

SUMMARY
BIN1, a member of the BAR adaptor protein family, is a significant late-onset Alzheimer disease risk factor. Here, we investigate BIN1 function in the brain using conditional knockout (cKO) models. Loss of neuronal Bin1 expression results in the select impairment of spatial learning and memory. Examination of hippocampal CA1 excitatory synapses reveals a deficit in presynaptic release probability and slower depletion of neurotransmitters during repetitive stimulation, suggesting altered vesicle dynamics in Bin1 cKO mice. Super-resolution and immunoelectron microscopy localizes BIN1 to presynaptic sites in excitatory synapses. Bin1 cKO significantly reduces synapse density and alters presynaptic active zone protein cluster formation. Finally, 3D electron microscopy reconstruction analysis uncovers a significant increase in docked and reserve pools of synaptic vesicles at hippocampal synapses in Bin1 cKO mice. Our results demonstrate a non-redundant role for BIN1 in presynaptic regulation, thus providing significant insights into the fundamental function of BIN1 in synaptic physiology relevant to Alzheimer disease.
Graphical Abstract
In Brief
BIN1 is a significant risk factor for late-onset Alzheimer disease. BIN1 has a general role in endocytosis and membrane dynamics in non-neuronal cells. De Rossi et al. show that BIN1 localizes to presynaptic terminals and plays an indispensable role in excitatory synaptic transmission by regulating neurotransmitter vesicle dynamics.
INTRODUCTION
Genome-wide association studies (GWAS) have identified BIN1 as a major susceptibility locus for late-onset Alzheimer disease (LOAD). BIN1, also known as Amphiphysin 2 (Amph2), is an adaptor protein that, among other roles, regulates membrane dynamics in the context of endocytosis and membrane remodeling (Prokic et al., 2014). Two alternative mechanisms have been proposed to link BIN1 with AD Tau pathology from work in cultured neurons: first, BIN1 can bind to Tau in the cytosol, and second, the function of BIN1 in endocytosis limits extracellular Tau uptake and pathology propagation (Calafate et al., 2016; Chapuis et al., 2013). Microglial BIN1 also has been recently found to influence the release of Tau in extracellular vesicles (Crotti et al., 2019). The loss of BIN1 expression influences AD b-amyloid pathogenesis in cultured neurons, but this does not appear to be the case in vivo (Andrew et al., 2019; Miyagawa et al., 2016). Among older individuals without dementia, carriers of the BIN1 rs744373 risk allele were found to have similar amyloid pathology but increased Tau pathology and significantly impaired memory performance (Franzmeier et al., 2019). The Bin1 gene undergoes complex alternative splicing to generate >10 tissue-specific and ubiquitous isoforms (De Rossi et al., 2016a; Prokic et al., 2014). Independent studies have reported a decrease in the longest BIN1 isoform 1, which contains a central clathrin-associated protein-binding region (CLAP domain), in the brains of individuals with LOAD (De Rossi et al., 2016a; Holler et al., 2014; McKenzie et al., 2017). Despite the burgeoning interest in deciphering the involvement of BIN1 in AD pathophysiology, fundamental information on its physiological function in the brain, especially in neurons and synapses, is still lacking.
BIN1 and its mammalian paralog, Amph1, are members of the BAR (Bin/Amphiphysin/Rversus) domain protein superfamily. Amph1 was discovered as a synaptic vesicle-associated protein enriched in nerve terminals (Lichte et al., 1992) and suggested to play a role in synaptic vesicle endocytosis based on interactions with the AP-2 adaptor protein, dynamin 1, and synaptojanin (Micheva et al., 1997). Amph1 knockout (KO) mice have reduced viability and cognitive impairments. Neurons cultured from Amph1 KO mice revealed defects in synaptic vesicle recycling (Di Paolo et al., 2002). Furthermore, stiff person syndrome-associated autoantibodies against Amph1 compromised endocytosis preferentially at inhibitory GABAergic synapses by altering synaptic vesicle dynamics (Geis et al., 2010; Werner et al., 2016).
The high sequence similarity between Amph1 and BIN1 and the ability of BIN1 to interact with Amph1, dynamin1, AP-2, clathrin, synaptojanin, and endophilin suggested a role for BIN1 at the presynapse (Prokic et al., 2014). A recent study reported altered AMPA receptor-mediated synaptic transmission in cultured cortical neurons following acute knockdown of BIN1 expression (Schürmann et al., 2019). Aside from this single report of an in vitro study, the BIN1 function in synaptic transmission has not been characterized. Here, we investigated the role of BIN1 at the synapse and in learning and memory using neuronal conditional KO (cKO) mice. We report the impairment of spatial memory in association with the loss of BIN1 expression in the hippocampus. Functional analysis of synaptic transmission in the CA1 of the hippocampus demonstrated impairment in neurotransmitter vesicle release at excitatory synapses of the cKO mice. Super-resolution direct stochastic optical reconstruction microscopy (dSTORM) and immunoelectron microscopy (immuno-EM) localized BIN1 to presynaptic sites in glutamatergic synapses. Confocal and stimulated emission depletion (STED) microscopy analysis of presynaptic morphology in cKO mice revealed a decrease in the density of presynaptic sites and the size of presynaptic protein clusters. Finally, analysis of 3D EM image stacks from hippocampal CA3-CA1 synapses revealed a significant increase in docked and reserve pools of synaptic vesicles in cKO mice. These results demonstrate the presynaptic localization and functional role for BIN1 that are highly relevant to AD pathophysiology.
RESULTS
Characterization of BIN1 Neuronal Expression in the Brain
We generated neuronal Bin1 cKO mice by crossing the Bin1fl/fl strain with the Syn1-Cre and Emx1-IRES-Cre driver lines to explore the function of BIN1 in neurons. The Syn1-Cre transgene is specific to neurons with high expression in the hippocampus (Zhu et al., 2001), and Emx1-Cre is expressed in excitatory neurons and glia in the cortex and hippocampus (Gorski et al., 2002). The loss of BIN1 in the Bin1fl/fl;Syn1-Cre (henceforth referred to as Syn mice) and Bin1fl/fl;Emx1-IRES-Cre (henceforth referred to as Emx mice) cKO models were confirmed by immunoblots and immunostaining. Both BIN1:H and BIN1:L isoforms were significantly decreased in different areas of the brain in the Syn cKO and the Emx models (Figures 1A and S1). The loss of BIN1 expression in the cKO mice did not affect the expression of the paralog of BIN1, Amph1, and several other proteins in the brain (Figure S2; Emx data not shown). Immunostaining of BIN1 using monoclonal antibody (mAb) 13463 under conditions that preferentially stain neuronal BIN1 (Andrew et al., 2019) revealed an almost complete loss of BIN1 in the hippocampus (Figure 1B). Microdissection immunoblot analysis confirmed a significant decrease in both the BIN1:H and BIN1:L isoforms (Figures 1C and S2), suggesting that hippocampal neurons express multiple isoforms of BIN1, including those lacking the brain-specific CLAP domain, as confirmed by immunoblotting using pAb B1415, an antibody that is specific for the longest BIN1 brain isoforms 1 and 2 (BIN1:H).
Figure 1. Loss of Neuronal BIN1 Expression in cKO Mice.
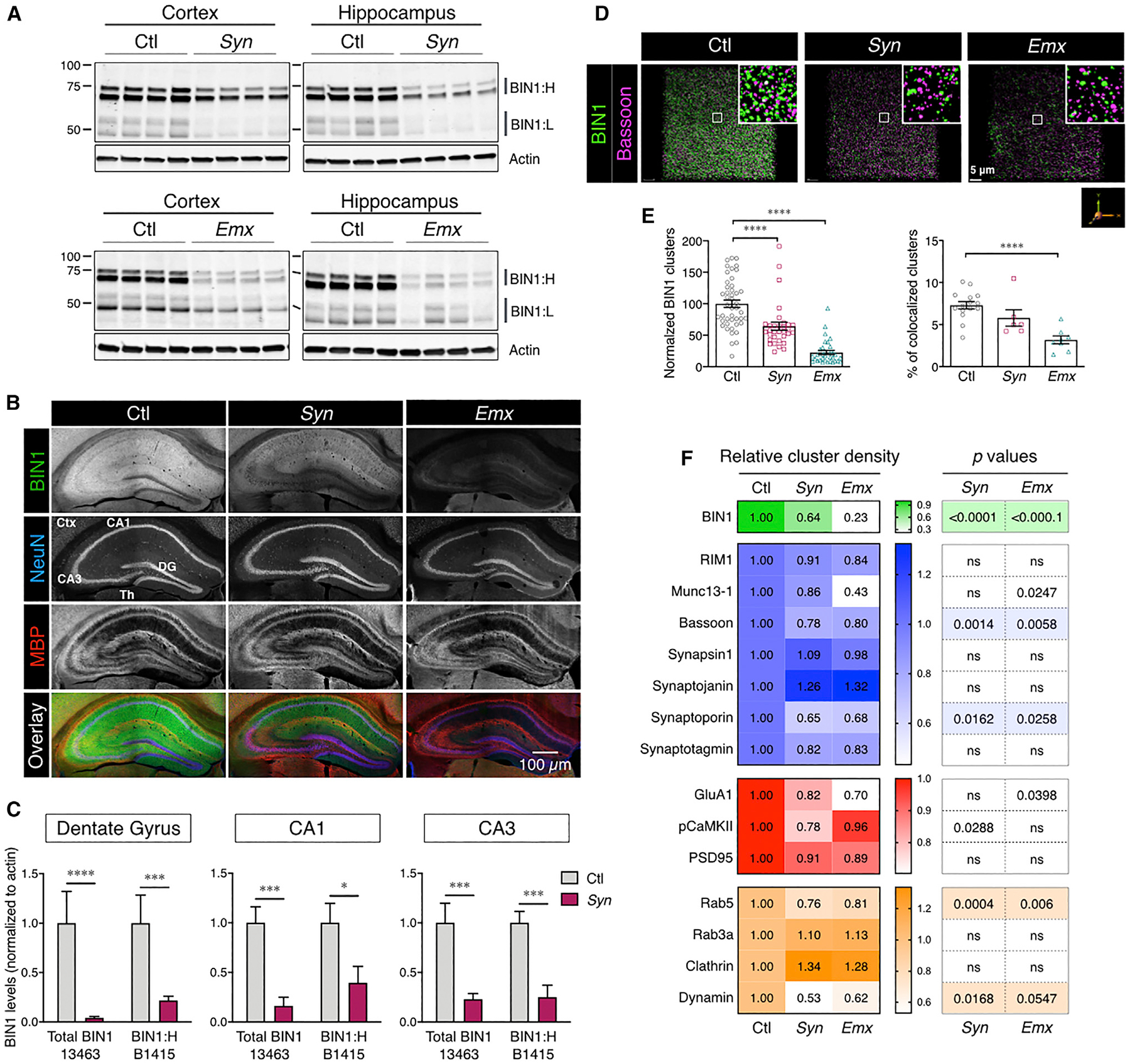
(A) Immunoblot analysis of BIN1 levels in the cortex and hippocampus of BIN1 cKO mice (polyclonal antibody [pAb] 14647).
(B) Confocal microscopy analysis of neuronal BIN1 expression in control (Ctl) and cKO (Syn and Emx) mice (mAb 13463; no epitope retrieval). Adjacent image panels acquired using a 10× objective were stitched together for the visualization of the entire hippocampus. Scale bar: 200 μm.
(C) Analysis of the diminution of BIN1 expression in the micro-dissected areas of the hippocampus of cKO mice. The means ± SEMs are plotted in each graph. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(D) Decreased presynaptic marker density in neuronal Bin1 cKO mice. Masks of BIN1 and Bassoon immunostaining in confocal images (see Method Details) were used to quantify synaptic marker densities from a block of 50 z stack images representing a depth of ~25 μm (96,193 mm3 volume).
(E) Decrease in BIN1 cluster density (left) in cKO mice (ANOVA: F(2, 107) = 50.7, p < 0.0001; Fisher’s LSD post hoc test: Ctl versus Syn and Ctl versus Emx p < 0.0001). Decrease of and BIN1-Bassoon colocalization, normalized to Bassoon cluster density (right) in Emx mice (ANOVA: F(2, 26) = 14.18, p < 0.0001; Fisher’s LSD post hoc test: Ctl versus Syn p = 0.0907 and Ctl versus Emx p < 0.0001).
(F) Heatmap representing mean changes in the density of different presynaptic markers (blue), postsynaptic markers (red), and presynaptic endocytic vesicle markers (orange).
See also Figures S1, S2, and S7 and Tables S1 and S2.
Impaired Recognition Memory and Spatial Learning in Mice Lacking Neuronal BIN1 Expression
We used multiple behavior tests to assess memory acuity, in which the hippocampus plays a major role. In the Y-maze test, a working memory task that engages neural circuits in the prefrontal cortex, hippocampus, basal forebrain, and septum (Lalonde, 2002), the loss of neuronal BIN1 expression did not affect total arm entries or spontaneous alternations (Figure 2A). In the novel object recognition test (NOR), a task that examines the communication between the hippocampus and the cortex in recognition memory (Winters et al., 2008), no difference was observed during the learning phase. Quantification of the interaction index during the memory recollection phase revealed a significantly impaired NOR performance in the Emx model but not in the Syn model (Ctl versus Syn p = 0.6411; Ctl versus Emx p = 0.0014). (Figure 2B). These data suggest that the loss of Bin1 in excitatory neurons in the forebrain led to an impairment in a task that assesses recognition memory performance.
Figure 2. Loss of Neuronal BIN1 Expression Impairs Spatial Learning and Memory.
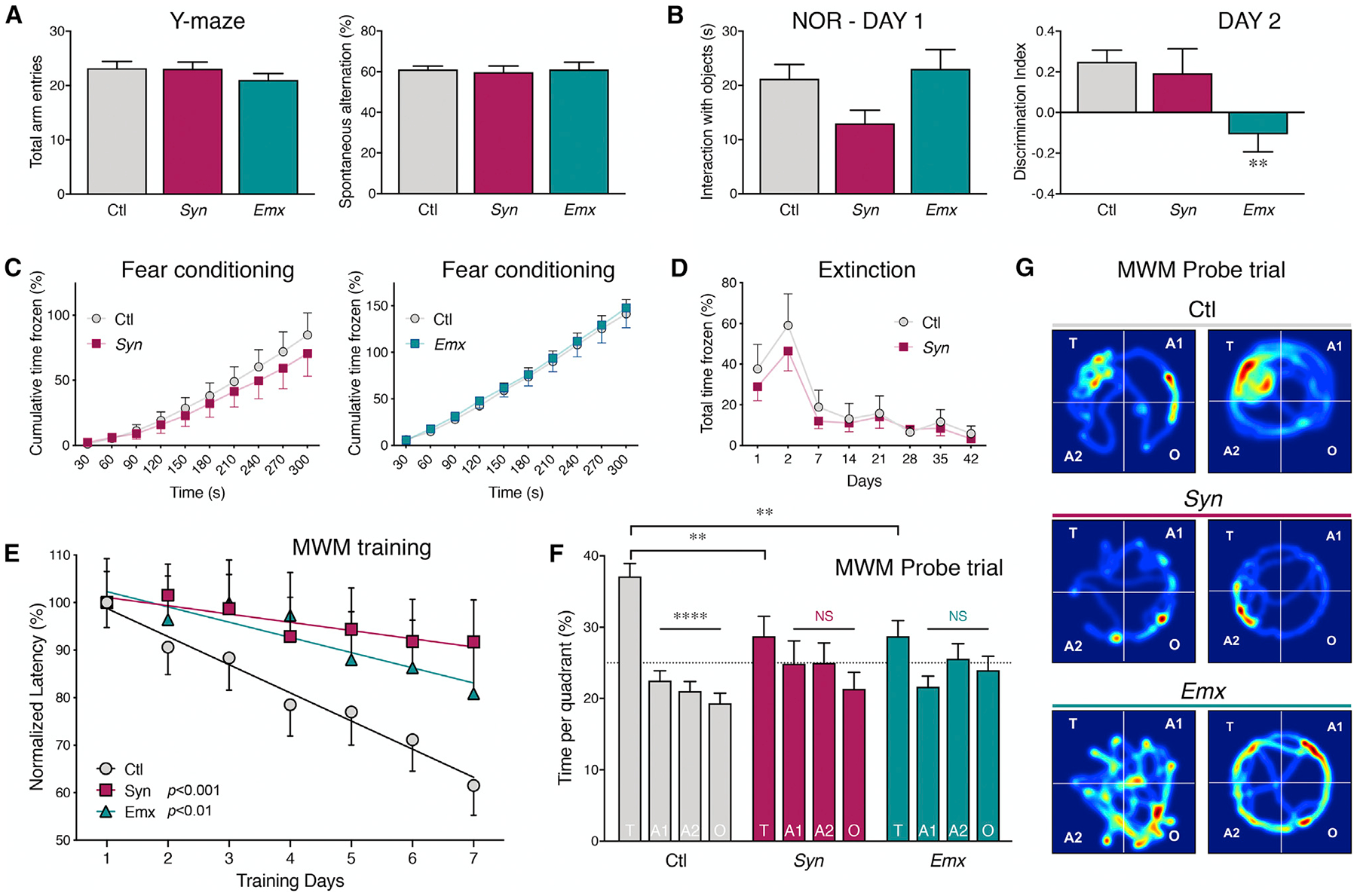
Neuronal cKO mice, 6 to 10 weeks old, and their littermate Ctl were subject to multiple behavioral tests.
