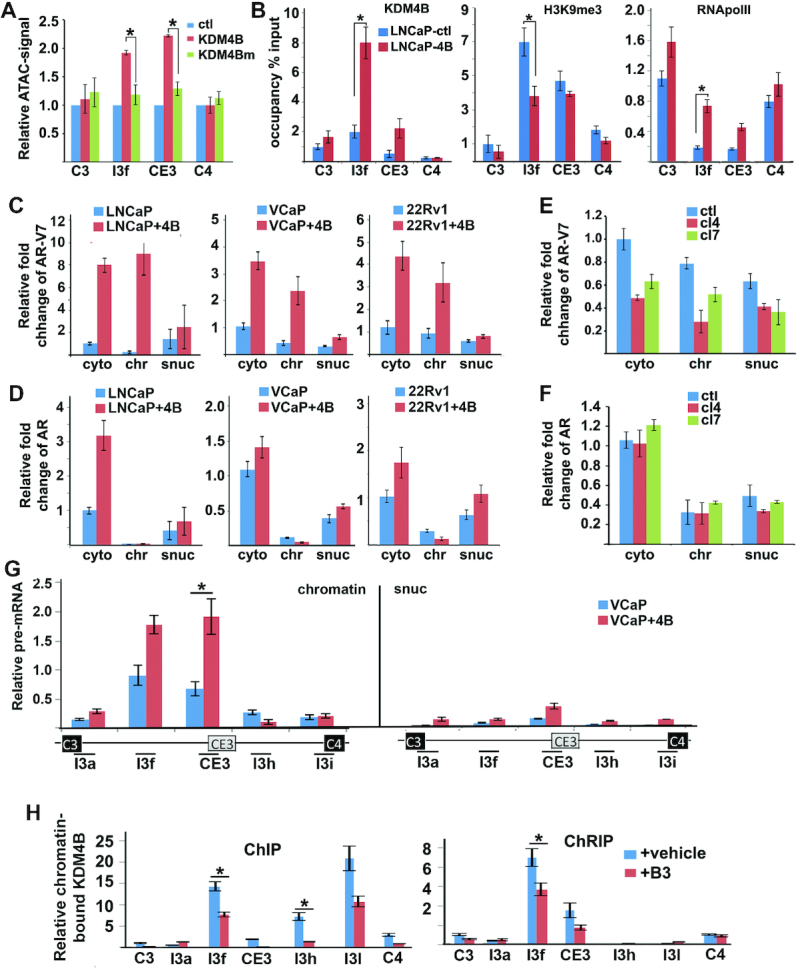
KDM4B regulates AR-V7 alternative splicing at chromatins. (A) ATAC-qPCR was used to assess open chromatin near CE3 locus in LNCaP cells transfected with control vector, KDM4B, or KDM4Bm. qPCR signals were expressed relative to vector transfected cells at each locus. (B) Chromatin occupancy of KDM4B, H3K9me3, and RNApolII around CE3 region in LNCaP-ctl and LNCaP-4B cells. (C–F) Relative cytosol (cyto), soluble nuclear (snuc), and chromatin-associated (chr) AR-V7 (C, D) and AR mRNAs (E, F) in LNCaP, VCaP, and 22Rv1 cells transfected with control vector or KDM4B (C, D), or 22Rv1-ctl or KDM4B KD cl4 and cl7 cells (E, F). RNAs were normalized against GAPDH and expressed again cytosol RNA in vector-transfected (C and D) or ctl cells (E and F). (G) Relative pre-mRNA near CE3 locus of AR in chromatin and soluble nuclear fractions of VCaP cells transfected with control vector or KDM4B. (H) Relative enrichment of KDM4B-bound chromatin DNA (left panel) and RNA (right panel) near the CE3 region in 22Rv1 cells treated with veh or B3. Values were expressed as % of input. qPCRs were performed using DNA from ChIP and cDNA from ChRIP cross the whole AR locus. Only representative peaks around CE3 are presented. (A–H) n = 3, mean ± SD, *P < 0.05.
