

Abstract
A highly regulated endoneurial microenvironment is required for normal axonal function in peripheral nerves and nerve roots, which structurally consist of an outer collagenous epineurium, inner perineurium consisting of multiple concentric layers of specialized epithelioid myofibroblasts that surround the innermost endoneurium, which consists of myelinated and unmyelinated axons embedded in a looser mesh of collagen fibers. Endoneurial homeostasis is achieved by tight junction-forming endoneurial microvessels that control ion, solute, water, nutrient, macromolecule and leukocyte influx and efflux between the bloodstream and endoneurium, and the innermost layers of the perineurium that control interstitial fluid component flux between the freely permeable epineurium and endoneurium. Strictly speaking, endoneurial microvascular endothelium should be considered the blood-nerve barrier (BNB) due to direct communication with circulating blood. The mammalian BNB is considered the second most restrictive vascular system after the blood-brain barrier (BBB) based on classic in situ permeability studies. Structural alterations in endoneurial microvessels or interactions with hematogenous leukocytes have been described in several human peripheral neuropathies; however major advances in BNB biology in health and disease have been limited over the past 50 years. Guided by transcriptome and proteome studies of normal and pathologic human peripheral nerves, purified primary and immortalized human endoneurial endothelial cells that form the BNB and leukocytes from patients with well-characterized peripheral neuropathies, validated by in situ or ex vivo protein expression studies, data are emerging on the molecular and functional characteristics of the human BNB in health and in specific peripheral neuropathies, as well as chronic neuropathic pain. These early advancements have the potential to not only increase our understanding of how the BNB works and adapts or fails to adapt to varying insult, but provide insights relevant to pathogenic leukocyte trafficking, with translational potential and specific therapeutic application for chronic peripheral neuropathies and neuropathic pain.
Keywords: Blood-Nerve Barrier, Chronic Neuropathic Pain, Endoneurium, Immune system, Inflammation, Junctional Complex, Leukocyte Trafficking, Peripheral Neuropathy
Human peripheral nerve anatomy
Human peripheral nerve and nerve roots serve to facilitate afferent and efferent communication between the central nervous system (brain and spinal cord) and the periphery (internal and external organs, such as the gastrointestinal tract, skin, heart, secretory organs and muscle) as required for normal physiological processes required for healthy body function. In order to understand how the human BNB works, it is imperative to understand peripheral nerve and nerve root structure and their vascular supply. Human peripheral nerves and nerve roots comprise of three structural compartments: the outer epineurium which consists of longitudinal arrays of collagen fibers that are important for maintaining structural integrity, the inner perineurium which consists of concentric layers of specialized cells and the innermost endoneurium which consists of a looser mesh of collagen fibers. A nerve fascicle consists of the endoneurium and its surrounding perineurium, initially described in 1876 (Figures 1A–B).(Mizisin and Weerasuriya, 2011; Olsson, 1990; Reina et al., 2000; Reina et al., 2003)
Figure 1. Peripheral nerve anatomy, including vascular supply.
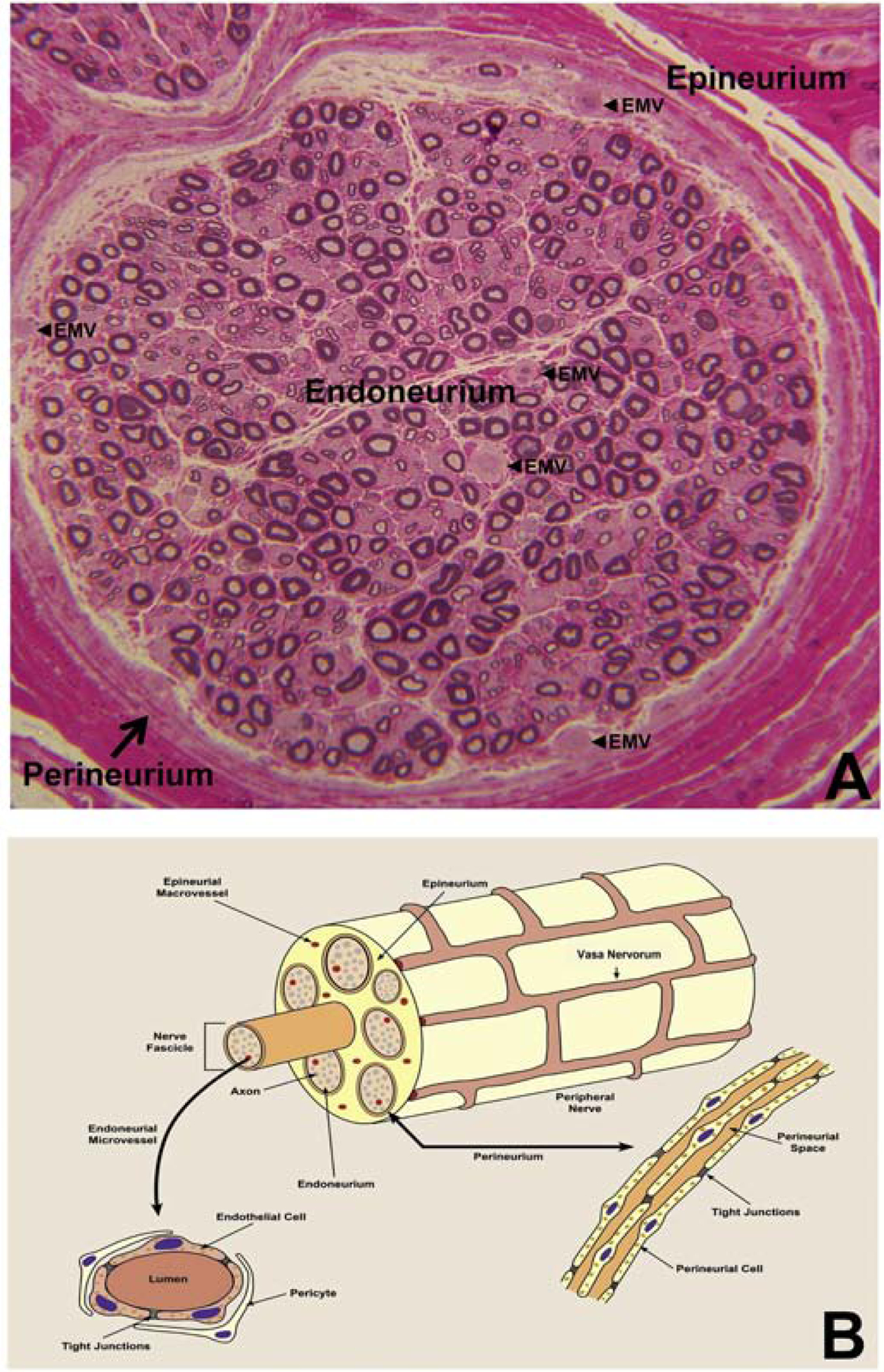
Digital light photomicrograph of an axial section of a normal adult sural nerve (plastic embedded semi-thin axial section stained with Toluidine Blue and counterstained with Basic Fuchsin) showing the three compartments in peripheral nerves, and endoneurial microvessels (EMV, black arrowheads) that form the BNB (A). This structural organization is further illustrated in (B), with individual nerves supplied by extrinsic vessels, called vasa nervorum, which form a vascular anastomoses and penetrate into the epineurium, resulting epineurial macrovessels. These macrovessels cross the perineurium, forming endoneurial microvessels. The anatomical organization of endoneurial microvessels and the perineurium is further illustrated, with restrictive tight junctions between the endothelial cells and innermost perineurial myofibroblasts respectively.
The epineurium consists of arteries, arterioles, venules and veins that are considered collectively as epineurial macrovessels. These macrovessels are derived from and communicate with the extrinsic vascular supply to individual peripheral nerves and nerve roots, and are referred to as the vasa nervorum. Lymphatic vessels are also present within the epineurium. Concentric layers of single-called epithelioid myofibroblasts form the perineurium (1–15 layers dependent on nerve diameter) that surround the endoneurium and form structures called fascicles. The perineurium houses smaller diameter blood vessels that transverse from the epineurium to the endoneurium and vice versa. The endoneurium predominantly consists of myelinated and unmyelinated axons, dependent on axonal size and function, which are aligned in the longitudinal axis of peripheral nerves and nerve roots, and segmentally supported by Schwann cells, the most prevalent cell type in peripheral nerves.(Bell and Weddell, 1984; Mizisin and Weerasuriya, 2011; Olsson, 1990; Reina et al., 2000; Reina et al., 2003) The endoneurium also consists of capillary-like microvessels that lack smooth muscle walls (Figure 1A–B), as well as rare resident leukocytes (macrophages and mast cells) and fibroblasts.(Bell and Weddell, 1984; Mizisin and Weerasuriya, 2011; Olsson, 1990; Reina et al., 2000; Reina et al., 2003)
The sciatic nerve is the largest nerve in mammals, comprising of 50–80 fascicles in adult humans in the mid-thigh region, and as many as 140 fascicles in the gluteal region (Reina et al., 2000; Sladjana et al., 2008; Yosef et al., 2010) and 1–4 fascicles in adult mice and rats.(Christensen and Tresco, 2015; Tanaka and Webster, 1991; Ubogu et al., 2012; Xia et al., 2010a, b; Yuan et al., 2014) The commonly studied human sural nerve, a pure sensory nerve that is one of the terminal branches of the sciatic nerve, typically consists of 8–10 fascicles in adults.(Ochoa and Mair, 1969a, b) It is important to recognize the rodent sciatic nerve typically consists of a thin epineurial layer with loose connective tissue,(Ubogu et al., 2012; Xia et al., 2010a, b; Yuan et al., 2014) in contrast with the more extensive and fibrous human epineurium. This significant structural difference between human and rodent peripheral nerves is important when extrapolating rodent in vivo or in situ experimental observations to human peripheral nerves, particularly with reference to nerve injury and local drug delivery (e.g. anesthetics and analgesics).
Identification and definition of the blood-nerve barrier
The importance of maintaining a highly regulated ionic microenvironment to facilitate peripheral nerve axonal impulse conduction was intuitive and led to the proposal of a BNB akin to the BBB. In vivo permeability studies performed in different animal species following intravenous administration of Evans blue albumin or fluoresceinated albumin or dextran or other macromolecules and nonelectrolytes demonstrated restriction within endoneurial microvessel lumens without extravasation into the endoneurium despite diffuse entry into the epineurium (which was in contrast with the diffuse lack of brain parenchymal entry), or low permeability coefficients, implying that restrictive interfaces exist within peripheral nerve and nerve root endoneurium. (Hultstrom et al., 1983; Olsson, 1966, 1968, 1971; Poduslo et al., 1994; Rechthand et al., 1987)
Subsequent ultrastructural assessment of human peripheral nerves showed that the impermeable endoneurial microvessels consisted of electron dense tight junction-forming endothelial cells that shared their basement membrane with surrounding cells called pericytes (that do not form intercellular contacts), lack fenestrations and possess very few 50–100 nm pinocytic vesicles (Figure 2A). This was in contrast with permeable epineurial macrovessels that contained a layer of endothelial cells that possessed fenestrations, and consisted of tunica media (smooth muscle layer) and tunica adventita (connective tissue layer) with varying thickness. Furthermore, the innermost concentric perineurial cells (i.e. closest to the endoneurium) were similarly connected by intercellular tight junctions, lacked fenestrations and possessed pinocytic vesicles (with higher density in the outermost layers). Thus, the internal microenvironment of the endoneurium was structurally deemed to be regulated by tight junction-forming endoneurial microvascular endothelial cells and the innermost perineurial epithelioid myofibroblast cells.(Bell and Weddell, 1984; Reina et al., 2000; Reina et al., 2003)
Figure 2. Endoneurial endothelial cell ultrastructure.
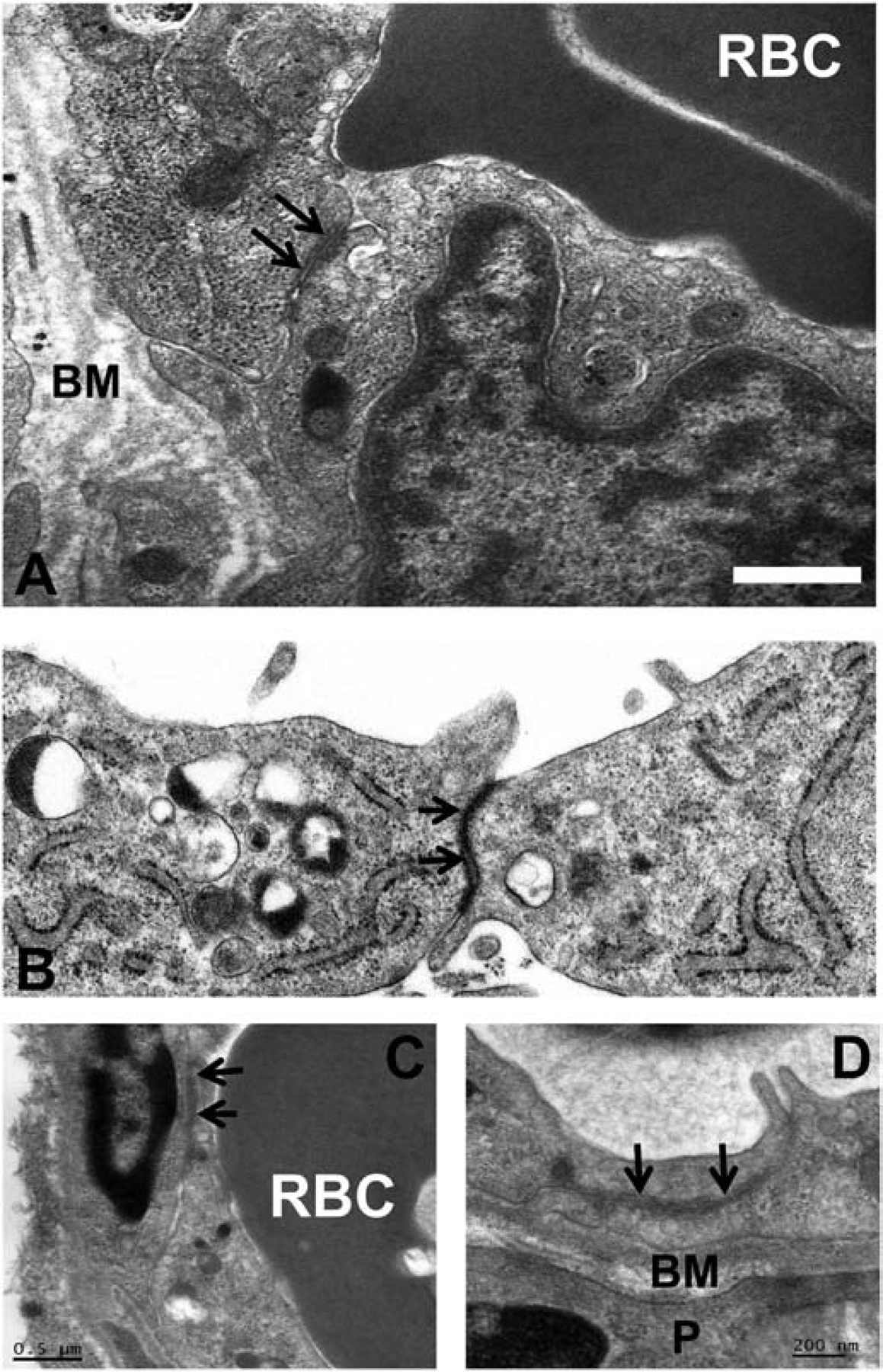
Digital electron ultramicrographs from an adult sural nerve (A) and cultured semipermeable rat-tail collagen coated Transwell™ inserts (B) shows human endoneurial endothelial cells with apically located electron-dense intercellular tight junctions (black arrows). Similar tight junctions are seen between endothelial cells in adult mouse sciatic nerve endoneurial microvessels (C and D). A red blood cell (RBC) is present in the lumen of the human and mouse endoneurial microvessels (A and C). BM indicates the basement membrane, shared between endoneurial endothelial cells and pericytes (P). Scale bar = 0.5 μm (A), or as indicated in figure (C, D).
Endoneurial endothelial cells are in direct contact with circulating blood (consisting of hematogenous leukocytes, nutrients, hormones and xenobiotics, to mention a few) while perineurial cells are in contact with epineurial and endoneurial interstitial fluid. Both cell types are necessary to maintain peripheral nerve homeostasis, as components of circulating blood and epineurial interstitial fluid derived from fenestrated epineurial macrovessels should not freely diffuse into the highly regulated endoneurium. Strictly speaking, however, endoneurial endothelial cells form the BNB while perineurial cells form a critical interface between the endoneurial and epineurial interstitial fluid compartments.
It is important to differentiate between endoneurial microvascular endothelial and perineurial cells in order to understand their unique characteristics and identify similarities relevant to endoneurial homeostasis influenced by their unique microenvironments. Since cellular cross-talk between the systemic immune system and peripheral nerves largely depends on naïve, recently activated or chronically activated circulating hematogenous leukocytes, it is important to understand the structural, molecular and functional characteristics of the human BNB in health in order to identify biologically relevant alterations and signaling perturbations that may occur in patients with peripheral neuropathies, as well as chronic neuropathic pain.
Molecular characterization of the human BNB in health
Basic knowledge of the structural, molecular and functional characteristics of the human BNB in health and disease is emerging, guided by data from the human BBB and studies performed on peripheral nerve biopsies in situ and primary and immortalized human endoneurial endothelial cells in vitro; however, our knowledge is far from complete. Structurally, human endoneurial endothelial cells that form the BNB possess electron-dense intercellular tight junctions in situ and in vitro (Figures 2A–B).(Reina et al., 2003; Yosef et al., 2010) In vitro, these tight intercellular membrane contacts consist of occludin, members of the claudin family such as claudin-5 as well as cytoplasmic adaptor molecules (i.e. molecules that link the cytoskeleton to cell membranes) such as members of the zona occludens (ZO) family such as ZO-1 and ZO-2 (also known as tight junction proteins 1 and 2 respectively), based on immunocytochemistry of confluent cultures,(Shimizu et al., 2011; Shimizu et al., 2012; Ubogu, 2013b; Yosef et al., 2010) while claudin-5 and ZO-1 had been previously demonstrated on human endoneurial microvessels in situ.(Kanda et al., 2004; Palladino et al., 2017; Pummi et al., 2004) Data has emerged over the past 15 years of the importance of the intercellular junctional complex, consisting of tight, adherens and gap junctions and their associated adaptor proteins and interacting cytoskeletal components for normal specialized endothelial and epithelial cell function.(Cichon et al., 2014; Dejana et al., 2009; Hartsock and Nelson, 2008; Muller, 2003; Palladino et al., 2017; Sluysmans et al., 2017; Stamatovic et al., 2016)
Our recent work elucidating the normal adult human BNB transcriptome based on conserved transcripts expressed by early- and late-passage primary human endoneurial endothelial cells (pHEndECs) and laser-capture microdissected endoneurial microvessels from four histologically normal adult sural nerve biopsies (freely accessible via the National Center for Biotechnology Information Gene Expression Omnibus [GEO] series accession number GSE107574: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107574) demonstrated transcript expression of 133 intercellular junctional complex molecules (22 tight junction or junction-associated, 45 adherens junction or junction-associated and 52 cell junction-associated or adaptor molecules), with in situ endoneurial endothelial microvessel protein expression of α1 catenin (CTNNA1), cadherin-5 (CHD5), cadherin-6 (CDH6), claudin-4 (CLDN4), claudin-5 (CLDN5), crumbs cell polarity complex component lin-7 homolog A, gap junction protein A1, multiple PDZ domain crumbs cell polarity complex component, protocadherin-1, vezatin, ZO-1 and zyxin demonstrated by indirect fluorescent immunohistochemistry.(Ouyang et al., 2019; Palladino et al., 2017)
This complexity suggests that significant molecular redundancy may exist to maintain a structurally normal BNB due to its essential homeostatic role in normal peripheral nerve function, or these molecules have other functions in endoneurial microvessels. Expression patterns of these junctional complex proteins in normal peripheral nerves would provide insights into their potential functions in health, guided by studies performed in neural and non-neural derived endothelial cells and other species. Restrictive intercellular tight-junction formation is a critical observation that differentiates endoneurial microvascular endothelial cells from epineurial macrovascular endothelial cells in human peripheral nerves. Interestingly, indirect immunohistochemistry in normal adult peripheral nerve demonstrated expression of commonly cited restrictive junctional complex molecules in epineurial macrovessels that do not form tight junctions (e.g. CLDN5 and ZO-1),(Kanda et al., 2004; Ouyang et al., 2019) emphasizing the importance of critically evaluating human peripheral nerves in situ to determine biological relevance prior to performing in vitro model studies.
Using confocal microscopy, we ascertained that CLDN4 is most likely associated with BNB tight junctions and CDH5 associated with adherens junctions, with CTNNA1 (a member of the α catenin family of adaptor molecules, that link cellular membranes to the cytoskeleton in order to maintain structural integrity),(Vite et al., 2015) proposed as important for linking the F-actin cytoskeleton with adherens junctions. Neither ZO-1 nor CLDN5 demonstrated expression patterns to suggest adaptor or tight junction complex localization in normal adult endoneurial microvessels, but appear as scaffolding proteins that link endothelial cell membranes to cytoplasmic components, including the cytoskeleton (Figure 3). Further studies are ongoing to further characterize the molecular components of the normal adult BNB using high resolution microscopy.
Figure 3. Human BNB Junctional complex.
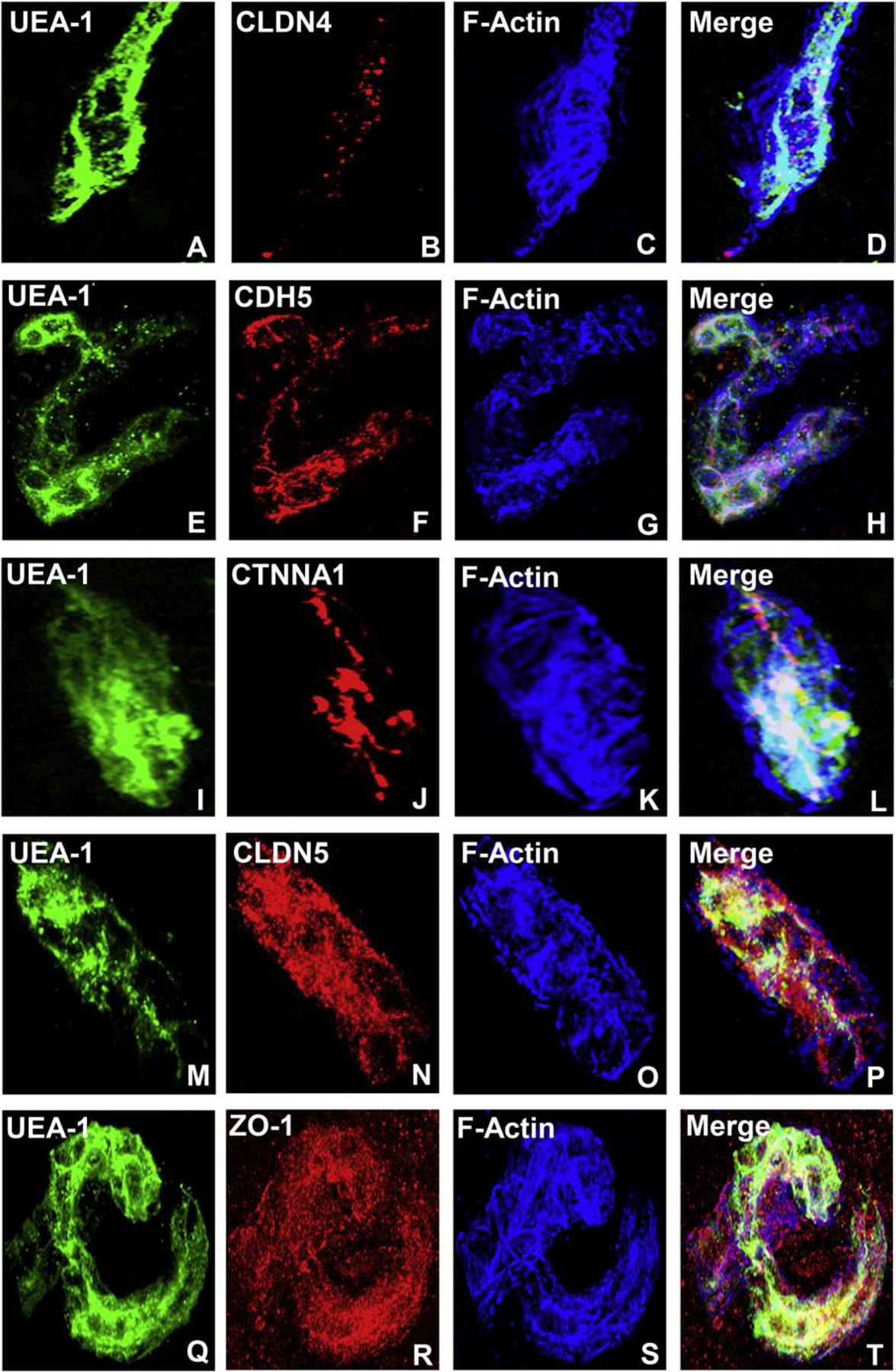
Digital high resolution indirect immunohistochemistry confocal photomicrographs of normal adult endoneurial microvessels stained with Ulex Europaeus Agglutinin-1 (UEA-1) to detect endothelial cell membranes (green; A, E, I, M and Q) and Rhodamine Phalloidin to detect endoneurial microvessel F-actin cytoskeleton (pseudocolor blue; C, G, K, O and S) were generated to determine cellular localization of hypothesized junctional complex molecules (red) claudin 4 (CLDN4, B), cadherin 5 (CDH5, F), α−1 catenin (CTNNA1, J), claudin 5 (CLDN5, N) and zona occludens-1 (ZO-1, R). The punctuate CLDN4 expression and membrane co-localization suggest that this molecule is an essential component of BNB tight junctions (D). The linear, continuous CDH5 expression and membrane co-localization are consistent with an adherens junction protein (H). The plaque-like linear CTNNA1 expression and strong membrane and cytoskeletal co-localization suggest a role as an adaptor molecule that links the endoneurial microvessel membrane to the cytoskeleton. Both CLDN5 (P) and ZO-1 (T) demonstrate diffuse microvessel expression with additional strong continuous membrane and cytoskeletal co-localization, implying that they are not structural components of BNB tight junctions, but may have essential roles in the overall structural integrity (e.g. adaptor or scaffolding protein) of normal human adult endoneurial microvessels.
Understanding intrinsic and extrinsic influences on the BNB in healthy peripheral nerves is important to future efforts to restore function in diseased nerves or alter function for anesthetic purposes. Endoneurial endothelial cells express specific mitogen receptors such as glial-derived neurotrophic factor (GDNF; GFRα1), vascular endothelial growth factor (VEGF; VEGFR2), basic fibroblast growth factor (bFGF; FGFR1) transforming growth factor-β (TGF-β; TGFRI/II) and glucocorticoids (GR) in vitro and in situ,(Palladino et al., 2017; Reddy et al., 2013; Shimizu et al., 2011; Shimizu et al., 2012; Yosef and Ubogu, 2012b, 2013) implying that autocrine or paracrine mitogen secretion by endothelial cells, Schwann cells, pericytes, mast cells or endoneurial fibroblasts could regulate BNB composition and function in health. Schwann cells, the glial cells of the peripheral nervous system present in the endoneurium, have been shown to secrete GDNF in vitro and in vivo,(Naveilhan et al., 1997; Trupp et al., 1995) and GDNF has been demonstrated to influence restrictive human BNB characteristics in vitro at low nanomolar concentrations in a dose-dependent manner via RET-tyrosine kinase-mitogen activated protein kinase (MAPK) signaling.(Dong et al., 2018; Dong and Ubogu, 2018; Yosef and Ubogu, 2012b) This suggests that GDNF is an essential paracrine regulator of BNB formation that may also have an important role during BNB formation during development and maintenance in health, with some redundancy demonstrated in vitro by other less efficacious mitogens, such as bFGF.