(A) Y-maze test shows no significant difference between the groups in the number of arm entries (left) or spontaneous alternation behavior (ANOVA F(2, 78) = 0.07933, p = 0.9238; n = 44 Ctl, 18 Syn, and 19 Emx mice) (right).
(B) In the novel object recognition test, no significant difference between groups was observed during the interaction with the objects on day 1 (ANOVA: F(2, 101) = 2.419, p = 0.0942; n = 56 Ctl, 23 Syn, and 25 Emx mice). The discrimination index is plotted at right. ANOVA (F(2, 97) = 5.560, p = 0.0052; n = 56 Ctl, 19 Syn, and 25 Emx mice) showed no significant difference between Ctl and Syn (Fisher’s LSD post hoc test, p = 0.6411) and a significant difference between Ctl and Emx (Fisher’s LSD post hoc test, p = 0.0014).
(C) Contextual fear conditioning test shows no difference between cKO mice and their respective littermate Ctl in the cumulative time frozen (Ctl versus Syn: t27 = 0.5876, p = 0.5617, n = 15 Ctl and 14 Syn mice; Ctl versus Emx: t32 = 0.2483, p = 0.8055, n = 21 Ctl and 13 Emx mice; two-tailed Student’s t test).
(D) No statistical difference was observed in fear conditioning extinction between Syn mice and their littermate Ctl (Ctl versus Syn p > 0.24 at all times; n = 19 Ctl and 18 Syn mice; multiple t test analysis).
(E) Morris water maze test shows spatial learning defects in Bin1 cKO mice performance over the 7 days of training. There was a significant effect of genotype on the rate of learning (raw latency to find the platform; [F(2, 107) = 19.501, p < 0.0005]; n = 54 Ctl, 28 Syn, and 27 Emx mice). The performance of Ctl mice significantly improved during training as compared with Syn (p < 0.0005) and Emx mice (p = 0.004).
(F) In the probe trial, Bin1 cKO mice spent significantly less time in the target quadrant as compared with Ctl mice (F(2, 105) = 5.497, p = 0.0054). The percentage of time spent in each quadrant is plotted. T, target; A1 and A2, adjacent; O, opposite quadrant. ANOVA analysis (F(6, 424) = 2.930, p = 0.0082); post hoc Tukey’s multiple comparisons for Ctl mice: T versus A1, p < 0.0001; T versus A2, p < 0.0001; T versus O, p < 0.0001; no significant difference between the quadrants was recorded for Syn and Emx mice.
(G) Representative heatmaps of average search during the probe trial for Ctl and cKO groups.
See also Figure S3.
We then tested whether BIN1 is required for behavioral tasks that engage the amygdala and hippocampal circuits using a fear-conditioning paradigm (Kim and Jung, 2006). No difference in the cumulative freezing time was observed in Bin1 cKO mice as compared to littermate Ctl (Figure 2C). These results suggested that mice are able to associate a specific environment with the aversive stimulus in the absence of BIN1 expression. Moreover, both Ctl and Syn mice showed the same fear extinction pattern when mice were returned once per week in the contextual chamber without shock (Figure 2D), confirming normal amygdala-dependent learning.
Finally, we tested spatial learning memory using the Morris water maze and observed a clear difference between Ctl mice and both neuronal Bin1 cKO models. Over the 7-day training period, Ctl mice significantly reduced the time to find the platform (>80% success of finding the platform in 3 consecutive days), indicative of a successful learning process. However, the two cKO models failed to show a definite improvement in the latency to find the platform during the acquisition phase (the latency in seconds to find platform day 1 versus day 7: Ctl 74.57 ± 3.9 versus 45.86 ± 4.65, p < 0.0001; Syn 80.51 ± 5.26 versus 73.86 ± 7.10, p = 0.449; Emx 68.33 ± 6.3 versus 55.23 ± 7.12, p = 0.1439, two-way ANOVA with Fisher’s least significant difference [LSD] post hoc test). The Emx cohort was observed to have faster mobility and longer swim distance. When the swim distance was included as a covariate in the linear mixed effects model, there was a significant effect of genotype on the rate of learning (latency to find the platform; F(2, 107) = 19.501, p < 0.0005) (Figure 2E). We also examined the explorative strategies that mice used to find the platform after 3 to 4 days of training (Graziano et al., 2003). The Ctl mice scanned the arena using a random search or circling strategy over the initial days and then switched to self-orienting and approaching target strategy (5–20 s escape latency) or direct finding (1–10 s) in the following days. However, most of the cKO mice did not change their strategy from random searching (40–120 s) or scanning (15–60 s), indicating that the capacity for spatial learning is limited in the absence of BIN1 expression. In the probe trial, Ctl mice spent more time in the target area compared to the other quadrants. In contrast, the neuronal Bin1 cKO mice spent the same amount of time in each quadrant (the percentage of time spent in target versus opposite: Ctl 37.11 ± 1.81 versus 19.33 ± 1.43, p < 0.0001; Syn 28.76 ± 2.75 versus 21.36 ± 2.31, p > 0.05; Emx 28.75 ± 2.18 versus 23.99 ± 1.94, p > 0.05, two-way ANOVA with Tukey’s multiple comparisons test) (Figure 1F). These results suggest a significant deficit in spatial learning memory in Bin1 cKO mice. All of the mice performed equally in the visible platform test, and the swimming parameters were identical for all of the genotypes (Figures S3A and S3B). Thus, the failure to complete the task was not related to a physical problem but was due to a spatial memory deficit. We tested motor impairment in Syn and Emx models using rotarod tests and DigiGait analyses (Figures S3C–S3G). All of the measured parameters showed no difference between Bin1 cKO and Ctl mice. These data suggest a role for BIN1 in memory formation and consolidation.
Absence of Bin1 Causes a Deficit in the Presynaptic Vesicular Release
Given the effect of Bin1 deletion on learning and memory, we hypothesized that the loss of BIN1 in neurons results in an alteration of synaptic function. First, we compared the input/output (I/O) curve of synaptic responses in the CA1 of Ctl and Syn mice to examine whether there are any gross alterations in synaptic transmission. Analysis of the data using a linear mixed effects model showed that there was a significant interaction between stimulation intensity and genotype, suggesting that synaptic responses in slices from Syn mice at different stimulation intensities were different from those of Ctl mice (F(9, 283) = 3.435, p < 0.0005; n = 16 Ctl and 18 Syn). However, the overall effect of the genotype was not significant (F(1, 32) = 0.671, p = 0.42) (Figure 3A). Second, we examined short-term plasticity induced by short trains of stimuli to determine whether there was any alteration in presynaptic release probability (Klemmer et al., 2011; Lou et al., 2012; Zucker and Regehr, 2002). During a train of 10 synaptic stimuli given at 1 Hz, field excitatory postsynaptic potential (fEPSP) responses were potentiated to a significantly higher magnitude in Syn mice than in the Ctl group, suggesting that the release probability of CA1 synapses was lower in Syn mice (F(1, 27) = 8.74, p < 0.01; n = 15 Ctl and 14 Syn, two-way ANOVA) (Figure 3B). In separate voltage clamp recordings of miniature excitatory postsynaptic currents (mEPSCs) we observed a significantly reduced frequency of synaptic events in Syn mice, consistent with reduced release at individual terminals (0.77 ± 0.11 versus 0.49 ± 0.07 Hz, p = 0.038, Mann-Whitney test) (Figure 3C). There was no significant difference in the amplitude of the quantal events between Ctl and Syn mice (21.6 ± 0.7 versus 21.5 ± 0.9 pA, p = 0.93, Mann-Whitney test) (Figure 3D). This latter finding in CA1 neuronal postsynaptic response in intact slices is in contrast to the effect of acute knockdown of BIN1 expression in cultured cortical neurons, in which a reduction in mEPSC amplitudes was observed (Schürmann et al., 2019). To further examine whether there is an alteration in the presynaptic vesicular release probability in Bin1 cKO mice, we analyzed other proxies of the release probability in the whole-cell recording configuration. Paired pulse facilitation (PPF) with various interstimulus intervals (ISIs) was not significantly different between Ctl and Syn mice (PPF with ISI = 20 ms: Ctl, 1.63 ± 0.07 versus Syn, 1.90 ± 0.11; 40 ms: Ctl, 1.55 ± 0.06 versus Syn, 1.66 ± 0.07; 60 ms: Ctl, 1.40 ± 0.06 versus Syn, 1.48 ± 0.07; 80 ms: Ctl, 1.35 ± 0.04 versus Syn, 1.42 ± 0.06; F(1, 26) = 2.851, p = 0.103; n = 16 Ctl and 12 Syn, two-way ANOVA). In contrast to this, analysis of the coefficient of variation (CV) of EPSCs determined a significantly larger CV in Syn mice than in Ctl mice (Ctl, 0.21 ± 0.02 versus Syn, 0.27 ± 0.02, p = 0.013, Mann-Whitney test), which would indicate a lower synaptic release probability in Syn mice. Despite the lack of difference in PPF between the genotypes, our results from recording in the CA1 of the hippocampus by three measures of release probability indicate that there is a significant reduction in presynaptic vesicular release probability, whereas postsynaptic properties of CA1 synapses are not perturbed in Syn mice.
Figure 3. Bin1 Deletion Causes an Altered Presynaptic Function.
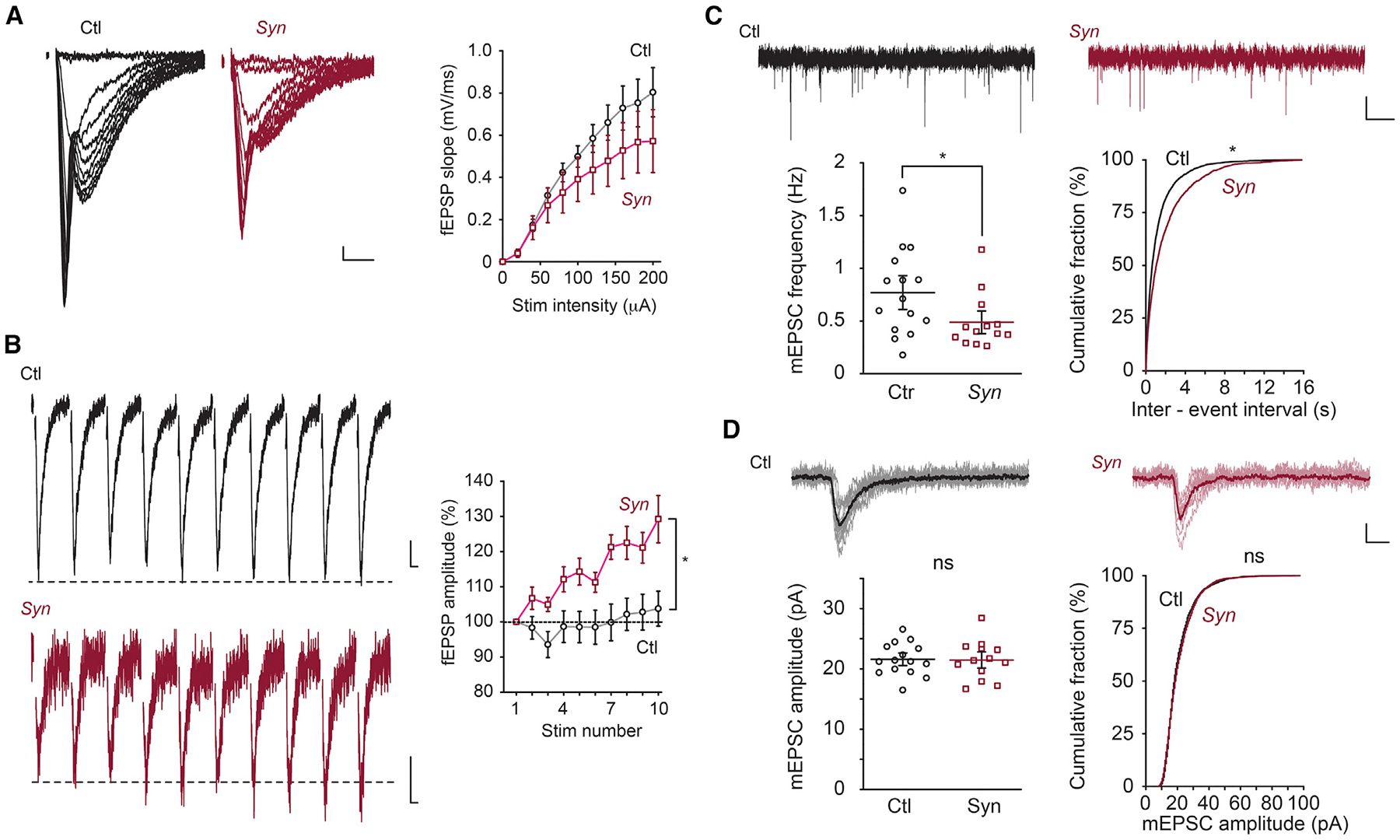
(A) I/O curve of fEPSPs. Representative fEPSP traces (left) and grouped data for average slope (right) in WT (Ctl) and Bin1 cKO (Syn) mice. There is a significant interaction between stimulation intensity and genotype; p < 0.0005. Calibration: 10 ms, 0.1 mV.
(B) Short-term plasticity (1 Hz). Representative fEPSP traces (left) and grouped data (right) in Ctl and Syn mice. Calibration: 10 ms, 0.1 mV.
(C) mEPSC frequency. Representative mEPSC traces (top) and group data for average frequency (bottom left) and cumulative plot of the interspike interval (bottom right) in Ctl and Syn mice. Calibration: 2 s, 10 pA.
(D) mEPSC amplitude. Representative individual mEPSC traces. Ten representative events and the averaged trace were presented as lighter and darker lines, respectively (top). Grouped data for average amplitude (bottom left) and cumulative plot of amplitude (bottom right) in Ctl and Syn mice. Calibration: 10 ms, 10 pA; 15–18 slices from 4 Ctl mice and 13–18 slices from 4 Syn mice were used; ns p ≥ 0.05.