We performed cell fractionation and isolated RNAs from cytosol (cyto), soluble nuclear (snuc), and chromatin fractions (chr) (Supplementary Figure S6C). AR-V7 mRNA in the chromatin fraction (chr-AR-V7) was preferentially upregulated in KDM4B OE PCa cells compared to chr-AR mRNA (Figure 6C versus D). Conversely, KDM4B KD in 22Rv1 cells downregulated chr-AR-V7 mRNA and had little effect on chr-AR mRNA (Figure 6E and F, respectively). We also estimated levels of pre-mRNA of AR near the CE3 region in VCaP cells transfected with or without KDM4B. A significant amount of AR-V7 pre-RNA, quantified by qRT-PCR with primers in the intron regions flanking CE3, were found in the chromatin fraction compared to that in the soluble nuclear fraction (Figure 6G), suggesting that the majority of alternative splicing of CE3 occurred at chromatin. Downregulation of chr-AR-V7 was also observed in KDM4B KD 22Rv1 cells compared to 22Rv1-ctl cells (Supplementary Figure. S6D). We split KDM4B-chromatin immunoprecipitates into two parts after reversing the cross-link for ChIP and chromatin RNA-immunoprecipitation (ChRIP). Strong KDM4B-ChRIP peaks (Figure 6H, right panel, I3f and CE3) were observed near the 5′ss of CE3 but not C4, suggesting that KDM4B preferentially regulates the usage of CE3. A KDM4B-ChIP peak was also observed, which coincides with the KDM4B-ChRIP peak at the I3f position (Figure 6H, left panel), suggesting a coupling of chromatin DNA and RNA by KDM4B. The KDM4B inhibitor B3 inhibited KDM4B binding to both the chromatin and chromatin-associated RNA (Figure 6H).
KDM4B regulates global alternative splicing of genes involved in prostatic neoplasms
We performed RNA-seq for LNCaP-ctl and LNCaP-4B cells to investigate whether KDM4B regulates alternative splicing globally. A total of 13,160 novel transcripts were identified from LNCaP-ctl and LNCaP-4B cells that included 3455 coding transcripts and 9705 non-coding transcripts. RNA-seq also identified 3282 novel isoforms and 173 novel genes. Using rMAT (35) we identified 5836 differentially spliced events that can be classified into five different types, including skipped/included exon (SE), alternative 5′splicing site (A5SS), alternative 3′splicing site (A3SS), mutually exclusive exons (MXE), and retained introns (RI) (Figure 7A and Supplementary Figure S7A). Despite the large number of differentially spliced events between LNCaP-ctl and LNCaP-4B cells, there were only 1018 genes whose expression was affected (239 upregulated/779 downregulated, >1.5-fold). Many of the genes have isoforms that were implicated in tumorigenesis, including KMT2C/MLL3. KMT2C is a member of the COMPASS family of histone H3 lysine 4 (H3K4) metyltransferases (36). Methylation of H3K4 is an open chromatin mark associated with active gene transcription. We identified four isoforms of KMT2C in the UCSD genome-wide database: three short ones (uc003wkx, uc003wky and uc003wkz) containing a cryptic exon ce between constitutive exons c44 and c45 and one long isoform (uc003wla) without the ce (Supplementary Figure S7B). qRT-PCR with primers in ce and flanking c44 and c45 of KMT2C confirmed that KDM4B promoted ce inclusion in KDM4B-overexpressing LNCaP cells (Figure 7B). KDM4B KD in 22Rv1 cells downregulated ce inclusion (Figure 7C). Analysis of the TCGA database revealed that isoform uc003wky is upregulated in tumors (Supplementary Figure S6C) and its expression correlated with that of KDM4B significantly (Figure 7D, Supplementary Figure S7D). X-tile analysis (37) indicated that higher KMT2C isoform uc003wky had poor prognosis in PCa (Figure 7E) whereas the expression level of uc003wla predicted no difference in recurrence-free rate (Figure 7F). These data suggested that KDM4B promotes generation of alternatively spliced oncogenic isoforms of KMT2C.
Figure 7.