Although direct proof is lacking from the developing BNB in neonatal human peripheral nerves, it is interesting to speculate that endoneurial-resident Schwann cells are necessary to transform fenestrated blood vessels penetrating from the epineurium into restrictive tight junction-forming microvessels within the endoneurium via a GDNF paracrine effect. Endoneurial Schwann cell GDNF secretion could also induce innermost perineurial cell tight junction specialization. Guided by prior knowledge of GDNF signaling, data from the human BNB transcriptome and our quantitative proteomic and confirmatory immunoblot data derived from confluent pHEndEC membrane fractions following GDNF treatment in vitro,(Allen et al., 2013; Dong and Ubogu, 2018; Encinas et al., 2001; He et al., 2008; Ibanez and Andressoo, 2017; Jing et al., 1996; Naveilhan et al., 1997; Palladino et al., 2017; Takahashi, 2001; Yosef and Ubogu, 2012b) our early preliminary studies suggests that GDNF upregulates pHEndEC membrane expression of junctional complex molecules cortactin (CTTN; an actin-binding protein that can serve as an adapter for focal adhesion or junctional complex formation, and is phosphorylated by SRC kinase),(Schnoor et al., 2018; Tehrani et al., 2007) CTNNA1 and CLDN4 during restrictive junction formation in vitro.
Guided by our published human pHEndEC quantitative proteomic database that identified specific pHEndEC membrane fraction-enriched proteins following GDNF treatment relative to basal cultures,(Dong and Ubogu, 2018) we performed western blots on these protein extracts using specific vendor validated antibodies and standard protocols. We observed significantly increased expression of CTTN, phosphorylated CTTN, CTNNA1 and CLDN4 following GDNF treatment compared to basal untreated pHEndECs. There was no difference in pCTTN/CTTN ratio seen comparing GDNF-treated and untreated cultures, supporting the notion that an endogenous, GDNF-independent kinase (presumed to be SRC kinase) was responsible for CTTN phosphorylation following GDNF-induced CTTN upregulation in vitro (Figures 4A–B). SEC31A (a component of the coat protein II vesicular transport complex that is regulated by MAPK kinase signaling and highly expressed at the human BNB based on transcript analyses)(Farhan et al., 2010; Palladino et al., 2017) was significantly downregulated (~20% of basal) in our pHEndEC membrane protein extracts after restrictive junctional formation (Figures 4A–B), suggesting membrane dissociation and cycling back to the cytosol. Figure 5 depicts our proposed hypothesis on the molecular determinants and signaling processes involved in GDNF-mediated BNB formation. Further studies are ongoing to better characterize the molecular regulation of the human BNB during development, guided by our transcriptome and proteome data with network analyses. Preliminary functional studies on restrictive BNB junctional complex formation using validated specific small interfering RNA (siRNA) sequences will be discussed later in this review (see section on BNB physiology).
Figure 4. Human BNB Junctional complex.
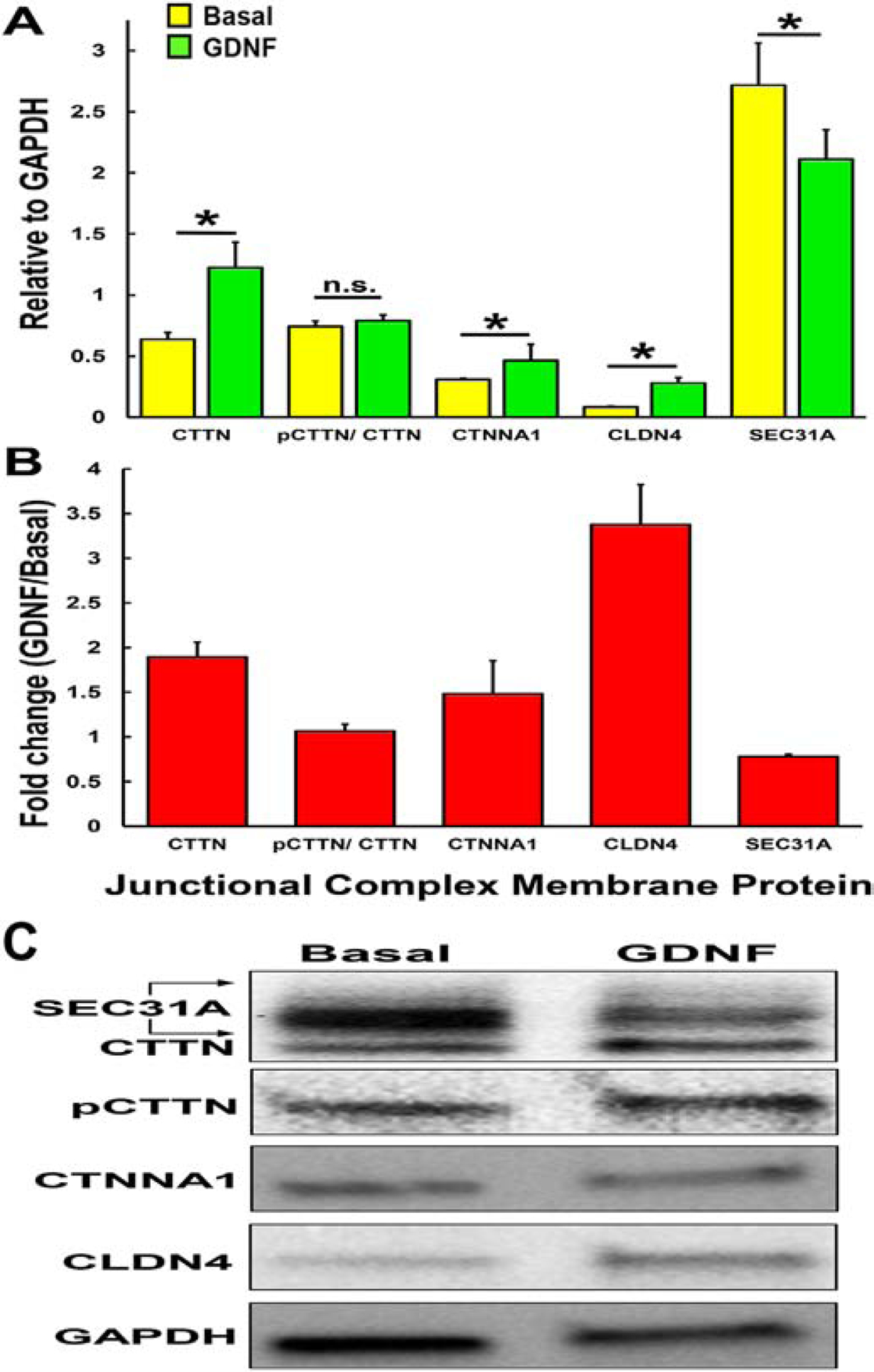
Bar histograms demonstrate the effect of exogenous human recombinant GDNF on human BNB tight junctional complex protein expression after 48 hours in vitro. Western blot of confluent pHEndEC membrane protein extracts was performed following SDS-PAGE, using standard protocols with specific validated primary and secondary antibodies. Semi-quantitative expression relative to GAPDH for basal (yellow) and GDNF-treated (green) pHEndECs (A) and GDNF-induced fold-change (B) are shown for each protein, with the ratio of pCTTN/ CTTN shown to indicate whether GDNF modulates pCTTN expression independent of CTTN. * indicates p<0.05, n.s. = not significant. Error bars indicate standard errors of means (N=3). A representative digital western blot image showing GDNF-induced human BNB junctional complex protein expression at 48 hours in vitro is shown, with reduced membrane SEC31A (C)
Figure 5. Human BNB junction complex formation: The GDNF-CREB1-SRC-CTTN/CTNNA1/CLDN4-SEC31A hypothesis.
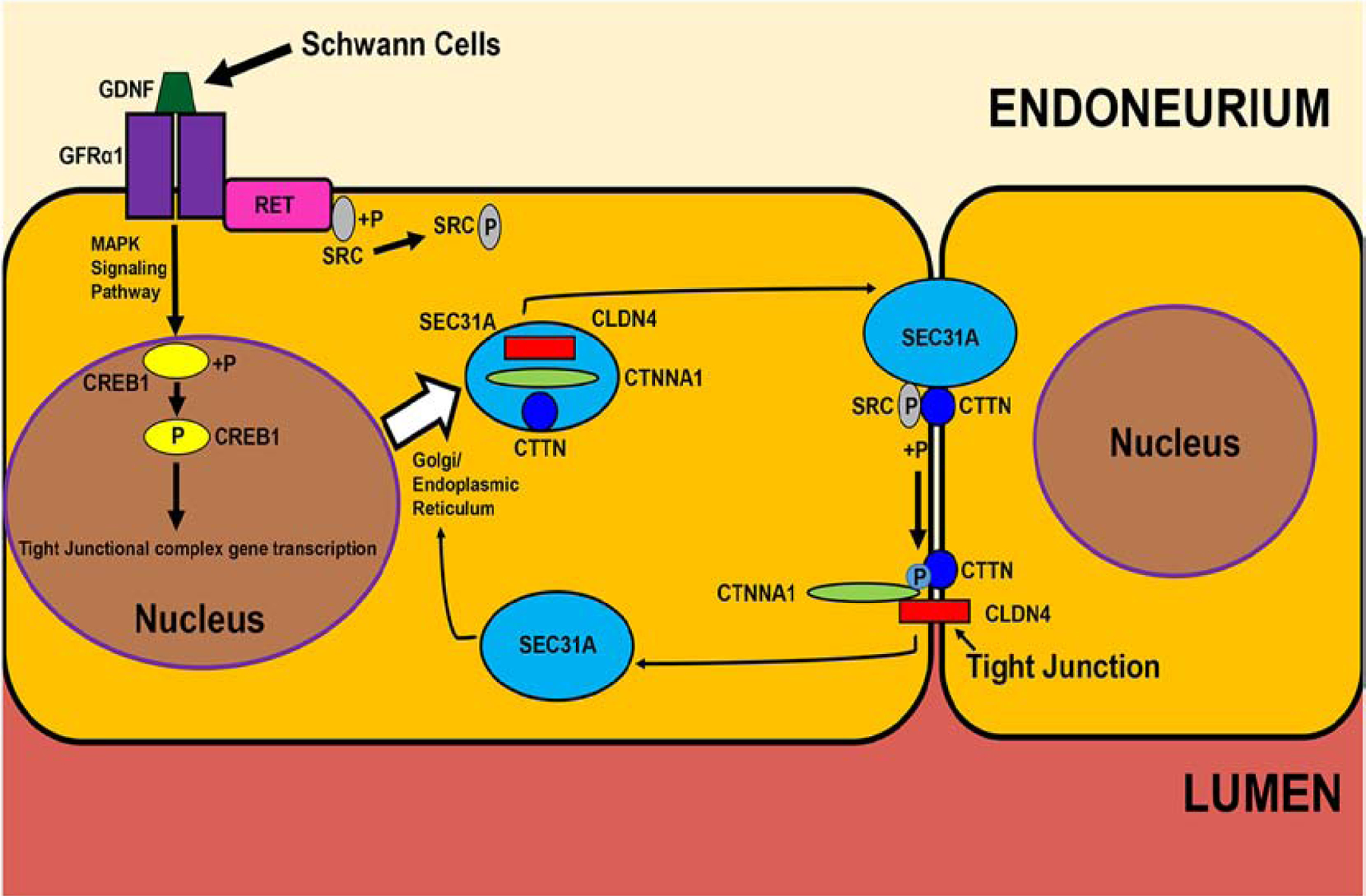
Guided by the human BNB transcriptome and in vitro pHEndEC membrane proteome, this figure illustrates a restrictive GDNF-mediated BNB forming pathway downstream of GFRα1-RET-MAPK signaling, including specialized junctional complex protein transport for further study. +P indicates a phosphorylation and P indicates an activated phosphorylated protein.
In addition to the restrictive junctional complex, specialized influx and efflux transporters that regulate ionic, water, molecule, nutrient, drug and xenobiotic entry into or removal from peripheral nerve endoneurium exist at the human BNB, influencing the endoneurial microenvironment. In fact, it could be argued that specialized transporters dynamically regulate endoneurial homeostasis by rapidly adapting to fluctuations in systemic blood molecular composition and variations in peripheral nerve metabolic activity under normal physiological states. Known transporters, based on human in vitro or in situ BNB observational studies include alkaline phosphatase, glucose transporter-1 (also known as solute carrier [SLC] 2A1), monocarboxylate transporter-1 (also known as SLC16A1), creatine transporter (also known as SLC6A8), large amino acid transporter-1 (also known as SLC7A5), γ-glutamyl transpeptidase and p-glycoprotein (also known as ATP-binding cassette [ABC] B1),(Abe et al., 2012; Allt and Lawrenson, 2000; Bell and Weddell, 1984; Froehner et al., 1988; Muona et al., 1993; Orte et al., 1999; Palladino et al., 2017; Yosef and Ubogu, 2013; Yosef et al., 2010)
Our recent work deducing the human BNB transcriptome demonstrated 509 transporter transcripts, including 196 members of the SLC transport family, 76 cation channel, 33 members of the ABC transport family, 14 zinc transporter, 13 anion channel, 4 solute carrier organic transporter and 3 aquaporin (AQP) molecules. Selected transporters, ABCA8, ABCB1, AQP1, SLC1A1, SLC2A1, SLC3A2, SLC5A6, SLC16A1 and SLC19A2 were demonstrated on BNB-forming endoneurial endothelial cells in normal human sural nerve biopsies by indirect immunohistochemistry in situ.(Palladino et al., 2017) Although there are major similarities, structural differences and molecular heterogeneity in BNB composition exists between different species,(Bell and Weddell, 1984; Latker et al., 1987) limiting the degree of extrapolation feasible to the human BNB from animal studies.
Our in situ normal adult peripheral nerve immunohistochemistry study interestingly showed that p-glycoprotein (ABCB1) and SLC1A1, also known as excitatory amino acid transporter-3 that actively transports glutamate across plasma membranes, (Mahringer and Fricker, 2016; Nalecz, 2017; Palladino et al., 2017) were the only transporters from the selected group restricted to endoneurial microvessels, implying specific roles in BNB function. We are performing high resolution microscopy studies guided by our BNB transcriptome database to determine BNB-restricted transporter and their luminal or abluminal localization profiles in normal endoneurial microvessels as a prerequisite for functional studies.
Human BNB Physiology
Transendothelial Electrical Resistance
The human BNB, similar to other specialized tight-junction forming microvascular systems such as the BBB, blood-retina barrier and blood-testis barrier, is expected to possess high transendothelial electrical resistance (TEER), low permeability to solutes and macromolecules and low transendothelial water flux (hydraulic conductivity). In support of this, comparative animal studies have determined that the BNB is the second most restrictive microvascular tissue barrier in mammals, after the BBB.(Poduslo et al., 1994; Rechthand et al., 1987) Unlike the human BBB, supported by the glia limitans (which consists of astrocyte and microglial foot processes), there is no physical support of the BNB by Schwann cells based on ultrastructural observations. It is debatable whether endoneurial microvascular pericytes (that lack intercellular junctions and share a basement membrane with endoneurial endothelial cells) are specialized to provide a physiological paracrine molecular effect on the human BNB or serve as smooth muscle-like cells that primarily control endoneurial microvessel diameter in response to changes in volume and shear forces, or both. Myelin, formed by concentric layers of a single Schwann cell membrane, is divided to compacted and non-compacted areas by autotypic junctions. The exact function of Schwann cell autotypic junctions is not fully understood; however they may separate the outer membrane and the endoneurium space (in which molecules can freely diffuse after crossing the BNB) from the compact myelin and also provide mechanical structural integrity for each axonal segment.(Peltonen et al., 2013)
The human BNB TEER in vivo is unknown; however, it is expected to >1500 Ω.cm2, based on BBB data.(Cohen-Kashi Malina et al., 2009; Lippmann et al., 2014; Lippmann et al., 2012; Wang et al., 2017) Similarly, specific permeability coefficients and hydraulic conductivity are also unknown; although some work has been published in other mammalian and non-mammalian species, measuring peripheral nerve endoneurial solute permeability and interstitial fluid flux following intravenous electrolyte and tracer injections in harvested nerves at specific time intervals.(Poduslo et al., 1994; Poduslo et al., 1988; Rechthand and Rapoport, 1987; Rechthand et al., 1987) Human BNB mean TEER has been measured to be as high as ~180 Ω.cm2 in confluent cultures by a voltohmmeter applying a direct current across Transwell™ inserts and as high as ~900 Ω when recorded in specialized culture wells with gold electrodes using a fixed alternating current at 4000 Hz via electrical-cell impedance sensing (ECIS).(Dong and Ubogu, 2018; Ubogu, 2013b; Yosef and Ubogu, 2013; Yosef et al., 2010) The effect of near physiologic hemodynamic flow and shear forces on restrictive junction formation has been established for the human BBB in vitro(Cucullo et al., 2011; Santaguida et al., 2006) and would be expected for the human BNB as well, although not formally tested yet.
We had previously ascertained the importance of GDNF in enhancing in vitro human BNB TEER via RET-tyrosine kinase-MAPK signaling.(Dong and Ubogu, 2018; Yosef and Ubogu, 2012b) GDNF-GFRα1-RET complex also activates SRC (a kinase associated with cytoskeletal and intercellular contact organization) (Roskoski, 2015) by phosphorylation independent of MAPK kinase signaling, as deemed necessary for neuronal survival.(Encinas et al., 2001) Guided by our recent BNB transcriptome and pHEndEC membrane fraction proteomic work, we sought to determine the molecular importance of selected proposed junctional complex molecules and signaling kinases on BNB TEER in vitro by gene silencing with specific siRNA sequences. Using validated adherent cell transfection protocols with HiPerfect® Transfection reagent (Qiagen Catalog # 301704), we administered siRNAs against human cyclic AMP [CAMP] Responsive Element Binding Protein 1 (CREB1; Qiagen catalog # GS1385), CTTN (Qiagen catalog # GS2017), CTNNA1 (Qiagen catalog # GS1495), CLDN4 (Qiagen catalog # GS1364) and negative control siRNA (Qiagen catalog # SI03650318) and Bosutinib (a cell permeable specific SRC kinase inhibitor, SKI-606, Selleckchem, catalog # S1014) to GDNF-treated confluent pHEndEC cell cultures for 48 hours after serum withdrawal (to induce endothelial cell detachment and disruption to endothelial intercellular contacts, with untreated (basal) pHEndEC cultures undergoing serum withdrawal as negative controls.
Our preliminary work implies a role for CREB1, CTTN, SRC kinase, CTNNA1 and CLDN4 in the GDNF-mediated increase in human BNB TEER in vitro over 48 hours, measured by ECIS. We observed that SRC kinase inhibition caused persistent TEER decline irrespective of GDNF, suggesting a non-redundant role in human BNB tight junction formation in vitro, or effects on cell survival (Figure 6). Guided by our work and published work of others,(Allen et al., 2013; Dong and Ubogu, 2018; Encinas et al., 2001; He et al., 2008; Ibanez and Andressoo, 2017; Jing et al., 1996; Naveilhan et al., 1997; Palladino et al., 2017; Takahashi, 2001; Yosef and Ubogu, 2012b) we speculate that GDNF, following RET-GFRα1-MAPK signaling, activates transcription factor CREB1 which induces BNB tight junctional complex CTTN, CTNNA1 and CLDN4 gene transcription, with SEC31A contributing to transporting translated proteins to intercellular membranes. GDNF (via RET) or other mitogens, could independently activate SRC kinase which phosphorylates CTTN, providing the essential scaffold for organized CTNNA1 binding. CTNNA1 interacts with CLDN4 which forms restrictive tight junctions (Figure 5). We are performing studies to confirm this interesting hypothesis, guided by molecular and signaling network data derived from our published human BNB transcriptome.
Figure 6. Molecular determinants of the in vitro human BNB.
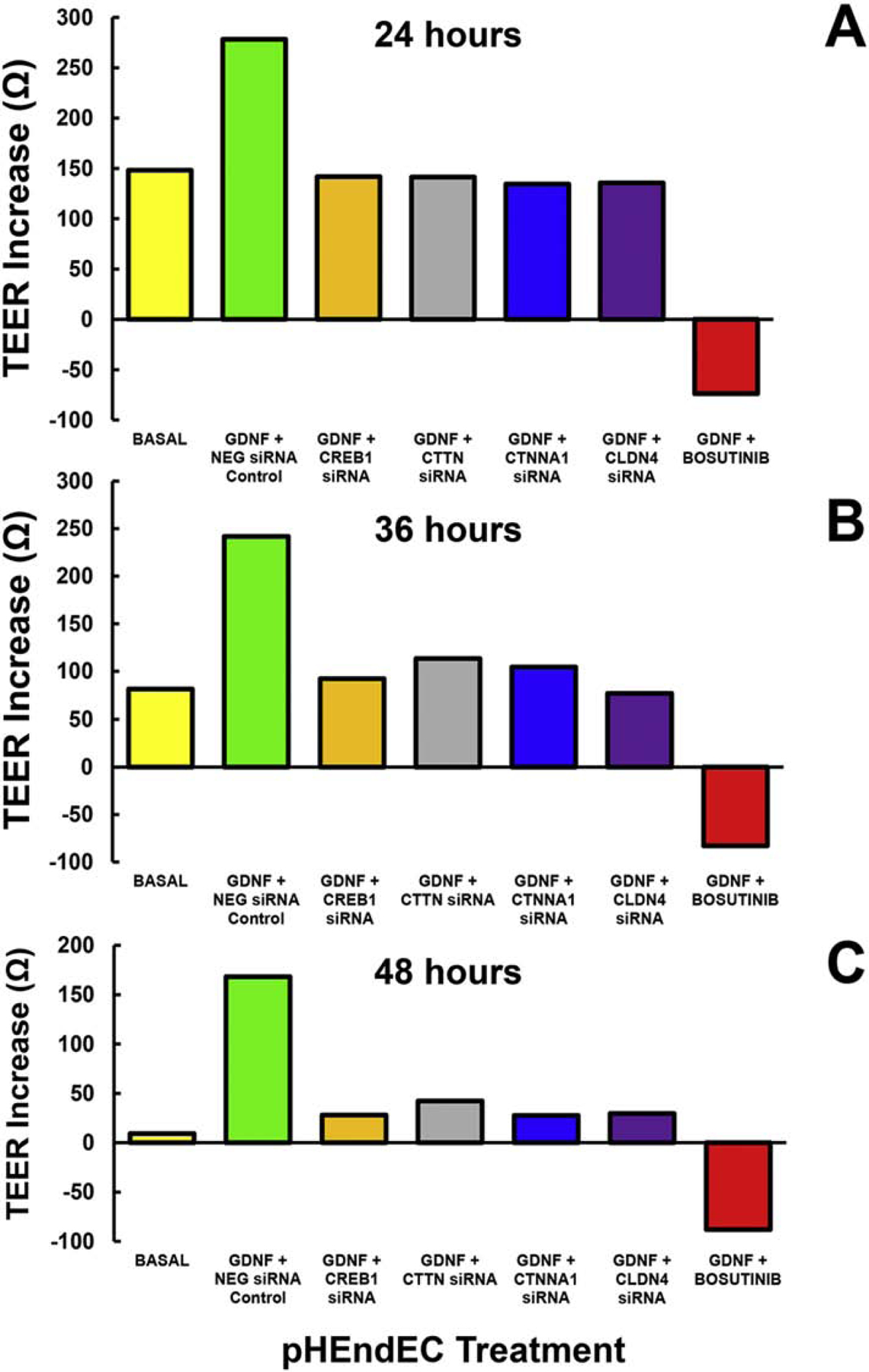
ECIS was performed to continuously measure pHEndEC TEER following initial plating for 5 days and 48 hours after serum withdrawal (to induce endothelial cell detachment). Following treatment with GDNF (1 ng/mL) and transfection with validated specific siRNA to inhibit gene transcription or SRC kinase blockade using a Bosutinib, TEER increase was calculated for each well by subtracting the mean lowest TEER during the 1st hour after serum withdrawal from mean maximal resistance at predefined time points, with appropriate negative controls. Histograms demonstrate averages for each time point (A: 24 hours, B: 36 hours, and C: 48 hours) in this single experiment performed in duplicate for each experimental condition.
Solute Permeability
Solute permeability, a measure of molecular translocation across apical and basal cellular membranes, is used to evaluate molecular flux between the luminal and abluminal surfaces of cultured endothelial cells.(Dong and Ubogu, 2018; Komarova and Malik, 2010; Ubogu, 2013b; Yosef and Ubogu, 2012b, 2013; Yosef et al., 2010; Zihni et al., 2016) Molecules could undergo passive diffusion or active transport through or between cells based on size, ionic composition, hydrophilicity, lipophilicity, and the presence of selective or specific membrane transporters or receptors. Studying molecules that are not deemed to undergo facilitated active transport aids demonstrate restrictive solute permeability to passive diffusion expected of tight junction-forming intercellular junctions. Solute permeability to sodium fluorescein (molecular mass 376 Da) and large molecular mass 70 KDa fluoresceinated dextran (dextran-70-FITC) across primary and immortalized human endoneurial endothelial cells that form the BNB is typically <5% of input concentration at 15 minutes using static Transwell™ systems in vitro, with higher values (~3–15 fold) seen with sodium fluorescein when directly compared to dextran-70-FITC using the same batch of endothelial cells in concurrent experiments.(Ubogu, 2013b; Yosef and Ubogu, 2012b, 2013; Yosef et al., 2010)
As described previously, we have published the paracrine effect of GDNF in reducing solute permeability at the in vitro human BNB via RET-tyrosine kinase-MAPK signaling pathways and have hypothesized that this biological effect is related to F-actin cytoskeletal organization with more continuous intercellular contacts and restrictive junction formation that limits molecular transport between adjacent endoneurial endothelial cells based on our histological observations.(Dong and Ubogu, 2018; Yosef and Ubogu, 2012b) It is plausible that restricted in situ human BNB molecular permeability may occur due to limited receptor-mediated or caveolae-driven transcytosis. Caveolin-1 (CAV1), a major protein component of caveolae, is highly expressed by normal adult BNB-forming endoneurial microvascular endothelial cells in situ,(Ouyang et al., 2019; Palladino et al., 2017) suggesting the importance of caveolae-mediated transcytosis in normal BNB function. Comparative in situ studies between endoneurial microvessels and epineurial macrovessels guided by the human BNB transcriptome and in vitro human BNB membrane proteome should provide insights into the biologically relevant molecular determinants of endoneurial solute/ macromolecular permeability in healthy adult nerves.
Hydraulic Conductivity
Endoneurial water regulation influences concentrations of ions that are tightly regulated (e.g. Na+ and K+) for normal peripheral axonal signal transduction. Our human BNB transcriptome work identified three water channels, or aquaporins, AQP1, AQP3 and AQP11, as potentially relevant to regulating peripheral nerve water flux in healthy adults.(Palladino et al., 2017) Based on our immunohistochemistry studies, AQP1 was expressed by endoneurial microvessels, epineurial macrovessels, perineurial cells and pericytes, implying that it may serve as a generalized water transport channel in multiple peripheral nerve cell types, as required for normal metabolism and physiology, rather than a specific water influx or efflux regulator at the BNB.(Ouyang et al., 2019) We have measured human BNB transendothelial water flux under the influence of hydrostatic pressure, otherwise known as hydraulic conductivity, in vitro (~2.0 ×10−7 cm/s/cm H2O) using a customized Transwell™ diffusion chamber-bubble tracker system.(Helton et al., 2017) Consistent with prior observations, the human BNB was the 2nd most restrictive human or mammalian microvascular endothelial cell type after the BBB in terms of water flux.(Helton et al., 2017; Poduslo et al., 1994; Rechthand and Rapoport, 1987; Rechthand et al., 1987) Guided by in situ observations, the biologically relevant molecular determinants and signaling pathways for endoneurial water regulation can be deduced using our published diffusion chamber-bubble tracker system, further enhancing our knowledge of normal adult human BNB physiology.
