

Abstract
Inhibition of the poly(ADP-ribose) polymerase (PARP) family of enzymes has become an attractive therapeutic strategy in oncology and beyond; however, chemical tools to profile PARP engagement in live cells are lacking. Herein, we report the design and application of PARPYnD, the first photoaffinity probe (AfBP) for PARP enzymes based on triple PARP1/2/6 inhibitor AZ9482, which induces multipolar spindle (MPS) formation in breast cancer cells. PARPYnD is a robust tool for profiling PARP1/2 and is used to profile clinical PARP inhibitor olaparib, identifying several novel off-target proteins. Surprisingly, while PARPYnD can enrich recombinant PARP6 spiked into cellular lysates and inhibits PARP6 in cell-free assays, it does not label PARP6 in intact cells. These data highlight an intriguing biomolecular disparity between recombinant and endogenous PARP6. PARPYnD provides a new approach to expand our knowledge of the targets of this class of compounds and the mechanisms of action of PARP inhibitors in cancer.
Introduction
Poly(ADP-ribose) polymerases (PARPs) represent an important enzyme family that catalyzes protein ADP-ribosylation, transferring one or more units of ADP-ribose from an NAD+ cofactor to specific biomolecular substrates.1 Of these 17 human enzymes, PARPs 1 and 2 and the tankyrases (PARP5a/5b) can catalyze transfer of longer/branched chains of poly(ADP-ribose) (PARylation), while the remaining family members transfer a single unit of ADP-ribose to their molecular targets (mono ADP-ribosylation, MARylation), with the exception of PARP13 which appears to be catalytically inactive.2,3
PARPs are implicated in a variety of cellular metabolic processes including DNA-damage repair,4 transcriptional regulation,5 cell death,6 immune responses,7 and mitotic spindle formation.8 In particular, PARP1 is essential in repairing DNA single- and double-strand breaks, and pharmacological inhibition of PARP1 with agents such as olaparib (Figure 1A) has been demonstrated to be an effective synthetic lethal strategy in certain tumors deficient in homologous recombination-mediated DNA repair.9 Since the approval of olaparib in 2014, multiple PARP1 inhibitors have been introduced in the clinic including niraparib, talazoparib, and rucaparib, and there is growing interest in targeting other members of the PARP family.10
Figure 1.

A photoaffinity-based probe (AfBP) PARPYnD was designed and synthesized for target profiling of AZ9482 and AZ0108. (A) Top: structures of clinical PARP1/2 inhibitor olaparib and MPS-inducing agents AZ0108 and AZ9482, the latter of which was diversified into the AfBP used in this study. Bottom: table showing the biochemical and biophysical parameters associated with olaparib, AZ9482, AZ0108, and PARPYnD.11,12 Table footnote a: data generated in this work, experiments performed in triplicate (±SEM); graphical analysis can be seen in Figure 2, and Supporting Information Figures S1 and S2 for multipolar spindle (MPS) induction data, PARP binding data, and cytotoxicity data (MDA-MB-468 cells), respectively. Table footnote b: cytotoxicity value represents a GI50 value previously generated in MDA-MB-468 cells.12 (B) Schematic of the photoaffinity labeling (PAL) workflow used for target profiling. The gray ball represents the recognition element of the probe that is designed based on the parent compound.
We recently reported a triple PARP1/2/6 inhibitor, AZ0108 (Figure 1A), which elicited therapeutic effects in breast cancer models in vivo by generating a cytotoxic multipolar spindle (MPS) phenotype in cancer cells but not in somatic tissue.11,12 The mechanism of action of AZ0108 is proposed to be via cellular inhibition of PARP6 MARylation of downstream substrates, one of which was identified as checkpoint kinase 1 (CHK1). MARylation of CHK1 by PARP6 was proposed to regulate the phosphorylation of CHK1; AZ0108 treatment prevented CHK1 MARylation and induced hyperphosphorylation of CHK1, contributing to MPS formation and dysregulation of the cell cycle. However, AZ0108 also displayed toxicity in vivo, the molecular basis of which is currently undefined,11 limiting pharmacological evaluation of AZ0108. Furthermore, while substantial in vitro evidence of the mechanism of action has been presented, bona fide target engagement of PARP6 by AZ0108 has not been reported in intact cells, and limited literature reports on alternative pharmacological modulators of PARP6 have restricted orthogonal validation of this mechanism of action.13−15 Therefore, generation of a complete cellular target profile of AZ0108 would be beneficial for both target validation and off-target identification.
Photoaffinity labeling (PAL) is a robust and increasingly popular strategy to profile the cellular interactome of a molecule with a noncovalent binding profile (Figure 1B).16−18 The methodology utilizes a photoaffinity-based probe (AfBP) that retains the binding profile and phenotypic properties of the parent compound but is modified with a photoreactive group activated with a specific wavelength of light to generate a covalent bond directed by the probe’s reversible, noncovalent interaction(s). Incorporation of a bioorthogonal handle allows downstream attachment of a capture reagent to probe tagged proteins via bioorthogonal ligation chemistry, resulting in an AfBP.17 The reporter can include a fluorophore for gel-based visualization of the probe interactome, or an enrichment handle such as biotin allowing for de novo target identification by tandem mass spectrometry or specific target interrogation by immunoblot.19 Ideally the photoaffinity group and bioorthogonal handle should be small and structurally discrete, allowing retention of biological mode of action, cell permeability, and live cell target protein profiling.