KDM4B targets global alternative splicing. (A) RNA-seq analysis of LNCaP-ctl and LNCaP-4B cells revealed global differential alternatively spliced events between the two cell lines. (B, C) The differential alternative splicing target KMT2C was confirmed by qRT-PCR, showing that inclusion of cassette exon (ce) of KMT2C is upregulated in LNCaP-4B cells (B) and downregulated in KDM4B KD 22Rv1 cells (C). The ratios of KMT2C isoforms with ce (C44-ce, ce-C45) over isoforms without ce (c44-c45) or over calnexin (CANX) in LNCaP-4B cells are expressed relative to those in LNCaP-ctl (B) or control siRNA transfected (C) cells. The alternative splicing pattern of ce is illustrated below the qRT-PCR graph. n = 4, mean ± SD. (D) Spearman's correlation coefficient analysis between mRNAs of KDM4B and KMT2C-uc003wky isoform from the TCGA database. (E, F) X-tile plots of mRNA expression of KMT2C short (E) and long isoform (F). The optimum cut point (arrow head) was automatically selected by the X-tile program according to the highest χ2-value defined by Kaplan–Meier survival analysis and log-rank test. Coloration of the plot represents the strength of the association at each division, ranging from low (dark, black) to high (bright, red or green). Red represents inverse association between marker expression and survival, whereas green represents direct association between marker expression and survival. The expression data of E and F are from the TCGA database.
DISCUSSION
In this study, we identified a novel mechanism by which epigenetic regulator KDM4B regulates alternative splicing in PCa cells in response to androgen deprivation. We showed that KDM4B promotes AR-V7 expression in PCa cells by regulating the alternative splicing of AR. RNA splicing is regulated by splicing regulatory elements (SREs) and trans-acting factors that bind SREs. SREs consist of splice site signals and splice enhancers and repressors. Alternative exons are normally associated with weak splice site signals that render partial selection by core components of the splicing machinery. The function of SREs and trans-acting factors near the alternative exon is to change the relative amount of core splicing machinery between the alternative exon and the competing constitutive exons through recruitment, leading to exon inclusion or skipping (38). Our data suggest that KDM4B could act as a scaffold and trans-acting splicing factor in response to oncogenic signals in context of this threshold model (Figure 8): androgen-deprivation could activate PKA that phosphorylates KDM4B, eliciting its binding to SF3B3 (Figure 5), a component of U2 snRNP. KDM4B may bind RNA near the alternative exon (Figure 3A and D) and recruit spliceosome to the nearby cryptic exon, and/or bind the chromatin via TRIM28 (Figure 4C) and tether the splicing machinery to the chromatin. KDM4B may use its demethylase activity to remove the heterochromatin mark H3K9me3, opening up the chromatin to expose alternative exons (Figure 6A and B). As a result of this signaling-dependent interaction of KDM4B with spliceosome and binding of KDM4B to the chromatin and RNA near the alternative exon, KDM4B could stabilize the spliceosome near the alternative exon and promote its inclusion. It is noted that the clinical relevance of AR-V7 as a causative factor for CRPC remains controversial since AR-V7 and AR are co-expressed. AR-V7 maybe a by-product of the increased transcription of the AR gene and simply reflects a mechanism for rapid induction of AR-FL expression by ADT (21,39–41). The novel alternative splicing mechanism identified here suggests that AR-V7 can be generated by KDM4B under conditions that promote castration resistance rather than a non-specific read-through by-product, providing supportive evidence for AR-V7 as a potent driver of recurrent CRPC. JMJD1A was recently shown to promote AR-V7 (18). The mechanism by which JMJD1A regulates AR-V7 alternative splicing is not known. Analysis of JMJD1A expression with that of AR and AR-V7 in both TGCA and SU2C databases indicated that although JMJD1A expression correlated with that of AR significantly in primary tumors, it did not correlate with that of AR or AR-V7 in mCRPC (Supplementary Figure S1K–N), suggesting that JMJD1A-regulated AR-V7 splicing is likely a result of read-through of enhanced AR transcription in mCRPC and further supporting the active role of KDM4B in alternative splicing.
Figure 8.