Physiological leukocyte trafficking at the human BNB: Immune surveillance and leukocyte-peripheral nerve cross-talk
Hematogenous leukocyte trafficking across microvascular endothelium in vivo (based on intravital microscopy) or in vitro under flow is a sequential coordinated process that attracts leukocytes from circulating blood to the endothelial cell luminal surface. This is mediated by specific chemoattractant molecules such as chemokines [‘chemotactic cytokines’] bound to glycosaminoglycans on the endothelium and leukocyte-selective chemokine receptors. The next step is leukocyte rolling, mediated by endothelium-expressed selectins and their glycoproteins or carbohydrate moiety counterligands expressed on leukocytes. This leads to firm leukocyte arrest and haptotaxis on the endothelial cell surface that is also mediated by chemokines and chemokine receptors, and followed by integrin activation and firm adhesion (via conformation change in leukocyte integrin heterodimer from an inactive non-binding molecule to an active, high affinity binding molecule to selective endothelial cell adhesion molecules). These changes facilitate leukocyte flattening with formation of pseudopodia, leukocyte transmigration via the paracellular (i.e. through intercellular junctions) or transcellular (i.e. through endothelial cells) routes followed by basement membrane disruption at the abluminal surface (via secretion of specific matrix metalloproteases) required for complete passage into tissues.(Ley et al., 2007; Man et al., 2007; Mempel et al., 2004; Muller, 2014; Pai et al., 2012; Teixeira et al., 2010; Zenaro et al., 2013)
Our in vitro data using a flow-dependent leukocyte-BNB trafficking model also demonstrated this sequential process (also known as the multi-step paradigm of leukocyte trafficking) using healthy adult peripheral blood mononuclear leukocytes (PBMLs) and confluent pHEndECs under basal culture conditions, implying that this process may occur at the normal healthy adult BNB.(Dong et al., 2017; Greathouse et al., 2016; Yosef and Ubogu, 2012a) The presence of rare endoneurial macrophages, mast cells and T-lymphocytes in normal human peripheral nerve endoneurium based on histological observations implies some physiological crosstalk between the systemic immune compartment and peripheral nerves facilitated, at least in part, by interactions with BNB-forming endoneurial microvessels.
Our human BNB transcriptome database supports the expression of Human Leukocyte Antigen (HLA, or Major Histocompatibility Complex [MHC]) Class I and II molecules in normal healthy and inflamed endoneurial microvessels in situ,(Mitchell et al., 1991; Palladino et al., 2017; Pollard et al., 1987; Pollard et al., 1986) suggesting that the human BNB may directly participate in peripheral nerve innate and adaptive immune responses. This is a reasonable assertion, as leukocytes would have to interact with endoneurial microvessels prior to endoneurial entry, and subsequently engage with Schwann cells that have been shown to act as facultative antigen-presenting cells.(Hartlehnert et al., 2017; Meyer Zu Horste et al., 2010a; Meyer zu Horste et al., 2010b; Mitchell et al., 1991; Weiss et al., 2016) Furthermore, specific chemokine transcripts were expressed by the normal healthy adult BNB. These include CCL2, CCL14, CCL28, CXCL3, CXCL12, CXCL16 and CX3CL1.(Palladino et al., 2017) Based on the expression of known signaling receptors on specific human leukocyte subpopulations, these chemokines could facilitate the interaction of hematogenous monocytes (CCL2, CCL14, CX3CL1), T-lymphocytes (CCL2, CX3CL1), natural killer T-cells (CXCL16) and neutrophils (CXCL3) with endoneurial microvascular cells during normal immunosurveillance or part of an early immune response to injury, while CXCL12 and CCL28 may be important for endoneurial endothelial cell migration during development and vascular repair.(Bachelerie et al., 2014)
Endoneurial microvascular endothelial cells also express selectins (e.g. P-selectin, E-selectin) and cell adhesion molecules (e.g. intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), fibronectin Type III connecting segment) under basal conditions in situ or in vitro,(Palladino et al., 2017; Yosef et al., 2010) that were upregulated or underwent alternative splicing following stimulus with physiological concentrations of pro-inflammatory cytokines tissue necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) in vitro.(Yosef and Ubogu, 2012a) The constitutive expression of these cell adhesion molecules known to facilitate leukocyte adhesion and transmigration supports the notion the endoneurial microvessels participate in crosstalk between subsets of circulating leukocytes from the systemic immune compartment and peripheral nerves.
Direct human in situ evidence for peripheral nerve immunosurveillance is limited,(Griffin et al., 1990) and testing this hypothesis poses challenges. The multilayered peripheral nerve structural organization (with its longitudinal array of epineurial collagen fibers, concentric layers of perineurial myofibroblasts, loose irregular meshwork of endoneurial collagen fibers), size of peripheral nerves (e.g. compared to the cerebral cortex), low endoneurial microvessel density (in the setting of a high density of myelinated and unmyelinated axons) provide a major challenge to mammalian animal model intravital BNB microscopy studies that may have some translational potential to in situ human BNB leukocyte trafficking.(Greathouse et al., 2016) Studying peripheral nerve immune responses in conditional knockout transgenic mice provides an avenue to further comprehend physiological crosstalk between the systemic immune compartment and peripheral nerves.
The human BNB in disease: current concepts and hypotheses
Structural and functional changes at the BNB associated with peripheral neuropathies: General Principles
Increased permeability of, or leukocyte trafficking at the human BNB, commonly cited as a “BNB breakdown”, has been pathologically associated with peripheral nerve autoimmune disorders such as Guillain-Barre syndrome (GBS, typically the acute inflammatory demyelinating polyradiculoneuropathy [AIDP] variant) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). This commonly held view point implies that extrinsic factors induce loss of restrictive human BNB function or disruption of intercellular tight junctions, with the endoneurial microvessels being relatively passive in these disorders.
Our recent data shows restrictive junction component complexity and possible redundancy at the human BNB,(Palladino et al., 2017) implying that loss or downregulation of a single tight junction forming molecule or reduction in TEER or increase in solute permeability demonstrated in vitro following administration of sera from GBS or CIDP patients (Kanda, 2013; Kanda et al., 2003; Shimizu et al., 2014) may be an insufficient structural or functional change at the human BNB in vivo. In support of this, physiological cytokine stimulus of confluent primary human endoneurial endothelial cells grown on Transwell™ inserts with TNF-α and IFN-γ over a 100-fold range did not alter TEER in vitro.(Yosef and Ubogu, 2012a) Ultrastructural examination of endoneurial microvessels within the inflammatory milleu from adult patients with GBS and CIDP demonstrated intact electron dense intercellular tight junctions, with similar electron-dense contacts between infiltrating leukocytes and endothelial cells (Figure 7).(Bosetti et al., 2016; Dong et al., 2016) These observations should provide the impetus to better understand the biological underpinnings of the structural and functional alterations at the human BNB during peripheral neuropathies relative to healthy nerves.
Figure 7. BNB tight junction ultrastructure in GBS (AIDP) and CIDP.
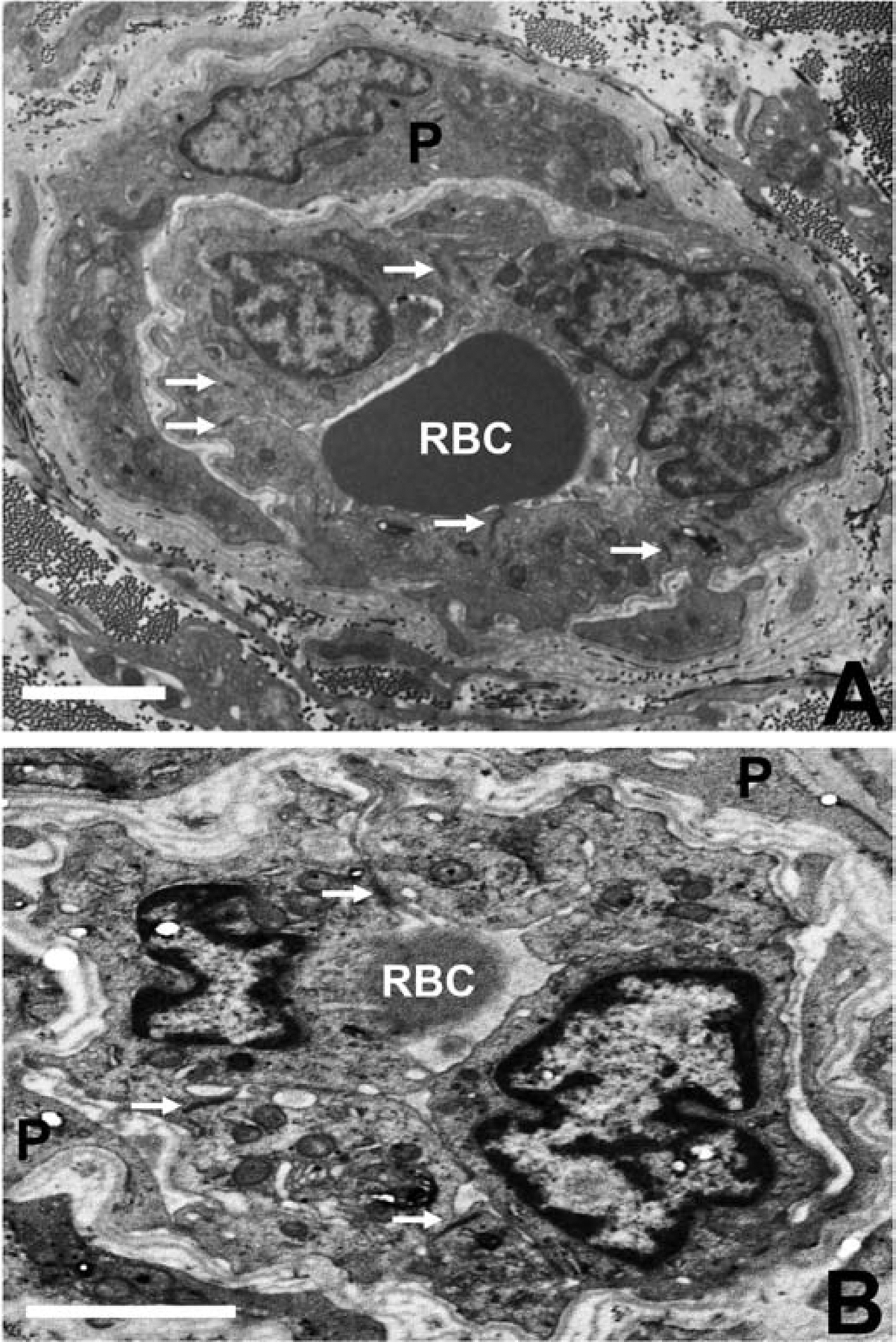
Composite digital electron ultramicrographs demonstrate structurally intact, apical membrane localized, electron dense intercellular tight junctions (solid white arrows) between endoneurial endothelial cells within the inflammatory milieu in severely affected GBS (A) and CIDP (B) patient sural nerve biopsies. RBC indicates luminal red blood cells and P indicates pericytes and their cytoplasmic projections surrounding these endoneurial microvessels. Scale bar = 2.5 μm.
Endoneurial microvessel wall thickening/ basement membrane duplication (Figures 8A–C, E and F) has been described in association with chronic peripheral neuropathies such as diabetes mellitus, CIDP and peripheral nerve vasculitis (which typically affects epineurial arteries or arterioles and rarely involves endoneurial capillary-like vessels with resultant endoneurial ischemia).(Bosetti et al., 2016; Eames and Lange, 1967; Richner et al., 2018; Ubogu, 2015) We have also observed this in our transgenic GDNF mice following sciatic nerve crush injury associated with delayed restoration of BNB function, measured as an impermeability to horseradish peroxidase (Figure 8G). The functional implications of the basement membrane alterations are undetermined; however, these may reflect adaptive responses to chronic and persistent endothelial cell or pericyte pro-inflammatory cytokine exposure or secretion, or hypoxia/ ischemia/ oxidative stress as a compensatory means of maintaining BNB functional integrity. We have recently performed some preliminary BNB junctional complex molecule confocal microscopy using sural nerves of chronic peripheral neuropathy patients (see BNB Dysfunction and Chronic Neuropathic Pain section).
Figure 8. Endoneurial microvessel wall thickening/ basement membrane duplication.
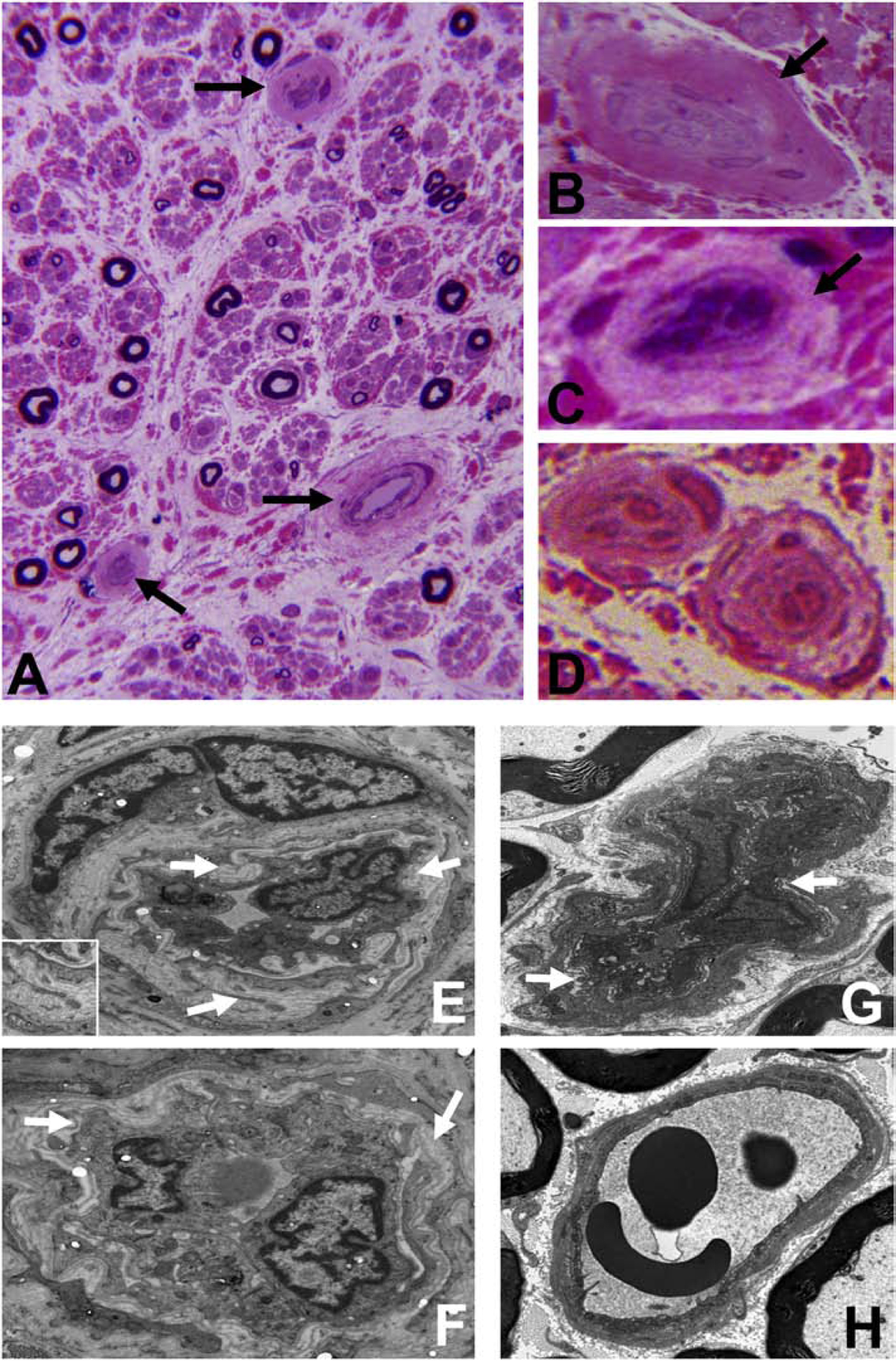
Composite digital light photomicrographs of an axial section of a CIDP patient sural nerve biopsy (plastic embedded semi-thin axial section stained with Toluidine Blue and counterstained with Basic Fuchsin) showing endoneurial microvessel wall thickening (black arrows) at low magnification (A) and at higher magnification in another CIDP patient (B). These changes are also seen in a vasculitic neuropathy patient with chronic neuropathic pain (C). These changes are in contrast to thin walls seen in normal adult endoneurial microvessels (D). Composite digital ultramicrographs of the sural nerve biopsy from a CIDP patient (E and F) show endoneurial microvessel basement membrane duplication (white arrows). This is shown at higher magnification in the insert in E. This is also observed in sciatic nerve endoneurial microvessels in transgenic GDNF wildtype mice following non-transecting crush injury followed by SRC kinase inhibition to impede BNB functional recovery (G). A normal sciatic nerve endoneurial microvessel from the contralateral uninjured side of the same transgenic GDNF wildtype mouse is shown for structural comparison (H).
BNB endothelial-leukocyte interactions in inflammatory peripheral neuropathies
It is unresolved whether in genetically susceptible individuals, systemic immune system activation (e.g. by infections, minor surgery or trauma) with primary attack of peripheral nerves and nerve roots (through the process of ‘molecular mimicry’)(Dalakas, 2015; Mathey et al., 2015) or endogenous activation of the peripheral nerve innate immune system (e.g. by viruses)(Ziganshin et al., 2016) with secondary selective adaptive immune system activation, or both are responsible for tissue-specific autoimmunity. It is also unclear whether suspected circulating polyclonal anti-myelin protein, anti-axonal nodal protein, anti-ganglioside or anti-glycolipid autoantibodies can cross the human BNB in vivo. However, a pathologic hallmark of several inflammatory peripheral neuropathies is the infiltration of hematogenous leukocyte subpopulations in peripheral nerves and nerve roots, as observed in situ on well-defined patient nerve biopsies.(Ubogu, 2015) Using a flow-dependent leukocyte-BNB trafficking model in vitro, untreated GBS and CIDP patient PBMLs firmly adhere to the surface of confluent pHEndECs and undergo paracellular transmigration at higher rates that normal healthy donor PBMLs in vitro,(Dong et al., 2017; Yosef and Ubogu, 2012a) supporting the notion that leukocyte trafficking at the human BNB is pathogenically relevant to these immune-mediated peripheral neuropathies.
The expression of HLA Class II molecules, interleukin 1-beta (IL-1β), IFN-γ, TNF-α, CCL2, CXCL10 and ICAM-1 on endoneurial endothelial microvessels has been described in GBS patient nerve biopsies. Similarly, HLA-DR, interleukin-2 (IL-2), IFN-γ, TNF-α, CXCL10 and ICAM-1 have also been expressed at the human BNB in situ in CIDP patient nerve biopsies at higher levels compared to control nerves, supporting the notion that local activation of the adaptive immune response at the BNB may be pathogenically significant in GBS and CIDP.(Kieseier et al., 2002; Lindenlaub and Sommer, 2003; Mathey et al., 1999; Matsumuro et al., 1994; Mitchell et al., 1991; Orlikowski et al., 2003; Pollard et al., 1987; Pollard et al., 1986; Putzu et al., 2000; Steck et al., 2011; Van Rhijn et al., 2000) Peripheral nerve vasculitis is typically associated with leukocyte infiltration of epineurial macrovascular endothelium walls, rather than direct involvement with endoneurial microvessels that form the BNB. However, strong expression of HLA Class I and class II molecules on affected vascular endothelial cells has been described, typically associated with prominent CD4+ and fewer CD8+ T-lymphocytes and CD68+ macrophages.(Bosboom et al., 2001; Collins et al., 2013; Engelhardt et al., 1993; Leppert et al., 1999; Lindenlaub and Sommer, 2003; Van Rhijn et al., 2000)
Expression of cluster of differentiation (CD) 58 (also known as lymphocyte function-associated antigen-3; a cell adhesion molecule typically expressed on antigen-presenting cells such as macrophages and binds to CD2 on T-lymphocytes) and CD86 (a protein expressed on antigen-presenting cells that provides costimulatory signals necessary for T cell activation and survival) on affected vascular endothelial cells have also been described, with the former also expressed by Schwann cells.(Van Rhijn et al., 2000) Variable focal expression of hypoxia-inducing factors (HIFs), HIF-1α, HIF-1β, HIF-2α, as well as VEGF and its receptor VEGFR, and erythropoietin receptor was seen on endoneurial microvessels in a small percentage of vasculitic neuropathy patient nerve biopsies at higher rates than control sural nerve biopsies.(Oka et al., 2007; Probst-Cousin et al., 2010) These studies suggest that human BNB abnormalities may contribute to vasculitic neuropathy pathogenesis or represent hypoxic-ischemic adaptive changes that could provide insights into signaling mechanisms that may guide the specific therapeutic development for these peripheral neuropathies.
Our recent work elucidating the normal adult BNB transcriptome provided molecular targets putatively involved in normal restrictive barrier function in peripheral nerves.(Palladino et al., 2017) Validation of these proposed molecules and their associated signaling networks, as well as future single cell transcriptomics and proteomics studies could provide avenues to more comprehensively elucidate molecular changes at the human BNB in situ associated with peripheral neuropathies. Such work would increase our comprehension of disease pathogenesis and help devise targeted efficacious molecular therapies, as well as prevent consequential chronic neuropathic pain, a very prevalent condition seen in peripheral neuropathy patients for which current therapies are suboptimal. The following sections illustrate our hypotheses and recent advancements on the importance of the human BNB in peripheral neuropathies and chronic neuropathic pain, guided by transcriptomics, proteomics and indirect immunohistochemistry of well-characterized adult patient nerve biopsies, with some supportive data from animal models of peripheral nerve injury associated with chronic reflexive nociception.
BNB in GBS (AIDP variant)
AIDP (the most common GBS variant seen in the Western hemisphere) is characterized pathologically by predominantly monocytes and less commonly T- and B-lymphocyte infiltration into peripheral nerve and nerve root endoneurium with macrophage-mediated demyelination. (Hall et al., 1992; Hughes et al., 1992; Kiefer et al., 2001; Meyer zu Horste et al., 2007; Prineas, 1981; Schmidt et al., 1996; Ubogu, 2015) Recognizing that endoneurial endothelial cells provide a critical interface between circulating leukocytes and peripheral nerve endoneurium, we isolated, purified and characterized adult pHEndECs and developed a flow-dependent in vitro human BNB model to better understand pathogenic leukocyte trafficking in peripheral nerves. Guided by our in vitro studies that demonstrated the importance of leukocyte integrin CD11b (also known as αM-integrin or Mac-1) and ICAM-1 interactions in GBS patient leukocyte trafficking at the human BNB under hydrodynamic forces mimicking in vivo capillary flow rates,(Yosef and Ubogu, 2012a) We observed clusters of infiltrating CD11b+ leukocytes directly interacting with endoneurial microvessels as well as foci of endoneurial CD11b+ leukocytes associated with disrupted Schwann cell membrane organization (which typically results in demyelination of large and small myelinated axons) in untreated GBS patient sural nerve biopsies (Figure 9).(Dong et al., 2016) Interestingly, real-time leukocyte trafficking at the in vitro human BNB occurred via the paracellular route and we observed paracellular leukocyte trafficking in situ by electron microscopy, with focal electron dense contacts present between endoneurial endothelial cells and infiltrating GBS patient leukocytes during transmigration.(Dong et al., 2016; Yosef and Ubogu, 2012a)
Figure 9. CD11b leukocyte infiltration and demyelination in GBS (AIDP).
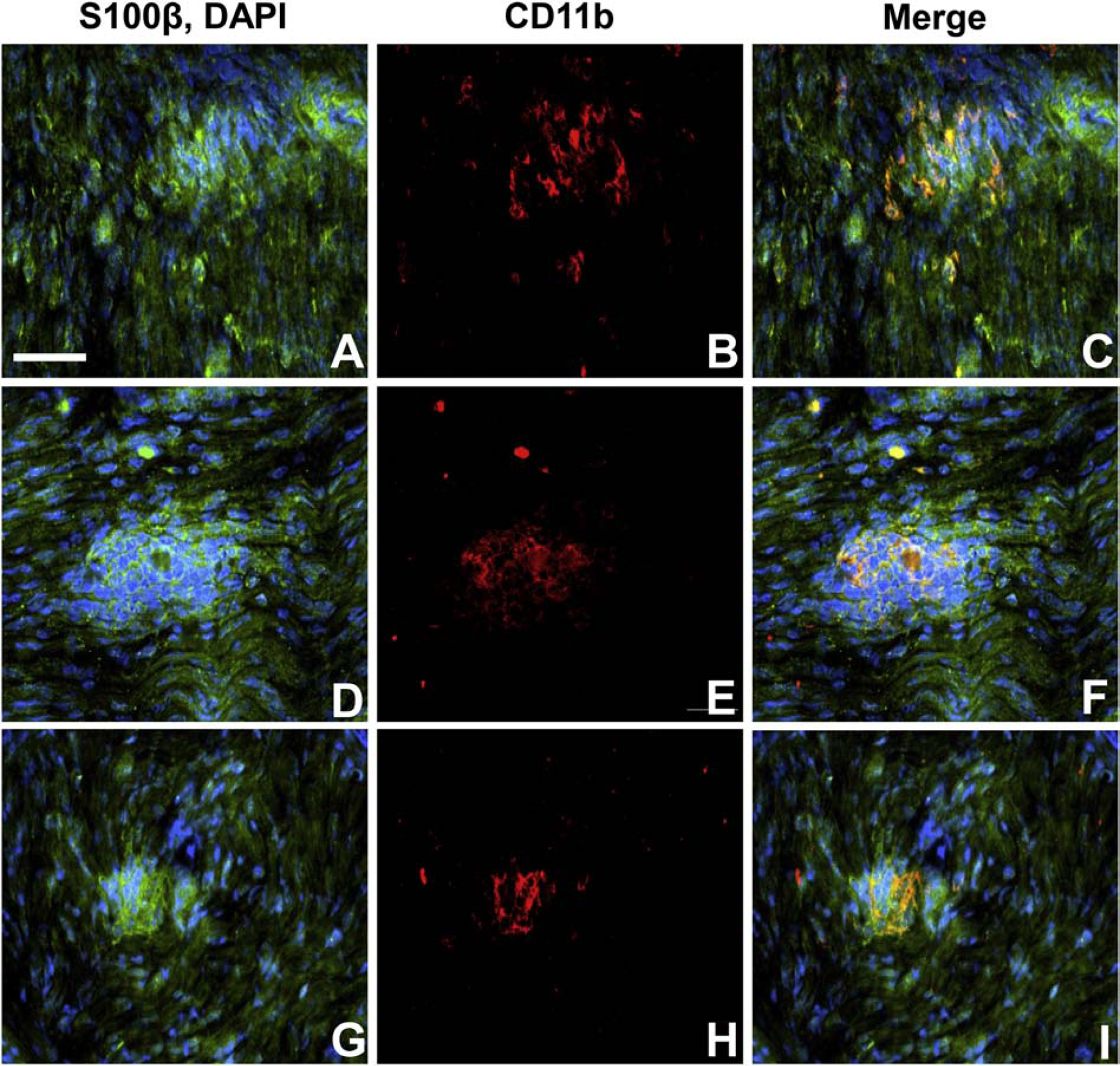
Digital indirect fluorescent photomicrographs of longitudinal sections from the sural nerves from 3 untreated adult patients with AIDP stained with S100β (green) to detect myelinating Schwann cells and membranes associated with axons (with DAPI [blue] detecting nuclei; A, D and G), show clusters of infiltrated endoneurial CD11b+ leukocytes (red; B, E and H) associated with disrupted Schwann cell membrane organization (C, F and I). Scale bar = 50 μm.
Our initial work on CD11b expression on circulating GBS (AIDP variant) leukocytes using flow cytometry demonstrated increased CD11b density expression on CD14+ monocytes, increased number of CD11b+ CD19+ B-lymphocytes and CD3+ CD8+ T-lymphocytes, and reduced numbers of CD3+ CD4+ T-lymphocytes (associated with class switch with higher CD3+ CD4+: CD3+ CD8+ T-cell ratio in GBS patients as seen in disorders associated with T-helper cell activation) compared to age- and sex-matched adult leukocytes.(Dong et al., 2016) We proposed that CD11b-mediated leukocyte trafficking is a potential therapeutic target for AIDP and monitoring leukocyte CD11b expression by flow cytometry could serve as a biomarker of disease activity. We subsequently performed whole exome shotgun sequencing (also known as RNA-Seq) on peripheral blood mononuclear leukocytes from GBS patients and observed an ~2.5-fold increase in CD11b transcript expression (normalized based on Fragments Per Kilobase of transcript per Million [FPKM]) compared to controls, further supporting our prior CD11b inferences in pathogenic GBS leukocyte trafficking at the human BNB and the role of RNA-Seq as an appropriate screening tool for potential determinants of pathogenic leukocyte trafficking in peripheral nerve inflammation.(Dong et al., 2016)
BNB in CIDP (classic variant)
Similar to the AIDP variant of GBS, CIDP is pathologically characterized by predominant infiltration of monocytes and T-lymphocytes, and less commonly B-lymphocytes into peripheral nerve and nerve root endoneurium with macrophage-mediated demyelination.(Dalakas, 2015; Mathey et al., 2015; Matsumuro et al., 1994; Rizzuto et al., 1998a; Schmidt et al., 1996; Sommer and Toyka, 2011; Ubogu, 2015) This supports an important pathogenic role for activated hematogenous leukocyte infiltration into peripheral nerves and nerve roots across the BNB, although the precise trigger(s) for autoimmune attack and lack of self-tolerance are undetermined.(Bouchard et al., 1999; Dong et al., 2017; Rizzuto et al., 1998b) As mentioned previously, we have observed structurally intact electron dense endothelial cell junctions and basement membrane duplication in endoneurial microvessels of CIDP patient peripheral nerve biopsies associated with perivascular or infiltrated leukocytes.(Bosetti et al., 2016) This provides an impetus to better understand pathogenic alterations at the BNB in CIDP patients that could guide specific therapeutic development.