Herein, we describe the design and synthesis of a novel AfBP PARPYnD for phthalazinone PARP inhibitors. We show that PARPYnD retains an MPS phenotype and in vitro PARP binding profile comparable to parent compounds AZ9482 and AZ0108. PARPYnD enriches PARP1 following crosslinking in intact cells by both proteomics and immunoblot, and PARP1 and PARP2 binding can be efficiently competed in cellulo by AZ9482, AZ0108, and olaparib in proteomics studies. PARPYnD thus constitutes the first cell-active AfBP for the PARP protein class, and we demonstrate its utility in profiling novel off-target interactions of both AZ0108 and the clinical PARP inhibitor olaparib. Furthermore, we show that PARPYnD can be applied to probe the potential mode of action of PARP6 inhibitors, generating novel hypotheses for future investigation.
Results and Discussion
Lead compound AZ9482 was found to be a potent MPS-inducing agent (Figure 1A) discovered via a high-content screen of the AstraZeneca collection of phthalazinone-based NAD+ mimetics.12 Due to the poor pharmacokinetic properties of AZ9482, we previously performed a lead optimization study which resulted in AZ0108, a molecule with comparable cellular effects to AZ9482 (Figure 1A) but with greatly improved in vivo characteristics; however, we chose to adopt the more synthetically tractable AZ9482 scaffold over AZ0108 as the basis for a novel AfBP. While there is currently no structural information available for PARP6, we previously crystallized AZ9482 with PARP5a (Figure 2A), and given the structural similarity of the NAD+-binding pocket throughout the family,1 this model was used to design an AfBP. As with all olaparib-like molecules, the AZ9482 phthalazinone core mimics the adenine of NAD+ and is essential for PARP family binding; this moiety was retained in our AfBP design, along with the central phenyl ring present in all potent compounds in our published series. During the pharmacokinetic optimization of AZ9482, we demonstrated that altering the pyridine group is tolerated in MPS assays, and while the 3-nitrile is a key backbone hydrogen bond acceptor, the 5-position (Figure 2A, red star) represents a convenient point from which to build out into a solvent channel with a small photocrosslinkable clickable moiety.12,20 This resulted in probe PARPYnD (Figure 1A), the synthesis of which is described in the Supplementary Methods section; briefly, commercially available 2-(piperazin-1-yl)pyridine-3-carbonitrile was Boc protected and brominated, and a copper-catalyzed amination21 was used to install the key aniline group that allowed coupling of the photoaffinity molecule.18 Boc deprotection and coupling to the phthalazinone core generated AfBP PARPYnD.
Figure 2.

Validation of PARPYnD as a suitable probe for parent molecule profiling. (A) Crystal structure of parent molecule AZ9482 bound in the NAD+-binding pocket of PARP5a (PDB ID: 5ECE), with key interactions highlighted in orange. The red star highlights the solvent exposed position at which modification is expected to minimally perturb the binding of a probe into this pocket of the PARP enzymes. (B) Quantification of the percentage of mitotic cells with MPS phenotype (>2 spindle poles per cell) after treatment with AZ9482 (N = 2) and PARPYnD (N = 2), both at 41 nM, versus DMSO (N = 4); double asterisk (**) represents raw P value <0.001 in unpaired Student’s t test; raw data found in the Supporting Information, Extended Data S1. (C) PARP6 activity assay: recombinant GST-tagged PARP6 was incubated with biotinylated NAD+ and varying concentrations of PARPYnD. GST-PARP6 auto-MARylation was measured by immunoblotting against NeutrAvidin-HRP; decreased signal with increasing PARPYnD concentration indicated catalytic inhibition of PARP6. (D) Gel and Western blot analysis of live cells labeled with PARPYnD and ligated to AzTB (Figure S3A) with/without cotreatment with various competitor molecules. In-gel fluorescence was used to qualitatively assess TAMRA-tagged proteins, and streptavidin-based enrichment and immunoblot analysis was used to validate specific targets. I = input (whole lysate); P = pull down (enriched fraction); > indicates competition seen on gel, validated as PARP1 by immunoblot. Uncropped gels and immunoblots associated with all figures can be found in Figure S7.