Deletion of Bin1 Causes Slower Vesicle Depletion
The results from the hippocampal slices described above contrast with recently reported data from cultured cortical neurons in which the knockdown of BIN1 expression altered AMPA receptor (AMPAR) expression and surface localization at the postsynaptic site (Schürmann et al., 2019). Amphiphysins are thought to be involved in synaptic vesicle recycling by interacting with presynaptic endocytic proteins (Calafate et al., 2016; Prokic et al., 2014; Wigge et al., 1997). Therefore, we examined whether the loss of BIN1 expression affects synaptic vesicle depletion during sustained presynaptic stimulation, as reported in mutant mice for other presynaptic proteins (Chung et al., 2010; Cremona et al., 1999; Di Paolo et al., 2002; Ferguson et al., 2007; Lou et al., 2012; Milosevic et al., 2011; Vargas et al., 2014). Repetitive trains of stimuli were applied at a frequency of 20 Hz for 60 s, and fEPSP synaptic responses were recorded. The amplitude of the fEPSP depressed more slowly during the train in Syn mice than in Ctl mice (p < 0.05 for depletion constants for 50%, 30%, 20%, and 10%, n = 14 Ctl and 16 Syn, Mann-Whitney test) (Figures 4A–4C). One possible explanation for the slower rate of depression of synaptic responses is by an increased size of the vesicular pool to maintain synaptic strength. Therefore, we calculated the size of the readily releasable pool (RRP) in Syn mice by measuring the cumulative fEPSPs during the stimulus trains (Fernandes et al., 2015; Fioravante and Regehr, 2011; Zucker and Regehr, 2002) (Figure 4D). This analysis assumes that the depression of the synaptic response reflects a depletion of the RRP, allowing an estimate to be made of the product of Nq (where N is the total number of releasable vesicles and q is the quantal size) from the cumulative fEPSP plot (Fernandes et al., 2015; Schneggenburger et al., 1999). Given that we had observed no difference in the mEPSC amplitude in Ctl and Syn mice (Figure 3D), it can be assumed that the quantal size q is constant between the groups. With this analysis, we did not find a statistically significant difference in Nq, suggesting that the RRP (N) is not grossly affected by Bin1 loss (Nq, 78.2 ± 18.1 versus 102.6 ± 16.3, p = 0.29, n = 14 Ctl mice and 16 Syn mice, Mann-Whitney test) (Figure 4E). We also examined the recovery time course of the synaptic responses after depletion. Low-frequency stimuli (2 Hz) were applied immediately after the depletion paradigms and the fEPSP response was measured. Again, we did not observe a significant difference in the recovery time constant between Ctl and Syn mice (1.6 ± 1.2 versus 6.5 ± 2.5 (slope of percentage of recovery per stimulus number), p = 0.19, n = 14 Ctl mice and 16 Syn mice, Mann-Whitney test) (Figures 4F–4H). The physiological characterization of synapses revealed a role for BIN1 in regulating presynaptic release probability, but Bin1 loss had no measurable effect on the RRP.
Figure 4. Bin1 Deletion Causes Altered Depression of the EPSP during Sustained Synaptic Trains.
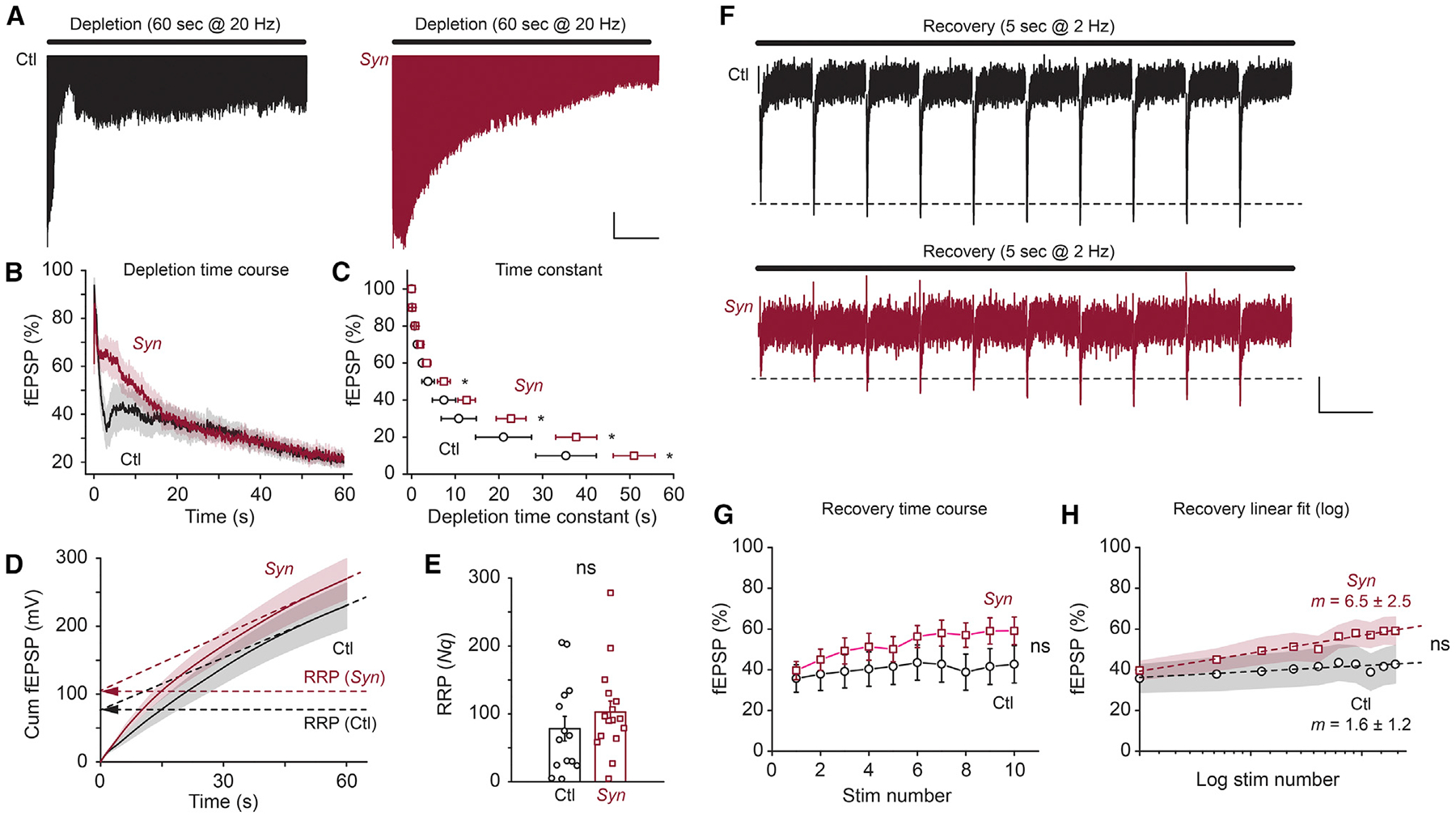
(A) Representative fEPSP traces during the depletion paradigm (20 Hz stimuli for 60 s) in Bin1 WT (Ctl) and cKO (Syn) mice. Calibration: 10 s, 0.1 mV.
(B) Grouped data for fEPSP depletion time course in Ctl and cKO mice.
(C) Grouped data for fEPSP depletion time constants for 90%–10% of the peak in Ctl and cKO mice.
(D) Grouped data for cumulative fEPSP amplitude during the depletion paradigm in Ctl and cKO mice. RRP is estimated from the y axis intersect of the linear regression fit.
(E) Grouped data for estimated RRP in Ctl and cKO mice.
(F) Representative fEPSP traces during the recovery paradigm (2 Hz stimuli for 5 s) in Ctl and cKO mice. Calibration: 500 ms, 0.1 mV.
(G) Grouped data for fEPSP recovery time course in Ctl and cKO mice.
(H) Grouped data for fEPSP recovery time course in logarithm fit in Ctl and cKO mice. The slope (m) of the linear fit is presented in each genotype; 14 slices from 3 Ctl mice and 16 slices from 4 Syn mice were used; ns, p ≥ 0.05.
BIN1 Localizes to Excitatory Presynaptic Sites
We performed immunogold EM analyses to determine the localization of BIN1 in CA3-CA1 synapses in the mouse brain using post-embedding immunogold EM on freeze-substituted tissue. Figure 5 shows representative images of BIN1 localization in a dendritic spine, a presynaptic excitatory bouton, and symmetric and asymmetric synapses (Figures 5A–5C). In the volume of tissue examined (578 excitatory pre- and postsynapses and 67 inhibitory boutons), 122 excitatory synapses were found to contain BIN1-immunogold particles as compared with 35 inhibitory presynaptic boutons. Only 35 of 578 postsynaptic sites were immunopositive. Thus, although BIN1 could be localized by immunogold particles in the postsynaptic compartment of axospinous synapses and symmetric and inhibitory presynaptic terminals, the frequency of labeling was highest in the presynaptic compartment of asymmetric, axospinous synapses. BIN1 was frequently found to be associated with large clusters of synaptic vesicles at axon terminals, but only occasionally located within the docking area of the active zone (Figures 5A–5C; see also Figure S4).
Figure 5. Investigation of BIN1 Synaptic Localization Using Immuno-EM and Ground-State Depletion (GSD) dSTORM Microscopy.
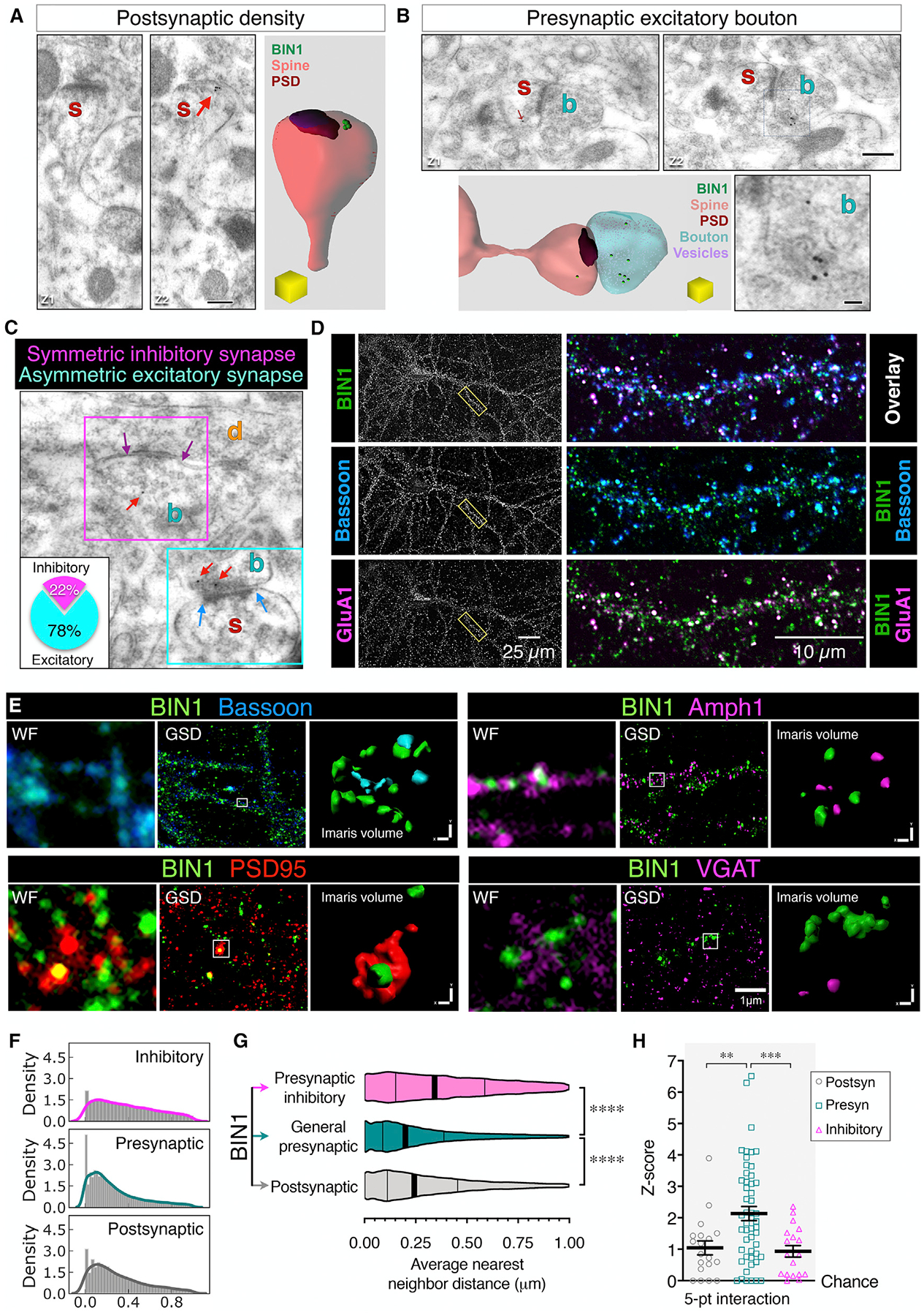
(A–C) Representative images of BIN1 immuno-EM localization in the mouse hippocampal CA1 region, showing BIN1-associated gold particles in the postsynaptic (A), general presynaptic (B), and inhibitory synapses (C). The inset in (C) indicates that of all of the synapses with BIN1-immunogold particles, 78% were excitatory terminals. Scale bars: 250 nm; enlarged inset, 50 nm.
(D) Representative confocal images of BIN1, Bassoon, and GluA1. Image overlays of the boxed region are shown at a higher magnification at right.
(E) Representative images of BIN1 (green) and markers for postsynaptic sites (PSD95; red), presynaptic sites (Bassoon; blue), and inhibitory presynaptic sites (Amph1 and VGAT; magenta). For each marker, the widefield image (WF), the super-resolution image (GSD), and the Imaris volume view (used for nearest-neighbor calculation) of the identical region are displayed.
(F) The density plot depicts the frequency of the distance between the BIN1 clusters and synaptic markers. The x axis indicates the binned DBSCAN average nearest-neighbor distance (microns) between BIN1 and synaptic markers.
(G) Violin plot of the average distance between BIN1 and its nearest neighbor. ANOVA analysis (F(2, 80700) = 1,332, p < 0.0001); BIN1-to-marker mean distance: general presynaptic markers = 0.2708 ± 0.001 μm; postsynaptic markers = 0.308 ± 0.002 μm; and presynaptic inhibitory markers = 0.3849 ± 0.002 μm). The vertical lines indicate the median and quartiles.
(H) DBSCAN analysis of randomization score to assess the specificity of BIN1 approximation to synaptic markers (5-point interaction; 50 points per cluster at 50-nm distance). The Z-score analysis showed a specific association of BIN1 with the synaptic markers; ANOVA analysis (F(8, 255) = 5.446, p < 0.0001). When the Z score equals 0, the association between BIN1 and the markers is not greater than a chance occurrence.
See also Figures S4–S6 and Table S1.
We further characterized BIN1 localization at the synapse in mature rat hippocampal primary neuronal cultures (Figures 5D and S5A). To precisely discriminate between presynaptic and postsynaptic localization of BIN1, we acquired super-resolution images at 20 nm pixel-size resolution using dSTORM (Figures S5B–S5D). Sub-diffraction resolution dSTORM images revealed a lack of enrichment of BIN1 in the dendritic spines (labeled with Ca2+/calmodulin-dependent protein kinase II [CaMKII]) or within the dendritic shaft (labeled with MAP2). We observed BIN1 in slender MAP2− processes, indicative of axonal localization of BIN1 (Figure S5C, white arrows). Moreover, BIN1 showed considerable overlap with Bassoon as compared with GluA1 or PSD95, suggesting a preponderance of presynaptic BIN1 localization (Figures 5E and S5). Our extensive data from immuno-EM and super-resolution microscopy analysis of hippocampal neurons in mouse brain slices and culture contrast with a recent study that focused on BIN1 postsynaptic localization (Schürmann et al., 2019). However, it should be noted that the authors of the previous report observed an obvious localization of BIN1 at the presynaptic site, visualized as a strong colocalization between BIN1 and synapsin1 (Schürmann et al., 2019).