Our recent published work, guided by observations at the BBB relevant to multiple sclerosis,(Man et al., 2009) demonstrated a direct role for the alternatively spliced fibronectin variant expressed on endothelium, called connecting segment-1 (FN CS-1), which interacts with α4 integrin (CD49d, also known as very late antigen-4), in CIDP patient leukocyte trafficking across the human in vitro BNB. The relevance of FN CS-1-CD49d mediated pathogenic CIDP leukocyte trafficking at the BNB was further supported in vivo using a reliable CIDP mouse model, spontaneous autoimmune peripheral polyneuropathy in female CD86-deficient non-obese diabetic mice, which recapitulates features of severe chronic demyelinating neuritis, and the detection of FN CS-1 expressing microvessels and clusters of trafficking and infiltrated CD49d+ leukocytes in CIDP patient nerve biopsies.(Dong et al., 2017) In order to innovatively identify potential disease specific targets unique for CIDP, we performed RNA-Seq on PBMLs from untreated adult CIDP patients compared to healthy age- and sex-matched controls [normalized based on FPKM] to identify integrins that may facilitate chronic pathogenic leukocyte trafficking at the BNB.
We interestingly observed a ~6.9-fold increase in CD11d (also known as αD-integrin) transcript expression in CIDP patients compared to healthy adult controls (Table 1). CD11d is a less well characterized integrin that forms heterodimers with CD18 (also known as β2-integrin), similar to CD11b, with expression on myeloid-derived dendritic cells, monocytes and macrophages in humans and mice.(Miyazaki et al., 2014; Schittenhelm et al., 2017; Villani et al., 2017) There is recent human data also showing CD11d expression on subsets of natural killer, B-lymphocytes and γδ T-lymphocytes(Siegers et al., 2017) (which are expressed at higher levels in CIDP nerve biopsies compared to GBS and non-inflammatory neuropathies).(Winer et al., 2002) CD11d biology is not well understood. It may facilitate leukocyte trafficking (via binding to an undetermined endothelial counterligand in humans) or pro-inflammatory macrophage tissue retention in chronic inflammatory diseases, such as CIDP.(Aziz et al., 2017; Yakubenko et al., 2008)
Table 1. PBML integrin transcript expression in untreated CIDP patients vs. controls by RNA-Seq.
Comparative untreated CIDP patient PBMLs demonstrate high relative CD11d integrin transcript expression compared to age- and sex-matched controls, suggesting CD11d as potentially relevant to CIDP pathogenesis
| Gene | Common Name(s) | CONTROL FPKM | CIDP FPKM | Fold Δ (CIDP vs control) |
|---|---|---|---|---|
| ITGA2 | α2 integrin, CD49b | 0.81 | 0.47 | 0.58 |
| ITGA3 | α3 integrin, CD49c | 1.15 | 0.94 | 0.81 |
| ITGA4 | α4 integrin, CD49d | 51.90 | 61.10 | 1.18 |
| ITGA5 | α5 integrin, CD49e | 33.10 | 32.20 | 0.98 |
| ITGA6 | α6 integrin, CD49fB | 21.5 | 29.7 | 1.38 |
| ITGA7 | α7 integrin | 0.48 | 0.51 | 1.06 |
| ITGA10 | α10 integrin | 0.50 | 0.35 | 0.68 |
| ITGAD | αD integrin, CD11d | 0.14 | 0.98 | 6.91 |
| ITGAE | αE integrin, CD103 | 7.87 | 9.74 | 1.24 |
| ITGAL | αL integrin, CD11a | 144.00 | 181.00 | 1.25 |
| ITGAM | αM integrin, CD11b | 78.4 | 68.6 | 0.88 |
| ITGAV | αV integrin, CD51 | 2.49 | 2.68 | 1.08 |
| ITGAX | αX integrin, CD11c | 90.1 | 61.0 | 0.68 |
| ITGB1 | B1 integrin, CD29 | 123.4 | 116.5 | 0.94 |
| ITGB2 | β2 integrin, CD18 | 478.6 | 499.0 | 1.04 |
| ITGB3 | β3 integrin, CD61 | 10.1 | 6.39 | 0.63 |
| ITGB5 | β5 integrin | 3.13 | 2.09 | 0.67 |
| ITGB7 | β7 integrin | 61.7 | 61.6 | 1.00 |
In our preliminary studies, we also observed increased ex vivo CD11d protein expression on CD14+ monocytes, CD19+ B-lymphocytes and CD3+ CD4+ T-lymphocytes, with reduced expression on CD3+ CD8+ and CD3+ CD4− CD8− (which also encompass γδ T-cells) T-lymphocytes in CIDP patients compared to healthy controls using flow cytometry (Figure 10). Importantly, we observed clusters of CD11d+ CD68+ macrophages (Figures 11A–C), CD19+ B-cells (Figures 11G–I) and perivascular CD3+ T-cells (Figures 11M–O) in untreated adult CIDP patient sural nerve biopsies with rare CD11d+ CD68+ monocytes (Figures 11D–F) and CD19+ B-cells (Figures 11J–L) seen in the lumens of endoneurial microvessels (white lines showing external vessel wall borders). Rare CD11d+ CD3+ T-cells were seen in control nerve endoneurium only (Figures 11P–R). These data suggest that CD11d upregulation and possible selective BNB transmigration into peripheral nerves/ nerve roots may occur in CIDP. Further studies are ongoing to further determine the pathogenic role(s) of CD11d in CIDP using our flow-dependent leukocyte-BNB trafficking model and a reliable animal model, spontaneous autoimmune peripheral polyneuropathy in female CD86-deficient non-obese diabetic mice. This could lead to specific therapeutics or disease monitoring biomarkers in CIDP.
Figure 10. CD11d expression in CIDP Patient PBMLs.
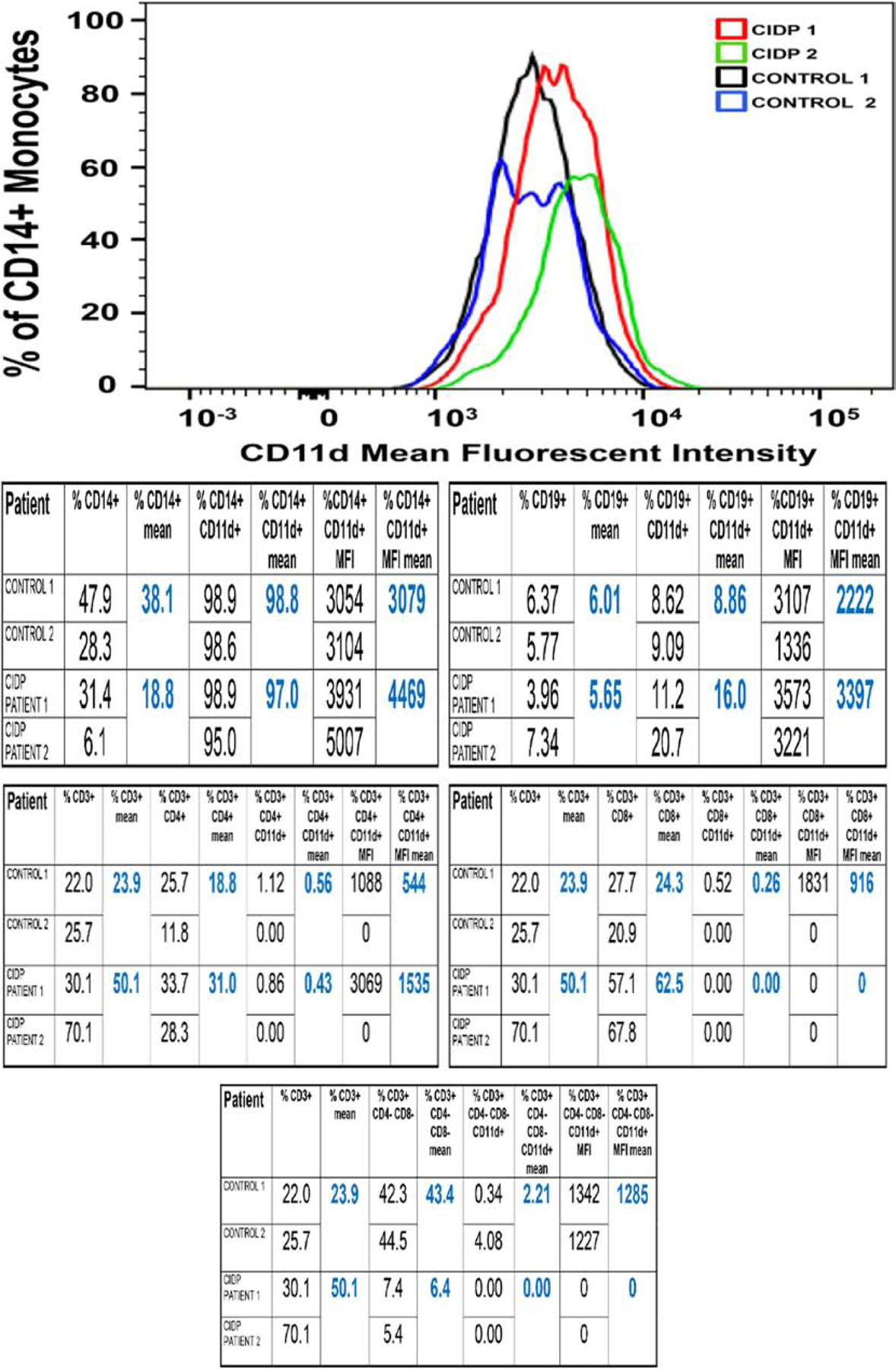
Merged FACS histograms showing CD14+ CD11d+ monocyte expression in 2 untreated CIDP patients (CIDP 1 and CIDP 2) compared to 2 age- and sex-matched controls (control 1 and control 2) show increased mean fluorescent intensity (MFI, a marker of receptor density) in the CIDP patients compared to controls. The tables show comparative CD11d expression data for CD14+ monocytes, CD19+ B-lymphocytes, as well as CD3+ CD4+, CD3+ CD8+ and CD3+ CD4− CD8− T-lymphocytes, suggesting a potential role in pathogenic leukocyte trafficking in CIDP.
Figure 11. CD11d expression in CIDP Patient sural nerve.
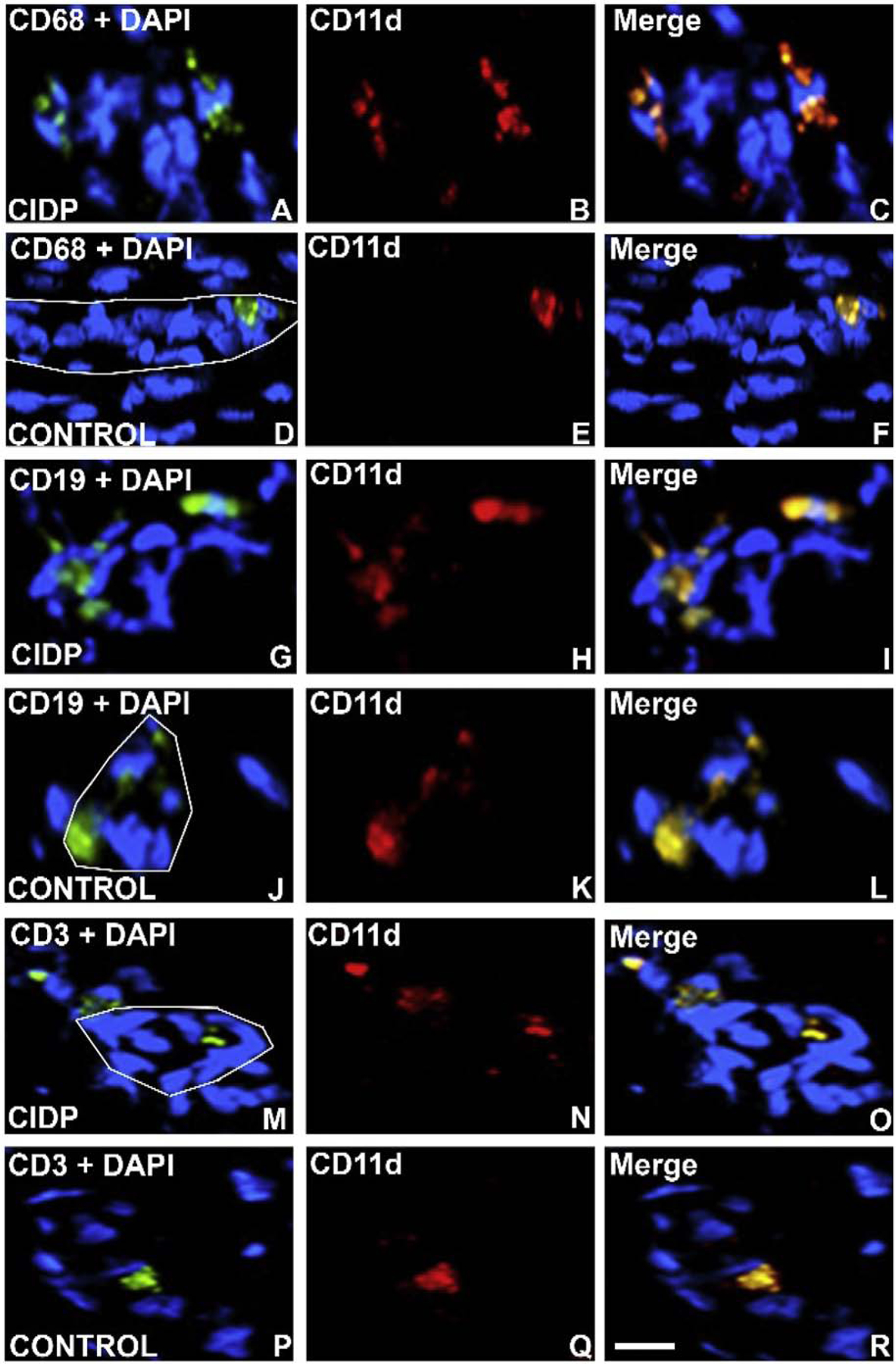
Digital indirect fluorescent photomicrographs of axial and longitudinal sections from 2 CIDP patients and age- and sex-matched adult controls stained with monocyte/ macrophage marker CD68, B-lymphocyte marker CD19 and T-lymphocyte marker CD3 (green) with nuclei stained with DAPI (blue; A, D, G, J, M, P) to co-localize with CD11d expression (red, B, E, H, K, N, Q), showing increased endoneurial expression in CD68+ CD11d+ monocytes/ macrophages (C), CD19+ CD11d+ B-lymphocytes (I) and CD3+ CD11d+ T-lymphocytes (O) compared to controls (F, L, R). The white lines depict the abluminal membrane of endoneurial microvessels in order to highlight adherent luminal CD11d+ leukocytes. Scale bar = 25 μm.
BNB in HIV Distal Sensory Neuropathy: The CCR5-CD11d-CD99L2 axis hypothesis
Human immunodeficiency virus (HIV)-associated peripheral nerve disorders are the most common neurological disorders associated with HIV infection.(Bacellar et al., 1994; Ellis et al., 2010; Evans et al., 2011; McArthur et al., 2005; Pardo et al., 2001; Schutz and Robinson-Papp, 2013; Simpson et al., 2006) Distal sensory neuropathy (DSN) is the most common HIV−associated peripheral neuropathy. HIV DSN is slowly progressive and associated with chronic refractory neuropathic pain.(Ellis et al., 2010; Evans et al., 2011; Pardo et al., 2001; Simpson et al., 2006; Verma, 2001) Despite significant advances in HIV outcomes and life expectancy with the use of combination antiretroviral therapy (cART) and the reduced administration of neurotoxic antiretroviral drugs, particularly in industrialized nations, the prevalence of DSN continues to rise.(2008; Bacellar et al., 1994; Ellis et al., 2010; Evans et al., 2011; Simpson et al., 2006) DSN affects ~60% of HIV+ patients in large cohorts (defined by 1 or more clinical signs of sensory dysfunction in a distal symmetrical pattern), with ~40% of affected patients reporting neuropathic pain.(Ellis et al., 2010) Peripheral nerves could serve as a sanctuary for HIV−infected leukocytes, in a similar way to the brain, supported by the observation that HIV−1 viral replication in peripheral nerves is rare, also as observed in the brain.(Jones et al., 2005; Nath, 2015)
The pathogenesis of HIV−associated peripheral neuropathy is not fully understood. Observational studies using sural nerves from affected patients implicate macrophage-mediated axonal injury in HIV DSN.(Pardo et al., 2001) Sequence analysis and infectious recombinant viruses containing peripheral nerve-derived membrane glycoprotein C2V3 sequences obtained from autopsy peroneal nerves from five HIV+ patients indicated a predominance of CCR5-dependent and macrophage tropic HIV−1 virus, implying an important role of CCR5+ macrophages in HIV DSN axonal degeneration.(Jones et al., 2005) It is highly plausible that chemokines and their G-protein coupled receptors actively participate in HIV DSN pathogenesis and in other forms of HIV−associated neuropathy by recruiting hematogenous CCR5 tropic HIV−infected leukocytes into peripheral nerves across the human BNB. Using our BNB model systems, transcript and protein analyses of well characterized HIV+ patient leukocytes and immunohistochemistry of HIV+ patient peripheral nerve biopsies compared to normal adult control specimens, we describe some of our preliminary observations pertaining to pathogenic leukocyte trafficking in HIV DSN that warrant further confirmation.
pHEndECs that form the BNB express high surface levels of CCR5 ligands, CCL4 and CCL5, under basal culture conditions in vitro based on a chemokine antibody array, with <1.5-fold increase following endothelial treatment with physiological concentrations of pro-inflammatory cytokines TNF-α and IFN-γ.(Yosef and Ubogu, 2012a) We have also observed CCL5 expression by the quiescent in vitro BNB at multiple cell passages by real-time quantitative polymerase chain reaction (rt-PCR) and western blot (Figures 12C and D respectively). Interestingly, we observed in situ transcript expression of CCR5 ligands CCL3, CCL4 or CCL5 by laser capture microdissected endoneurial microvessels in at least 2 of 4 normal adult peripheral nerves by RNA-Seq.(Palladino et al., 2017) These data suggest the redundancy, and relative importance of CCR5-mediated chemokine signaling at the normal adult human BNB. Coupled the published observations of predominance of CCR5-dependent and macrophage tropic HIV−1 virus predominance in HIV DSN, these data imply that CCR5+ monocytes from HIV+ patients may “hijack” constitutive immunosurveillence mechanisms to gain access into peripheral nerves, with subsequent axonal degeneration and chronic neuropathic pain.
Figure 12. CCL5 expression by the basal human in vitro BNB.
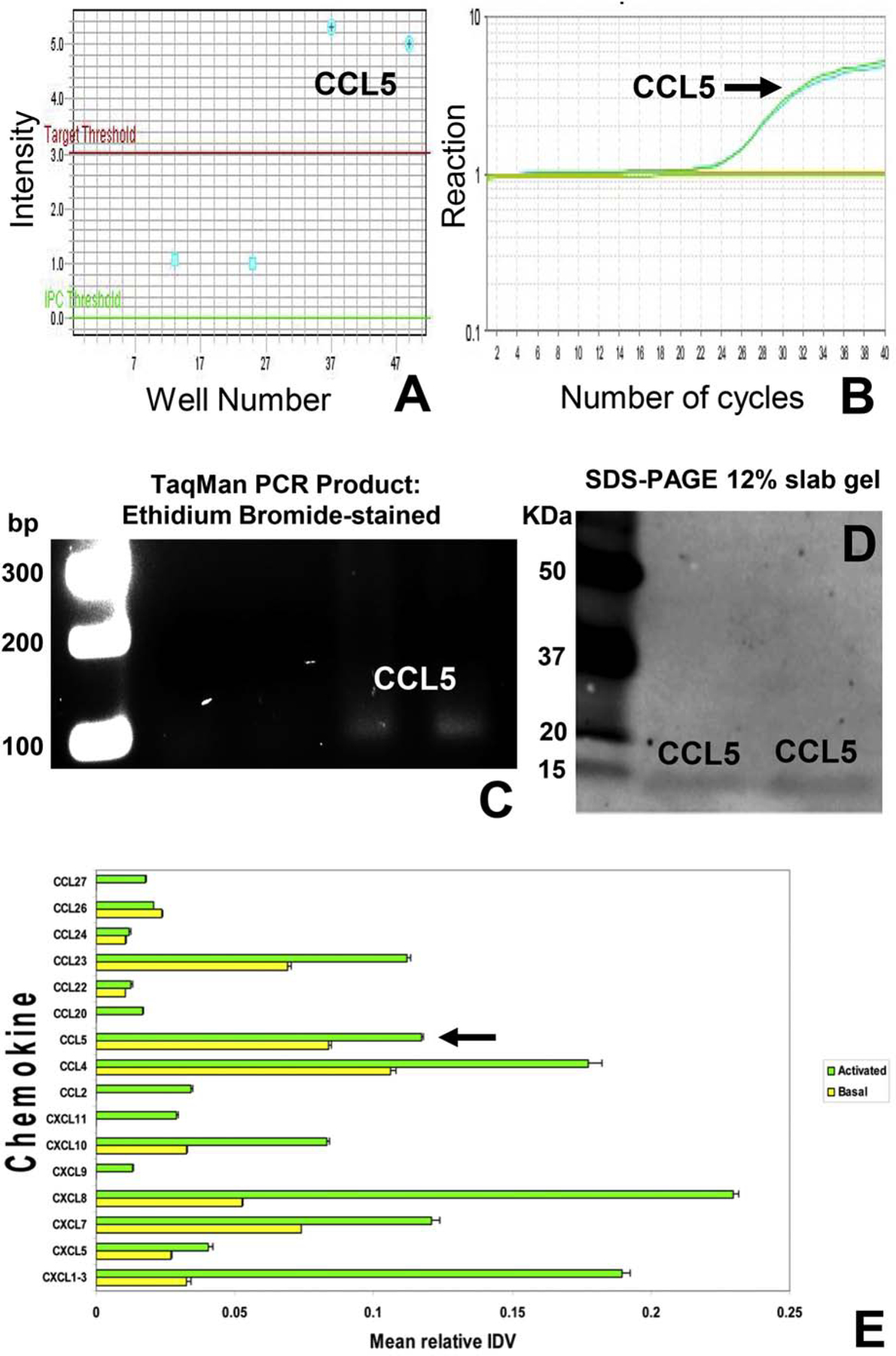
TaqMan rt-PCR was performed using complementary DNA obtained by reverse transcription of confluent cultured pHEndEC mRNA extracts 4–5 days after initial plating to detect CCL5 expression using specific validated primers. Expression was verified from 2 separate cultures via amplicon detection intensity above a predefined detection threshold (A), detection of PCR reaction with appropriate sigmoidal amplification with repetitive cycles (B) and verification of expected PCR product using ethidium bromide-stained gel electrophoresis (C, with numbers indicating base pair [bp] size markers), compared to negative controls that lack specific primers. CCL5 pHEndEC protein expression was confirmed in duplicate by western blot (most likely as a dimer of ~ 15 KDa) using a specific validated primary goat-anti human antibody following SDS-PAGE of total cytoplasmic and membrane protein extracts from confluent cultures, using standard protocols (D). Numbers indicate molecular weight of the protein standard markers, in KDa. Bar histograms from a human chemokine antibody array show chemokine expression by basal confluent pHEndECs (yellow) and 10 U/mL TNF-α and 20 U/mL IFN-γ cytokine treated pHEndECs for 24 hours in vitro, relative to internal array controls. CCL5 expression is shown with black arrow, with no significant upregulation following physiological cytokine stimulus.
To further validate CCR5+ leukocyte infiltration into peripheral nerves in HIV disease, we performed indirect immunohistochemistry on frozen axial and longitudinal sural nerve biopsy sections from a 53 year old HIV+ man (CD4 T-cell count 242, and viral load <20 copies) with clinical and histopathological evidence of DSN. We observed CD68+ CCR5+ monocytes/ macrophages in clusters within the endoneurium (Figures 13A–C), with several of these cells attached to an endoneurial microvessel in longitudinal section (Figure 13D), implying preferential adhesion and subsequent transmigration in HIV DSN. CD3+ CD4+ CCR5+ (Figure 13E) and CD3+ CD8+ CCR5+ T-cells (Figure 13F) were also observed but less commonly seen within the endoneurium compared to monocytes/ macrophages. This data provides some support to the hypothesis that CCR5+ monocytes may preferentially migrate across the human adult BNB in HIV DSN, utilizing endogenously expressed chemokine ligands on endoneurial microvessels.
Figure 13. CCR5 expression in HIV DSN Patient sural nerve: IHC.
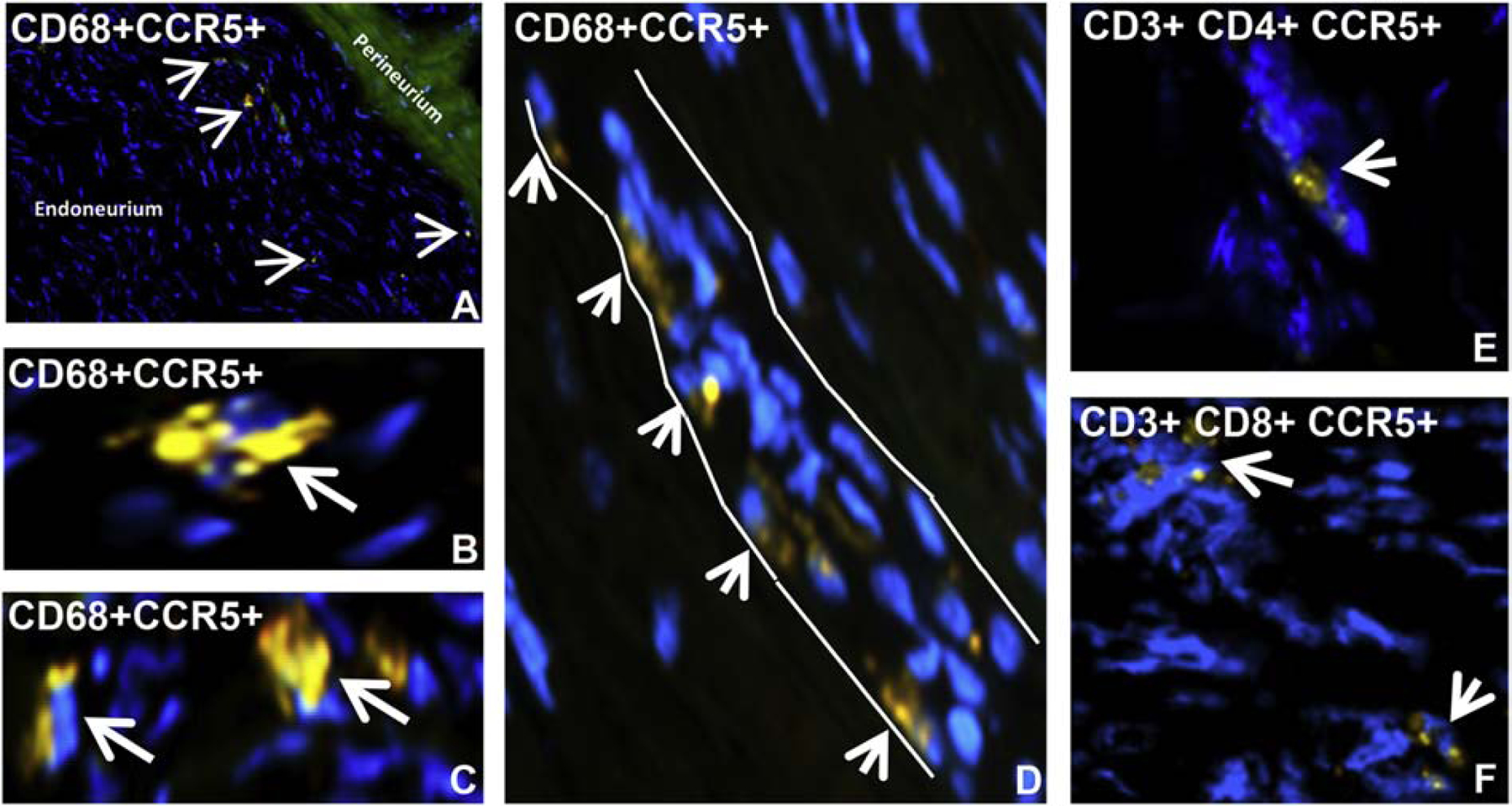
Merged digital indirect fluorescent photomicrographs of axial and longitudinal sections from a HIV+ patient with DSN show clusters of endoneurial CCR5+ CD68+ monocytes/macrophages at low (A) and higher magnification (B and C, white arrows), and an endoneurial microvessel (white outline) with multiple luminal adherent CCR5+ CD68+ monocytes (white arrow heads, D). Less common endoneurial clusters of CCR5+ CD4+ T-lymphocytes (E) and CCR5+ CD8+ T-lymphocytes (F) are also observed in this specimen.