PARPYnD was first tested for its MPS activity in the same fluorescence microscopy assay used in the original high-content library screen12 and was found to induce the MPS phenotype with comparable potency to AZ9482 (Figure 2B) or AZ0108.11 Furthermore, it possesses a similar profile to AZ9482 in fluorescence polarization competition-based binding assays with recombinant PARPs 1, 2, and 6 (Figure 1A, Figure S1), with PARPYnD being an even more potent PARP6 binder than AZ9482. PARPYnD also displayed similar cytotoxic properties to the parent compounds against the triple negative breast cancer cell line MDA-MB-468 previously used in AZ0108 target validation studies (Figure 1A, Figure S2).11 Finally, recombinant glutathione S-transferase (GST)-tagged PARP6 was incubated with a biotinylated analogue of NAD+ and increasing concentrations of PARPYnD. The reactions were separated by gel electrophoresis, transferred to nitrocellulose, and immunoblotted with NeutrAvidin-HRP (Figure 2C);22 dose-dependent inhibition of PARP6 auto-MARylation was observed, indicating that PARPYnD binds to PARP6 and inhibits its catalytic activity.
These data support the proposal that PARPYnD is an effective tool to interrogate the biomolecular profile of AZ9482 and AZ0108, and therefore, the molecule was taken forward to preliminary live cell profiling studies. MDA-MB-468 cells were cultured in 6-well plates and treated with either DMSO, PARPYnD alone, or cotreated with PARPYnD and either AZ9482, AZ0108, or olaparib at various concentrations (Figure 2D). After 3 h of treatment, cells were irradiated with 365 nm UV light, lysed, and subjected to copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) to an azido-TAMRA/biotin capture reagent previously reported by our laboratories (AzTB, Figure S3A).19 Qualitative assessment of PARPYnD-labeled proteins by in-gel fluorescence revealed a dose-dependent profile (Figure 2D, lanes 1–3), indicating robust labeling efficiency and cell permeability. PARPYnD competition against the parent compounds AZ9482 or AZ0108 was used to identify targets against the background of nonspecific probe interactions, characterized experimentally by dose-dependent reduction in band labeling intensity as a result of target-specific blockade of probe binding. One band (Figure 2D) appeared to be depleted in a dose-dependent manner by AZ9482, and to a lesser extent AZ0108, at around 110 kDa, corresponding to the molecular weight of PARP1. Further competition experiments with PARP1 inhibitor olaparib also resulted in depletion of this band (Figure 2D, lanes 10–12).
To validate target engagement with PARP1, the labeled proteome was enriched on streptavidin beads and the pull-down fraction interrogated by immunoblotting (Figure 2D).23,24 PARP1 was enriched by PARPYnD but not in the vehicle (DMSO) control, and dose-dependent reduction in enrichment by competition against AZ9482, AZ0108, or olaparib was in line with what was observed in-gel. These results demonstrate the utility of PARPYnD as the first AfBP for the PARP enzymes, providing a useful tool molecule that can assess bona fide target engagement for PARP1 by a molecule of interest in a live cell setting. Surprisingly, PARPYnD did not enrich PARP6 in a similar fashion (Figure 2D), even when cells were treated with up to 10 μM probe, a concentration >10-fold the IC50 of PARPYnD for PARP6 as determined by the in vitro binding assay (Figure S4). We hypothesized that antibody efficiency or low expression may render immunoblotting insufficient to detect PARP6, and so PARPYnD was taken on to unbiased proteomics studies.
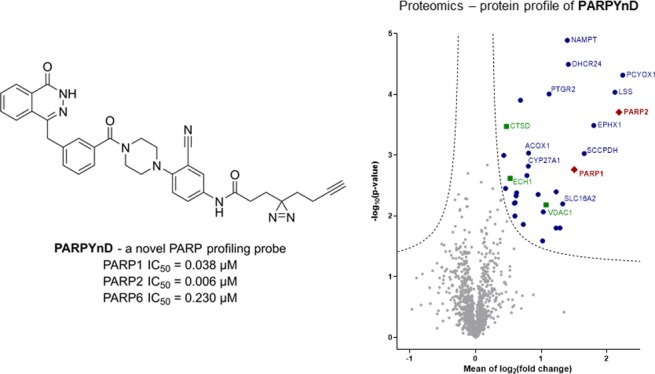
MDA-MB-468 cells were treated with either DMSO, PARPYnD alone (1 μM), or cotreated with PARPYnD (1 μM) and either AZ9482 (5 μM), AZ0108 (5 μM), or olaparib (5 μM). Cells were irradiated at 365 nm, lysed, and subjected to CuAAC with AzRB (Figure S3B), a biotin-containing reporter molecule with a trypsin-cleavable linker previously developed in our laboratories.25 Labeled proteins were enriched on NeutrAvidin agarose resin, reduced and alkylated, digested into constituent peptides using trypsin, and labeled with tandem mass tag (TMT) reagents for quantification. Samples were combined, fractionated, and analyzed by nanoscale liquid chromatography tandem mass spectrometry on a high-resolution QExactive orbitrap mass spectrometer (nanoLC-MS/MS). Comparing PARPYnD-treated samples to untreated samples revealed the probe–protein interaction profile (Figure 3A), the diverse enrichment of proteins corresponding to the variety of bands observed by in-gel fluorescence (Figure 2D). Enrichment of these proteins was found to be entirely dependent on UV irradiation and was not the result of any unanticipated covalent reactivity of PARPYnD (Figure S5E–G). Furthermore, we observed significant enrichment of PARP1, confirming the results from immunoblotting, and also PARP2. Of the other proteins enriched, many are well-known targets of the diazirine motif which previous work has shown tend to be strongly enriched regardless of the core probe scaffold.26 These include voltage-dependent anion-gated channel 1 (VDAC1), cathepsin D (CTSD), and enoyl-CoA hydratase 1 (ECH1); enrichment of these proteins provides a useful internal control for successful intracellular diazirine-based photoaffinity labeling. Competition studies confirmed that these proteins are targets of the probe rather than the parent molecules, since no significant depletion of these proteins was observed upon cotreatment with any competitor molecule (Figure 3B–D).