For unambiguous quantification, BIN1 localization was compared with general presynaptic markers (Bassoon, synapsin1, and synaptophysin), and inhibitory presynaptic (VGAT) or postsynaptic (AMPAR-GluA1, PSD95, and CaMKII) markers. Recent studies showed an enrichment and a role for Amph1, the paralog of BIN1, in GABAergic synapses (Geis et al., 2010; Werner et al., 2016). For this reason, we included Amph1 as a marker of inhibitory synapses in addition to VGAT. To precisely determine BIN1 localization in dSTORM images, we mapped the cluster locations of BIN1 and the synaptic markers and calculated the distances between the cluster centers using the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) (see Method Details). We found that a significantly higher proportion of BIN1 clusters were within 30, 40, and 50 nm of presynaptic clusters as compared with the proximity of BIN1 to inhibitory presynaptic or postsynaptic clusters (ANOVA, 30-nm threshold: F(2, 84) = 30.09, p < 0.0001; 40-nm threshold: F(2, 84) = 27.05, p < 0.0001, 50-nm threshold: F(2, 84) = 38.73, p < 0.0001) (Figure S6A). However, we note that there still were a sizeable number of BIN1 clusters that were located in nearby postsynaptic markers. We confirmed these results using Imaris software 3D reconstruction of dSTORM images and calculating the nearest-neighbor distance between all BIN1 and the synaptic marker-stained structures. For statistical relevance and to not bias the data, the distance analyses were performed using a 1,000-nm threshold. A frequency distribution analysis revealed that a large number of BIN1 clusters were localized within 250 nm from synapses (Figures 5F and 5G). The average distance comparison confirmed significant differences in the proximity of BIN1 with presynaptic markers as compared with the nearest-neighbor distance between BIN1 and postsynaptic or inhibitory presynaptic markers (p < 0.0001). Next, we calculated how the proportion of estimated synaptic and BIN1 clusters differed from the chance occurrence, by shifting all of the coordinates belonging to a single synaptic cluster by a common vector (see Method Details). This randomization analysis confirmed a significant association between BIN1 and general presynaptic markers (Figures 5H and S6C). The compelling results from immuno-EM and dSTORM analyses indicate that BIN1 is preferentially but not exclusively enriched at the excitatory presynaptic terminals in hippocampal neurons.
Reduced Synaptic Density in Bin1 cKO Mice
To determine whether the loss of BIN1 alters the levels of synaptic proteins, we analyzed synaptosome fractions prepared from hippocampal samples of cKO mice and their littermate Ctl. Higher levels of the brain-specific BIN1:H were recovered in the non-PSD fractions relative to PSD fractions (p < 0.0001), and there was a significant decrease in both fractions of cKO mice as compared with wild-type (WT) animals (t test, non-PSD fraction, BIN1:H p < 0.0001, PSD fraction, BIN1:H p = 0.0161; n = 3 Ctl and 3 Syn) (Figure S7A). BIN1:L isoforms were also recovered in the non-PSD synaptic fractions (p = 0.0205) and were significantly reduced in cKO mice (p = 0.033). The levels of postsynaptic marker proteins in hippocampal PSD fractions showed no difference in AMPAR phosphorylation (p831 or p845) or CaMKII activation (4-month sample: F(4, 55) = 0.739, p = 0.5694; n = 8–12 Ctl, 3–7 Syn, and 5 Emx) (Figure S7). We also found no statistical differences between the genotypes in the abundance of 10 presynaptic and vesicle-associated proteins (4-month samples: F(18, 150) = 0.4135, p = 0.9835; n = 9 Ctl, 4 Syn, and 5 Emx) (Figure S7C). These results indicate that the loss of neuronal BIN1 expression has little effect on the overall abundance of several synaptic proteins.
To detect more subtle changes in the localization of synaptic proteins, we performed confocal and super-resolution microscopy, focusing on the CA1 region of the hippocampus, to examine whether the number or the size of presynaptic and postsynaptic clusters were altered in cKO mice. As expected, there was a significant reduction in the number of BIN1 clusters in cKO mice as compared with Ctl mice (Figures 1D and 1E). In addition, we observed a significant reduction in Bassoon clusters that colocalized with BIN1 in Syn and Emx mice as compared to Ctl mice. Furthermore, our results showed a significant reduction in the cluster density of SNAP receptor (SNARE)-associated proteins Bassoon and Synaptoporin (Figure 1F; Table S2). The cluster density of Munc13–1 was also significantly decreased in Emx mice, but the smaller decrease observed in Syn mice did not reach statistical significance. In addition, we observed a significant decrease in the cluster density of proteins associated with internalization at the presynaptic site: dynamin and Rab5. We also examined the postsynaptic proteins and observed no change in the cluster density of PSD95 (Figure 1F). The levels of GluA1 and pCamKII (Thr286) were also reduced, but the decrease in GluA1 was statistically significant only in Emx mice, and the reduction of pCaMKII was significant only in Syn mice (Figure 1F; Table S2). Synaptoporin and Munc13–1 are involved in presynaptic vesicle regulation and SNARE complex association (He et al., 2017; Li et al., 2017; Singec et al., 2002), and Bassoon is an organizer of the docking area at the presynaptic site (Annamneedi et al., 2018; Gundelfinger et al., 2016). These results suggested that the loss of BIN1 leads to a potential defect in synaptic density and the organization of synaptic proteins, including SNARE proteins at the presynaptic sites.
We, therefore, performed super-resolution STED microscopy to precisely assess the cluster volumes of SNARE-associated proteins in the CA1 region of hippocampal sections. We observed a significant increase in the volume of Rab5+ and Rab3a+ clusters at presynaptic sites (Figures 6A and 6C). An earlier study reported an increase in Rab5-GFP puncta in cultured neurons, as observed by conventional microscopy, following the knockdown of BIN1 expression (Calafate et al., 2016). Histogram analysis was performed to reveal better how the loss of BIN1 in cKO brains alters the size distribution of Rab5+ clusters, as observed by super-resolution microscopy. This analysis revealed an overall increase in the volume of Rab5+ clusters in Syn and Emx mice (Figure 6D). STED microscopy analysis also disclosed a significant decrease in the cluster size of synaptotagmin-1 (p < 0.0001), a protein involved in presynaptic endocytosis and known to be associated with BIN1 (Yao et al., 2010). Furthermore, we observed a significant reduction in the volume of Munc13–1 clusters (p < 0.0001), a SNARE-associated protein, whose cluster density was also reduced in Emx mice. However, we observed a significant increase in the cluster volume of synaptophysin (p = 0.0003), an abundant synaptic vesicle membrane protein (Figure 6G; Table S3). Moreover, the cluster volume of synaptojanin (p < 0.0001), a protein involved in ultra-fast endocytosis at the presynaptic site (Watanabe et al., 2018), was increased in the Syn model. Since immunoblot analysis of synaptosomes indicated no difference in the overall abundance of presynaptic proteins (Figure S7), the results of STED analyses would denote a difference in the local protein density at the synapse or indicate a change in the size of the vesicles where they are detected. The above results suggest a role for BIN1 in the organization of SNARE-regulating proteins and presynaptic vesicle dynamics. At the postsynaptic site, an increase in pCaMKII cluster volumes was recorded in cKO mice as compared with Ctl animals (p < 0.0001). The volumes of PSD95 clusters remained unchanged, and the volumes for GluA1 clusters were significantly increased only in the Emx model (p < 0.0001) (Figures 6F and 6G; Table S3). These results correlate well with the electrophysiology observations described above and suggest a role for BIN1 in the clustered organization of presynaptic machinery, and likely, compensatory changes occurring at the postsynaptic sites. Altered organizations of the presynaptic site, in particular, crucial active zone proteins in Bin1 cKO mice, could account for the robust differences in release probability and vesicular depletion that we observed in CA1 synapses.
Figure 6. Decrease in the Volume of SNARE-Regulating Proteins in cKO Mice.
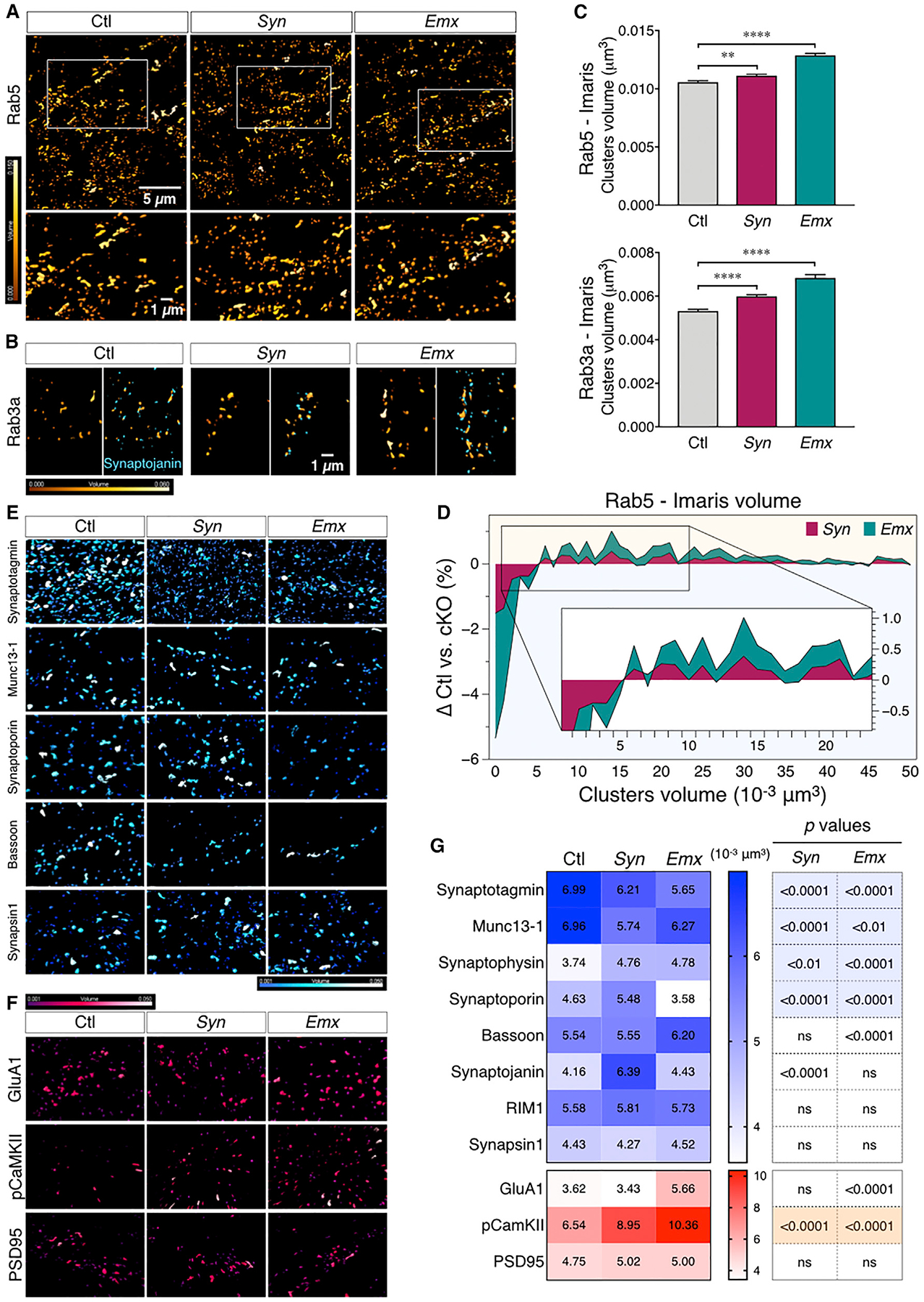
(A) Representative images of Rab5 cluster intensity-coded by volume size (Imaris). The boxed region is shown at higher magnification in the bottom panels.
(B) Rab3a clusters (left, coded by volume size) and two-color images (intensity-coded by volume) depicting the close proximity of Rab3a clusters to synaptojanin (right).
(C) Bar graphs representing the analysis of Rab5 and Rab3a cluster volumes in Ctl and cKO mice. ANOVA with Fisher’s LSD post hoc test revealed a significant increase in the cluster volume sizes for Rab5 (F(2, 47322) = 60.43, p < 0.0001, Ctl versus Syn p = 0.0018, Ctl versus Emx p < 0.0001) and Rab3a (F(2, 25397) = 50.55, p < 0.0001, Ctl versus Syn and Ctl versus Emx p < 0.0001).
(D) Stacked area plot of the difference in Rab5 cluster-size frequency distribution between Ctl and cKO. The difference in distribution was calculated as [(number of Rab5 clusters/volume)Ctl (number of Rab5 clusters/volume)cKO], with a bin size of 0.001 μm3. For clarity, only the clusters ranging from 0.001 to 0.05 μm3 are shown in the plot. The inset shows an enlarged area of the plot.
(E and F) Representative images of presynaptic (E, blue) and postsynaptic (F, red) protein clusters generated with Imaris and intensity-coded by volume (from 0.001 to 0.05 μm3).
(G) Heatmap representation of the mean variation of presynaptic and postsynaptic protein cluster volumes.
See also Figures S7 and S8 and Table S3.
To examine the possibility that the loss of BIN1 in cKO mice alters neurotransmitter vesicle properties and/or distribution, we used serial section conventional EM. An automatic tape-collecting ultramicrotome was used to generate serial ultra-thin sections, which were then imaged at nanoscale resolution to reconstruct the presynaptic and postsynaptic compartments of CA3-CA1 axospinous synapses from Ctl, Syn, and Emx mice in their entirety (Kasthuri et al., 2015). This approach allowed us to measure the size of the PSD as well as count the number of docked neurotransmitter vesicles and the reserve pool in each presynaptic terminal of Ctl and Bin1 cKO mice. We found that the number of docked vesicles was significantly elevated in the cKO mice, as compared to their Ctl counterpart (Figure 7). Furthermore, the reserve pool within the synaptic bouton was significantly elevated in the cKO mice, as compared with Ctl animals (Figure 7). Although there was no measurable difference in Nq calculated from electrophysiological measurements, the slower time constant for the depletion of synaptic events observed in CA1 pyramidal neurons from cKO mice during sustained presynaptic stimulation is consistent—or atleast not contradictory—with the EMdata. Finally, the PSD areas of the different mouse lines did not differ from one another (Figure 7), a finding that is in agreement with the enrichment of BIN1 in the presynaptic compartment and the absence of apparent postsynaptic changes in cKO neurons. In aggregate, the electrophysiology and 3D EM analysis of hippocampal synapses show that the lack of BIN1 expression affects synaptic transmission and suggest that BIN1 may be an important component of presynaptic vesicular dynamics.
Figure 7. Bin1 Deletion Increases Docked Neurotransmitter Vesicles and Reserve Pool but Does Not Affect Synapse Size.