Using cryopreserved PBMLs from a mean age- and sex-matched cohort of 36 HIV+ and 18 HIV− adults, we performed flow cytometry to determine CCR5 expression on circulating leukocytes to further evaluate our hypothesis on the role of CCR5 in HIV DSN. Our data showed that the mean total % of the pathogenic CD14+ CD16+ monocytes (13.8% vs 4.0%) and its CCR5+ subset (18.8% vs 9.6% of CD14+ CD16+ cells) were increased in HIV+ patients compared to HIV− controls, associated with a mean 354% increase in CCR5 mean fluorescent intensity (Figures 14A–B). No difference in mean % CCR5 expression in CD3+ CD4+ (19.0% vs 18.7%) or CD3+ CD8+ (28.5% vs 25.0%) T-lymphocytes was observed; however, there was a mean 191% and 230% increase in CCR5 mean fluorescent intensity respectively, implying increased CCR5 density per T-lymphocyte in HIV+ patients (Figures 14C–D).
Figure 14. PBML CCR5 expression in untreated HIV+ patient vs. HIV− controls.
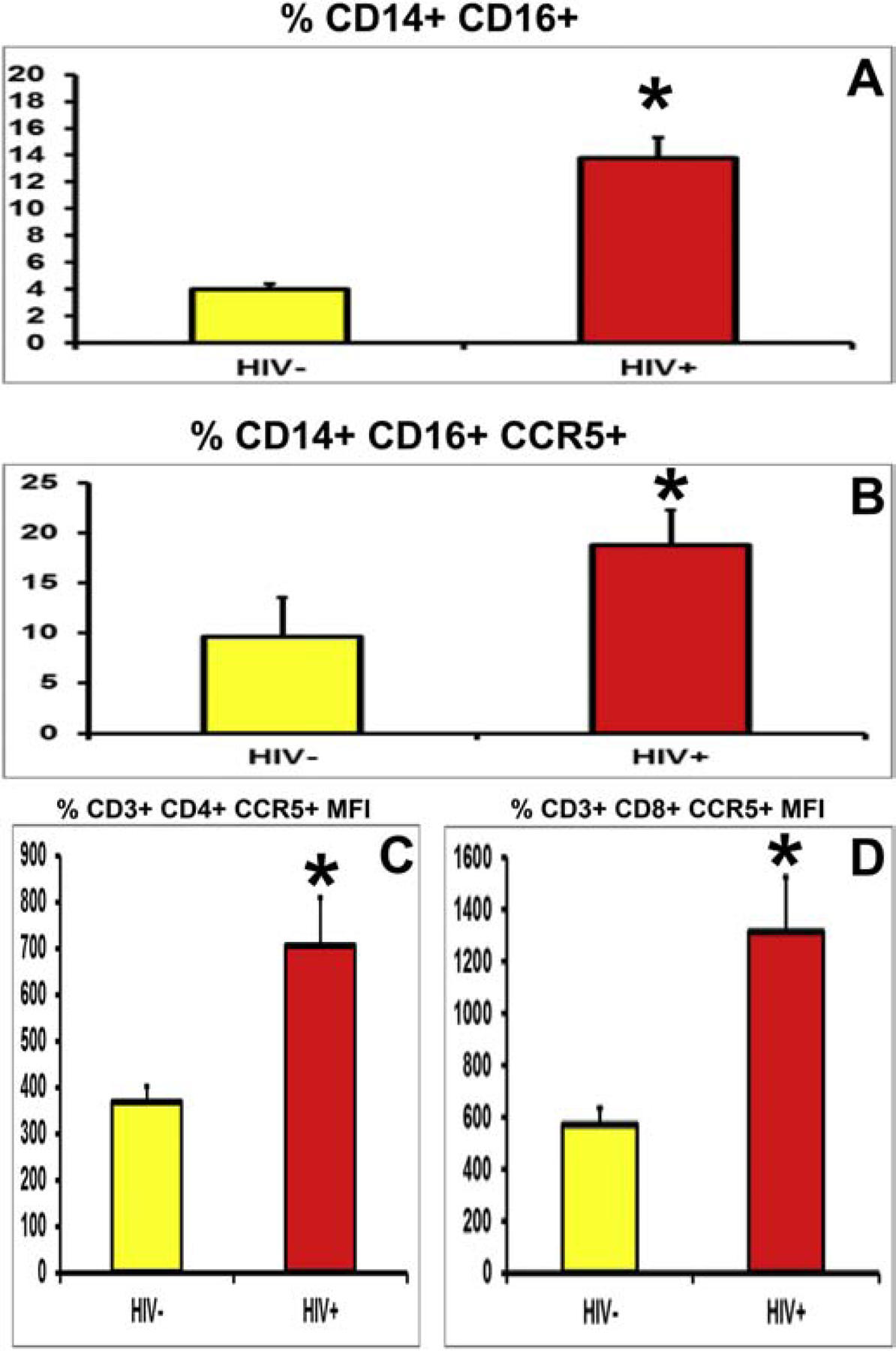
Comparative histograms of HIV+ cART-naïve patients and age- and sex-matched controls show increased mean % of CD14+ CD16+ (A) and CD14+ CD16+ CCR5+ (B) monocytes in HIV+ patients, as well as increased CCR5 mean fluorescent intensity (marker of receptor density per cell) in CD3+ CD4+ (C) and CD3+ CD8+ (D) T-lymphocytes, suggesting a potential role in pathogenic leukocyte trafficking in HIV−DSN.
Using our published flow-dependent leukocyte-human in vitro BNB model,(Greathouse et al., 2016; Ubogu, 2013a) we studied real-time leukocyte trafficking of a 38 year old HIV+, cART-naïve man (CD4 T-cell count 814, viral load 32,000) at the under basal culture conditions by time-lapse video microscopy, mimicking leukocyte trafficking into peripheral nerves relevant to early HIV DSN. To determine whether CCR5 signaling was required for HIV+ leukocyte trafficking, we used a specific function neutralizing mouse anti-human CCR5 IgG1 antibody (clone 2D7) and quantified leukocyte trafficking relative to uninhibited conditions as an adhesion/ migration index (AMI), as previously published.(Dong et al., 2017; Yosef and Ubogu, 2012a) A concentration of 0.1 μg/mL (100 ng/mL) maximally reduced HIV+ leukocyte AMI to 0.52 (mean of 22 cells vs. 42 cells in a under basal, uninhibited conditions), with no further reduction seen with higher antibody concentrations (Figure 15). This exciting preliminary data, coupled with our observations on CCR5 expression in HIV+ patient leukocytes in circulation and in HIV DSN peripheral nerves, as well as endogenous ligand expression at the BNB in vitro and in situ, support the notion that CCR5 may actively participate in pathogenic HIV+ leukocyte trafficking at the human BNB and entry into peripheral nerve endoneurium. Studies are ongoing to further validate this hypothesis.
Figure 15. CCR5 blockade during HIV+ PBML trafficking at the human in vitro BNB under flow.
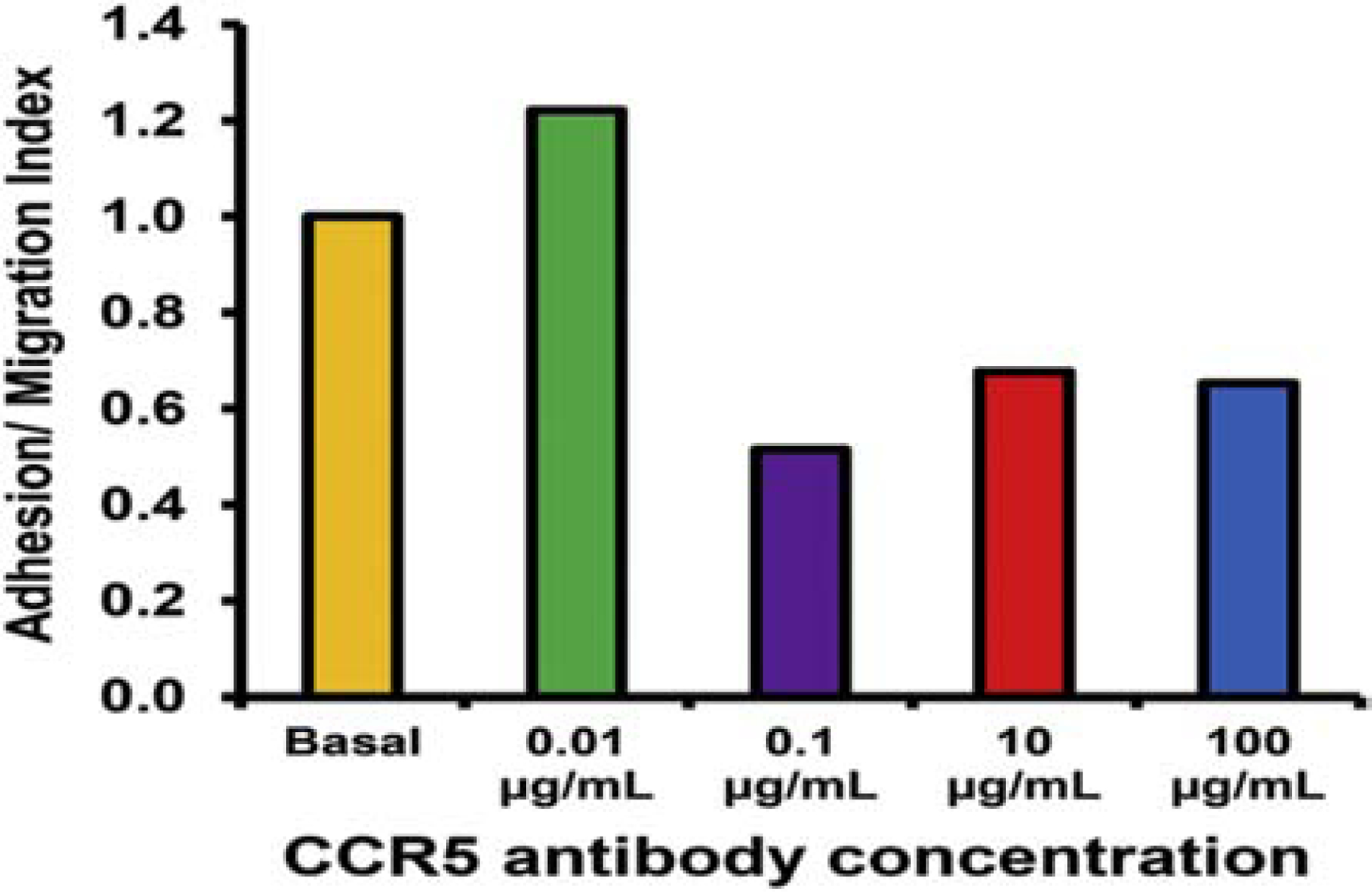
0.1 μg/mL of function neutralizing mouse anti-human CCR5 antibody maximally inhibited a single HIV+ cART-naïve PBML adhesion/ migration at the in vitro basal human BNB, mimicking conditions that may occur during the early stages of HIV infection, using our published leukocyte-BNB dynamic trafficking assay. Numbers are normalized to trafficking measured during basal PBML migration without antagonists or agonists.
As described with CIDP, we performed RNA-Seq to determine which integrin(s) could potentially drive pathogenic leukocyte trafficking in HIV DSN downstream of CCR5 signaling in cART-naïve HIV+ patients compared to age- and sex-matches controls. We observed a significant 22.3-fold increase in CD11d (also known as αD integrin) expression, with no significant comparative differences in other α and β integrins, so we hypothesized that CD11d (known to be expressed monocytes/macrophages, neutrophils and a subset of T-cells, B-cells, dendritic cells and natural killer cells, with roles on inflammation and tissue injury)(Miyazaki et al., 2014; Podolnikova et al., 2016; Siegers et al., 2017; Weaver et al., 2015) could drive HIV+ leukocyte trafficking at the blood-nerve barrier in HIV DSN. To further support of this hypothesis, we observed CD11d+ leukocytes actively migrating across endoneurial microvessels (Figures 16A–B) and present with peripheral nerve endoneurium (Figures 16 C–E) by indirect immunohistochemistry in the sural nerve biopsy of a patient with HIV DSN. As previously mentioned, the endothelial counterligand for CD11d is unknown, with constitutively expressed ICAM-1 or VCAM-1 being possible options in HIV DSN.
Figure 16. CD11d expression in HIV DSN Patient sural nerve.
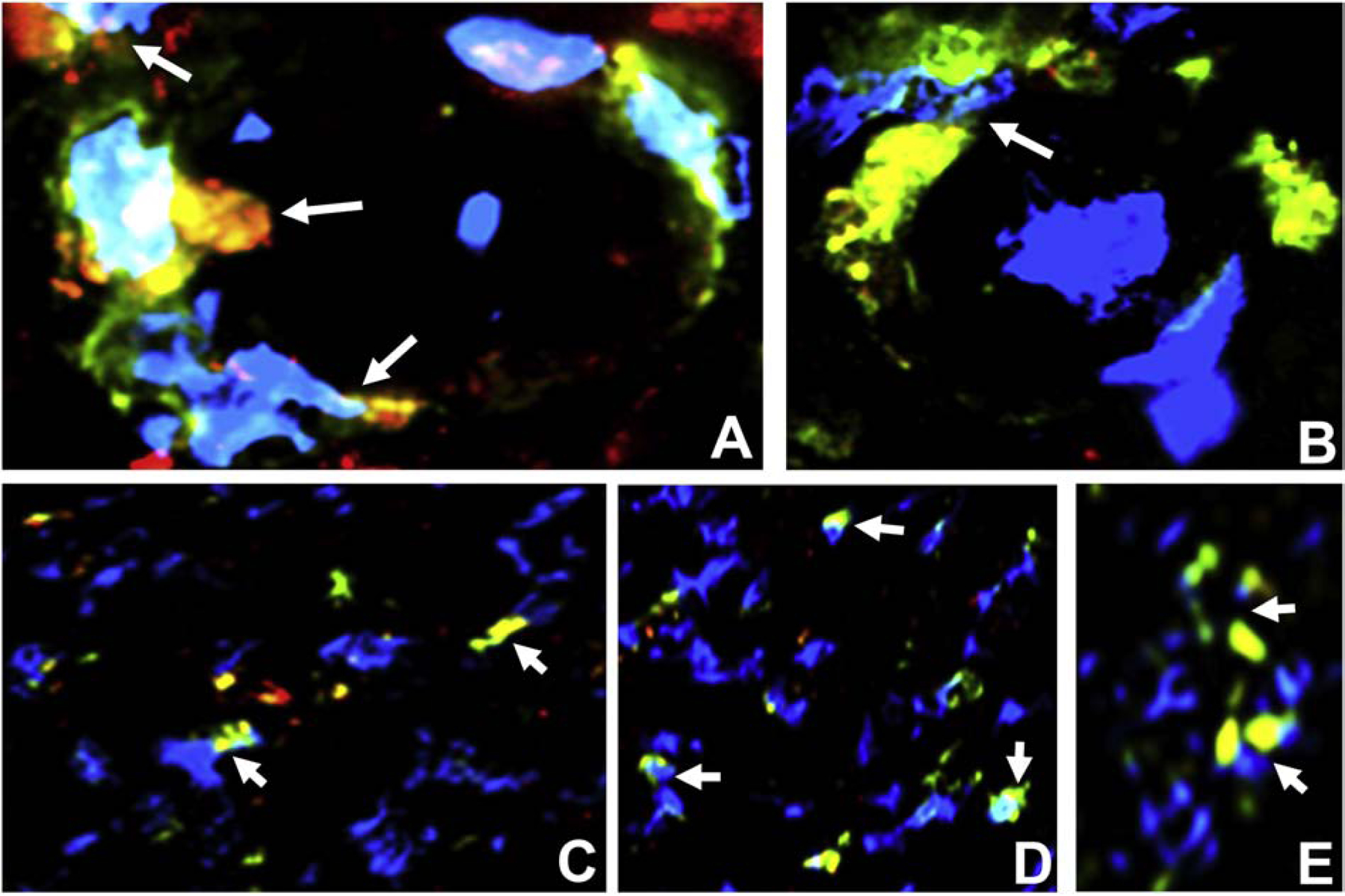
Merged digital indirect fluorescent photomicrographs of axial sections from a HIV+ patient with DSN show CD11d+ CD45+ leukocytes (white arrows) trafficking across endoneurial microvessels (A and B) at high magnification. CCR5+ CD68+ monocytes/macrophages (white arrows, A, B). Clusters of endoneurial CD11d+ CD45 leukocytes at different magnifications are also shown (C, D and E).
Using a proprietary mouse anti-human CD11d IgG1 antibody (clone CB6, Eli Lilly and Company), we performed flow cytometry to determine whether CD11d was preferentially expressed on CD14+ CD16+ CCR5+ monocytes, the hypothesized HIV DSN effector leukocyte, compared age- and sex-matched HIV− controls. Our initial studies showed increased % of circulating CD14+ CD16+ and CD14+ CD16+ CCR5+ monocytes in the HIV+ patients compared to the HIV− controls, with 100% CD11d+ expression in these CCR5+ monocytes cells (Figure 17), supporting notion that this integrin could participate in pathogenic HIV+ effector monocytes trafficking across the human BNB in HIV DSN. Our flow-dependent human in vitro BNB-leukocyte trafficking model provides an avenue to further study biologically relevant integrin signaling pathways at the BNB in HIV DSN, guided by our knowledge of the human BNB transcriptome.
Figure 17. PBML CD11d expression in untreated HIV+ patient vs. HIV− control.
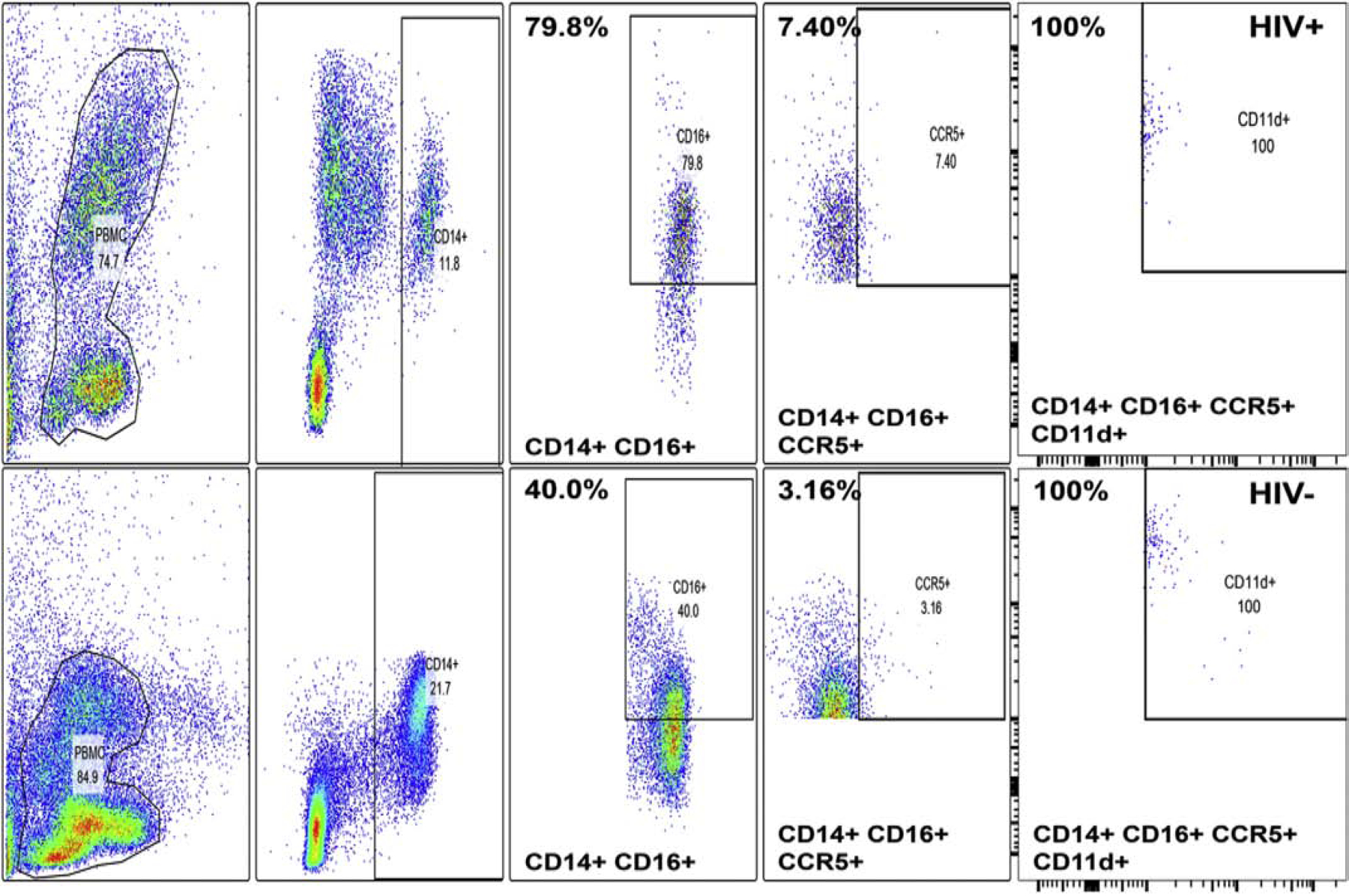
Comparative FACS gated color dot plots for cryopreserved PBMLs from HIV+ cART-naïve patient and age- and sex-matched HIV− control show increased relative expression of CD16+, CD16+ CCR5+ CD14+ monocytes in the HIV+ patient with 100% of CD14+ CD16+ CCR5+ cells being CD11d+ in both the patient and HICV control. Upper panels indicate HIV+ patient data while lower panels represent the HIV− control data, with the numbers showing % of the indicated subpopulation in each panel. These data suggest the HIV effector cell, CD14+ CD16+ monocytes may utilize CD11d integrin for leukocyte trafficking downstream of CCR5.
Ultrastructural observations of trafficking monocytes at the human BNB in situ showed electron dense leukocyte-endothelial cell contacts during paracellular diapedesis.(Dong et al., 2016) Molecules such as junctional adhesion molecules-A,-B and -C, platelet-endothelial cell adhesion molecule-1, CD99 and an evolutionally conserved related glycoprotein, CD99 Molecule Like 2 (CD99L2), are expressed on both leukocytes and endothelial cells, and have been implicated in leukocyte diapedesis via hemophilic (i.e. self-to-self) interactions.(Muller, 2003, 2009, 2011, 2014; Schenkel et al., 2007; Seelige et al., 2013) Based on our published human BNB transcriptome(Palladino et al., 2017) and RNA-Seq performed on PBMLs from HIV+ cART-treatment naïve patients and age- and sex-matched controls, we observed that CD99L2 transcript was reliably expressed both the human BNB and HIV+ leukocytes, implying a possible role in HIV+ trafficking at the BNB downstream of chemokine-driven, integrin-mediated leukocyte adhesion.
We observed focal CD99L2 expression on transmigrating HIV+ leukocyte filopodia and endoneurial microvessels in HIV DSN by indirect immunohistochemistry (Figures 18A–B), supporting our hypothesis that implies homophilic CD99L2 interactions may be important for HIV+ leukocyte transmigration at the BNB in DSN. Overall, our preliminary data supports the notion that the presumed pathogenic leukocyte effector cells, CD14+ CD16+ monocytes, utilize CCR5 for chemoattraction and binding to constitutively endoneurial endothelial cell expressed CCR5 ligands such as CCL4 and CCL5; activating CD11d that possibly binds to endothelial cell ICAM-1 or VCAM-1, followed by homophilic leukocyte-endothelial cell CD99L2 binding to drive migration across the BNB via the paracellular route in HIV DSN (Figure 19). This is the “CCR5-CD11d-CD99L2 axis” hypothesis that we plan to further study using our experimental model systems, not only to better understand pathogenic leukocyte trafficking into peripheral nerves in HIV, but to identify potential specific targets to treat HIV−DSN as well as elucidate mechanisms of peripheral nerve immunosurveillence.
Figure 18. CD99L2 expression during diapedesis in HIV DSN Patient sural nerve.
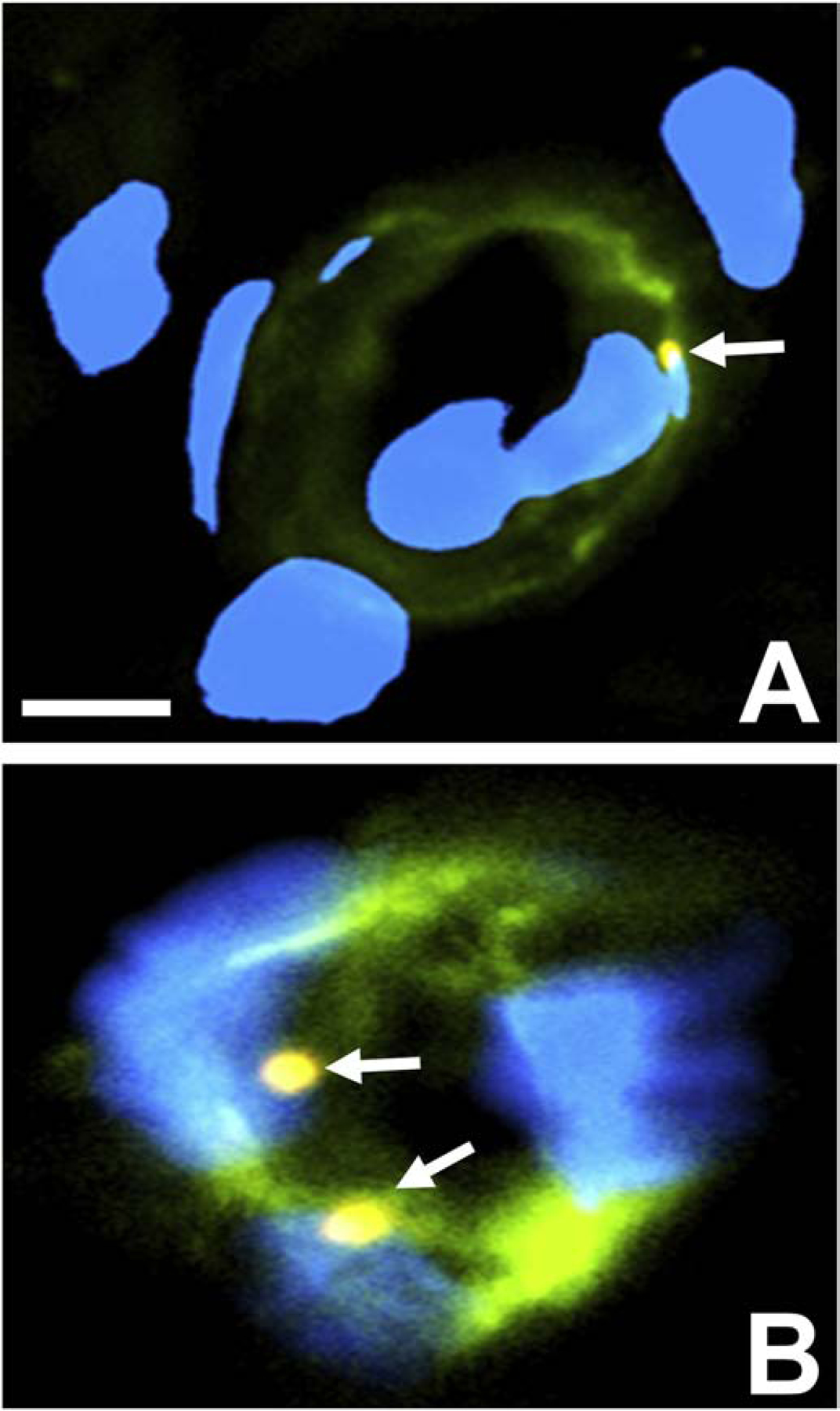
Merged digital indirect fluorescent high magnification photomicrographs of axial sections from a HIV+ patient with DSN show focal CD99L2 co-localization (white arrows, A and B) between infiltrating leukocytes and UEA-1+ endoneurial microvessels (green) with nuclei stained with DAPI (blue), implying a role in HIV+ leukocyte diapedesis associated with DSN. Scale bar = 5 μm.