Figure 3.

Target engagement profiles of PARPYnD, AZ9482, AZ0108, and olaparib. (A–D) Proteomics analysis of live cells labeled with PARPYnD and ligated to AzRB (Figure S3B) with/without cotreatment with parent competitor molecules. Tagged proteins were enriched on NeutrAvidin agarose, digested into peptides, and tandem mass tag (TMT) labeled for identification and quantification by LC-MS/MS. Volcano plots demonstrate enrichment (x axis) of one sample versus another and the associated significance (y axis), determined by pairwise Student’s t test (cut off: A, S0 = 0.1, false discovery rate (FDR) = 5%; B–D, S0 = 0.1, FDR = 15%). Red diamonds = PARP family, blue dots = other significantly enriched/depleted hits, green squares = known background photocrosslinking binders,26 gray dots = nonsignificant proteins. Significant hits are annotated with their gene names only when they are appear significantly enriched/depleted across more than one pairwise comparison, with the exception of the known background binders. Larger, fully annotated plots can be found in Figure S5A–D, and complete raw data can be found in the Supporting Information, Extended Data S2. (A) PARPYnD (1 μM) versus DMSO. (B) PARPYnD (1 μM) and AZ9482 (5 μM) versus PARPYnD (1 μM) only. (C) PARPYnD (1 μM) and AZ0108 (5 μM) versus PARPYnD (1 μM) only. (D) PARPYnD (1 μM) and olaparib (5 μM) versus PARPYnD (1 μM) only. (E) Transient overexpression of FLAG-PARP6 and attempted enrichment of the overexpressed protein and identification by Western blot. I = input (whole lysate); P = pull down (enriched fraction).
As expected, PARP1 and PARP2 labeling were significantly depleted by AZ9482, AZ0108, and olaparib (Figure 3B–D), demonstrating the utility of PARPYnD for assessing target engagement of purported PARP1/2 inhibitors in a live cell setting. Strikingly, several other proteins enriched by PARPYnD were outcompeted by both AZ0108 and olaparib and represent potentially important off-target interactions for both molecules. Among these identified proteins, nicotinamide phosphoribosyltransferase (NAMPT) stands out as an off-target for both AZ0108 and olaparib as it is a key enzyme in the biosynthetic pathway for NAD+ and a promising anticancer target in its own right, with drug discovery campaigns already reported in the literature.27,28 Proteins involved in sterol biosynthesis and metabolism are also significantly competed by both molecules, including lanosterol synthase (LSS),29 the NADPH-dependent reductase Δ(24)-sterol reductase (DHCR24),30 and the NAD(P)-independent oxidases prenylcysteine oxidase (PCYOX1)31 and mitochondrial sterol 26-hydroxylase (CYP27A1—engaged by AZ0108 only).32 These proteins represent novel off-targets for both AZ0108 and olaparib and, for the latter, have not been reported in previous protein profiling campaigns.33,34
PARP6 was not enriched in these proteomics experiments, confirming the results obtained from immunoblotting. To investigate whether the expression level of this protein precluded detection by Western blot and LC-MS/MS, a FLAG-tagged version of PARP6 was transiently overexpressed in MDA-MB-468 cells, and target engagement experiments with PARPYnD were repeated. After transfection with the corresponding construct, cells were treated with PARPYnD with/without competition, irradiated, lysed, and ligated to AzTB. The labeled proteome was once again incubated with streptavidin beads, and the enriched fraction was immunoblotted against the FLAG epitope, with no FLAG-PARP6 enrichment detected (Figure 3E). Given that PARPYnD binds strongly to PARP6 in in vitro binding assays, the failure of PARPYnD to engage the corresponding endogenous protein was unanticipated. To investigate this disparity, recombinant GST-tagged PARP6 used in in vitro binding assays was spiked into MDA-MB-468 lysates and the lysate treated with PARPYnD with or without competitor AZ0108, irradiated with 365 nm UV light and ligated to AzTB. In-gel fluorescence showed clear labeling of a band corresponding to the molecular weight of GST-PARP6 (98 kDa) which was depleted by PARP6 inhibitor AZ0108 (Figure 4). Confirming these results, the same samples were enriched on streptavidin and immunoblotted against PARP6, demonstrating significant enrichment of recombinant protein by PARPYnD compared to DMSO control, which could be depleted in the presence of AZ0108. Importantly, labeling of endogenous PARP6 in lysate lacking recombinant GST-PARP6 protein was again not observed, reinforcing the results from live cell experiments. Finally, to rule out the possibility that the GST-fusion tag influences binding of PARPYnD and AZ0108 to the recombinant protein, the same lysate-based photocrosslinking experiment was performed before and after cleavage of the GST tag from the protein using an internal HRV3C cleavage site present in the linker between PARP6 and GST tag (Figure S6). In both cases, labeling of the 71 kDa band corresponding to the intact PARP6 protein was demonstrated, showing that the GST tag is neither the site of probe binding, nor does it bias the conformation of PARP6 in such a way that artificially promotes binding. These results imply that PARPYnD can bind to and label recombinant but not endogenous PARP6, raising interesting questions regarding the activity and inhibitor binding affinity for PARP6 in cellulo.