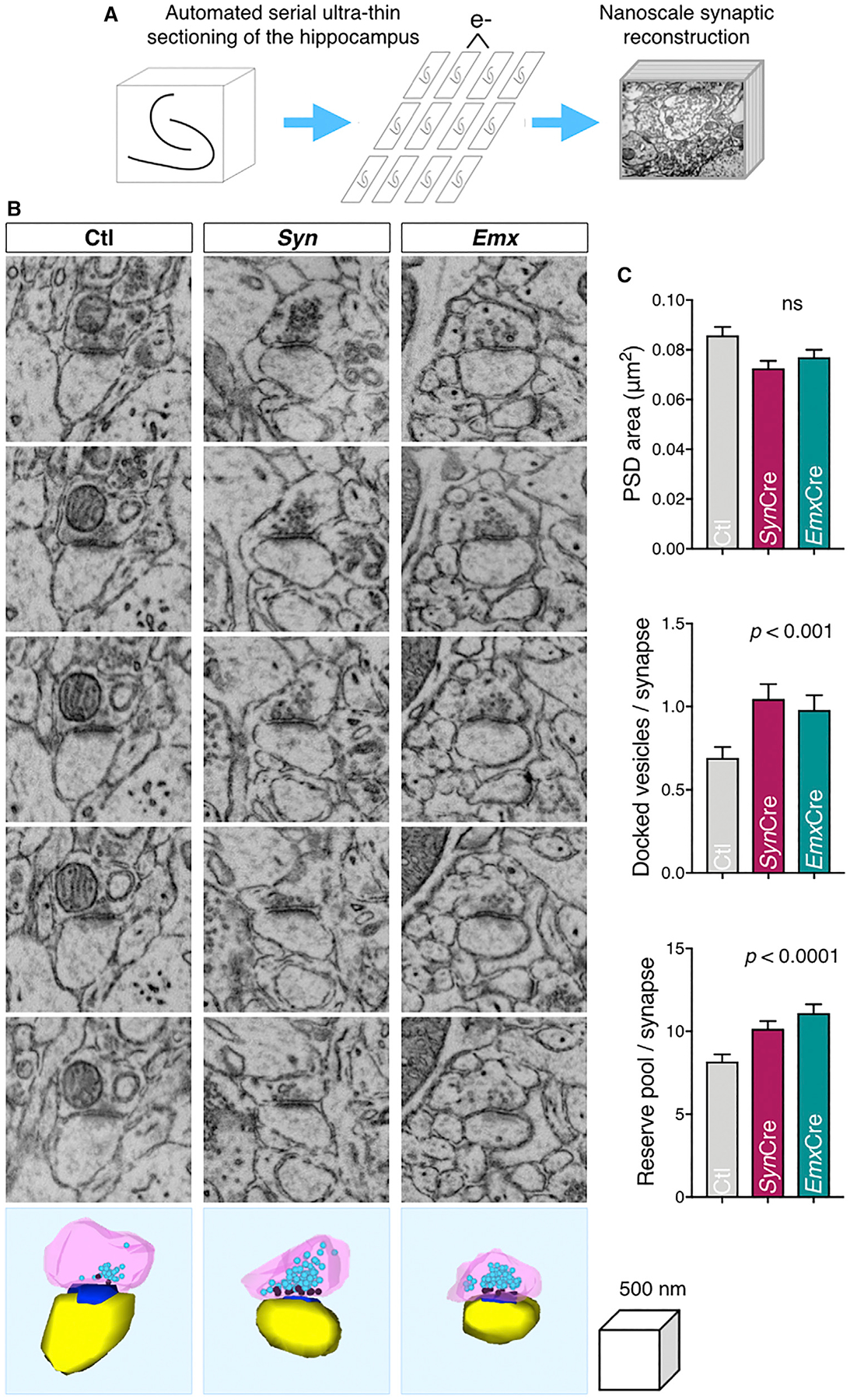
(A) Schematic representation of the tape-based pipeline for electron microscopic reconstruction of hippocampal synapses (Kasthuri et al., 2015).
(B) Electron micrographs of serial sections through representative synapses are shown along with a 3D reconstruction of the dendritic spine head (yellow), the postsynaptic density (PSD; blue), and the presynaptic terminal (pink) containing docked neurotransmitter vesicles (dark pink) and the reserve pool of vesicles (aqua).
(C) The PSD area was not significantly different among the three mouse genotypes, but the number of docked vesicles per synapse and the number of vesicles in the reserve pool was significantly higher among synapses in both cKO mouse strains as compared with Ctl by multivariate analysis of covariance. Multivariate analysis of variance (MANOVA) on PSD area: F(2, 436) = 0.001, p = 0.42; multivariate analysis of covariance (MANCOVA) on docked vesicles: F(2, 436) = 8.81, p = 0.0002; MANCOVA on reserve pool: F(2, 436) = 9.53, p = 0.00009.
See also Figure S8.
DISCUSSION
Despite the identification of BIN1 as the second most common susceptibility gene for LOAD, the underlying cellular mechanism by which BIN1 elevates the risk for AD remains poorly understood because the normal role of BIN1 in neurons has not been fully elucidated. Using behavioral and electrophysiological approaches, we characterized Bin1 cKO models to reveal that the loss of BIN1 expression in neurons leads to impaired spatial memory consolidation and deficits in synaptic transmission, particularly at the level of presynaptic vesicle release. Using super-resolution microscopy and immuno-EM analyses, we show that BIN1 localizes to presynaptic sites in excitatory synapses and document reduced synaptic density, altered presynaptic protein clustering, and neurotransmitter vesicle distribution in Bin1 cKO mice. These findings are highly relevant to synaptic dysfunction and cognitive decline in human LOAD because BIN1 has been implicated in memory impairments. For example, recent studies have uncovered a correlation between LOAD-associated BIN1 rs744373 single nucleotide polymorphism and memory impairment (Franzmeier et al., 2019) or poor performance in high-load working memory task and lower functional connectivity in healthy carriers (Zhang et al., 2015) and memory impairment in patients with temporal lobe epilepsy (Bungenberg et al., 2016).
BIN1 is an essential component of the endocytic machinery as evidenced by (1) the recruitment dynamics of BIN1 to clathrin-coated pits before scission (Taylor et al., 2011); (2) the critical function of the BIN1 SH3 domain in recruiting dynamin and regulating its self-assembly at the clathrin-coated pits (Owen et al., 1998); (3) BIN1 interaction with clathrin-coated pits-associated proteins Amph1, AP-2 complex, clathrin, synaptojanin 1, endophilin, and multiple other proteins involved in regulating endocytosis (Prokic et al., 2014). Moreover, the activation of the calcium-dependent phosphatase calcineurin coordinately dephosphorylates dynamin, synaptophysin, Amph1, and BIN1 during synaptic vesicle recycling (Marks and McMahon, 1998). However, the involvement of BIN1 in hippocampal synaptic transmission has not been experimentally assessed in vivo. By characterizing Bin1 cKO mice, we demonstrate that the ablation of BIN1 expression in neurons results in (1) reduced vesicular release probability concomitant with an increase in the number of docked vesicles; and (2) an increase in the number of reserve pools of synaptic vesicles, which is consistent with a slower depletion time course of synaptic responses during sustained activity in excitatory synapses in the CA1 region of the hippocampus. These results indicate an altered synaptic vesicle turnover in Bin1 cKO mice that could reflect a critical role of BIN1 in the regulation of both exocytic and endocytic mechanisms associated with synaptic transmission. The presynaptic changes in hippocampal synapses of Bin1 cKO mice are schematically represented in Figure S8.
In the pan-neuronal Syn cKO model, the frequency of Cre-mediated recombination varied in different brain regions, with the hippocampus showing the striking loss of BIN1 expression in all subregions. Consistent with lineage tracing and fluorescence reporter analyses using the Emx1-IRES-Cre line (Gorski et al., 2002), BIN1 expression was ablated in forebrain glutamatergic neurons and glia in the Emx1 cKO model. Both Syn and Emx1 cKO models displayed impaired spatial learning, whereas the Emx1 cKO showed an additional impairment in recognition memory. The latter finding likely represents the extent to which BIN1 expression is lost in the Emx1 cKO cortex as compared with Syn cKO mice (Figure 1). Of note, unlike the concomitant loss of BIN1 expression in Amph1 KO mice reported previously (Di Paolo et al., 2002), neither the Syn nor the Emx1 cKO model showed a diminution of Amph1 expression. Thus, the phenotypes we observe are entirely attributable to the primary loss of BIN1 expression. The selectivity of Emx1-IRES-Cre to target forebrain glutamatergic neurons suggests an essential role for BIN1 in excitatory synaptic transmission. This notion is experimentally supported by the enrichment of BIN1 at presynaptic sites observed by sub-diffraction-limited dSTORM microscopy and immuno-EM analyses. BIN1 localization is not exclusive at the presynaptic site, as we observed BIN1 immunofluorescence staining along axons and found occasional BIN1+ immunogold particles in the periphery of the postsynaptic density in hippocampal CA3-CA1 synapses.
Genetic deletion of endocytic proteins results in vesicular recycling deficits (Lou et al., 2012; Rizzoli and Betz, 2005). However, in contrast to our findings in Bin1 cKO mice, loss of endocytic proteins typically results in faster depletion of synaptic responses during sustained activity, which is a shared phenotype in mouse lines lacking synaptojanin1 (Cremona et al., 1999), dynamin1 (Ferguson et al., 2007), endophilins (Milosevic et al., 2011), and most important, Amph1 (Di Paolo et al., 2002). It is important to note that loss of Amph1 expression also results in a drastic reduction in BIN1, which may indicate that phenotypes observed in Amph1 KO neurons represent the effect of the loss of both Amph1 and BIN1 function (Di Paolo et al., 2002). Double KO mice lacking neuronal dynamin isoforms, dynamin1, and dynamin3, demonstrate slower vesicle depletion and reduced release probability similar to what we have observed in Bin1 cKO mice (Lou et al., 2012). However, unlike in dynamin double KO mice (Lou et al., 2012), there was no detectable alteration in the size of the functional RRP in Bin1 cKO mice (Figure 4). Therefore, it seems that the primary role of BIN1 in the presynapse is to regulate release probability. Based on the 3D EM data, the defect in vesicular release in Bin1 cKO mice appears to be in a step that follows initial vesicle docking. The intriguing finding that BIN1 regulates exocytosis of the synaptic vesicles does not rule out an additional role for BIN1 in presynaptic endocytic processes. Following the knockdown of BIN1 expression in cultured neurons, Rab5 is overactivated, leading to increased endocytic flux and suggesting that the BIN1 function limits the kinetics of endocytosis (Calafate et al., 2016). In agreement, our STED analyses revealed an increase in the size of Rab5+ clusters in the hippocampus of Bin1 cKO mice.
Paradoxically, a recently published study reported that BIN1 regulates postsynaptic trafficking and glutamatergic signaling in cultured cortical neurons (Schürmann et al., 2019). Despite the focus on postsynaptic BIN1, the results of the published study included presynaptic BIN1 localization documented by the immuno-EM analysis of cortical tissue and images showing the colocalization between synapsin1 and BIN1 in cultured cortical neurons, as observed by immunofluorescence labeling. Moreover, although the authors focused on changes in mEPSC amplitude that were found to be statistically significant in their electrophysiological characterization, acute BIN1 knockdown increased the average AMPA mEPSC inter-event interval by 2-fold, which was found to be non-significant due to a large spread in the data (Schürmann et al., 2019). In contrast to the findings reported by Schürmann et al. (2019), our investigation of Bin1 cKO mice reveals reduced synapse density and a significant role for BIN1 in synaptic vesicle dynamics, as well as no effect on quantal size based on a comprehensive characterization of multiple presynaptic parameters by hippocampal slice physiology. The presynaptic organization changes revealed by our investigation were confirmed by sophisticated 3D-EM reconstruction of hippocampal slices from two cKO models.
Our characterization of hippocampal synapses in Bin1 cKO mice using super-resolution STED microscopy uncovered a significant reduction in the cluster volume of two presynaptic proteins that are essential for presynaptic vesicle docking and neurotransmitter release, namely synaptotagmin-1 and Munc13–1 (Li et al., 2017; Sakamoto et al., 2018; Südhof and Rizo, 1996). Moreover, there was a significant increase in pCaMKII cluster volume, but not that of GluA1 or PSD95. The increase of pCaMKII cluster volume could be the result of a compensatory response to presynaptic changes or represent a specific role for BIN1 in postsynaptic sites in the brain. Thus, the findings reported here reveal that the loss of BIN1 expression in vivo has a more robust impact in the presynaptic terminal, leading to synaptic defect and memory impairments. We surmise that the reduction in the cluster volume of SNARE-related presynaptic proteins may contribute to the accumulation of docked vesicles and the lower release probability observed in CA1 synapses in Bin1 cKO mice. A range of synaptic dysfunction, including presynaptic vesicular defects, have been described in mouse models of AD. For example, presynaptic functions have been detailed for APP (Priller et al., 2006), Aβ (Fogel et al., 2014), PS1 (Barthet et al., 2018; Zhang et al., 2009), and APP/tau/PS1 (Chakroborty et al., 2019). Finally, a recent in-depth proteomic analysis found unbiased evidence to support presynaptic alterations that are more important than postsynaptic changes in the early stages of the disease in human AD ((Haytural et al., 2020).
Using cKO models, our study demonstrates the presynaptic BIN1 localization and a role for BIN1 in synaptic physiology that correlates with effects on learning and memory. BIN1 is known to interact with several proteins found in synaptic boutons, including Amph1, dynamin, clathrin, AP-2, endophilin, synaptojanin, and phospholipase D (Prokic et al., 2014). How BIN1 mechanistically participates in exocytosis at the presynaptic site and whether the dysregulation of the presynaptic vesicle dynamics is indirectly responsible for the altered cluster organization of SNARE-associated proteins in hippocampal neurons in Bin1 cKO are key questions to address in future studies. In understanding the involvement of BIN1 in LOAD, only correlative data are available thus far to link BIN1 expression or the presence of LOAD-associated BIN1 single nucleotide polymorphisms with Tau pathology and memory impairment (Franzmeier et al., 2019; Holler et al., 2014; Zhang et al., 2015). Large-scale analysis of transcriptomes and proteomes have identified an AD-associated decrease in BIN1 expression as part of the oligo-dendrocyte gene coexpression network dysfunction (McKenzie et al., 2017) and alternative splicing of BIN1 in AD (Johnson et al., 2018). With the revelation that BIN1 may be an essential component of presynaptic vesicular dynamics, our report opens new paths for future investigation on the precise role of BIN1 as a risk factor in AD pathophysiology.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Gopal Thinakaran (thinakaran@usf.edu).
This study did not generate new transgenic mouse lines. The antibody generated in this study is available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bin1fl/fl animals were obtained from Dr. George C. Prendergast (Lankenau Institute for Medical Research) (Chang et al., 2007). Syn1-Cre (JAX stock #003966) and Emx1-IRES-Cre (JAX stock #005628) lines were obtained from The Jackson Laboratory (Bar Harbor, ME). We crossed Bin1fl/fl strain with Syn1-Cre or Emx1-IRES-Cre driver lines to generate Bin1fl/fl;Syn1-Cre (Syn mice), Bin1fl/fl;Emx1-IRES-Cre (Emx mice), and Bin1fl/fl or Cre littermates (referred to as Ctl mice). Female mice carrying the Syn-Cre transgene were used in breeding to prevent possible germline ablation of Bin1. Mice were maintained in a C57BL/6J background. All mice were housed under standard conditions with free access to food and water. All procedures related to animal care and treatment conformed to the policies of the Institutional Animal Care and Use Committee at the University of Chicago.
METHOD DETAILS
Behavioral tests
Behavioral assays were conducted using 6- to 10-week-old neuronal Bin1 cKO mice and their littermate controls to test memory and motor functions. Male and female cohorts were used, but no sex differences were recorded, and the data presented in this study reports the effect of the genotype in both males and females. In addition, animals as young as P15 were subject to rotarod analysis. The number of animals used per genotype for individual experiments is indicated in the figure legends.