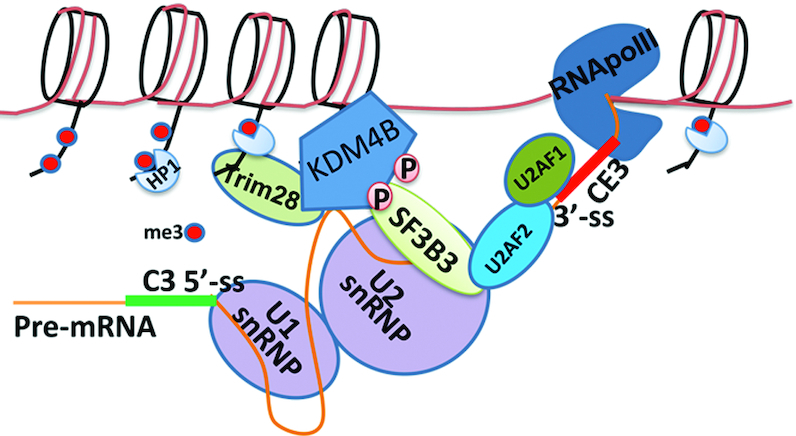
Mechanistic model showing how KDM4B might regulate alternative splicing of AR-V7 at chromatin. Androgen-deprivation promotes KDM4B phosphorylation by PKA, resulting its binding to splicing factor SF3B3. Consequently, KDM4B could recruit and stabilize the spliceosome near CE3 via binding to the chromatin and/or pre-mRNA near the alternative exon, promoting its inclusion.
Several histone lysine demethylases such as LSD1/KDM1A and KDM4D were also found to bind RNA (17,42). LSD1 binds lncRNA TERRA, which serves as a noncompetitive inhibitor of LSD1-catalyzed demethylation (42). KDM4D contains only the demethylase domain and lacks the C-terminal Tudor and PHD domains that are present in other KDM4 family members. KDM4D has been shown to bind RNA independently of its demethylase activity (17). We have shown that the RNA-binding domain of KDM4B overlaps with its demethylase domain. The structural determinants of KDM4B RNA-binding and catalytic activity remain to be determined and is a worthy avenue to be explored in the future.
We noted that even though KDM4B protein levels in LNCaP-4B cells is similar to that of VCaP cells, the upregulated AR-V7 expression in LNCaP-4B is still significantly lower than that of VCaP cells (Figure 1B). 22Rv1 and VCaP cells express high levels of AR-V7, which was attributed to AR gene rearrangement in 22Rv1 cells (10) and amplification in VCaP cells (43). Although the model we proposed in Figure 8 does not capture the aforementioned complexity of the AR gene, these mechanisms are not mutually exclusive since the chromatin (I3f) and cis-element (4BRBS) regulated by KDM4B are also present in 22Rv1 and VCaP cells albeit them being duplicated or amplified. Enhanced usage of CE3 by KDM4B during RNA splicing in 22rv1 and VCaP cells could add additional layer of AR-V7 regulation in these cells.
Although our study here focused on AR-V7, KDM4B likely has other genome-wide alternative splicing targets as revealed by RNA-seq (Figure 7). Since some of these targets such as KMT2C are implicated in tumorigenesis, they may mediate the oncogenic function of KDM4B as well. Interestingly, in addition to A3SS, by which KDM4B promotes CE3 inclusion, we also identified other KDM4B-regulated alternative splicing events including exon inclusion/skipping (SE) and mutually exclusive exon (MXE). How KDM4B carries out these alternative-splicing mechanisms and what determines the specificity of KDM4B remain to be determined. KDM4B is a large protein that contains multiple protein-interactive domains including PHD and Tudor domains. KDM4B interacts with a number of general splicing factors and RBPs via different regions (Figure 4D), which are likely to be part of KDM4B-regulated alternative splicing mechanisms. High KDM4B chromatin-occupancy and H3K9me3 peaks around alternative exons as in the case of CE3 may also be a determinant for the specificity of KDM4B. A large number of differential alternatively spliced events were identified between LNCaP-ctl and LNCaP-4B cells. Many of them may not be the direct targets of KDM4B. Overexpression of KDM4B likely results in changes of chromatin environment and gene expression of general splicing regulators, which in turn can change the splicing pattern. Nevertheless, given the tumorigenic function of KDM4B, KDM4B likely targets alternative splicing events that are specifically involved in tumorigenesis.