Figure 19: Leukocyte Trafficking in HIV DSN: The CCR5-CD11d-CD99L2 hypothesis.
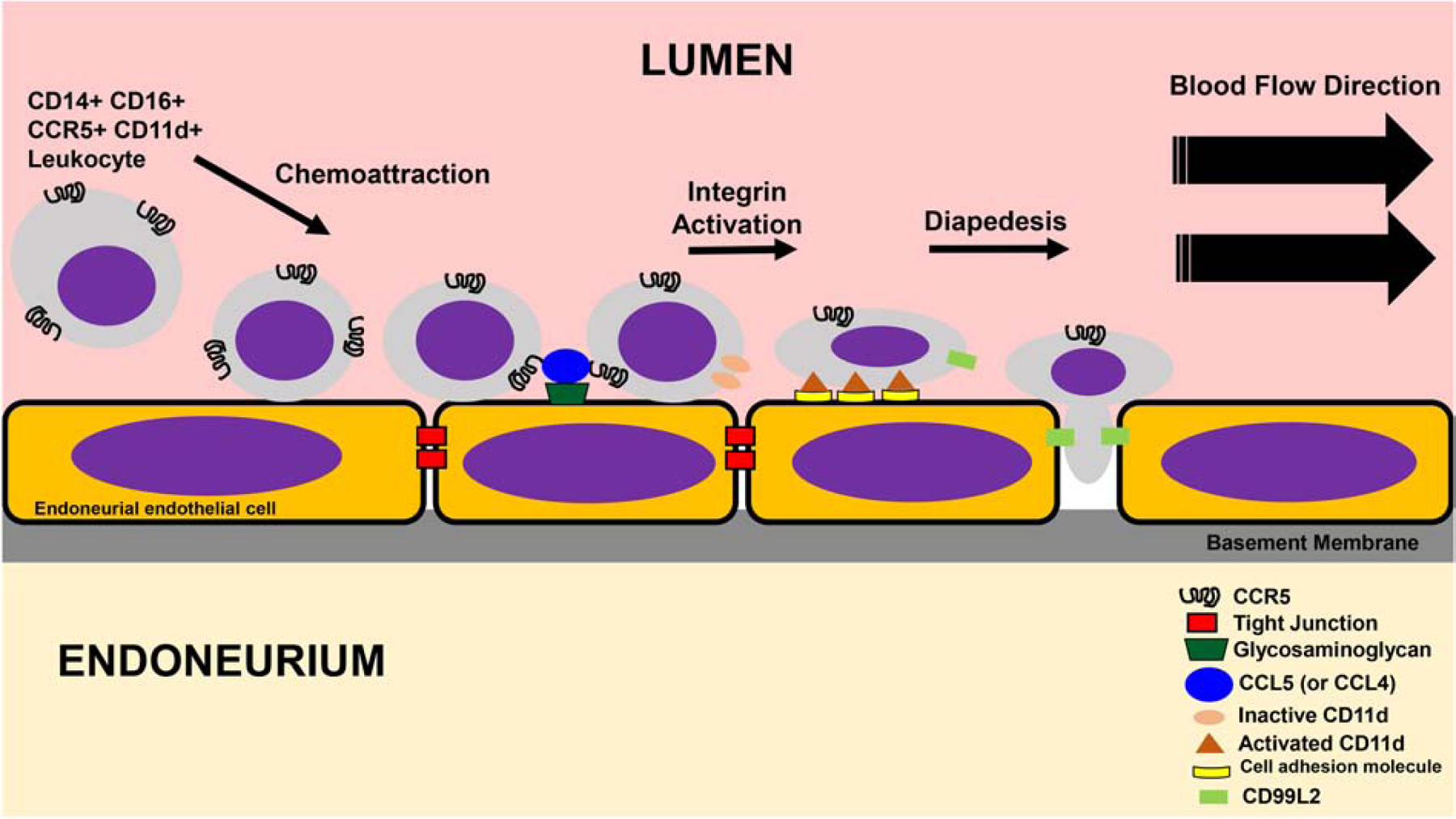
Guided by the multistep paradigm for leukocyte trafficking and emerging data on HIV−DSN, the figure illustrates chemoattraction of hematogenous CD14+ CD16+ CCR5+ monocytes to endoneurial microvascular endothelial cells that express CCL5 on the luminal (apical) surface membranes bound to glycosaminoglycans. CCR5-CCL5 binding results in a conformational change in CD11d integrin that converts it from a non-binding to a binding molecule (integrin activation) that induces firm leukocyte adhesion with flattening due to strong flow-resistant binding and interactions with an unknown endothelial surface cell adhesion molecule (? VCAM-1, ICAM-1). This is followed by paracellular diapedesis via CD99L2 hemophilic interactions between infiltrating leukocytes and endoneurial endothelial cells en route to the endoneurium to carry out effector functions.
BNB dysfunction and chronic neuropathic pain
Despite significant advances in biomedical research that have discovered molecular mediators and cellular pathways involved in nociception, there has been a disappointing lack of translation of these advances into safe and efficacious chronic neuropathic pain treatments for patients.(Yezierski and Hansson, 2018) For many of the current therapies, it is also unknown how penetrant these drugs are into the normal or diseased peripheral nerve and whether these drugs are sufficiently retained within nerves following systemic administration as required for sustained efficacy in controlling chronic neuropathic pain.
Studies have demonstrated the importance of non-neural cells in the initiation and persistence of chronic nociception in animal models with potential relevance to human diseases. These cells include mast cells, basophils, platelets, macrophages, neutrophils, endothelial cells, keratinocytes, and fibroblasts.(Basbaum et al., 2009) Although the direct contribution of peripheral nerve vascular dysfunction to neuropathic pain has not been comprehensively studied, evidence of microvascular disturbances has been reported in both humans experiencing chronic neuropathic pain and in neuropathic pain animal models. For example, in diabetic neuropathy, pathological changes result in endoneurial microvessel basement membrane thickening and reduced luminal diameter.(Richner et al., 2018; Yasuda and Dyck, 1987) These alterations were also observed in patients with nerve compression,(Mackinnon et al., 1986) vasculitic neuropathy and CIDP (see Figure 8).(Bosetti et al., 2016; Ubogu, 2015) Similar microvascular alterations have been observed in traumatic models of painful neuropathy.(Sommer et al., 1993; Sommer and Myers, 1996) Furthermore, vascular dysfunction has been proposed as a direct potential mechanism for some types of chronic pain. Restriction of blood flow and tissue ischemia results in complex regional pain syndrome-like symptoms in rats.(Coderre et al., 2004) In rodent models of nerve ischemia, photochemical damage to blood vessels in peripheral nerves generates neuropathic pain behavior.(Hao et al., 2000; Kupers et al., 1998)
A direct important role for BNB dysfunction in generating neuropathic pain, associated with increased endoneurial permeability for small and large molecules or accumulation of blood-borne substances and hematogenous inflammatory cells that are normally excluded from peripheral nerves, has been recently observed in the rodent partial sciatic nerve ligation and sciatic and infraorbital nerve chronic constriction injury models.(Lim et al., 2015; Lim et al., 2014; Maiuolo et al., 2019; Moreau et al., 2016; Moreau et al., 2017) Chronic aberrant signaling from diseased peripheral nerves is a plausible contributor to chronic neuropathic pain experienced by peripheral neuropathy patients.
In support of our in vitro human BNB studies on the molecular determinants and signaling pathways involved in restrictive barrier function, we have recently demonstrated that GDNF restores murine sciatic nerve BNB macromolecular impermeability within 14 days following non-transecting crush nerve injury using a Tamoxifen-inducible conditional GDNF knockout (GDNF CKO) transgenic mouse strain.(Dong et al., 2018) Restoring BNB function may be a necessary requisite to re-establish the tightly regulated microenvironment required for axonal regeneration and normal physiologic signal transduction in peripheral neuropathies, and as a consequence, contribute to the alleviation or cessation of chronic neuropathic pain. This hypothesis is supported by a study demonstrating restrictive BNB recovery preceding complete axonal regeneration by 6 weeks in a rat sciatic nerve crush injury model.(Bridge et al., 1994)
Guided by prior published GDNF research(He et al., 2008; Ibanez and Andressoo, 2017; Takahashi, 2001) and our published human BNB studies that have generated whole exome sequencing in situ and in vitro transcriptomes, in vitro tight and adherens junction rt-PCR array data as well as quantitative proteomic data of pHEndECs (whole cell homogenate, membrane protein and cytosolic protein fractions) under basal and GDNF-stimulated conditions, with network/ pathway identification analyses,(Dong and Ubogu, 2018; Palladino et al., 2017; Yosef and Ubogu, 2012b; Yosef et al., 2010) and our preliminary data implicating CREB1, CTTN, SRC kinase, CTNNA1 and CLDN4 in human BNB formation downstream of GDNF-RET-MAPK signaling (see Figures 3–6), we are performing further studies to determine the temporal and spatial relationship of these molecules in chronic peripheral neuropathies associated with neuropathic pain, supported by murine peripheral nerve injury models.
Using confocal microscopy following indirect fluorescent immunohistochemistry to visualize 40 μm-thick axial sections of a cryopreserved human sural nerve biopsy from a patient with primary vasculitic neuropathy with documented severe chronic neuropathic pain, we observed loss of CTNNA1 (Figures 20A–C) and CLDN4 (Figures 20 G–I) expression in endoneurial microvessels, in contrast to CTNNA1 plaque-like bands associated with endoneurial endothelial cell membranes and its F-actin cytoskeleton (Figures 20D–F), and punctate CLDN4 expression on endoneurial cell membranes (as expected of a tight junction protein) in histologically normal adult sural nerves (Figures 20J–L).This interesting observation speculates an association between junctional molecule loss, endoneurial microvessel structural alternations and chronic neuropathic pain that requires further study.
Figure 20. Loss of CTNNA1 and CLDN4 loss in endoneurial microvessels in the sural nerve of a patient with vasculitic neuropathy with chronic neuropathic pain.
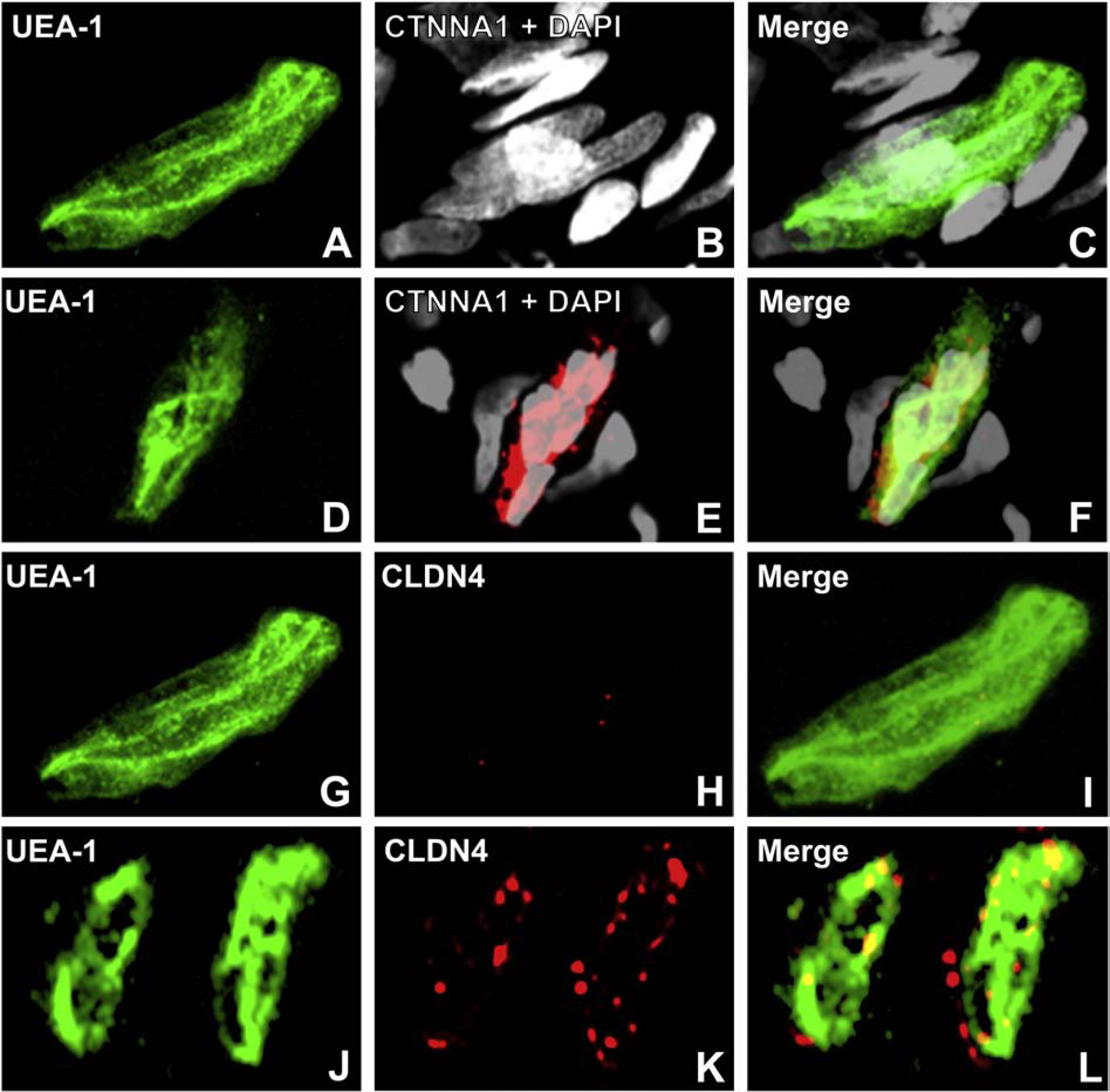
Digital high resolution indirect immunohistochemistry confocal micrographs of endoneurial microvessels from a patient with a vasculitic neuropathy and chronic neuropathic pain and age- and sex-matched normal adult control sural nerves stained with UEA-1 to detect endothelial cell membranes (green; A, D, G and J), and junctional complex molecules CTNNA1 (red, with nuclei stained with DAPI, pseudocolor grey, B and E) or CLDN4 (red, H and K), show loss of both endoneurial microvessel CTNNA1 (B and C) and CLDN4 (H and I) expression compared to the plaque-like linear CTNNA1 (E and F) and punctate CLDN4 (K and L, consistent with a tight junction protein) associated with normal endoneurial endothelial microvessel membranes. This observation suggests that molecular alterations of essential junctional complex proteins may occur in vasculitic neuropathy in association with chronic neuropathic pain.
As a means to experimentally evaluate chronic neuropathic pain in a murine axonal degeneration-regeneration model similar to chronic human peripheral neuropathies, we performed standardized validated reflexive neurobehavioral nociception tests of mechanical hypersensitivity, thermal hyperalgesia and cold allodynia in age- and sex-matched adult (>12 weeks old) GDNF wildtype (GDNF WT) and Tamoxifen-inducible GDNF CKO mice 4–6 weeks following right sciatic nerve crush injury, with left sciatic nerve Sham surgery serving as an internal control. As mentioned previously, our published work showed that GDNF restores BNB impermeability to a large macromolecule within 2 weeks after crush injury, with persistent permeability (implying chronic BNB dysfunction) observed following Tamoxifen-induced conditional gene knockout with abrogated GDNF expression.(Dong et al., 2018)
Mechanical hypersensitivity was studied by von Frey filament application using a simplified up-down method, (Bonin et al., 2014) thermal hyperalgesia was studied using the Hargreaves method (Hargreaves et al., 1988) and cold allodynia was studied using the cold plantar assay.(Brenner et al., 2012) Briefly, each mouse was placed in a Plexiglass chamber situated atop a Plexiglass platform and allowed to acclimate for an hour. A calibrated polypropylene monofilament (filaments numbered 2–9, North Coast Medical) was applied from below to the plantar surface of each hind paw and the force at which the hind paw was withdrawn recorded to study mechanical hypersensitivity. Starting with filament 5, the testing progressed up or down, such that a positive withdrawal response to a particular filament indicated use of the next lower value filament in the subsequent test, while a negative non-withdrawal response indicated use of the next higher filament. After each series of experiments, the von Frey filaments were calibrated and converted from paw withdrawal latencies to force (in grams).(Bonin et al., 2014) To evaluate thermal hyperalgesia, a moveable infrared generator connected to an automatic timing device (Ugo Basile) was placed below to the plantar surface of each hind paw and the paw withdrawal latency recorded (in seconds) after the stimulus application was measured. To evaluate cold allodynia, a cold probe consisting of a compacted powdered dry ice pellet was applied to each hind paw plantar surface and the paw withdrawal latency (in seconds) was manually recorded using a digital stopwatch.
The threshold for reflexive hind paw withdrawal was measured twice for each side and averaged to obtain a withdrawal threshold for each reflexive neurobehavioral test. For each mouse, we calculated the % of normal (left) withdrawal threshold for the crush injured (right) side and compared GDNF WT and GDNF CKO mice at different time points. We observed normalization of paw withdrawal thresholds for all modalities in GDNF WT mice ~4 weeks following sciatic nerve crush injury, with statistically significant lower mean % normal thresholds seen in GDNF CKO mice that persisted for up to 6 weeks post-injury based on one-tailed analysis of variance (Figures 21A–C). These data supports GDNF’s known analgesic effect on induced chronic nociception present in rodent peripheral nerve injury models,(Merighi, 2016) providing further rationale to evaluate whether specific molecules implicated in human BNB dysfunction are relevant to chronic neuropathic pain.
Figure 21. Chronic reflex nociception in transgenic GDNF mice following sciatic nerve crush injury.
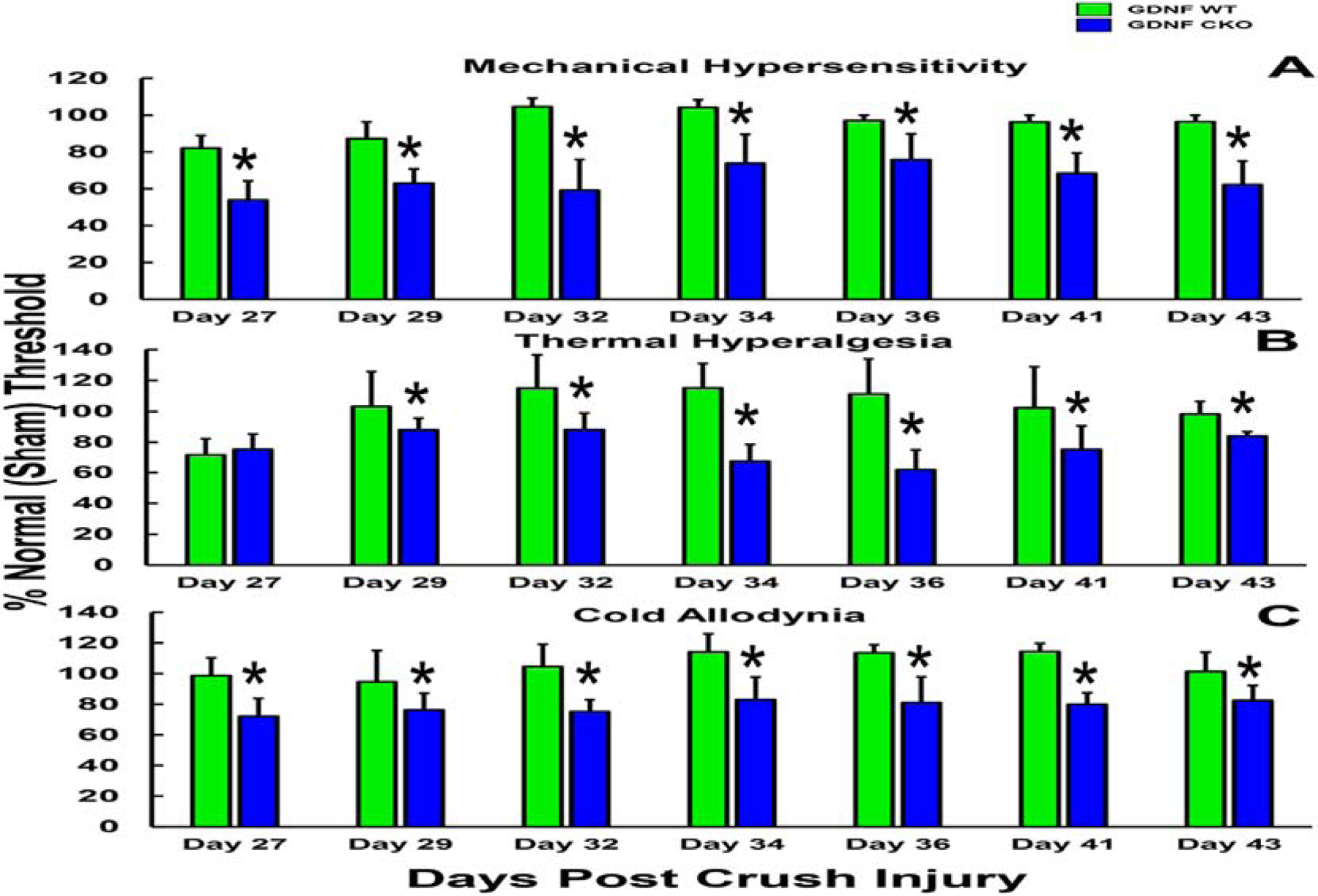
Blinded reflexive nociception tests demonstrate recovery in reflexive neurobehavioral nociception measures in GDNF WT mice (associated with restoration of restrictive BNB permeability function within 14 days after injury) by 29 days post-injury, indicated as equivalent mean % threshold (~100%) between the injured and uninjured (Sham) sciatic nerves, with significant persistent increased nociception, indicated a % mean reduction in normal withdrawal thresholds (normalized to the contralateral injured [Sham] sciatic nerve) in GDNF CKO mice (associated with delayed restoration of restrictive BNB permeability function) up until 6 weeks post-injury. Error bars indicate standard errors of the mean. * indicates p<0.05, N=4
In our preliminary studies, we performed sciatic nerve crush injury as published,(Dong et al., 2018) and harvested the sciatic nerves within 3 hours of crush injury (Day 0) and on Days 7 following crush injury and performed indirect fluorescent immunohistochemistry to detect phosphorylated cortactin (pCTTN), CTNNA1 and CLDN4 on endoneurial microvessels. We observed focal pCTTN expression on normal microvessels (Figures 22A–D), consistent with tight junction localization, with loss of expression in both GDNF WT (Figures 22E–H) and CKO mice (Figures 22I–L) on Day 0. pCTTN expression was partly restored in GDNF WT mice on Day 7 at a time of maximal sciatic nerve GDNF secretion(Dong et al., 2018) (Figures 22M–P). Persistent pCTTN loss was observed in GDNF CKO mice on Day 7 (Figures 22Q–T). CTNNA1 was expressed on normal mouse sciatic nerve endoneurial microvessels, as described in human sural nerves (Figures 23A–D). We observed loss of expression in both GDNF WT (Figures 23E–H) and GDNF CKO mice (Figures 23I–L) on Day 0. CTNNA1 expression was near completely restored in GDNF WT mice on Day 7 (Figures 23M–P), with persistent loss or low expression observed in GDNF CKO mice at this time point (Figures 23Q–T), suggesting its importance for the early structural reorganization of the restrictive BNB junctional complex.
Figure 22. pCTTN expression at the murine BNB in GDNF transgenic mice following sciatic nerve crush injury.
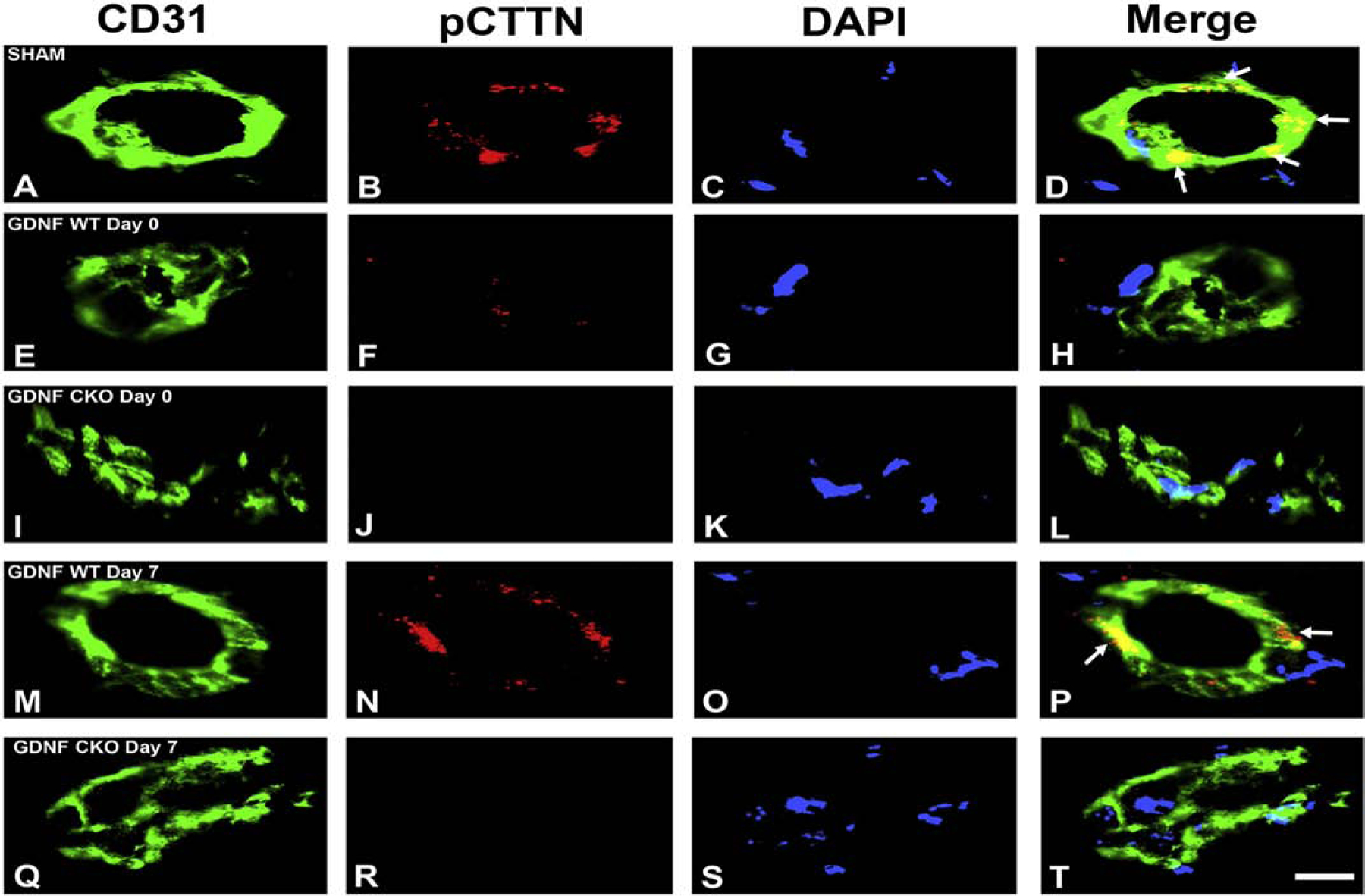
Digital indirect fluorescent photomicrographs of murine sciatic nerve endoneurial microvessels within 3 hours (Day 0) and 7 days (Day 7) after crush injury in GDNF WT and GDNF CKO mice are shown, with Sham indicating uninjured sciatic nerves. CD31 (green, endothelial cell marker, A, E, I, M and Q) and nuclear marker DAPI (blue, C, G, K, O and S) were performed to identify endoneurial microvessels. Punctuate membrane pCTTN expression is lost in both GDNF WT and CKO mice immediately after injury, with partial recovery seen in GDNF WT mice only on Day 7. N=2, Scale bar = 2.5 μm.