Y-Maze spontaneous alternation
Testing occurred in a Y-shaped maze with three white, opaque plastic arms at a 120° angle from each other. Each mouse was placed in the middle of the symmetrical Y-maze and allowed to explore freely through the maze during an 8-min session. The movements of the mice were recorded using a 180° fisheye camera (Yi 4K Action and Sports Camera, 12MP) to avoid manipulator-stress induced interference, and analyzed a posteriori. An entry occurred when all four limbs were within the arm. The sequence and the total number of arm entries were recorded. An alternation is defined as entries into all three arms on consecutive occasions. The percentage of alternations was calculated as: the number of triads containing entries into all three arms / maximum alternations) X 100. A minimum of 12 entries/mouse was required. The results were compared by ANOVA.
Novel object recognition (NOR)
NOR test was conducted in an open field arena (11.7” H X 15.7” W X 19.7” D) using objects that were similar in height and volume but were different in shape and appearance. During habituation, the animals were allowed to explore an empty arena. 24 h after habituation, mice were exposed to the familiar arena with two identical objects placed at an equal distance for a total period of 8 min. The next day, the mice were allowed to explore the open field in the presence of one familiar object (Old) and a novel object (Novel) to test long-term recognition memory. Mice that did not interact with the objects on day 1 were excluded from the study. The time spent exploring each object, and the discrimination index percentage were recorded and analyzed by ANOVA. The discrimination index was calculated as [(time with Novel object - time with Old object) / total time interaction].
Contextual fear conditioning
The experiment was performed using an Actimetrics fear conditioning chamber, and the footshock was delivered by a Coulbourn Instrument shocker. The mouse movements were recorded by FreezeFrame 4 software. On day 1, each mouse was placed in a conditioning chamber and allowed to explore. After a 2 min habituation period, a brief, mild foot shock (1.0 mA, 2 s) was delivered through a grid floor at the bottom of the cage. After an interval of 2 min, the animal was presented again with the foot shock to strengthen the association. The mouse remained in the chamber for an additional 3 min before it was returned to its home cage. On the following day, the animal was brought to the same conditioning chamber, and its movements were recorded with a video camera for 5 min to test for contextual conditioning. The data were analyzed using FreezeFrame 4 software, which automatically monitors baseline motor activity and expression of fear. For contextual fear conditioning extinction, the animals were returned to the conditioning chamber (no foot shock) 3, 7, 15, 21, 28, 35, and 42 days after the initial fear conditioning and their movements were monitored for 5 min (as above) to determine extinction of the fear memory. Multiple unpaired t tests were used to compare cumulating freezing time between genotypes, with multiple comparison adjustments made using the Holm-Sidak method.
Morris water maze spatial learning
The water maze apparatus (a 122 cm tank) was filled with room temperature water (22–25°C), which was made opaque by the addition of non-toxic, white tempera paint. The water temperature was monitored using a thermometer before beginning the trials. Several high-contrast visual cues were placed around the apparatus in the room. Mice were placed on the platform for 10 s prior to the first training trial of each day to reduce stress. Over a period of 7 days, mice were trained to swim to a 14 cm diameter circular clear Plexiglas platform submerged 1.5 cm beneath the surface of the water and invisible to the mice while swimming. If they did not find the platform within the 120 s trial limit, they were guided to the hidden platform. The platform location (positioned in the middle of the Target (T) quadrant) was selected randomly and kept constant for each cohort throughout the training. On each trial, the mouse was placed into the tank at one of three designated start points (excluding the T quadrant) in a semi-random order. To ensure that memory differences are not due to lack of task learning, each mouse was undergoing a daily five-trial session (120 s for each with a 1 min interval). 24 h after the last training trial, the retention of the spatial memory was assessed in a probe trial, which consisted of a 60 s free swim in the pool without the platform. An overhead-mounted video tracking system (EthoVision XT, v.8.5 and v.14) was used to monitor and quantify different parameters during the probe trial. The corrected time spent in each quadrant (total time per quadrant – the time of immobility in each quadrant) was measured, and the results were compared by ANOVA. Other swimming parameters (speed, mobility, immobility, and velocity) were analyzed to control for each mouse’s ability to swim. The following day, mice were submitted to a visible platform test to ensure no physical impairment influenced the performance during the test. The parameters were kept the same as for the training days, except that the location of the platform was indicated by a visible cue directly placed on the platform in the pool. The platform was placed at a different location than that during the probe trial to avoid any spatial memory-related success. The distance traveled to reach the platform was significantly longer in the Emx cohort (p < 001). Hence, the swim distance was included as a covariate and the training data were analyzed by a linear mixed effects model (SPSS, v. 21).,The probe trial data were analyzed using ANOVA.
Rotarod
Mice were examined for deficits in motor coordination and balance by the accelerating rotarod test and fixed rotarod test. Mice were placed on rotating drums (3-cm diameter) of an accelerating rotarod with 4 lanes (Harvard Apparatus). Adult mice were given 5 trials per day, with a 60 s inter-trial interval for 4 consecutive days. Younger mice were tested [5 trials per day, with a 60 s inter-trial interval] twice a week (P15 to 1 month) and then less frequently (i.e., once a week or once every two weeks) till approximately 4 months of age. The animal’s latency to fall and rotation speed when the fall occurs were recorded using an automated system. The data were analyzed by a linear mixed effects model (SPSS). Mice were also tested on the rotarod using fixed speeds of 5, 10, 15, or 20 rpm for a maximum of 60 s per trial. The latency to fall was recorded as above. Each mouse was given 3 trials per speed, with a 60 s inter-trial interval and at least 5 min rest between the different speeds (Carter et al., 2001).
DigiGait
The DigiGait trials were performed using the DigiGait Imaging System (Mouse Specifics, Inc.). Each animal was placed on a stationary treadmill, which was then accelerated to the test speeds of 20, 30, and 40 cm/s. At their first trials, mice were allowed to run on the treadmill by gradually increasing the speed of the treadmill. Ten trials were attempted at each speed, and the animals were rested at least 10 s between trials. The successful trials (in which the animal is able to stay on the treadmill for > 5 s) were counted and recorded. If a mouse failed to gain speed and run, the trial was repeated after 5 min of rest with the treadmill held at the next lower speed.
Immunoblot analysis
Hemibrain and sub-regions of the central nervous system were dissected and homogenized (20% weight/volume) in ice-cold lysis buffer [150 mM NaCl, 50 mM Tris-HCl pH 7.4, 0.5% NP-40, 0.5% sodium deoxycholate, 5 mM EDTA, 0.25 mM PMSF and protease inhibitor cocktail (Sigma)]. An equal volume of lysis buffer containing 2% SDS was then added to each sample and briefly sonicated. Protein samples were resolved on 4%–20% SDS-PAGE gels and analyzed by immunoblots using the antibodies listed in Table S1. Rabbit polyclonal antiserum B1415 was generated against a fusion protein containing the BIN1 CLAP domain (residues encoded by exons 14 and 15). The blots were developed with IR680 anti-rabbit and IR800 anti-mouse secondary antibodies and imaged with the Odyssey Infrared Imaging System (Li-COR Biosciences). Quantification was performed using Fiji software (Rueden et al., 2017).
Synaptosome fractionation
Synaptosome fractions were prepared following the protocol adapted from De Rossi et al. (2016b). Briefly, hippocampi were dissected from the brains of 4-month-old animals and resuspended in cold buffer containing 0.32 M sucrose and 10 mM HEPES at pH 7.4 and centrifuged twice (at 770 × g) to remove nuclei and large debris, followed by centrifugation at 12000 × g to obtain the synaptosome fraction. The synaptosomes were washed and pelleted in EDTA buffer to chelate calcium (4 mM HEPES, 1mM EDTA, pH 7.4, 20 min at 12000 × g). The synaptosomes were then incubated in a low-Triton buffer for 30 minutes on ice (20 mM HEPES, 100 mM NaCl, 0.5% Triton X-100, pH 7.2) and centrifuged at 12000 × g to separate the postsynaptic density fraction (PSD; pellet) from the non-postsynaptic density fraction (non-PSD; supernatant). Protein samples were resolved on 4%–20% SDS-PAGE gels and analyzed by immunoblots using the antibodies listed in Table S1. The blots were developed with IR680 anti-rabbit and IR800 anti-mouse secondary antibodies and imaged with the Odyssey Infrared Imaging System (Li-COR Biosciences). Quantification was performed using Fiji software (Rueden et al., 2017).
Immunofluorescence staining
Mice were anesthetized by isoflurane inhalation before perfusion with PBS containing 4% paraformaldehyde and 4% sucrose. Brains were harvested and post-fixed overnight in the same fixative and then stored at 4°C in PBS containing 30% sucrose. Fifty μm-thick coronal sections were cut on a cryostat and processed for free-floating immunofluorescence staining. Brain sections were incubated with the indicated primary antibodies (Table S1) for 48 h at 4°C, followed by secondary antibodies for 3–4 h at room temperature. The antibodies were diluted in a 1X Tris-buffered saline solution containing 10% donkey serum, 3% BSA, and 0.25% Triton X-100. Nuclei were labeled using Hoechst stain (Molecular Probes) before mounting the sections on slides with Vectashield mounting medium (VectorLabs) for confocal microscopy or Prolong Diamond (Life Technologies) for STED microscopy.
Electrophysiology
Acute hippocampal slices from 2–3-month-old Ctl and Syn mice were prepared as described previously (Nomura et al., 2016). Briefly, animals were deeply anesthetized with an intraperitoneal injection of xylazine (10 mg/kg) and ketamine (100 mg/kg). Mice were transcardially perfused with an ice-cold sucrose artificial cerebrospinal fluid (ACSF) solution containing (in mM): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2, and 4 MgCl2, including 10 μM DL-APV and 100 μM kynurenate, equilibrated with 95% O2 and 5% CO2. Horizontal sections (350 μm-thick) were made in the same ice-cold sucrose ACSF on a Leica Vibratome (Leica Microsystems, Inc). Slices were transferred to, and incubated in, a recovery chamber containing the same sucrose ACSF at 30 – 32°C for ~30 min, then the solution was gradually exchanged for a recovery ACSF containing (in mM): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 1 CaCl2, and 2 MgCl2, including 10 μM DL-APV and 100 μM kynurenate at room temperature. After at least 1.5 h of recovery, slices were transferred to a recording chamber and visualized using Dodt contrast optics. Slices were continuously perfused with normal ACSF containing (in mM): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2, equilibrated with 95% O2 and 5% CO2. For extracellular recordings, electrodes were manufactured from borosilicate glass pipettes and had tip resistances of 3 – 5 MΩ when filled with regular ACSF. Extracellular field excitatory postsynaptic potentials (fEPSPs) were elicited by an extracellular tungsten bipolar electrode placed in the Stratum Radiatum. For whole-cell recordings, recording electrodes were filled with an internal solution containing (in mM) 75 Cs-methanesulfonate, 60 CsCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 GTP-Na2, 10 Na2-phosphocreatine, 10 TEA, 5 QX-314. Cells were voltage clamped at −70 mV for EPSC recordings. EPSCs were pharmacologically isolated by the treatment of the slices with the GABAA receptor antagonist picrotoxin (PTX) (50 μM). The coefficient of variation (CV) of EPSCs was defined as the ratio of the standard deviation to the mean of the amplitude of EPSCs at given stimulation intensity in each cell. Miniature EPSCs (mEPSCs) were recorded in the presence of the Na+ channel blocker tetrodotoxin (TTX) (1 μM) in the extracellular solution. Data were collected and analyzed using pClamp 10 (Molecular Devices, Sunnyvale, CA) or MiniAnalysis (Synaptosoft Inc.) software. The recordings were discarded when the series resistance changed by > 20% during experiments.
Depletion plots were constructed by normalizing the responses to the amplitude of the peak response during each train (60 s at 20 Hz) (Vargas et al., 2014). Depletion time constants for the fEPSP amplitude of 90 – 10% (10% increment) were measured in each train. Data were collected and compared at each point between genotypes. Recovery plots were generated by normalizing the responses to the 1st responses of the depletion trains and were fit logarithmically to the equation y = ln (mx) + b where m and b represent the slope and the y axis intersect obtained in each recording (5 s at 2 Hz) (Vargas et al., 2014). m was measured in each recording, and collected data were compared between genotypes. The readily releasable pool (RRP) was estimated by analyses of the cumulative amplitudes of the fEPSPs during trains of stimuli used for the depletion paradigm. The RRP was quantified as Nq, where N and q represent the total number of releasable vesicles and the quantal size of synaptic responses, respectively. Nq can be estimated from the zero-time y axis intersect of the linear regression fit to the cumulative fEPSP amplitude plot (Fernandes et al., 2015; Fioravante and Regehr, 2011). Nq was analyzed in each recording, and grouped data were compared between genotypes. Statistical analyses were conducted with GraphPad Prism 8, SPSS, and Origin Pro 9.0 software. Two sample comparisons were made using the Kolmogorov–Smirnov (K-S) test or Mann-Whitney test. For multiple comparisons, repeated two-way analysis of variance (ANOVA) followed by post hoc Bonferroni correction or a linear mixed effects model was employed. Differences were considered significant when p < 0.05. Data are shown as mean ± SEM.
Direct Stochastic Optical Reconstruction Microscopy (dSTORM)
Super-resolution images were acquired on a Leica SR Ground State Depletion 3D / 4 color TIRFM microscope with an Andor iXon Ultra EM-CCD camera (Andor Technology PLC). DIV18–21 rat primary neurons were fixed for 20 min in 4% PFA and 4% sucrose. Primary antibodies were incubated overnight at 4% in PBS containing 10% donkey serum, 3% BSA, and 0.25% Triton X-100. Secondary antibodies were incubated at RT for 3 h in the same buffer. After 3 washes in PBS, the cells were re-fixed with 4% PFA for 5 min. The coverslips were then washed over a period of 2 days at 4°C in PBS to remove the non-specific binding of the secondary antibodies. Coverslips were mounted extemporarily in a monoethanolamine (MEA) solution to limit oxidation of the fluorophores during image acquisition. The areas of capture were blindly selected by direct observation in DIC. Images were acquired using a 160X (NA 1.43) objective in the TIRF mode North direction with a penetration of 200 nm. Far-red channels (Alexa 647 or 660) were acquired using a 642 nm laser and 664LP filter. Red channels (Alexa 568 or 555) were acquired using a 532 nm laser and a 582–636 nm bandpass filter. Green channel (Alexa 488) was acquired using a 488 nm laser and a 503–547 nm bandpass filter. Images were acquired using GSD 3D astigmatism mode in the Leica LAS-X suite software. The irradiation intensity was adjusted until the single-molecule detection reached a frame correlation < 0.25. Detection particle threshold was defined between 20–60 photons depending on the marker and adjusted to obtain a number of events per frame between 0 and 25. The exposure was maintained at 7 ms, and the EM gain was set at 300. The power of depletion and acquisition was defined for each marker and kept constant during acquisition. The number of localizations collected was maintained constant per markers and between experiments. At least 4 independent cultures or coverslips were imaged per marker.
GSD Super-resolution image processing and analysis
Raw GSD images were processed using a custom-made macro in Fiji to remove background by subtraction of a running median of frames (300 renewed every 300 frames) and subtracting the previously processed image once the background was removed (Hoogendoorn et al., 2014). A blur (0.7-pixel radius) per slice prior to median subtraction was applied to reduce the noise. Finally, a 3D Gaussian blur (0.7-pixel radius) was applied to decrease noise further and improve fitting certainty. These images were then processed using Thunderstorm plugin in ImageJ (Ovesný et al., 2014). Blink detection was performed using a Wavelet filter (B-spline, order 3/scale2.0). The molecules were localized using the centroid of connected components, and the peak intensity threshold was determined per marker/dye to maintain an XY uncertainty < 50. Sub-pixel localization of molecules was performed using PSF elliptical Gaussian (3D astigmatism) and least squared fitting methods with a fitting radius of 5 pixels and an initial sigma of 1.6 pixels. 3D location was mapped from −500 nm to +500 nm from the focus point.