Figure 4.

Successful engagement of recombinant but not endogenous PARP6 in lysate-based crosslinking experiments. In-gel fluorescence (top) and immunoblot (bottom) analysis of lysates labeled with PARPYnD and ligated to AzTB with/without cotreatment with AZ0108. Left panel—lysates were spiked with recombinant GST-PARP6 before treatment. Ligated samples were further enriched on streptavidin beads and all samples analyzed as above: > GST-PARP6 (98 kDa); * PARP6 (71 kDa). More recombinant protein is labeled/enriched compared to vehicle (DMSO) control, where residual signal can be observed through nonspecific interaction of the recombinant protein with the beads. No enrichment of endogenous PARP6 is observed.
Conclusion
In this work we have developed PARPYnD as the first photoaffinity-based probe (AfBP) for the PARP family and have shown that it can effectively label PARP1/2 in a live cell setting, highlighting this AfBP as a useful tool molecule to validate target engagement of PARP inhibitors in cellulo. Previous studies using the label-free approach of the cellular thermal shift assay (CETSA) have similarly measured target engagement of PARP1 by drug molecules in live cell systems;35,36 photoaffinity labeling with PARPYnD complements this work while additionally being able to identify and quantify interactions of PARP inhibitors with other proteins. In particular, we highlight that PARPYnD is able to covalently engage several non-PARP proteins in intact cells, and competitive affinity-based protein profiling experiments have identified several of these proteins as novel off-targets of olaparib and AZ0108. Further investigation is required to demonstrate that these proteins are truly engaged by PARPYnD and are not enriched due to an orthogonal interaction between the protein and PARP1/2 that brings the identified protein in close proximity to the photoaffinity element of the probe while bound to the PARP enzyme. However, this is unlikely since none of the identified proteins reported in Figure 3 are known binding partners of PARP1 or PARP2.37,38 These proteins may therefore represent novel biomolecular interactions for phthalazinone-based PARP inhibitors,33,34 and engagement of these targets may be significant for the polypharmacology of phthalazinone-based PARP inhibitors in the clinic, leading to deeper understanding of toxicology or novel indications. It has been discussed that the off-target interactions identified may have an impact on sterol metabolism, and further network analysis37 demonstrated that off-target binding to CYP27A1 and NAMPT may affect the peroxisome proliferator-activated receptor (PPAR) axis and therefore disrupt lipid biosynthesis and metabolism.39 This could be of importance when addressing questions of safety and efficacy of these compounds.
Furthermore, examining the full list of proteins labeled by the probe but not necessarily depleted in competition experiments (Figure S5A), many are known binders of nucleoside analogues, suggesting that PARPYnD may have potential as a tool to profile nucleoside mimicking drugs outside of the PARP inhibitor family.40 These include NAD(P) binding proteins prostaglandin reductase (PTGR2), NADPH-cytochrome P450 reductase (POR), squalene synthase (FDFT1), retinal dehydrogenase (ALDH1A1); Coenzyme A binding proteins peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), carnitine O-palmitoyltransferase 2, mitochondrial (CPT2), nuclear receptor coactivator 3 (NCOA3); and ATP binding proteins adenosine kinase (ADK), methionine adenosyltransferase 2 subunit beta (MAT2B), and ribosomal protein S6 kinase alpha-1 (RPS6KA1).
AZ0108 has been reported to be a cellular PARP6 inhibitor, and this activity is thought to be responsible for MPS formation and subsequent cytotoxicity.11 However, despite the fact that PARPYnD efficiently enters cells (based on PARP1/2 labeling), generates an MPS phenotype, and inhibits PARP6 in vitro with comparable potency to the parent compounds, it fails to bind or photocrosslink to endogenous PARP6, even when transiently overexpressed. This suggests that cellular PARP6 inhibition may not be the only mechanism that contributes to MPS induction by these molecules. Furthermore, PARPYnD can label recombinant but not endogenous PARP6, implying that access to the PARP6 NAD+ binding site may be restricted in the endogenous protein, for example as a result of endogenous post-translational modification of PARP6, or complex formation with another biomolecule, that restricts compound binding. Given the current lack of structural and biophysical characterization of PARP6, further work is required to understand the molecular basis for regulated access to the NAD+-binding site.