Figure 23. CTNNA1 expression at the murine BNB in GDNF transgenic mice following sciatic nerve crush injury.
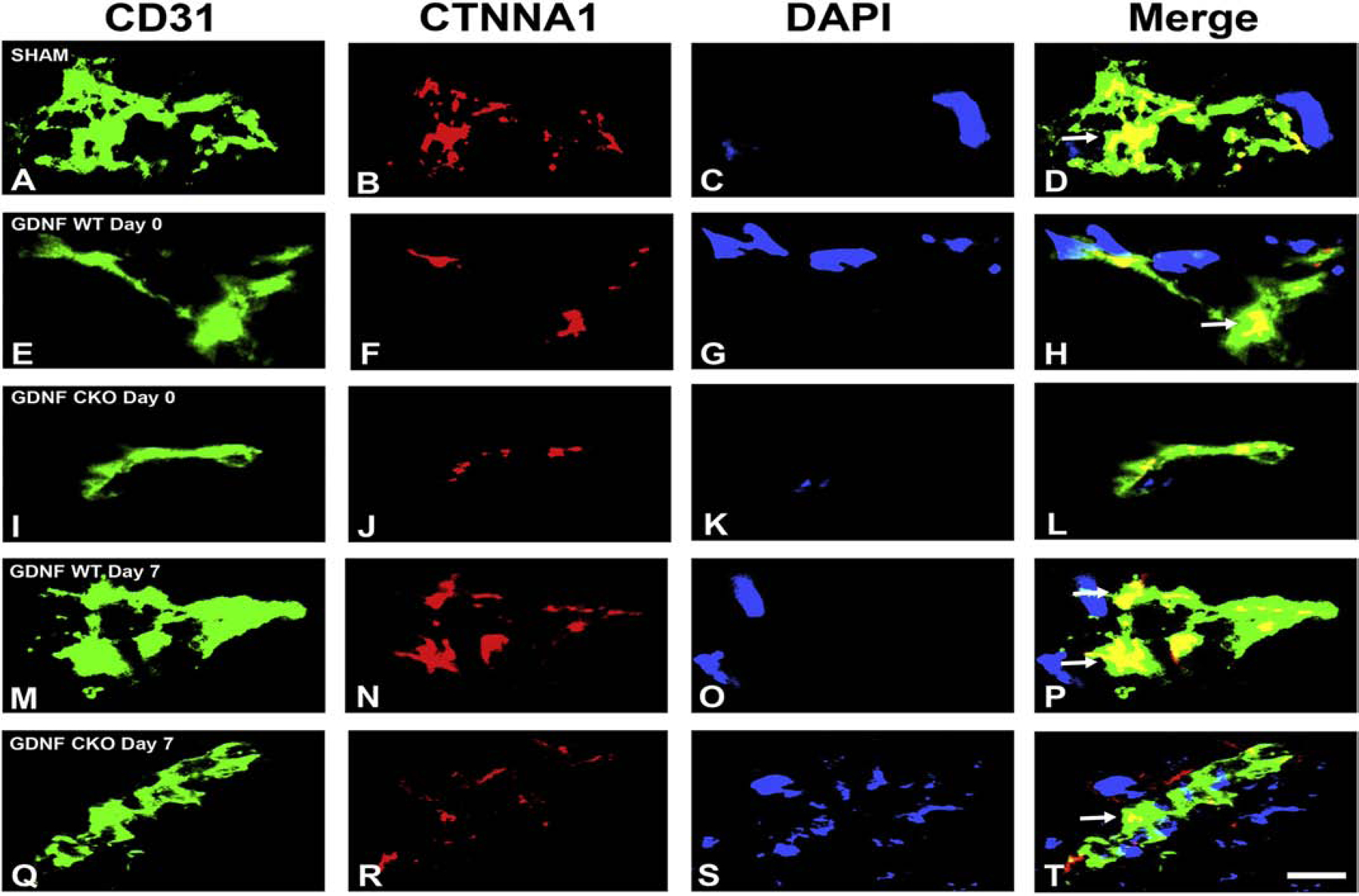
Digital indirect fluorescent photomicrographs of murine sciatic nerve endoneurial microvessels within 3 hours (Day 0) and 7 days (Day 7) after crush injury in GDNF WT and GDNF CKO mice are shown, with Sham indicating uninjured sciatic nerves. CD31 (green, endothelial cell marker, A, E, I, M and Q) and nuclear marker DAPI (blue, C, G, K, O and S) were performed to identify endoneurial microvessels. Plaque-like linear membrane CTNNA1 expression is partly lost in both GDNF WT and CKO mice immediately after injury, with near complete recovery seen in GDNF WT mice and partial recovery in GDNF CKO mice on Day 7, implying an important role in restoring endoneurial homeostasis after injury. N=2, Scale bar = 2.5 μm.
Similar to human sural nerves, focal CLDN4 expression was observed on normal mouse sciatic nerve endoneurial vessels in a pattern consistent with tight junctions (Figures 24A–D). We observed complete loss of CLDN4 expression in both GDNF WT (Figures 24E–H) and GDNF CKO mice (Figures 24I–L) on Day 0, with partly restored focal expression observed in GDNF WT mice on Day 7 (Figures 24M–P). Persistent CLDN4 loss was observed in GDNF CKO mice on Day 7 (Figures 24Q–T). Based on our published data that showed near complete BNB functional recovery at Day 7 in GDNF WT mice with persistent high permeability in GDNF CKO mice,(Dong et al., 2018) these data suggest that pCTTN and CLDN4 may serve as important structural components of the BNB with CTNNA1 serving as an essential adapter necessary for functional BNB recovery that is partially expressed via GDNF-dependent and independent pathways following peripheral nerve injury in mice.
Figure 24. CLDN4 expression at the murine BNB in GDNF transgenic mice following sciatic nerve crush injury.
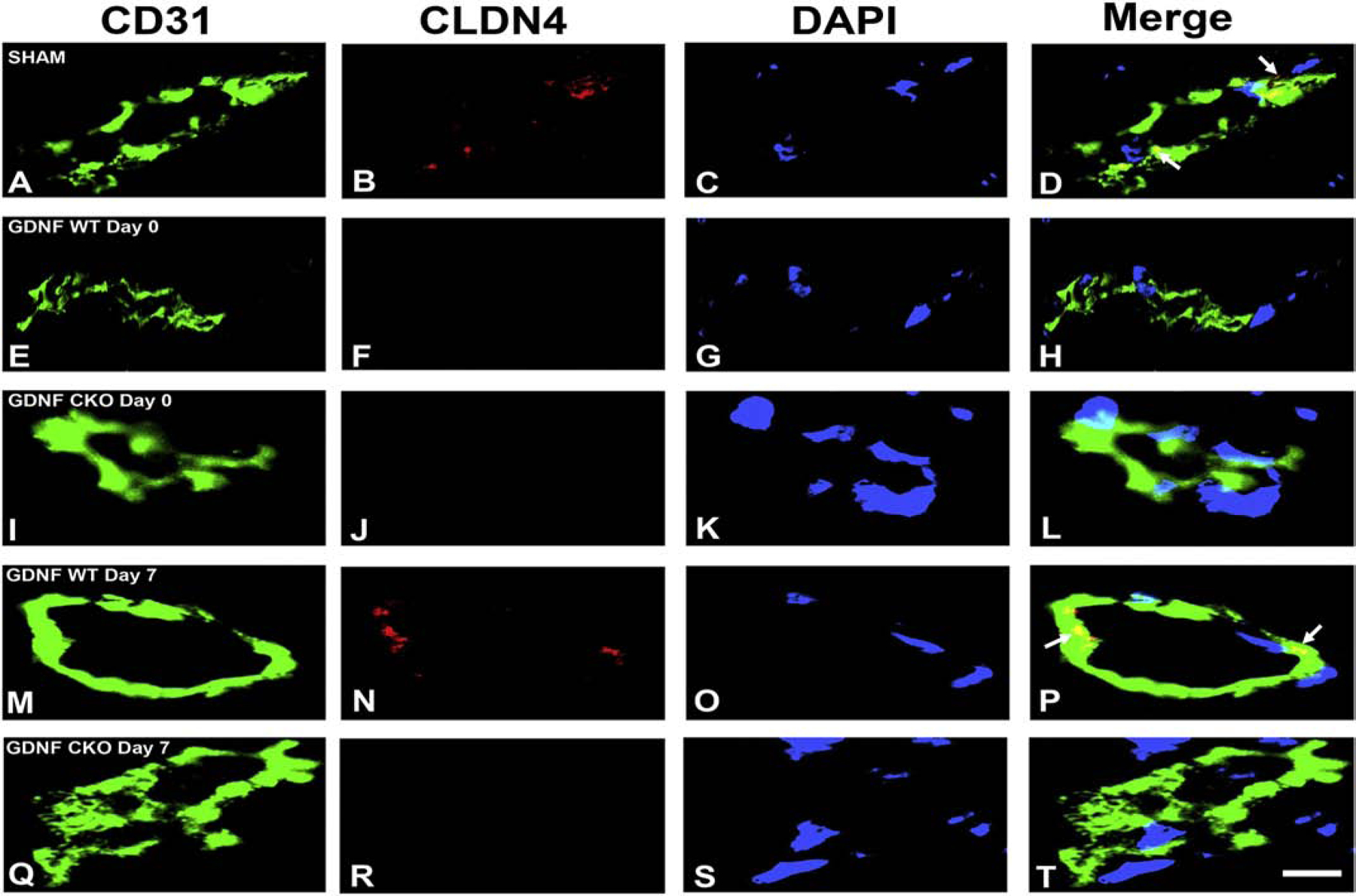
Digital indirect fluorescent photomicrographs of murine sciatic nerve endoneurial microvessels within 3 hours (Day 0) and 7 days (Day 7) after crush injury in GDNF WT and GDNF CKO mice are shown, with Sham indicating uninjured sciatic nerves. CD31 (green, endothelial cell marker, A, E, I, M and Q) and nuclear marker DAPI (blue, C, G, K, O and S) were performed to identify endoneurial microvessels. Punctuate membrane CLDN4 expression is lost in both GDNF WT and CKO mice immediately after injury, with partial recovery seen in GDNF WT mice only on Day 7. N=2, Scale bar = 2.5 μm.
Using electron microscopy, we frequently observed permeable endoneurial microvessels with disorganized endothelial cells and basement membrane duplication in GDNF WT mice treated with a specific cell permeable SRC kinase inhibitor, Bosutinib, for 5 days (days 2–6) following sciatic nerve crush injury (Figure 25), similar to microvascular changes seen in chronic peripheral neuropathy patients (see Figures 8E–H).(Bosetti et al., 2016; Mackinnon et al., 1986; Ubogu, 2015; Yasuda and Dyck, 1987) Our preliminary studies showed that SRC kinase inhibition resulted in more permeable microvessels in GDNF WT mice on both Days 7 and 14 compared to untreated mice (Figures 25I and J), supporting an important early role for SRC kinase in GDNF-mediated restoration of murine BNB function. Studies are ongoing to longitudinally evaluate BNB molecular expression and relevant signaling pathways, BNB dysfunction/repair and correlation with chronic nociception.
Figure 25. Role of SRC kinase in restoring structure and restrictive murine BNB permeability in GDNF transgenic mice following sciatic nerve crush injury.
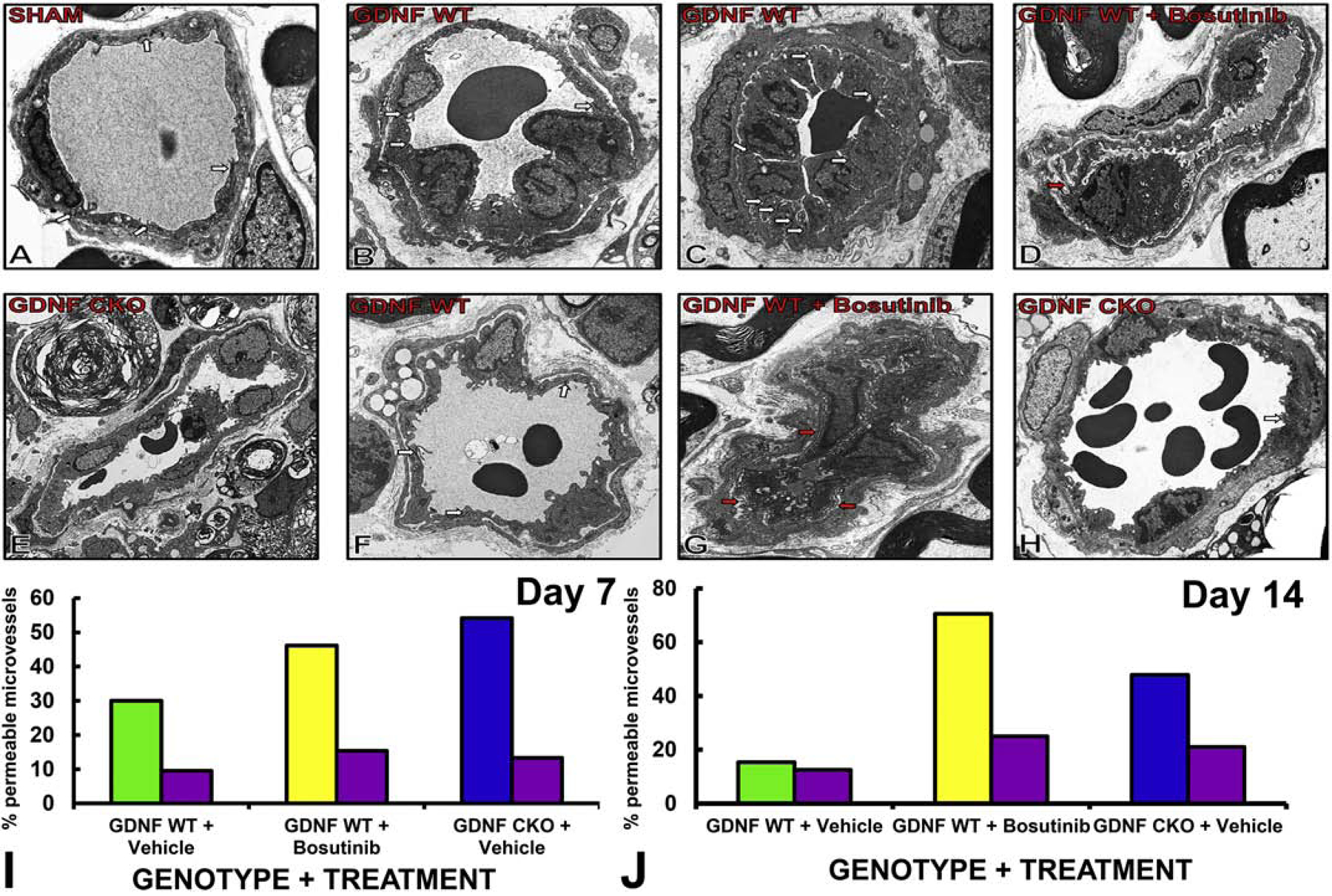
Axial digital ultramicrographs show electron-dense inter-cellular tight junctions (white arrows) in normal, horseradish peroxidase-impermeable endoneurial microvessels in Sham surgery control nerves (A). On Day 7 after crush injury, tight junctions are commonly seen in GDNF WT mice (B), even in microvessels with endothelial cell proliferation (C). Structurally disorganized permeable microvessels are commonly seen in GDNF WT mice treated with SRC kinase inhibitor, Bosutinib, with basement membrane duplication (red arrow, D). Structurally organized permeable microvessels that lack tight junctions are commonly seen in GDNF CKO mice (E). On Day 14, more intact endoneurial microvessels with tight junctions (white arrows) are seen in GDNF WT mice (F). Permeable microvessels with disorganized endothelial cells and basement membrane duplication (red arrows) are commonly seen in GDNF WT mice treated with Bosutinib (G). More permeable microvessels are seen in GDNF CKO mice, with less frequent tight junctions (white arrow, H) compared to GDNF WT mice. Original magnification 4500–7000X. Bar histograms show sciatic nerve BNB permeability to horseradish peroxidase, quantified as the % of permeable microvessels in each nerve by electron microscopy, with data compared with the uninjured contralateral Sham surgery nerve (purple) and the different experimental groups on Day 7 (I) and Day 14 (J). SRC kinase inhibitor, Bosutinib abrogated the expected GDNF-driven restoration of BNB impermeability following nerve injury on days 7 and 14, with the expected delayed in GDNF CKO mice also observed as previously published. N=1 (quantified in duplicate with a mean of 17 microvessels/ mouse evaluated).
Conclusions
The human BNB, formed by endoneurial microvascular endothelial cells, is a critical interface between circulating blood and the innermost compartment of peripheral nerves and nerve roots. The BNB is required for physiologic homeostasis, including presumed immune-neural crosstalk in health, with structural, molecular and functional alterations observed in several peripheral neuropathies and chronic neuropathic pain. The molecular determinants and signaling pathways responsible for normal BNB function are incompletely understood; however significant advances are being made, guided by observational data obtained from well characterized human peripheral nerve biopsies. Non-biased bioinformatics analyses of transcriptome and proteome data derived from BNB-forming human endoneurial endothelial cells in situ and in vitro in order to establish biologically relevant molecules and their interacting networks/ signaling pathways, have provided novel insights into the essential structural and functional characteristics of the adult human BNB in health, and alterations or adaptations in specific peripheral neuropathies and chronic neuropathic pain. In addition to comprehensively understanding how the human BNB works and adapts or fails to adapt to insult, a major goal of our dedicated efforts is to discover molecular targets for disease-specific therapy in peripheral neuropathies and chronic neuropathic pain that take into account the unique biology of the human BNB.
Acknowledgements and Funding
Special thanks to past and current employees of the Shin J Oh Muscle and Nerve Histopathology Laboratory, the University of Alabama at Birmingham (UAB) for processing human tissue and generating histopathology slides from which digital photomicrographs are shown, and current and past members and collaborators of the Neuromuscular Immunopathology Research Laboratory (NIRL) for insightful discussions, and experimental work, particularly digital photomicrographs and ultramicrographs of human cells and tissues and mouse tissues. Access to HIV+ patient and HIV− control PBMLs was provided through the UAB Center for AIDS Research (CFAR) and the CFAR Network of Integrated Clinical Systems (CNICS). Digital ultramicrographs were generated by Beth Weeks at EMLabs, Inc. (Birmingham, AL) and special thanks to the staff in the Civitan International Research Center and UAB High Resolution Microscopy Core facilities for technical assistance with confocal microscopy. Work described from the NIRL was supported by National Institutes of Health (NIH) Grants R21 NS073702 (2011-2014), R21 NS078226 (2012-2015), R01 NS075212 (2012-2018) and a Creative and Novel Ideas in HIV Research Subaward P30 AI27767 (2012-2015), as well as institutional support from the Department of Neurology, the University of Alabama at Birmingham. The content is solely the responsibility of the author and does not necessarily represent the official views of UAB or NIH.
Abbreviations:
- ABC
ATP-binding cassette
- AIDP
acute inflammatory demyelinating polyradiculoneuropathy
- AQP
aquaporin
- BBB
blood-brain barrier
- bFGF
basic fibroblast growth factor
- BNB
blood-nerve barrier
- CAMP
cyclic adenosine monophosphate
- c-ART
combination antiretroviral therapy
- CAV
caveolin
- CD
cluster of differentiation
- CD99L2
CD99 Like 2
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- CLDN
claudin
- CREB
CAMP Responsive Element Binding Protein
- CTNNA1
α−1 catenin
- CTTN
cortactin
- DSN
distal sensory neuropathy
- ECIS
electrical-cell impedance sensing
- FACS
fluorescence activated cell sorting
- FGFR
fibroblast growth factor receptor
- FITC
fluorescein isothiocyanate
- FN-CS1
fibronectin connecting segment-
- FPKM
Fragments Per Kilobase per Million
- GBS
Guillain-Barré syndrome
- GDNF
glial-derived neurotrophic factor
- GFRα1
GDNF Family Receptor Alpha 1
- GR
glucocorticoid receptor
- HIF
hypoxia-inducing factor
- HIV
human immunodeficiency virus
- HLA
human leukocyte antigen
- ICAM-1
intercellular adhesion molecule-1
- IFN
interferon
- IL
interleukin
- MAPK
mitogen activated protein kinase
- MHC
Major Histocompatibility Complex
- PBMLs
peripheral blood mononuclear leukocytes
- PCR
polymerase chain reaction
- pCTTN
phosphorylated cortactin
- PDZ
Postsynaptic density protein, Drosophila disc large tumor suppressor, and ZO-1
- pHEndEC
primary human endoneurial endothelial cell
- RET
“rearranged upon transformation”
- RNA
ribonucleic acid
- SDS-PAGE
sodium dodecyl sulfate- protein agarose gel electrophoresis
- SEC
secretory
- siRNA
small interfering RNA
- rt-PCR
real time PCR
- SLC
solute carrier
- SRC
sarcoma
- TEER
transendothelial electrical resistance
- TGF-β
transforming growth factor-β
- TGFR
TGF Receptor
- UEA-1
Ulex Europaeus Agglutinin-1
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial cell growth factor
- VEGFR
VEGF Receptor
- ZO
zona occludens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 2008. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 372, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe M, Sano Y, Maeda T, Shimizu F, Kashiwamura Y, Haruki H, Saito K, Tasaki A, Kawai M, Terasaki T, Kanda T, 2012. Establishment and characterization of human peripheral nerve microvascular endothelial cell lines: a new in vitro blood-nerve barrier (BNB) model. Cell Struct Funct 37, 89–100. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK, 2013. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther 138, 155–175. [DOI] [PubMed] [Google Scholar]
- Allt G, Lawrenson JG, 2000. The blood-nerve barrier: enzymes, transporters and receptors--a comparison with the blood-brain barrier. Brain Res Bull 52, 1–12. [DOI] [PubMed] [Google Scholar]
- Aziz MH, Cui K, Das M, Brown KE, Ardell CL, Febbraio M, Pluskota E, Han J, Wu H, Ballantyne CM, Smith JD, Cathcart MK, Yakubenko VP, 2017. The Upregulation of Integrin alphaDbeta2 (CD11d/CD18) on Inflammatory Macrophages Promotes Macrophage Retention in Vascular Lesions and Development of Atherosclerosis. J Immunol 198, 4855–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacellar H, Munoz A, Miller EN, Cohen BA, Besley D, Selnes OA, Becker JT, McArthur JC, 1994. Temporal trends in the incidence of HIV−1-related neurologic diseases: Multicenter AIDS Cohort Study, 1985–1992. Neurology 44, 1892–1900. [DOI] [PubMed] [Google Scholar]
- Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, Horuk R, Sparre-Ulrich AH, Locati M, Luster AD, Mantovani A, Matsushima K, Murphy PM, Nibbs R, Nomiyama H, Power CA, Proudfoot AE, Rosenkilde MM, Rot A, Sozzani S, Thelen M, Yoshie O, Zlotnik A, 2014. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 66, 1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D, 2009. Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MA, Weddell AG, 1984. A descriptive study of the blood vessels of the sciatic nerve in the rat, man and other mammals. Brain : a journal of neurology 107 (Pt 3), 871–898. [DOI] [PubMed] [Google Scholar]
- Bonin RP, Bories C, De Koninck Y, 2014. A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Molecular pain 10, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosboom WM, Van den Berg LH, Mollee I, Sasker LD, Jansen J, Wokke JH, Logtenberg T, 2001. Sural nerve T-cell receptor Vbeta gene utilization in chronic inflammatory demyelinating polyneuropathy and vasculitic neuropathy. Neurology 56, 74–81. [DOI] [PubMed] [Google Scholar]
- Bosetti F, Galis ZS, Bynoe MS, Charette M, Cipolla MJ, Del Zoppo GJ, Gould D, Hatsukami TS, Jones TL, Koenig JI, Lutty GA, Maric-Bilkan C, Stevens T, Tolunay HE, Koroshetz W, Small Blood Vessels: Big Health Problems” Workshop, P., 2016. “Small Blood Vessels: Big Health Problems?”: Scientific Recommendations of the National Institutes of Health Workshop. J Am Heart Assoc 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Lacroix C, Planté V, Adams D, Chedru F, Guglielmi J, Said G, 1999. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology 52, 498–503. [DOI] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, Gereau R.W.t., 2012. A novel behavioral assay for measuring cold sensation in mice. PloS one 7, e39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge PM, Ball DJ, Mackinnon SE, Nakao Y, Brandt K, Hunter DA, Hertl C, 1994. Nerve crush injuries--a model for axonotmesis. Exp Neurol 127, 284–290. [DOI] [PubMed] [Google Scholar]
- Christensen MB, Tresco PA, 2015. Differences Exist in the Left and Right Sciatic Nerves of Naive Rats and Cats. Anat Rec (Hoboken) 298, 1492–1501. [DOI] [PubMed] [Google Scholar]
- Cichon C, Sabharwal H, Ruter C, Schmidt MA, 2014. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2, e944446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Xanthos DN, Francis L, Bennett GJ, 2004. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain 112, 94–105. [DOI] [PubMed] [Google Scholar]
- Cohen-Kashi Malina K, Cooper I, Teichberg VI, 2009. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res 1284, 12–21. [DOI] [PubMed] [Google Scholar]
- Collins MP, Arnold WD, Kissel JT, 2013. The neuropathies of vasculitis. Neurologic clinics 31, 557–595. [DOI] [PubMed] [Google Scholar]
- Cucullo L, Marchi N, Hossain M, Janigro D, 2011. A dynamic in vitro BBB model for the study of immune cell trafficking into the central nervous system. J Cereb Blood Flow Metab 31, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, 2015. Pathogenesis of immune-mediated neuropathies. Biochimica et biophysica acta 1852, 658–666. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM, 2009. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res 335, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Greathouse KM, Beacham RL, Palladino SP, Helton ES, Ubogu EE, 2017. Fibronectin connecting segment-1 peptide inhibits pathogenic leukocyte trafficking and inflammatory demyelination in experimental models of chronic inflammatory demyelinating polyradiculoneuropathy. Exp Neurol 292, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Helton ES, Zhou P, Ouyang X, d’Anglemont de Tassigny X, Pascual A, Lopez-Barneo J, Ubogu EE, 2018. Glial-derived neurotrophic factor is essential for blood-nerve barrier functional recovery in an experimental murine model of traumatic peripheral neuropathy. Tissue Barriers 6, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Palladino SP, Helton ES, Ubogu EE, 2016. The pathogenic relevance of alphaM-integrin in Guillain-Barre syndrome. Acta neuropathologica 132, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Ubogu EE, 2018. GDNF enhances human blood-nerve barrier function in vitro via MAPK signaling pathways. Tissue Barriers 6, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames RA, Lange LS, 1967. Clinical and pathological study of ischaemic neuropathy. Journal of neurology, neurosurgery, and psychiatry 30, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Rosario D, Clifford DB, McArthur JC, Simpson D, Alexander T, Gelman BB, Vaida F, Collier A, Marra CM, Ances B, Atkinson JH, Dworkin RH, Morgello S, Grant I, 2010. Continued high prevalence and adverse clinical impact of human immunodeficiency virus-associated sensory neuropathy in the era of combination antiretroviral therapy: the CHARTER Study. Arch Neurol 67, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Tansey MG, Tsui-Pierchala BA, Comella JX, Milbrandt J, Johnson EM Jr., 2001. c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J Neurosci 21, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt A, Lorler H, Neundorfer B, 1993. Immunohistochemical findings in vasculitic neuropathies. Acta neurologica Scandinavica 87, 318–321. [DOI] [PubMed] [Google Scholar]
- Evans SR, Ellis RJ, Chen H, Yeh TM, Lee AJ, Schifitto G, Wu K, Bosch RJ, McArthur JC, Simpson DM, Clifford DB, 2011. Peripheral neuropathy in HIV: prevalence and risk factors. AIDS 25, 919928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan H, Wendeler MW, Mitrovic S, Fava E, Silberberg Y, Sharan R, Zerial M, Hauri HP, 2010. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J Cell Biol 189, 997–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehner SC, Davies A, Baldwin SA, Lienhard GE, 1988. The blood-nerve barrier is rich in glucose transporter. J Neurocytol 17, 173–178. [DOI] [PubMed] [Google Scholar]
- Greathouse KM, Palladino SP, Dong C, Helton ES, Ubogu EE, 2016. Modeling leukocyte trafficking at the human blood-nerve barrier in vitro and in vivo geared towards targeted molecular therapies for peripheral neuroinflammation. J Neuroinflammation 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Stoll G, Li CY, Tyor W, Cornblath DR, 1990. Macrophage responses in inflammatory demyelinating neuropathies. Annals of neurology 27 Suppl, S64–68. [DOI] [PubMed] [Google Scholar]
- Hall SM, Hughes RA, Atkinson PF, McColl I, Gale A, 1992. Motor nerve biopsy in severe Guillain-Barre syndrome. Annals of neurology 31, 441–444. [DOI] [PubMed] [Google Scholar]
- Hao JX, Blakeman KH, Yu W, Hultenby K, Xu XJ, Wiesenfeld-Hallin Z, 2000. Development of a mouse model of neuropathic pain following photochemically induced ischemia in the sciatic nerve. Exp Neurol 163, 231–238. [DOI] [PubMed] [Google Scholar]
- Hartlehnert M, Derksen A, Hagenacker T, Kindermann D, Schafers M, Pawlak M, Kieseier BC, Meyer Zu Horste G, 2017. Schwann cells promote post-traumatic nerve inflammation and neuropathic pain through MHC class II. Sci Rep 7, 12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ, 2008. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochimica et biophysica acta 1778, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Jiang J, Kokkinaki M, Golestaneh N, Hofmann MC, Dym M, 2008. Gdnf upregulates c-Fos transcription via the Ras/Erk1/2 pathway to promote mouse spermatogonial stem cell proliferation. Stem Cells 26, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton ES, Palladino S, Ubogu EE, 2017. A novel method for measuring hydraulic conductivity at the human blood-nerve barrier in vitro. Microvasc Res 109, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes R, Atkinson P, Coates P, Hall S, Leibowitz S, 1992. Sural nerve biopsies in Guillain-Barre syndrome: axonal degeneration and macrophage-associated demyelination and absence of cytomegalovirus genome. Muscle & nerve 15, 568–575. [DOI] [PubMed] [Google Scholar]
- Hultstrom D, Malmgren L, Gilstring D, Olsson Y, 1983. FITC-Dextrans as tracers for macromolecular movements in the nervous system. A freeze-drying method for dextrans of various molecular sizes injected into normal animals. Acta neuropathologica 59, 53–62. [DOI] [PubMed] [Google Scholar]
- Ibanez CF, Andressoo JO, 2017. Biology of GDNF and its receptors - Relevance for disorders of the central nervous system. Neurobiol Dis 97, 80–89. [DOI] [PubMed] [Google Scholar]
- Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis JC, Hu S, Altrock BW, Fox GM, 1996. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell 85, 1113–1124. [DOI] [PubMed] [Google Scholar]
- Jones G, Zhu Y, Silva C, Tsutsui S, Pardo CA, Keppler OT, McArthur JC, Power C, 2005. Peripheral nerve-derived HIV−1 is predominantly CCR5-dependent and causes neuronal degeneration and neuroinflammation. Virology 334, 178–193. [DOI] [PubMed] [Google Scholar]
- Kanda T, 2013. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. Journal of neurology, neurosurgery, and psychiatry 84, 208–212. [DOI] [PubMed] [Google Scholar]
- Kanda T, Numata Y, Mizusawa H, 2004. Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. Journal of neurology, neurosurgery, and psychiatry 75, 765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Yamawaki M, Mizusawa H, 2003. Sera from Guillain-Barre patients enhance leakage in blood-nerve barrier model. Neurology 60, 301–306. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Kieseier BC, Stoll G, Hartung HP, 2001. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol 64, 109–127. [DOI] [PubMed] [Google Scholar]
- Kieseier BC, Tani M, Mahad D, Oka N, Ho T, Woodroofe N, Griffin JW, Toyka KV, Ransohoff RM, Hartung HP, 2002. Chemokines and chemokine receptors in inflammatory demyelinating neuropathies: a central role for IP-10. Brain : a journal of neurology 125, 823–834. [DOI] [PubMed] [Google Scholar]
- Komarova Y, Malik AB, 2010. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Kupers R, Yu W, Persson JK, Xu XJ, Wiesenfeld-Hallin Z, 1998. Photochemically-induced ischemia of the rat sciatic nerve produces a dose-dependent and highly reproducible mechanical, heat and cold allodynia, and signs of spontaneous pain. Pain 76, 45–59. [DOI] [PubMed] [Google Scholar]
- Latker CH, Shinowara NL, Miller JC, Rapoport SI, 1987. Differential localization of alkaline phosphatase in barrier tissues of the frog and rat nervous systems: a cytochemical and biochemical study. J Comp Neurol 264, 291–302. [DOI] [PubMed] [Google Scholar]
- Leppert D, Hughes P, Huber S, Erne B, Grygar C, Said G, Miller KM, Steck AJ, Probst A, Fuhr P, 1999. Matrix metalloproteinase upregulation in chronic inflammatory demyelinating polyneuropathy and nonsystemic vasculitic neuropathy. Neurology 53, 62–70. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S, 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7, 678–689. [DOI] [PubMed] [Google Scholar]