To analyze the localization of BIN1 and synaptic marker clusters, we used the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) data clustering algorithm (Ester et al., 1996; Yue et al., 2004). In this method, a coordinate belongs to a cluster when a minimum number of neighbors required to form a dense region (minPts) exists within a defined distance ε. We set minPts to 3 and ε to 50 nm, although values of minPts between 2 and 4, and values of ε between 50 and 100 nm produced qualitatively similar results. We discarded clusters that contained fewer than 50 coordinates, or if the diameter (as measured between the two furthest apart coordinates) was less than 25 nm. Furthermore, to ensure we only analyzed clusters that were confined to a single synaptic bouton, we discarded clusters with diameters greater than 1000 nm. For BIN1 and synaptic marker clusters, we calculated the centroid of each cluster and then calculated the distance between each BIN1 cluster and each synaptic marker cluster. We considered two clusters to be in close proximity if the distance between their centroids was less than 30 nm, and calculated “experimental colocalization” as the proportion of BIN1 clusters found with a synaptic marker cluster within 30 nm distance, averaged across all the samples; since it is a proportion, the values range from 0–1. We also estimated the cluster proximity by setting the distance as 40 nm or 50 nm. We next measured how the proportion of estimated synaptic and BIN1 clusters differed from chance. To do so, we shifted all coordinates belonging to a single synaptic cluster by a common vector, in which the x, y, and z components of this common vector were randomly sampled from a uniform distribution from −250 to 250 nm. We randomly shifted all synaptic clusters and then re-calculated the proportion of synaptic clusters that were co-located with a BIN1 cluster. We repeated this process 100 times to obtain a null distribution of colocalization proportions. We then calculated the z-score between the experimental colocalization proportion and the null distribution:
where epsilon = 1e-4 (results were qualitatively similar for different values of ε). Negative z-scores were set to zero.
To confirm these results, images were also analyzed using Bitplane Imaris software v.9.2 (Andor Technology PLC). Volumes for each marker were generated using smooth surfaces with details set up at 0.005. The diameter of the largest sphere was set up at 0.75 μm. A threshold background subtraction method was used to create the surface, and the threshold was calculated and applied to all the images of the same experiment. Surfaces were then filtered by setting up the number of voxels > 10 and area between 0.01–1 μm2. The nearest neighbor distance was processed using the integrated distance transformation tool in Imaris. Distances were then organized and statistically analyzed using median comparison and ANOVA with Fisher’s LSD post hoc test. The nearest neighbor distances greater than 1000 nm were excluded to facilitate calculating the frequency distribution of clusters located within 500 nm from each side of the synaptic cleft. A mask of the spines was generated based on CaMKII staining using smooth surfaces with details set up at 0.1. Imaris filament tracing was performed for MAP2 and BIN1.
Confocal image acquisition and quantification
Confocal images were acquired on Leica SP5 or Leica SP8 microscopes using 10X (NA 0.4), 20X (NA 0.75), 40X (NA 1.25), 60X (NA 1.4), or 100X (NA 1.4) objectives. For Figure 1B, four tiled images each were acquired using a 10X objective and stitched using LAS-AF software (Leica). Images acquired on a Leica SP5 using a 100X (NA1.4) objective were used to quantify the density of synaptic markers. Images were acquired in the CA1 region ~50 μm from the soma at the bifurcation of the apical dendrite of pyramidal cells, using the same parameters for Ctl and cKO animals. Z-stacks of fifty 2048 × 2048-pixel images were acquired at a scan zoom of 2.5, yielding 30.28 nm pixels.
Confocal microscopy data were quantified from at least 2 image stacks acquired from each of two immunostained sections per animal in at least 3 animals per genotype. Confocal Z stacks were deconvolved using Huygens Professional software (Scientific Volume Imaging) and analyzed using the built-in particle analysis function in Fiji (Rueden et al., 2017). The size of the particles was defined according to previously published studies (MacGillavry et al., 2013; Nair et al., 2013). To assess the number of clusters, images were thresholded (same threshold per marker and experiment), and a binary mask was generated. As image pixel size was 0.000917 μm2, a low size threshold of 0.01 μm diameter was applied to eliminate individual pixels from the quantification. A high pass threshold of 0.4 μm diameter was applied to avoid the quantification of non-clustered structures. For the analysis, the number of clusters per stack was summed. The density was normalized by the control of each experiment to eliminate variability between experiments. The densities were compared by one-way ANOVA with Fisher’s LSD post hoc test. The Spots Colocalization plugin ComDet v.0.3.7 was used to analyze the % colocalization between BIN1 and Bassoon clusters, utilizing the counting mask generated by the cluster quantification. The detection of particles was set up independently for each channel (channel 1: Bassoon; channel 2: BIN1). The default parameters were applied to quantify the colocalization: particle size 3, intensity threshold 3, the maximum distance between colocalized particles 5 pixels (= 150 nm). The results were expressed as the % of Bassoon clusters colocalizing with BIN1 clusters.
Super-resolution STED image acquisition and quantification
Super-resolution STED images of BIN1 and synaptic markers were acquired on a Leica SP8 3D, 3-color gated STED laser scanning confocal microscope. Images were acquired in the CA1 area, at the bifurcation of the apical dendrite of pyramidal cells. A 775 nm depletion laser was used to deplete both 647 and 594 dyes, and a 592 nm laser was used for the depletion of 488 dyes. The powers used for depletion lasers, the excitation laser parameters, and the gating parameters necessary to obtain STED resolution were assessed on control sections, and the same parameters were applied for sections from Ctl and cKO mice. 3 μm-thick Z stacks of 1248 × 1248-pixel images at 50 nm step size were acquired at 8 kHz bidirectional scan rate with a line averaging of 32 and 2–4 frame accumulation, using a 100X (1.45) objective with a digital zoom factor of 5, yielding 18.64 nm pixels.
STED microscopy data were quantified from at least 2 image stacks acquired from each of two immunostained sections per animal in at least 3 animals per genotype. The STED images were deconvolved using Huygens Professional software using the gated 3D-STED module (Scientific Volume Imaging). The images were analyzed using Imaris software. Each image was cropped 3D X = 50 to 1200 and Y = 50 to 1200 and Z = 1 to 60 to obtain a 21.5 μm × 21.5 μm × 3 μm block to avoid edge artifacts. Volumes for each marker were generated using smooth surfaces with details set up at 0.01 μm. The diameter of the largest sphere was set up at 0.75 μm. Threshold background subtraction methods were used to create the surface, and the threshold was calculated with control images and applied to all the images of the same experiment. Surfaces were then filtered by setting up the number of voxels > 10 and area between 0.01–1 μm2. The volume of each surface was then exported and analyzed by ANOVA with Fisher’s LSD post hoc test. Each surface was then color-coded for the volume in Imaris using the same scale in both control and cKO images for the purpose of illustration. The extent of colocalization between two proteins was determined by calculation of the nearest neighbor distance using the integrated distance transformation tool in Imaris. Distances were then organized and statistically analyzed using ANOVA with Fisher’s LSD post hoc test.
Post-embedding immunogold EM
Reagents used for EM were from Electron Microscopy Sciences (Hatfield, PA) unless otherwise noted. Tissue was processed as described previously (Neuman et al., 2015). Briefly, mice were perfused transcardially with 4% paraformaldehyde/0.5% glutaraldehyde, after which the hippocampus was dissected and sectioned into 300 μm-thick slices. Slivers containing CA1 stratum radiatum were isolated from these sections and then plunge-frozen in liquid propane (−184°C), and then processed using freeze-substitution, first stained en bloc in 1.5% uranyl acetate at −90°C, then infiltrated with Lowicryl HM20 (Electron Microscopy Sciences) at progressively escalated temperatures, and finally cured with ultraviolet light at 0°C. Ribbons of serial sections (20–55) were then mounted on formvar-coated, carbon-coated nickel slotted grids, and immunolabeled using pAb BSH3 (10 μg/ml) overnight at 4°C, followed by secondary immunogold staining with 1:20 dilution of 10-nm immunogold particles (Ted Pella, Inc.). Images were acquired with a Zeiss Sigma SE-EM in STEM mode with an accelerating voltage of 29 kV at 1.8 nm/pixel.
Conventional electron microscopy
Large-volume electron microscopy datasets from Ctl and cKO mouse brains were performed as previously described (Hua et al., 2015; Kasthuri et al., 2015). Briefly, mice were anesthetized and transcardially perfused with buffered aldehydes (2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer). The brain was dissected out and post-fixed overnight at 4°C in a similar fixative solution as described above. The brain was then coronally sectioned at 500 mm thickness using a vibratome, and the CA1 region of the hippocampus was removed. Tissues were stained with heavy metals (Hua et al., 2015), dehydrated, infiltrated with epon resin, and cured in an oven at 60°C for 48 h. The cured block was trimmed, and then 200–300 ultra-thin sections (55 nm) from CA1 stratum radiatum were collected on Kapton tape using the automatic tape-collecting ultramicrotome (ATUM). The serial sections were imaged using the backscattered electron detector on Gemini SEM 300 (Carl Zeiss) equipped with ATLAS 5 software (Fibics) (Kasthuri et al., 2015). The high-resolution (6 nm pixel size) images were aligned using trackEM2 and analyzed using ImageJ. A total of 444 asymmetric axospinous synapses were analyzed, with measured parameters being PSD area, number of docked vesicles, and the number of vesicles in the reserve pool (both of the latter in the presynaptic terminal). PSD area was estimated as the product of section thickness (55 nm) and the sum, total length of the PSD. Docked and reserve pool vesicles were defined using the parameters as defined in Schikorski and Stevens (1997). Briefly, docked vesicles were those neurotransmitter vesicles making contact with the presynaptic active zone, immediately apposed to the PSD. Reserve pool vesicles were all other vesicles present in the presynaptic terminal in sections harboring a PSD profile on the dendritic spine head. Quantifications were performed blinded with respect to genotype, and the results were statistically analyzed by multivariate analysis of covariance.
QUANTIFICATION AND STATISTICAL ANALYSIS
The software used for behavior analysis, electrophysiology, image quantification are listed under each section above. Statistical analyses were conducted with GraphPad Prism 8, SPSS v.21, and Origin Pro 9.0 software. Two sample comparisons were made using the Kolmogorov–Smirnov (K-S) test or Mann-Whitney test. Analysis of variance (ANOVA), two-way ANOVA, or repeated two-way ANOVA were used for multiple comparisons. In some experiments, the data was analyzed using MANOVA, MANCOVA, or a linear mixed effects model, as stated under Results or figure legnds. Individual tests and post hoc analyses can be found in the Results and figure legends. ANOVA F statistic, degrees of freedom between groups (DFn) and within groups (DFd), and the p value are listed in results, figure legends, or supplementary tables. The mean ± SEM are plotted in each graph. Differences were considered significant when p < 0.05. The bar graphs depict mean ± SEM and the violin plots depict the median and quartiles. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND CODE AVAILABILITY
The code generated during this study are publically available at https://github.com/nmasse/Storm-Microscopy-Analysis
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-BIN1 (clone EPR13463) | AbCam | Cat# ab182562 |
| Rabbit polyclonal anti-BIN1 | Protein Tech | Cat# 14647–1-AP; RRID:AB_2243396 |
| Rabbit polyclonal anti-BIN1 BSH3 | Thinakaran Lab | De Rossi et al., 2016a |
| Rabbit polyclonal anti-BIN1 B1415 | Thinakaran Lab | This paper |
| Mouse monoclonal anti-Actin (clone 2D4H5) | Protein Tech | Cat# 66009-I-Ig; RRID:AB_2687938 |
| Mouse monoclonal anti-Amph1 (clone 8) | Santa Cruz | Cat# SC21710; RRID:AB_673386 |
| Rabbit polyclonal anti-APLP1 CT11 | Thinakaran Lab | Thinakaran et al., 1998 |
| Rabbit polyclonal anti-APLP2 CT12 | Thinakaran Lab | Thinakaran et al., 1995 |
| Guinea Pig polyclonal anti-Bassoon | Synaptic Systems | Cat# 141–004; RRID:AB_2290619 |
| Mouse monoclonal anti-CaMKII (clone 6G9) | Santa Cruz | Cat# SC32288; RRID:AB_626787 |
| Mouse monoclonal anti-CASK (clone K56A/50) | NeuroMab | Cat# O14936; RRID:AB_2750669 |
| Mouse monoclonal anti-CNPase (clone 11–5B) | Millipore | Cat# MAB326; RRID:AB_2082608 |
| Rabbit polyclonal anti-Flotillin-2 | Thinakaran Lab | Meckler et al., 2010 |
| Mouse monoclonal anti-GluA1(clone N355/1) | NeuroMab | Cat# P19490; RRID:AB_2750785 |
| Mouse monoclonal anti-MAP2 (clone HM-2) | Sigma | Cat# M4403; RRID:AB_477193 |
| Mouse monoclonal anti-MBP (clone SMI-94) | Covance | Cat# SMI-94R; RRID:AB_510039 |
| Mouse monoclonal anti-Mint2 | BD Biosciences | Cat# M76120 |
| Rabbit polyclonal anti-Munc13–1 | Synaptic Systems | Cat# 126–102; RRID:AB_887734 |
| Mouse monoclonal anti-NeuN (clone A60) | Chemicon | Cat# MAB377; RRID:AB_2298772 |
| Rabbit monoclonal anti-pCaMKII (clone D21E4) | Cell Signaling | Cat# 12716S; RRID:AB_2713889 |
| Rabbit polyclonal anti-pGluA1 −831 | Millipore | Cat# AB5847, RRID:AB_92077 |
| Rabbit polyclonal anti-pGluA1 −845 | Millipore | Cat# AB5849; RRID:AB_92079 |
| Mouse monoclonal anti-PSD95 (clone K28/43) | UC Davis | Cat# K28/43; RRID:AB_2750659 |
| Mouse monoclonal anti-Rab3a (Clone9) | Santa Cruz | Cat# Sc-136050; RRID:AB_2300792 |
| Rabbit monoclonal anti-Rab5 (clone C8B1) | Cell Signaling | Cat# 3547S; RRID:AB_2300649 |
| Rabbit polyclonal anti-RIM1α | Synaptic Systems | Cat# 140–003; RRID:AB_887774 |
| Mouse monoclonal anti-Synapsin1 (clone 46.1) | Synaptic Systems | Cat# 106 011BT; RRID:AB_2271476 |
| Rabbit polyclonal anti-Synaptojanin1 | Synaptic Systems | Cat# 145–103; RRID:AB_887820 |
| Mouse monoclonal anti-Synaptophysin1 (clone SVP38) | Sigma | Cat# S5768; RRID:AB_477523 |
| Rabbit polyclonal anti-Synaptoporin | Synaptic Systems | Cat# 102–003; RRID:AB_2619748 |
| Chicken polyclonal anti-Synaptotagmin1 | Synaptic Systems | Cat# 105–106; RRID:AB_2661782 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J Bin1fl/fl | Dr. George C. Prendergast | Lankenau Institute for Medical Research |
| Mouse: B6.Cg-Tg(Syn1-cre)671Jxm/J | The Jackson Laboratory | JAX stock #003966, RRID:IMSR_JAX:003966 |
| Mouse: B6.129S2-Emx1tm1(cre)Krj/J | The Jackson Laboratory | JAX stock #005628, RRID:IMSR_JAX:005628 |
| Software and Algorithms | ||
| FreezeFrame 4 software | Coulbourn | https://www.coulbourn.com/product_p/act-100a.htm |
| EthoVision XT, v. 8.5 and v.14 | Noldus | https://www.noldus.com/ethovision-xt, RRID:SCR_000441 |
| GraphPad Prism 8 | Graphpad software | https://www.graphpad.com/scientific-software/prism/, RRID:SCR_002798 |
| SPSS® software platform v. 21 | IBM | https://www.ibm.com/analytics/spss-statistics-software, RRID:SCR_002865 |
| Origin Pro 9.0 software | Origin Lab | https://www.originlab.com/, RRID:SCR_014212 |
| Odyssey Infrared Imaging System v. 3.0.30 | LI-COR Biosciences | RRID:SCR_013430 |
| MiniAnalysis 6.0.3 | Synaptosoft, Inc. | http://www.synaptosoft.com/MiniAnalysis/, RRID:SCR_002184 |
| pClamp 10 | Molecular devices | https://www.moleculardevices.com/products/software/pclamp.html, RRID:SCR_011323 |
| Leica LAS-X suite software | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/, RRID:SCR_013673 |
| FIJI ImageJ 1.52 | Rueden et al., 2017 | https://imagej.net/Fiji, RRID:SCR_002285 |
| Spots colocalization (ComDet) | ImageJ plugin | https://imagej.net/Spots_colocalization_(ComDet) |
| Thunderstorm | ImageJ plugin | https://github.com/zitmen/thunderstorm, RRID:SCR_016897 |
| Python v. 3.6.8 | Python.org | https://www.python.org/, RRID:SCR_008394 |
| SciPy v. 1.2.1 | SciPy.org | https://www.scipy.org/, RRID:SCR_008058 |
| Bitplane Imaris software v. 9.2 | Oxford instrument | https://imaris.oxinst.com/packages, RRID:SCR_007370 |
| ATLAS 5 software | Fibics / Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/atlas.html |
| TrackEM2 | ImageJ plugin | https://imagej.net/TrakEM2 |
| dSTORM analysis code | This paper | https://github.com/nmasse/Storm-Microscopy-Analysis |
Highlights.