In summary, our data provide the first evidence of a direct crosstalk between the chromatin-modifying enzyme KDM4B and the spliceosome, suggesting a novel mechanism of epigenetic regulation of alternative splicing. Given the role of KDM4B and AR-V7 in promoting CPRC, the mechanistic link between them will provide new insight into future therapeutic strategy against CRPC.
DATA AVAILABILITY
Genomic data has been deposited in GEO(GSE130620).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Min Kim at Bioinformatics core of UT Southwestern for the data analysis using SU2C dataset and proteomics core of UT Southwestern for the mass spectrometry.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81725016 and 81872094 to J.H.L., in part]; National Institute of Health (NIH), Cancer Prevention and Research Institute of Texas (CPRIT) and Department of Defense (DOD) [RO1HL109471, RP120717-AC, RP120717-P1, PC150152 to Z.P.L.]; CPRIT and DOD [RP120717-AC, RP120717-C, PC150152P2 to J.T.H.]; NIH [R01CA215063 to Z.P.L., J.T.H., J.-M.A.]; CPRIT [RP120717-P3 to J.R., RP120717-P4 to J.-M.A., RP150596 to Bioinformatics Core Facility at UT Southwestern Medical Center]; Welch Foundation [AT-1595 to J.-M.A., I-1304 to J.R.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S. et al.. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012; 150:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J.. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008; 40:1413–1415. [DOI] [PubMed] [Google Scholar]
- 3. Sveen A., Kilpinen S., Ruusulehto A., Lothe R.A., Skotheim R.I.. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016; 35:2413–2427. [DOI] [PubMed] [Google Scholar]
- 4. Li Y., Chan S.C., Brand L.J., Hwang T.H., Silverstein K.A., Dehm S.M.. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013; 73:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu C., Armstrong C., Zhu Y., Lou W., Gao A.C.. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. 2016; 7:32210–32220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo J., Attard G., Balk S.P., Bevan C., Burnstein K., Cato L., Cherkasov A., De Bono J.S., Dong Y., Gao A.C. et al.. Role of androgen receptor variants in prostate cancer: Report from the 2017 mission androgen receptor variants meeting. Eur. Urol. 2017; 73:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharp A., Coleman I., Yuan W., Sprenger C., Dolling D., Rodrigues D.N., Russo J.W., Figueiredo I., Bertan C., Seed G. et al.. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest. 2019; 129:192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu L.L., Xie N., Sun S., Plymate S., Mostaghel E., Dong X.. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014; 33:3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y., Alsagabi M., Fan D., Bova G.S., Tewfik A.H., Dehm S.M.. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011; 71:2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brand L.J., Dehm S.M.. Androgen receptor gene rearrangements: new perspectives on prostate cancer progression. Curr. Drug Targets. 2013; 14:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shin S., Janknecht R.. Diversity within the JMJD2 histone demethylase family. Biochem. Biophys. Res. Commun. 2007; 353:973–977. [DOI] [PubMed] [Google Scholar]
- 12. Fodor B.D., Kubicek S., Yonezawa M., O'Sullivan R.J., Sengupta R., Perez-Burgos L., Opravil S., Mechtler K., Schotta G., Jenuwein T.. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006; 20:1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Young L.C., Hendzel M.J.. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem. Cell Biol. 2013; 91:369–377. [DOI] [PubMed] [Google Scholar]
- 14. Berry W.L., Janknecht R.. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013; 73:2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chin Y.W., Han S.Y.. KDM4 histone demethylase inhibitors for anti-cancer agents: a patent review. Expert Opin. Ther. Pat. 2015; 25:135–144. [DOI] [PubMed] [Google Scholar]