In summary, PARPYnD constitutes a novel and useful probe for on- and off-target profiling of PARP inhibitor interactions and has been used to identify the need for future investigation of the purported mechanism of action of these compounds and associated off-target pharmacology. More generally, this work highlights the importance of assessing target engagement in a biological system at the earliest stages of the drug discovery and development pipeline, and photoaffinity labeling with an AfBP provides a complementary approach to uncover the interactome of a molecule in intact, living cells.
Acknowledgments
The authors would like to thank the Francis Crick Institute’s Cell Services for providing the MDA-MB-468 cells, A. Coulson for maintaining the Imperial College Chemistry Department Tissue Culture facility, L. Haigh for managing the Imperial College Chemistry Department Mass Spectrometry suite, and P. Haycock for managing the Imperial College Chemistry Department NMR facility. This work was funded jointly by AstraZeneca and the Engineering and Physical Sciences Research Council (CHBBC/G98108).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00963.
The authors declare the following competing financial interest(s): This work was partially funded by AstraZeneca, and P.H., P.P., J.S.S., and J.W.J. are all employees of AstraZeneca. E.W.T. is a Director and shareholder of Myricx Pharma Ltd.
Notes
The data underpinning this study are available upon request from the corresponding author.
Supplementary Material
References
- Gibson B. A.; Kraus W. L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424. 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Vyas S.; Matic I.; Uchima L.; Rood J.; Zaja R.; Hay R. T.; Ahel I.; Chang P. (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 5, 4426. 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-S.; Jividen K.; Spencer A.; Dworak N.; Ni L.; Oostdyk L. T.; Chatterjee M.; Kuśmider B.; Reon B.; Parlak M.; Gorbunova V.; Abbas T.; Jeffery E.; Sherman N. E.; Paschal B. M. (2017) Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 66, 503–516. 10.1016/j.molcel.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Vyas A.; Kassab M. A.; Singh A. K.; Yu X. (2017) The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 45, 8129–8141. 10.1093/nar/gkx565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus W. L.; Hottiger M. O. (2013) PARP-1 and gene regulation: Progress and puzzles. Mol. Aspects Med. 34, 1109–1123. 10.1016/j.mam.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Fatokun A. A.; Dawson V. L.; Dawson T. M. (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J. C.; Duhnam-Ems S.; Vake C. La; Cruz A. R.; Moore M. W.; Caimano M. J.; Velez-Climent L.; Shupe J.; Krueger W.; Radolf J. D. (2009) Activation of Human Monocytes by Live Borrelia burgdorferi Generates TLR2-Dependent and -Independent Responses Which Include Induction of IFN-β. PLoS Pathog. 5, e1000444 10.1371/journal.ppat.1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P.; Coughlin M.; Mitchison T. J. (2005) Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 7, 1133–1139. 10.1038/ncb1322. [DOI] [PubMed] [Google Scholar]
- Lord C. J.; Ashworth A. (2017) PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R.; Liu Z.; Kraus W. L. (2017) PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126. 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Grosskurth S. E.; Cheung T.; Petteruti P.; Zhang J.; Wang X.; Wang W.; Gharahdaghi F.; Wu J.; Su N.; Howard R. T.; Mayo M.; Widzowski D.; Scott D. A.; Johannes J. W.; Lamb M. L.; Lawson D.; Dry J. R.; Lyne P. D.; Tate E. W.; Zinda M.; Mikule K.; Fawell S. E.; Reimer C.; Chen H. (2018) Pharmacological Inhibition of PARP6 Triggers Multipolar Spindle Formation and Elicits Therapeutic Effects in Breast Cancer. Cancer Res. 78, 6691–6702. 10.1158/0008-5472.CAN-18-1362. [DOI] [PubMed] [Google Scholar]
- Johannes J. W.; Almeida L.; Daly K.; Ferguson A. D.; Grosskurth S. E.; Guan H.; Howard T.; Ioannidis S.; Kazmirski S.; Lamb M. L.; Larsen N. A.; Lyne P. D.; Mikule K.; Ogoe C.; Peng B.; Petteruti P.; Read J. A.; Su N.; Sylvester M.; Throner S.; Wang W.; Wang X.; Wu J.; Ye Q.; Yu Y.; Zheng X.; Scott D. A. (2015) Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg. Med. Chem. Lett. 25, 5743–5747. 10.1016/j.bmcl.2015.10.079. [DOI] [PubMed] [Google Scholar]