The loss of BIN1 in neurons leads to impaired spatial memory consolidation
Neuronal Bin1 cKO mice have deficits in excitatory synaptic transmission
BIN1 regulates presynaptic vesicular release in hippocampal excitatory synapses
The results highlight a non-redundant role for BIN1 in presynaptic regulation
ACKNOWLEDGMENTS
We thank Xiaoxi Zhuang for advice on behavioral experiments and statistical analysis. This work was supported by National Institutes of Health grants AG054223 and AG056061 (to G.T.), AG017139 and K02 AG050767 (to D.A.N.), and R01MH099114 (to A.C.); by the Cure Alzheimer’s Fund (G.T.); by the Alzheimer’s Association (P.D.R.); and by fellowships from the BrightFocus Foundation (P.D.R.) and the Illinois Department of Public Health (R.J.A.). Fluorescence imaging was performed at the Integrated Microscopy Core Facility at the University of Chicago (supported by grant S10OD010649).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.026.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andrew RJ, De Rossi P, Nguyen P, Kowalski HR, Recupero AJ, Guerbette T, Krause SV, Rice RC, Laury-Kleintop L, Wagner SL, and Thinakaran G (2019). Reduction of the expression of the late-onset Alzheimer’s disease (AD) risk-factor BIN1 does not affect amyloid pathology in an AD mouse model. J. Biol. Chem 294, 4477–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annamneedi A, Caliskan G, Müller S, Montag D, Budinger E, Angenstein F, Fejtova A, Tischmeyer W, Gundelfinger ED, and Stork O (2018). Ablation of the presynaptic organizer Bassoon in excitatory neurons retards dentate gyrus maturation and enhances learning performance. Brain Struct. Funct 223, 3423–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthet G, Jordà-Siquier T, Rumi-Masante J, Bernadou F, Müller U, and Mulle C (2018). Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat. Commun 9, 4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungenberg J, Surano N, Grote A, Surges R, Pernhorst K, Hofmann A, Schoch S, Helmstaedter C, and Becker AJ (2016). Gene expression variance in hippocampal tissue of temporal lobe epilepsy patients corresponds to differential memory performance. Neurobiol. Dis 86, 121–130. [DOI] [PubMed] [Google Scholar]
- Calafate S, Flavin W, Verstreken P, and Moechars D (2016). Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 17, 931–940. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Morton J, and Dunnett SB (2001). Motor coordination and balance in rodents. Curr. Protoc. Neurosci Chapter 8, 12. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Hill ES, Christian DT, Helfrich R, Riley S, Schneider C, Kapecki N, Mustaly-Kalimi S, Seiler FA, Peterson DA, et al. (2019). Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol. Neurodegener 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MY, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, Muller AJ, and Prendergast GC (2007). Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 67, 7605–7612. [DOI] [PubMed] [Google Scholar]
- Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, et al. ; GERAD Consortium (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X, and Kavalali ET (2010). Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J. Neurosci 30, 1363–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Lüthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. [DOI] [PubMed] [Google Scholar]
- Crotti A, Sait HR, McAvoy KM, Estrada K, Ergun A, Szak S, Marsh G, Jandreski L, Peterson M, Reynolds TL, et al. (2019). BIN1 favors the spreading of Tau via extracellular vesicles. Sci. Rep 9, 9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rossi P, Buggia-Prévot V, Clayton BL, Vasquez JB, van Sanford C, Andrew RJ, Lesnick R, Botté A, Deyts C, Salem S, et al. (2016a). Predominant expression of Alzheimer’s disease-associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol. Neurodegener 11, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rossi P, Harde E, Dupuis JP, Martin L, Chounlamountri N, Bardin M, Watrin C, Benetollo C, Pernet-Gallay K, Luhmann HJ, et al. (2016b). A critical role for VEGF and VEGFR2 in NMDA receptor synaptic function and fear-related behavior. Mol. Psychiatry 21, 1768–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, and De Camilli P (2002). Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 33, 789–804. [DOI] [PubMed] [Google Scholar]
- Ester M, Kriegel HP, Sander H, and Xu X (1996). A Density-Based Algorithm for Discovering Clusters in Large Spatial Datasets with Noise. In Proceedings of the 2nd International Conference on KDD, pp. 1232–1239. [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, et al. (2007). A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 316, 570–574. [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, and Contractor A (2015). Epac2 Mediates cAMP-Dependent Potentiation of Neurotransmission in the Hippocampus. J. Neurosci 35, 6544–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, and Regehr WG (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol 21, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O’Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, et al. (2014). APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep. 7, 1560–1576. [DOI] [PubMed] [Google Scholar]
- Franzmeier N, Rubinski A, Neitzel J, and Ewers M; Alzheimer’s Disease Neuroimaging Initiative (ADNI) (2019). The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun 10, 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis C, Weishaupt A, Hallermann S, Grünewald B, Wessig C, Wultsch T, Reif A, Byts N, Beck M, Jablonka S, et al. (2010). Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 133, 3166–3180. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano A, Petrosini L, and Bartoletti A (2003). Automatic recognition of explorative strategies in the Morris water maze. J. Neurosci. Methods 130, 33–44. [DOI] [PubMed] [Google Scholar]
- Gundelfinger ED, Reissner C, and Garner CC (2016). Role of Bassoon and Piccolo in Assembly and Molecular Organization of the Active Zone. Front. Synaptic Neurosci 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haytural H, Mermelekas G, Emre C, Nigam SM, Carroll SL, Winblad B, Bogdanovic N, Barthet G, Granholm AC, Orre LM, et al. (2020). The proteome of the dentate terminal zone of the perforant path indicates presynaptic impairment in Alzheimer disease. Mol. Cell. Proteomics 19, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He E, Wierda K, van Westen R, Broeke JH, Toonen RF, Cornelisse LN, and Verhage M (2017). Munc13–1 and Munc18–1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat. Commun 8, 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler CJ, Davis PR, Beckett TL, Platt TL, Webb RL, Head E, and Murphy MP (2014). Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis 42, 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendoorn E, Crosby KC, Leyton-Puig D, Breedijk RM, Jalink K, Gadella TW, and Postma M (2014). The fidelity of stochastic single-molecule super-resolution reconstructions critically depends upon robust background estimation. Sci. Rep 4, 3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Laserstein P, and Helmstaedter M (2015). Large-volume en-bloc staining for electron microscopy-based connectomics. Nat. Commun 6, 7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Yin L, Thambisetty M, Troncoso JC, Lah JJ, Levey AI, and Seyfried NT (2018). Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vázquez-Reina A, Kaynig V, Jones TR, et al. (2015). Saturated Reconstruction of a Volume of Neocortex. Cell 162, 648–661. [DOI] [PubMed] [Google Scholar]
- Kim JJ, and Jung MW (2006). Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci. Biobehav. Rev 30, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemmer P, Meredith RM, Holmgren CD, Klychnikov OI, Stahl-Zeng J, Loos M, van der Schors RC, Wortel J, de Wit H, Spijker S, et al. (2011). Proteomics, ultrastructure, and physiology of hippocampal synapses in a fragile X syndrome mouse model reveal presynaptic phenotype. J. Biol. Chem 286, 25495–25504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R (2002). The neurobiological basis of spontaneous alternation. Neurosci. Biobehav. Rev 26, 91–104. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang S, Li T, Zhu L, Xu Y, and Ma C (2017). A Stimulation Function of Synaptotagmin-1 in Ternary SNARE Complex Formation Dependent on Munc18 and Munc13. Front. Mol. Neurosci 10, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichte B, Veh RW, Meyer HE, and Kilimann MW (1992). Amphiphysin, a novel protein associated with synaptic vesicles. EMBO J. 11, 2521–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Fan F, Messa M, Raimondi A, Wu Y, Looger LL, Ferguson SM, and De Camilli P (2012). Reduced release probability prevents vesicle depletion and transmission failure at dynamin mutant synapses. Proc. Natl. Acad. Sci. USA 109, E515–E523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Song Y, Raghavachari S, and Blanpied TA (2013). Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron 78, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks B, and McMahon HT (1998). Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr. Biol 8, 740–749. [DOI] [PubMed] [Google Scholar]
- McKenzie AT, Moyon S, Wang M, Katsyv I, Song WM, Zhou X, Dammer EB, Duong DM, Aaker J, Zhao Y, et al. (2017). Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer’s disease. Mol. Neurodegener 12, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckler X, Roseman J, Das P, Cheng H, Pei S, Keat M, Kassarjian B, Golde TE, Parent AT, and Thinakaran G (2010). Reduced Alzheimer’s disease b-amyloid deposition in transgenic mice expressing S-palmitoylation-deficient APH1aL and nicastrin. Journal of Neuroscience 30, 16160–16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Kay BK, and McPherson PS (1997). Synaptojanin forms two separate complexes in the nerve terminal. Interactions with endophilin and amphiphysin. J. Biol. Chem 272, 27239–27245. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O’Toole E, Ferguson S, Cremona O, and De Camilli P (2011). Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa T, Ebinuma I, Morohashi Y, Hori Y, Young Chang M, Hattori H, Maehara T, Yokoshima S, Fukuyama T, Tsuji S, et al. (2016). BIN1 regulates BACE1 intracellular trafficking and amyloid-b production. Hum. Mol. Genet 25, 2948–2958. [DOI] [PubMed] [Google Scholar]
- Nair D, Hosy E, Petersen JD, Constals A, Giannone G, Choquet D, and Sibarita JB (2013). Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci 33, 13204–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman KM, Molina-Campos E, Musial TF, Price AL, Oh KJ, Wolke ML, Buss EW, Scheff SW, Mufson EJ, and Nicholson DA (2015). Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct. Funct 220, 3143–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Oyamada Y, Fernandes HB, Remmers CL, Xu J, Meltzer HY, and Contractor A (2016). Subchronic phencyclidine treatment in adult mice increases GABAergic transmission and LTP threshold in the hippocampus. Neuropharmacology 100, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesný M, K rí zek P, Borkovec J, Svindrych Z, and Hagen GM (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DJ, Wigge P, Vallis Y, Moore JD, Evans PR, and McMahon HT (1998). Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 17, 5273–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, and Herms J (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci 26, 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokic I, Cowling BS, and Laporte J (2014). Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. (Berl.) 92, 453–463. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, and Betz WJ (2005). Synaptic vesicle pools. Nat. Rev. Neurosci 6, 57–69. [DOI] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, Takikawa K, Asanuma D, Namiki S, and Hirose K (2018). Synaptic weight set by Munc13–1 supramolecular assemblies. Nat. Neurosci 21, 41–49. [DOI] [PubMed] [Google Scholar]
- Schikorski T, and Stevens CF (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci 17, 5858–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, and Neher E (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23, 399–409. [DOI] [PubMed] [Google Scholar]
- Schürmann B, Bermingham DP, Kopeikina KJ, Myczek K, Yoon S, Horan KE, Kelly CJ, Martin-de-Saavedra MD, Forrest MP, Fawcett-Patel JM, et al. (2019). A novel role for the late-onset Alzheimer’s disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol. Psychiatry 9 10.1038/s41380-019-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singec I, Knoth R, Ditter M, Hagemeyer CE, Rosenbrock H, Frotscher M, and Volk B (2002). Synaptic vesicle protein synaptoporin is differently expressed by subpopulations of mouse hippocampal neurons. J. Comp. Neurol 452, 139–153. [DOI] [PubMed] [Google Scholar]
- Südhof TC, and Rizo J (1996). Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron 17, 379–388. [DOI] [PubMed] [Google Scholar]
- Taylor MJ, Perrais D, and Merrifield CJ (2011). A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 9, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Kitt CA, Roskams AJ, Slunt HH, Masliah E, von Koch C, Ginsberg SD, Reed RR, Price DL, and Sisodia SS (1995). Distribution of an APP homolog, APLP2, in the mouse olfactory system: a potential role for APLP2 in axogenesis. Journal of Neuroscience 15, 6314–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Regard JB, Bouton CML, Harris CL, Price DL, Borchelt DR, and Sisodia SS (1998). Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiology of Disease 4, 438–453. [DOI] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, and Chandra SS (2014). Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci 34, 9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Mamer LE, Raychaudhuri S, Luvsanjav D, Eisen J, Trimbuch T, Sohl-Kielczynski B, Fenske P, Milosevic I, Rosenmund C, and Jorgensen EM (2018). Synaptojanin and Endophilin Mediate Neck Formation during Ultrafast Endocytosis. Neuron 98, 1184–1197.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Pauli M, Doose S, Weishaupt A, Haselmann H, Grünewald B, Sauer M, Heckmann M, Toyka KV, Asan E, et al. (2016). Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain 139, 365–379. [DOI] [PubMed] [Google Scholar]
- Wigge P, Vallis Y, and McMahon HT (1997). Inhibition of receptor-mediated endocytosis by the amphiphysin SH3 domain. Curr. Biol 7, 554–560. [DOI] [PubMed] [Google Scholar]
- Winters BD, Saksida LM, and Bussey TJ (2008). Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci. Biobehav. Rev 32, 1055–1070. [DOI] [PubMed] [Google Scholar]
- Yao J, Nowack A, Kensel-Hammes P, Gardner RG, and Bajjalieh SM (2010). Cotrafficking of SV2 and synaptotagmin at the synapse. J. Neurosci 30, 5569–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue SH, Li P, Guo JD, and Zhou SG (2004). Using Greedy algorithm: DBSCAN revisited II. J. Zhejiang Univ. Sci 5, 1405–1412. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, and Shen J (2009). Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yu JT, Li J, Wang C, Tan L, Liu B, and Jiang T (2015). Bridging Integrator 1 (BIN1) Genotype Effects on Working Memory, Hippocampal Volume, and Functional Connectivity in Young Healthy Individuals. Neuropsychopharmacology 40, 1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, and Parada LF (2001). Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 15, 859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu. Rev. Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated during this study are publically available at https://github.com/nmasse/Storm-Microscopy-Analysis