- 16. Duan L., Rai G., Roggero C., Zhang Q.J., Wei Q., Ma S.H., Zhou Y., Santoyo J., Martinez E.D., Xiao G. et al.. KDM4/JMJD2 Histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-Regulated genes. Chem. Biol. 2015; 22:1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zoabi M., Nadar-Ponniah P.T., Khoury-Haddad H., Usaj M., Budowski-Tal I., Haran T., Henn A., Mandel-Gutfreund Y., Ayoub N.. RNA-dependent chromatin localization of KDM4D lysine demethylase promotes H3K9me3 demethylation. Nucleic Acids Res. 2014; 42:13026–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fan L., Zhang F., Xu S., Cui X., Hussain A., Fazli L., Gleave M., Dong X., Qi J.. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E4584–E4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alam M.S. Proximity Ligation Assay. Curr. Protoc. Immunol. 2018; 123:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wuarin J., Schibler U.. Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell Biol. 1994; 14:7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G. et al.. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patel R.K., Jain M.. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012; 7:e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim T.D., Jin F., Shin S., Oh S., Lightfoot S.A., Grande J.P., Johnson A.J., van Deursen J.M., Wren J.D., Janknecht R.. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J. Clin. Invest. 2016; 126:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aksoy B.A., Gao J., Dresdner G., Wang W., Root A., Jing X., Cerami E., Sander C.. PiHelper: an open source framework for drug-target and antibody-target data. Bioinformatics. 2013; 29:2071–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho Y., Dehm S.M.. Androgen receptor rearrangement and splicing variants in resistance to endocrine therapies in prostate cancer. Endocrinology. 2017; 158:1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deeble P.D., Cox M.E., Frierson H.F. Jr, Sikes R.A., Palmer J.B., Davidson R.J., Casarez E.V., Amorino G.P., Parsons S.J.. Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 2007; 67:3663–3672. [DOI] [PubMed] [Google Scholar]
- 29. Jones S.E., Palmer T.M.. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal. 2012; 24:1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarwar M., Sandberg S., Abrahamsson P.A., Persson J.L.. Protein kinase A (PKA) pathway is functionally linked to androgen receptor (AR) in the progression of prostate cancer. Urol. Oncol. 2014; 32:25. [DOI] [PubMed] [Google Scholar]
- 31. Merkle D., Hoffmann R.. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: cross-talk with the androgen receptor. Cell Signal. 2011; 23:507–515. [DOI] [PubMed] [Google Scholar]
- 32. Sang M., Hulsurkar M., Zhang X., Song H., Zheng D., Zhang Y., Li M., Xu J., Zhang S., Ittmann M. et al.. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget. 2016; 7:45171–45185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y., Zheng D., Zhou T., Song H., Hulsurkar M., Su N., Liu Y., Wang Z., Shao L., Ittmann M. et al.. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018; 9:4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bunch H., Calderwood S.K.. TRIM28 as a novel transcriptional elongation factor. BMC Mol. Biol. 2015; 16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y.. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sze C.C., Shilatifard A.. MLL3/MLL4/COMPASS Family on epigenetic regulation of enhancer function and cancer. Cold Spring Harb. Perspect. Med. 2016; 6:a026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Camp R.L., Dolled-Filhart M., Rimm D.L.. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin. Cancer Res. 2004; 10:7252–7259. [DOI] [PubMed] [Google Scholar]
- 38. Fu X.D., Ares M. Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014; 15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henzler C., Li Y., Yang R., McBride T., Ho Y., Sprenger C., Liu G., Coleman I., Lakely B., Li R. et al.. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 2016; 7:13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu R., Lu C., Mostaghel E.A., Yegnasubramanian S., Gurel M., Tannahill C., Edwards J., Isaacs W.B., Nelson P.S., Bluemn E. et al.. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012; 72:3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watson P.A., Chen Y.F., Balbas M.D., Wongvipat J., Socci N.D., Viale A., Kim K., Sawyers C.L.. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirschi A., Martin W.J., Luka Z., Loukachevitch L.V., Reiter N.J.. G-quadruplex RNA binding and recognition by the lysine-specific histone demethylase-1 enzyme. RNA. 2016; 22:1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Makkonen H., Kauhanen M., Jaaskelainen T., Palvimo J.J.. Androgen receptor amplification is reflected in the transcriptional responses of Vertebral-Cancer of the Prostate cells. Mol. Cell Endocrinol. 2011; 331:57–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data has been deposited in GEO(GSE130620).