- Qi G.; Kudo Y.; Tang B.; Liu T.; Jin S.; Liu J.; Zuo X.; Mi S.; Shao W.; Ma X.; Tsunematsu T.; Ishimaru N.; Zeng S.; Tatsuka M.; Shimamoto F. (2016) PARP6 acts as a tumor suppressor via downregulating Survivin expression in colorectal cancer. Oncotarget 7, 18812–18824. 10.18632/oncotarget.7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. Y.; Wang K.; Vermehren-Schmaedick A.; Adelman J. P.; Cohen M. S. (2016) PARP6 is a Regulator of Hippocampal Dendritic Morphogenesis. Sci. Rep. 6, 18512. 10.1038/srep18512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncel H.; Tanaka S.; Oka S.; Nakai S.; Fukutomi R.; Okamoto M.; Ota T.; Kaneko H.; Tatsuka M.; Shimamoto F. (2012) PARP6, a mono(ADP-ribosyl) transferase and a negative regulator of cell proliferation, is involved in colorectal cancer development. Int. J. Oncol. 41, 2079–2086. 10.3892/ijo.2012.1652. [DOI] [PubMed] [Google Scholar]
- Heal W. P.; Dang T. H. T.; Tate E. W. (2011) Activity-based probes: discovering new biology and new drug targets. Chem. Soc. Rev. 40, 246–257. 10.1039/C0CS00004C. [DOI] [PubMed] [Google Scholar]
- Lapinsky D. J.; Johnson D. S. (2015) Recent developments and applications of clickable photoprobes in medicinal chemistry and chemical biology. Future Med. Chem. 7, 2143–2171. 10.4155/fmc.15.136. [DOI] [PubMed] [Google Scholar]
- Parker C. G.; Galmozzi A.; Wang Y.; Correia B. E.; Sasaki K.; Joslyn C. M.; Kim A. S.; Cavallaro C. L.; Lawrence R. M.; Johnson S. R.; Narvaiza I.; Saez E.; Cravatt B. F. (2017) Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541. 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broncel M.; Serwa R. A.; Ciepla P.; Krause E.; Dallman M. J.; Magee A. I.; Tate E. W. (2015) Multifunctional reagents for quantitative proteome-wide analysis of protein modification in human cells and dynamic profiling of protein lipidation during vertebrate development. Angew. Chem., Int. Ed. 54, 5948–5951. 10.1002/anie.201500342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. J.; Qian W.; Biswas K.; Yuan C.; Amegadzie A.; Liu Q.; Nixey T.; Zhu J.; Ncube M.; Rzasa R. M.; Chavez F.; Chen N.; DeMorin F.; Rumfelt S.; Tegley C. M.; Allen J. R.; Hitchcock S.; Hungate R.; Bartberger M. D.; Zalameda L.; Liu Y.; McCarter J. D.; Zhang J.; Zhu L.; Babu-Khan S.; Luo Y.; Bradley J.; Wen P. H.; Reid D. L.; Koegler F.; Dean C.; Hickman D.; Correll T. L.; Williamson T.; Wood S. (2013) Discovery of 2-methylpyridine-based biaryl amides as γ-secretase modulators for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 23, 6447–6454. 10.1016/j.bmcl.2013.09.041. [DOI] [PubMed] [Google Scholar]
- Hanthorn J. J.; Valgimigli L.; Pratt D. A. (2012) Preparation of Highly Reactive Pyridine- and Pyrimidine-Containing Diarylamine Antioxidants. J. Org. Chem. 77, 6908–6916. 10.1021/jo301013c. [DOI] [PubMed] [Google Scholar]
- Hutin D., Grimaldi G., and Matthews J. (2018) Methods to Study TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP) Mono-ADP-Ribosyltransferase Activity. In ADP-ribosylation and NAD+ Utilising Enzymes, pp 109–124, Humana, New York, NY. [DOI] [PubMed] [Google Scholar]
- Albrow V. E.; Grimley R. L.; Clulow J.; Rose C. R.; Sun J.; Warmus J. S.; Tate E. W.; Jones L. H.; Storer R. I.; Nigg E. A.; Brunak S.; Mann M.; Plotnikov A. N.; Vedadi M.; Arrowsmith C. H.; Kruidenier L.; Reid R. A.; Burkhart W.; Turunen B. J.; Rong J. X.; Wagner C.; Moyer M. B.; Wells C.; Hong X.; Moore J. T.; Williams J. D.; Soler D.; Ghosh S.; Nolan M. A. (2016) Design and development of histone deacetylase (HDAC) chemical probes for cell-based profiling. Mol. BioSyst. 12, 1781–1789. 10.1039/C6MB00109B. [DOI] [PubMed] [Google Scholar]
- Kallemeijn W. W.; Lueg G. A.; Faronato M.; Hadavizadeh K.; Goya Grocin A.; Song O.-R.; Howell M.; Calado D. P.; Tate E. W. (2019) Validation and Invalidation of Chemical Probes for the Human N-myristoyltransferases. Cell Chem. Biol. 26, 892–900. 10.1016/j.chembiol.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storck E. M.; Morales-Sanfrutos J.; Serwa R. A.; Panyain N.; Lanyon-Hogg T.; Tolmachova T.; Ventimiglia L. N.; Martin-Serrano J.; Seabra M. C.; Wojciak-Stothard B.; Tate E. W. (2019) Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat. Chem. 11, 552–561. 10.1038/s41557-019-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner P.; Heydenreuter W.; Stahl M.; Korotkov V. S.; Sieber S. A. (2017) A Whole Proteome Inventory of Background Photocrosslinker Binding. Angew. Chem., Int. Ed. 56, 1396–1401. 10.1002/anie.201605993. [DOI] [PubMed] [Google Scholar]