- Lim TK, Shi XQ, Johnson JM, Rone MB, Antel JP, David S, Zhang J, 2015. Peripheral nerve injury induces persistent vascular dysfunction and endoneurial hypoxia, contributing to the genesis of neuropathic pain. J Neurosci 35, 3346–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim TK, Shi XQ, Martin HC, Huang H, Luheshi G, Rivest S, Zhang J, 2014. Blood-nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain 155, 954–967. [DOI] [PubMed] [Google Scholar]
- Lindenlaub T, Sommer C, 2003. Cytokines in sural nerve biopsies from inflammatory and non-inflammatory neuropathies. Acta neuropathologica 105, 593–602. [DOI] [PubMed] [Google Scholar]
- Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV, 2014. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep 4, 4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV, 2012. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol 30, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon SE, Dellon AL, Hudson AR, Hunter DA, 1986. Chronic human nerve compression--a histological assessment. Neuropathology and applied neurobiology 12, 547–565. [DOI] [PubMed] [Google Scholar]
- Mahringer A, Fricker G, 2016. ABC transporters at the blood-brain barrier. Expert Opin Drug Metab Toxicol 12, 499–508. [DOI] [PubMed] [Google Scholar]
- Maiuolo J, Gliozzi M, Musolino V, Carresi C, Nucera S, Macri R, Scicchitano M, Bosco F, Scarano F, Ruga S, Zito MC, Oppedisano F, Mollace R, Paone S, Palma E, Muscoli C, Mollace V, 2019. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S, Tucky B, Bagheri N, Li X, Kochar R, Ransohoff RM, 2009. alpha4 Integrin/FN-CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. Journal of neuroimmunology 210, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S, Ubogu EE, Ransohoff RM, 2007. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain pathology 17, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathey EK, Park SB, Hughes RA, Pollard JD, Armati PJ, Barnett MH, Taylor BV, Dyck PJ, Kiernan MC, Lin CS, 2015. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. Journal of neurology, neurosurgery, and psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathey EK, Pollard JD, Armati PJ, 1999. TNF alpha, IFN gamma and IL-2 mRNA expression in CIDP sural nerve biopsies. Journal of the neurological sciences 163, 47–52. [DOI] [PubMed] [Google Scholar]
- Matsumuro K, Izumo S, Umehara F, Osame M, 1994. Chronic inflammatory demyelinating polyneuropathy: histological and immunopathological studies on biopsied sural nerves. Journal of the neurological sciences 127, 170–178. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A, 2005. Neurological complications of HIV infection. Lancet Neurol 4, 543–555. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Scimone ML, Mora JR, von Andrian UH, 2004. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol 16, 406–417. [DOI] [PubMed] [Google Scholar]
- Merighi A, 2016. Targeting the glial-derived neurotrophic factor and related molecules for controlling normal and pathologic pain. Expert Opin Ther Targets 20, 193–208. [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste G, Hartung HP, Kieseier BC, 2007. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nature clinical practice. Neurology 3, 198–211. [DOI] [PubMed] [Google Scholar]
- Meyer Zu Horste G, Heidenreich H, Lehmann HC, Ferrone S, Hartung HP, Wiendl H, Kieseier BC, 2010a. Expression of antigen processing and presenting molecules by Schwann cells in inflammatory neuropathies. Glia 58, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zu Horste G, Heidenreich H, Mausberg AK, Lehmann HC, ten Asbroek AL, Saavedra JT, Baas F, Hartung HP, Wiendl H, Kieseier BC, 2010b. Mouse Schwann cells activate MHC class I and II restricted T-cell responses, but require external peptide processing for MHC class II presentation. Neurobiol Dis 37, 483–490. [DOI] [PubMed] [Google Scholar]
- Mitchell GW, Williams GS, Bosch EP, Hart MN, 1991. Class II antigen expression in peripheral neuropathies. Journal of the neurological sciences 102, 170–176. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Vieira-de-Abreu A, Harris ES, Shah AM, Weyrich AS, Castro-Faria-Neto HC, Zimmerman GA, 2014. Integrin alphaDbeta2 (CD11d/CD18) is expressed by human circulating and tissue myeloid leukocytes and mediates inflammatory signaling. PloS one 9, e112770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizisin AP, Weerasuriya A, 2011. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta neuropathologica 121, 291–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau N, Mauborgne A, Bourgoin S, Couraud PO, Romero IA, Weksler BB, Villanueva L, Pohl M, Boucher Y, 2016. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 157, 827–839. [DOI] [PubMed] [Google Scholar]
- Moreau N, Mauborgne A, Couraud PO, Romero IA, Weksler BB, Villanueva L, Pohl M, Boucher Y, 2017. Could an endoneurial endothelial crosstalk between Wnt/beta-catenin and Sonic Hedgehog pathways underlie the early disruption of the infra-orbital blood-nerve barrier following chronic constriction injury? Molecular pain 13, 1744806917727625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, 2003. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol 24, 327–334. [DOI] [PubMed] [Google Scholar]
- Muller WA, 2009. Mechanisms of transendothelial migration of leukocytes. Circulation research 105, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, 2011. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6, 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, 2014. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol 184, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muona P, Jaakkola S, Salonen V, Peltonen J, 1993. Expression of glucose transporter 1 in adult and developing human peripheral nerve. Diabetologia 36, 133–140. [DOI] [PubMed] [Google Scholar]
- Nalecz KA, 2017. Solute Carriers in the Blood-Brain Barier: Safety in Abundance. Neurochem Res 42, 795–809. [DOI] [PubMed] [Google Scholar]
- Nath A, 2015. Eradication of human immunodeficiency virus from brain reservoirs. J Neurovirol 21, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveilhan P, ElShamy WM, Ernfors P, 1997. Differential regulation of mRNAs for GDNF and its receptors Ret and GDNFR alpha after sciatic nerve lesion in the mouse. Eur J Neurosci 9, 1450–1460. [DOI] [PubMed] [Google Scholar]
- Ochoa J, Mair WG, 1969a. The normal sural nerve in man. I. Ultrastructure and numbers of fibres and cells. Acta neuropathologica 13, 197–216. [DOI] [PubMed] [Google Scholar]
- Ochoa J, Mair WG, 1969b. The normal sural nerve in man. II. Changes in the axons and Schwann cells due to ageing. Acta neuropathologica 13, 217–239. [DOI] [PubMed] [Google Scholar]
- Oka N, Kawasaki T, Mizutani K, Sugiyama H, Akiguchi I, 2007. Hypoxia-inducible factor 1alpha may be a marker for vasculitic neuropathy. Neuropathology : official journal of the Japanese Society of Neuropathology 27, 509–515. [DOI] [PubMed] [Google Scholar]
- Olsson Y, 1966. Studies on vascular permeability in peripheral nerves. I. Distribution of circulating fluorescent serum albumin in normal, crushed and sectioned rat sciatic nerve. Acta neuropathologica 7, 1–15. [DOI] [PubMed] [Google Scholar]
- Olsson Y, 1968. Topographical differences in the vascular permeability of the peripheral nervous system. Acta neuropathologica 10, 26–33. [DOI] [PubMed] [Google Scholar]
- Olsson Y, 1971. Studies on vascular permeability in peripheral nerves. IV. Distribution of intravenously injected protein tracers in the peripheral nervous system of various species. Acta neuropathologica 17, 114–126. [DOI] [PubMed] [Google Scholar]
- Olsson Y, 1990. Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol 5, 265–311. [PubMed] [Google Scholar]
- Orlikowski D, Chazaud B, Plonquet A, Poron F, Sharshar T, Maison P, Raphael JC, Gherardi RK, Creange A, 2003. Monocyte chemoattractant protein 1 and chemokine receptor CCR2 productions in Guillain-Barre syndrome and experimental autoimmune neuritis. Journal of neuroimmunology 134, 118–127. [DOI] [PubMed] [Google Scholar]
- Orte C, Lawrenson JG, Finn TM, Reid AR, Allt G, 1999. A comparison of blood-brain barrier and blood-nerve barrier endothelial cell markers. Anat Embryol (Berl) 199, 509–517. [DOI] [PubMed] [Google Scholar]
- Ouyang X, Dong C, Ubogu EE, 2019. In situ molecular characterization of endoneurial microvessels that form the blood-nerve barrier in normal human adult peripheral nerves. Journal of the peripheral nervous system : JPNS 24, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai S, Danne KJ, Qin J, Cavanagh LL, Smith A, Hickey MJ, Weninger W, 2012. Visualizing leukocyte trafficking in the living brain with 2-photon intravital microscopy. Front Cell Neurosci 6, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino SP, Helton ES, Jain P, Dong C, Crowley MR, Crossman DK, Ubogu EE, 2017. The Human Blood-Nerve Barrier Transcriptome. Sci Rep 7, 17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo CA, McArthur JC, Griffin JW, 2001. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst 6, 21–27. [DOI] [PubMed] [Google Scholar]
- Peltonen S, Alanne M, Peltonen J, 2013. Barriers of the peripheral nerve. Tissue Barriers 1, e24956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolnikova NP, Kushchayeva YS, Wu Y, Faust J, Ugarova TP, 2016. The Role of Integrins alphaMbeta2 (Mac-1, CD11b/CD18) and alphaDbeta2 (CD11d/CD18) in Macrophage Fusion. Am J Pathol 186, 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Berg CT, 1994. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A 91, 5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Dyck PJ, 1988. Increase in albumin, IgG, and IgM blood-nerve barrier indices in human diabetic neuropathy. Proc Natl Acad Sci U S A 85, 4879–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JD, Baverstock J, McLeod JG, 1987. Class II antigen expression and inflammatory cells in the Guillain-Barre syndrome. Annals of neurology 21, 337–341. [DOI] [PubMed] [Google Scholar]
- Pollard JD, McCombe PA, Baverstock J, Gatenby PA, McLeod JG, 1986. Class II antigen expression and T lymphocyte subsets in chronic inflammatory demyelinating polyneuropathy. Journal of neuroimmunology 13, 123–134. [DOI] [PubMed] [Google Scholar]
- Prineas JW, 1981. Pathology of the Guillain-Barre syndrome. Annals of neurology 9 Suppl, 6–19. [DOI] [PubMed] [Google Scholar]
- Probst-Cousin S, Neundorfer B, Heuss D, 2010. Microvasculopathic neuromuscular diseases: lessons from hypoxia-inducible factors. Neuromuscular disorders : NMD 20, 192–197. [DOI] [PubMed] [Google Scholar]
- Pummi KP, Heape AM, Grenman RA, Peltonen JT, Peltonen SA, 2004. Tight junction proteins ZO-1, occludin, and claudins in developing and adult human perineurium. J Histochem Cytochem 52, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Putzu GA, Figarella-Branger D, Bouvier-Labit C, Liprandi A, Bianco N, Pellissier JF, 2000. Immunohistochemical localization of cytokines, C5b-9 and ICAM-1 in peripheral nerve of Guillain-Barre syndrome. Journal of the neurological sciences 174, 16–21. [DOI] [PubMed] [Google Scholar]
- Rechthand E, Rapoport SI, 1987. Regulation of the microenvironment of peripheral nerve: role of the blood-nerve barrier. Prog Neurobiol 28, 303–343. [DOI] [PubMed] [Google Scholar]
- Rechthand E, Smith QR, Rapoport SI, 1987. Transfer of nonelectrolytes from blood into peripheral nerve endoneurium. Am J Physiol 252, H1175–1182. [DOI] [PubMed] [Google Scholar]
- Reddy CL, Yosef N, Ubogu EE, 2013. VEGF-A165 potently induces human blood-nerve barrier endothelial cell proliferation, angiogenesis, and wound healing in vitro. Cellular and molecular neurobiology 33, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina MA, Lopez A, Villanueva MC, de Andres JA, Leon GI, 2000. [Morphology of peripheral nerves, their sheaths, and their vascularization]. Rev Esp Anestesiol Reanim 47, 464–475. [PubMed] [Google Scholar]
- Reina MA, Lopez A, Villanueva MC, De Andres JA, Maches F, 2003. [The blood-nerve barrier in peripheral nerves]. Rev Esp Anestesiol Reanim 50, 80–86. [PubMed] [Google Scholar]
- Richner M, Ferreira N, Dudele A, Jensen TS, Vaegter CB, Goncalves NP, 2018. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front Neurosci 12, 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto N, Morbin M, Cavallaro T, Ferrari S, Fallahi M, Galiazzo Rizzuto S, 1998a. Focal lesions area feature of chronic inflammatory demyelinating polyneuropathy (CIDP). Acta neuropathologica 96, 603–609. [DOI] [PubMed] [Google Scholar]
- Rizzuto N, Morbin M, Cavallaro T, Ferrari S, Fallahi M, Galiazzo Rizzuto S, 1998b. Focal lesions area feature of chronic inflammatory demyelinating polyneuropathy (CIDP). Acta Neuropathol 96, 603–609. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr., 2015. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res 94, 9–25. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L, 2006. Side by side comparison between dynamic versus static models of blood-brain barrier in vitro: a permeability study. Brain Res 1109, 1–13. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, Dufour EM, Chew TW, Sorg E, Muller WA, 2007. The murine CD99-related molecule CD99-like 2 (CD99L2) is an adhesion molecule involved in the inflammatory response. Cell Commun Adhes 14, 227–237. [DOI] [PubMed] [Google Scholar]
- Schittenhelm L, Hilkens CM, Morrison VL, 2017. beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front Immunol 8, 1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Toyka KV, Kiefer R, Full J, Hartung HP, Pollard J, 1996. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barre syndrome and chronic inflammatory demyelinating neuropathy. Muscle & nerve 19, 474–487. [DOI] [PubMed] [Google Scholar]
- Schnoor M, Stradal TE, Rottner K, 2018. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol 28, 79–98. [DOI] [PubMed] [Google Scholar]
- Schutz SG, Robinson-Papp J, 2013. HIV−related neuropathy: current perspectives. HIV AIDS (Auckl) 5, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelige R, Natsch C, Marz S, Jing D, Frye M, Butz S, Vestweber D, 2013. Cutting edge: Endothelial-specific gene ablation of CD99L2 impairs leukocyte extravasation in vivo. J Immunol 190, 892–896. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Abe MA, Maeda T, Ohtsuki S, Terasaki T, Kanda T, 2011. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J Cell Physiol 226, 255–266. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T, 2012. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem Res 37, 401–409. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sawai S, Sano Y, Beppu M, Misawa S, Nishihara H, Koga M, Kuwabara S, Kanda T, 2014. Severity and patterns of blood-nerve barrier breakdown in patients with chronic inflammatory demyelinating polyradiculoneuropathy: correlations with clinical subtypes. PloS one 9, e104205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegers GM, Barreira CR, Postovit LM, Dekaban GA, 2017. CD11d beta2 integrin expression on human NK, B, and gammadelta T cells. J Leukoc Biol 101, 1029–1035. [DOI] [PubMed] [Google Scholar]
- Simpson DM, Kitch D, Evans SR, McArthur JC, Asmuth DM, Cohen B, Goodkin K, Gerschenson M, So Y, Marra CM, Diaz-Arrastia R, Shriver S, Millar L, Clifford DB, Group AAS, 2006. HIV neuropathy natural history cohort study: assessment measures and risk factors. Neurology 66, 1679–1687. [DOI] [PubMed] [Google Scholar]
- Sladjana UZ, Ivan JD, Bratislav SD, 2008. Microanatomical structure of the human sciatic nerve. Surg Radiol Anat 30, 619–626. [DOI] [PubMed] [Google Scholar]
- Sluysmans S, Vasileva E, Spadaro D, Shah J, Rouaud F, Citi S, 2017. The role of apical cell-cell junctions and associated cytoskeleton in mechanotransduction. Biol Cell 109, 139–161. [DOI] [PubMed] [Google Scholar]
- Sommer C, Galbraith JA, Heckman HM, Myers RR, 1993. Pathology of experimental compression neuropathy producing hyperesthesia. Journal of neuropathology and experimental neurology 52, 223–233. [DOI] [PubMed] [Google Scholar]
- Sommer C, Myers RR, 1996. Vascular pathology in CCI neuropathy: a quantitative temporal study. Exp Neurol 141, 113–119. [DOI] [PubMed] [Google Scholar]
- Sommer C, Toyka K, 2011. Nerve biopsy in chronic inflammatory neuropathies: in situ biomarkers. Journal of the peripheral nervous system : JPNS 16 Suppl 1, 24–29. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV, 2016. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 4, e1154641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck AJ, Kinter J, Renaud S, 2011. Differential gene expression in nerve biopsies of inflammatory neuropathies. Journal of the peripheral nervous system : JPNS 16 Suppl 1, 30–33. [DOI] [PubMed] [Google Scholar]
- Takahashi M, 2001. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev 12, 361–373. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Webster HD, 1991. Myelinated fiber regeneration after crush injury is retarded in sciatic nerves of aging mice. J Comp Neurol 308, 180–187. [DOI] [PubMed] [Google Scholar]
- Tehrani S, Tomasevic N, Weed S, Sakowicz R, Cooper JA, 2007. Src phosphorylation of cortactin enhances actin assembly. Proc Natl Acad Sci U S A 104, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MM, Vilela MC, Soriani FM, Rodrigues DH, Teixeira AL, 2010. Using intravital microscopy to study the role of chemokines during infection and inflammation in the central nervous system. Journal of neuroimmunology 224, 62–65. [DOI] [PubMed] [Google Scholar]
- Trupp M, Ryden M, Jornvall H, Funakoshi H, Timmusk T, Arenas E, Ibanez CF, 1995. Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol 130, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu EE, 2013a. Chemokine-dependent signaling pathways in the peripheral nervous system. Methods Mol Biol 1013, 17–30. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, 2013b. The molecular and biophysical characterization of the human blood-nerve barrier: current concepts. J Vasc Res 50, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu EE, 2015. Inflammatory neuropathies: pathology, molecular markers and targets for specific therapeutic intervention. Acta neuropathologica 130, 445–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu EE, Yosef N, Xia RH, Sheikh KA, 2012. Behavioral, electrophysiological, and histopathological characterization of a severe murine chronic demyelinating polyneuritis model. Journal of the peripheral nervous system : JPNS 17, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rhijn I, Van den Berg LH, Bosboom WM, Otten HG, Logtenberg T, 2000. Expression of accessory molecules for T-cell activation in peripheral nerve of patients with CIDP and vasculitic neuropathy. Brain : a journal of neurology 123 (Pt 10), 2020–2029. [DOI] [PubMed] [Google Scholar]
- Verma A, 2001. Epidemiology and clinical features of HIV−1 associated neuropathies. J Peripher Nerv Syst 6, 8–13. [DOI] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, 2017. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite A, Li J, Radice GL, 2015. New functions for alpha-catenins in health and disease: from cancer to heart regeneration. Cell Tissue Res 360, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YI, Abaci HE, Shuler ML, 2017. Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol Bioeng 114, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, Brown A, 2015. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp Neurol 271, 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Taschner-Mandl S, Bileck A, Slany A, Kromp F, Rifatbegovic F, Frech C, Windhager R, Kitzinger H, Tzou CH, Ambros PF, Gerner C, Ambros IM, 2016. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 64, 2133–2153. [DOI] [PubMed] [Google Scholar]
- Winer J, Hughes S, Cooper J, Ben-Smith A, Savage C, 2002. gamma delta T cells infiltrating sensory nerve biopsies from patients with inflammatory neuropathy. Journal of neurology 249, 616–621. [DOI] [PubMed] [Google Scholar]
- Xia RH, Yosef N, Ubogu EE, 2010a. Clinical, electrophysiological and pathologic correlations in a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. Journal of neuroimmunology 219, 54–63. [DOI] [PubMed] [Google Scholar]
- Xia RH, Yosef N, Ubogu EE, 2010b. Selective expression and cellular localization of pro-inflammatory chemokine ligand/receptor pairs in the sciatic nerves of a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. Neuropathology and applied neurobiology 36, 388–398. [DOI] [PubMed] [Google Scholar]
- Yakubenko VP, Belevych N, Mishchuk D, Schurin A, Lam SC, Ugarova TP, 2008. The role of integrin alpha D beta2 (CD11d/CD18) in monocyte/macrophage migration. Exp Cell Res 314, 2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Dyck PJ, 1987. Abnormalities of endoneurial microvessels and sural nerve pathology in diabetic neuropathy. Neurology 37, 20–28. [DOI] [PubMed] [Google Scholar]
- Yezierski RP, Hansson P, 2018. Inflammatory and Neuropathic Pain From Bench to Bedside: What Went Wrong? J Pain 19, 571–588. [DOI] [PubMed] [Google Scholar]
- Yosef N, Ubogu EE, 2012a. alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol 227, 3857–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Ubogu EE, 2012b. GDNF restores human blood-nerve barrier function via RET tyrosine kinase-mediated cytoskeletal reorganization. Microvasc Res 83, 298–310. [DOI] [PubMed] [Google Scholar]
- Yosef N, Ubogu EE, 2013. An immortalized human blood-nerve barrier endothelial cell line for in vitro permeability studies. Cellular and molecular neurobiology 33, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Xia RH, Ubogu EE, 2010. Development and characterization of a novel human in vitro blood-nerve barrier model using primary endoneurial endothelial cells. Journal of neuropathology and experimental neurology 69, 82–97. [DOI] [PubMed] [Google Scholar]
- Yuan F, Yosef N, Lakshmana Reddy C, Huang A, Chiang SC, Tithi HR, Ubogu EE, 2014. CCR2 gene deletion and pharmacologic blockade ameliorate a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. PloS one 9, e90463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Rossi B, Angiari S, Constantin G, 2013. Use of imaging to study leukocyte trafficking in the central nervous system. Immunol Cell Biol 91, 271–280. [DOI] [PubMed] [Google Scholar]
- Ziganshin RH, Ivanova OM, Lomakin YA, Belogurov AA Jr., Kovalchuk SI, Azarkin IV, Arapidi GP, Anikanov NA, Shender VO, Piradov MA, Suponeva NA, Vorobyeva AA, Gabibov AG, Ivanov VT, Govorun VM, 2016. The Pathogenesis of the Demyelinating Form of Guillain-Barre Syndrome (GBS): Proteo-peptidomic and Immunological Profiling of Physiological Fluids. Mol Cell Proteomics 15, 2366–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zihni C, Mills C, Matter K, Balda MS, 2016. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17, 564–580. [DOI] [PubMed] [Google Scholar]