- Wang W.; Elkins K.; Oh A.; Ho Y.-C.; Wu J.; Li H.; Xiao Y.; Kwong M.; Coons M.; Brillantes B.; Cheng E.; Crocker L.; Dragovich P. S.; Sampath D.; Zheng X.; Bair K. W.; O’Brien T.; Belmont L. D. (2014) Structural basis for resistance to diverse classes of NAMPT inhibitors. PLoS One 9, e109366 10.1371/journal.pone.0109366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Bauer P.; Baumeister T.; Buckmelter A. J.; Caligiuri M.; Clodfelter K. H.; Han B.; Ho Y.-C.; Kley N.; Lin J.; Reynolds D. J.; Sharma G.; Smith C. C.; Wang Z.; Dragovich P. S.; Oh A.; Wang W.; Zak M.; Gunzner-Toste J.; Zhao G.; Yuen P.; Bair K. W. (2013) Structure-Based Identification of Ureas as Novel Nicotinamide Phosphoribosyltransferase (Nampt) Inhibitors. J. Med. Chem. 56, 4921–4937. 10.1021/jm400186h. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Chen X.-J.; Zhu J.; Xi Y.-B.; Yang X.; Hu L.-D.; Ouyang H.; Patel S. H.; Jin X.; Lin D.; Wu F.; Flagg K.; Cai H.; Li G.; Cao G.; Lin Y.; Chen D.; Wen C.; Chung C.; Wang Y.; Qiu A.; Yeh E.; Wang W.; Hu X.; Grob S.; Abagyan R.; Su Z.; Tjondro H. C.; Zhao X.-J.; Luo H.; Hou R.; Jefferson J.; Perry P.; Gao W.; Kozak I.; Granet D.; Li Y.; Sun X.; Wang J.; Zhang L.; Liu Y.; Yan Y.-B.; Zhang K. (2015) Lanosterol reverses protein aggregation in cataracts. Nature 523, 607–611. 10.1038/nature14650. [DOI] [PubMed] [Google Scholar]
- Waterham H. R.; Koster J.; Romeijn G. J.; Hennekam R. C.; Vreken P.; Andersson H. C.; FitzPatrick D. R.; Kelley R. I.; Wanders R. J. (2001) Mutations in the 3β-hydroxysterol Δ24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet. 69, 685–694. 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschantz W. R.; Zhang L.; Casey P. J. (1999) Cloning, expression, and cellular localization of a human prenylcysteine lyase. J. Biol. Chem. 274, 35802–35808. 10.1074/jbc.274.50.35802. [DOI] [PubMed] [Google Scholar]
- Mast N.; Anderson K. W.; Lin J. B.; Li Y.; Turko I. V.; Tatsuoka C.; Bjorkhem I.; Pikuleva I. A. (2017) Cytochrome P450 27A1 Deficiency and Regional Differences in Brain Sterol Metabolism Cause Preferential Cholestanol Accumulation in the Cerebellum. J. Biol. Chem. 292, 4913–4924. 10.1074/jbc.M116.774760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic C. E.; Wright G.; Remsing Rix L. L.; Kim W.; Kuenzi B. M.; Luo Y.; Watters J. M.; Koomen J. M.; Haura E. B.; Monteiro A. N.; Radu C.; Lawrence H. R.; Rix U. (2016) Proteome-wide Profiling of Clinical PARP Inhibitors Reveals Compound-Specific Secondary Targets. Cell Chem. Biol. 23, 1490–1503. 10.1016/j.chembiol.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowska A.; Thomson D. W.; Vappiani J.; Werner T.; Mueller K. M.; Dittus L.; Krause J.; Muelbaier M.; Bergamini G.; Bantscheff M. (2016) A Modular Probe Strategy for Drug Localization, Target Identification and Target Occupancy Measurement on Single Cell Level. ACS Chem. Biol. 11, 2541–2550. 10.1021/acschembio.6b00346. [DOI] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Shaw J.; Dale I.; Hemsley P.; Leach L.; Dekki N.; Orme J. P.; Talbot V.; Narvaez A. J.; Bista M.; Martinez Molina D.; Dabrowski M.; Main M. J.; Gianni D. (2019) Positioning High-Throughput CETSA in Early Drug Discovery through Screening against B-Raf and PARP1. SLAS Discovery Adv. Life Sci. R&D 24, 121–132. 10.1177/2472555218813332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Mering C. v. (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata L.; Briganti L.; Peluso D.; Perfetto L.; Iannuccelli M.; Galeota E.; Sacco F.; Palma A.; Nardozza A. P.; Santonico E.; Castagnoli L.; Cesareni G. (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–861. 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J.; Moller D. E. (2002) The Mechanisms of Action of PPARs. Annu. Rev. Med. 53, 409–435. 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium T. (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699–2699. 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.