

Abstract
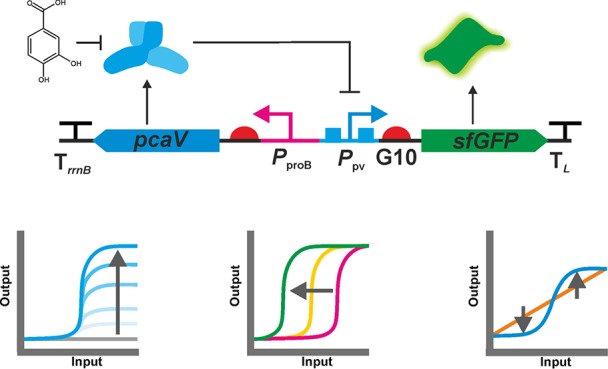
Whole cell biosensors are genetic systems that link the presence of a chemical, or other stimulus, to a user-defined gene expression output for applications in sensing and control. However, the gene expression level of biosensor regulatory components required for optimal performance is nonintuitive, and classical iterative approaches do not efficiently explore multidimensional experimental space. To overcome these challenges, we used a design of experiments (DoE) methodology to efficiently map gene expression levels and provide biosensors with enhanced performance. This methodology was applied to two biosensors that respond to catabolic breakdown products of lignin biomass, protocatechuic acid and ferulic acid. Utilizing DoE we systematically modified biosensor dose–response behavior by increasing the maximum signal output (up to 30-fold increase), improving dynamic range (>500-fold), expanding the sensing range (∼4-orders of magnitude), increasing sensitivity (by >1500-fold), and modulated the slope of the curve to afford biosensors designs with both digital and analogue dose–response behavior. This DoE method shows promise for the optimization of regulatory systems and metabolic pathways constructed from novel, poorly characterized parts.
Keywords: design of experiments, definitive screening design, whole cell biosensors, protocatechuic acid, ferulic acid
A whole cell biosensor is a biosynthetic system of cellular components designed to convert a stimulus (e.g., the presence of chemical, a change in osmolality or redox state) into a measurable cellular response. Biosensors enable fast, simple sensing of small molecule effectors through measurement of a relatively easy quantifiable output. Compared to the low or medium throughput of standard chemical analytical techniques, biosensors can be constructed that allow easy, high-throughput assessment of the stimuli in question. Allosteric transcription factors (aTFs) have been widely co-opted for biosensing applications for the detection of a wide range of stimuli.1−4 Repression based aTFs bind their cognate promoter-operator in the absence of a specific effector, thus inhibiting transcription. Binding of the effector molecule to the aTF induces a conformational change, causing a loss of DNA binding and derepression leading to the expression of a reporter gene, such as gfp. Biosensors have been exploited to control protein expression, monitor metabolism, identify novel genes in metagenomics libraries, and have found application in biotechnological and biomedical sensing, and diagnostic devices.5−7
Following initial construction of a biosensor system, further iterative refinement is often required to achieve a highly performing system in its new genetic context. Several parameters must be optimized for good biosensor performance: output in the OFF-state (leakiness) should be minimized to allow accurate measurements at low signal levels; output in the ON-state (reporter expression level) should be maximized to allow signal detection in the presence of background noise and to achieve high levels of gene expression for sensing and control applications; dynamic range is the ratio of the system’s ON and OFF states and a high dynamic range allows more confident “hit” identification due to a high signal-to-noise ratio. Additionally, for certain applications the sensitivity, the sensing range, and specificity of a biosensor should be considered. For primary screening applications biosensors should display high sensitivity to permit analyte detection at low levels (<μM), and allow binary (yes/no) classification of positive hits. For secondary screening applications biosensors that respond over a wide range of inducer concentrations would allow clustering of primary hits and separation into different subgroups based on analyte concentration. For biotransformation and other diagnostic applications the biosensor must also be highly specific to minimize false detection from analytes with closely related chemical structures or properties. Generic biosensor design and engineering rules are currently lacking, which limits the broader adoption of biosensors in sensing and control applications.2 Biosensors have been optimized using directed evolution, mechanistic modeling, and rational engineering.8−14 While these approaches have been successful in elucidating biosensors, they often require resource intensive, iterative directed evolution using technically challenging selection methods, or the use of well characterized sensory elements. Structured, multivariate experimentation, and statistical modeling have not been previously applied to this problem but could offer rapid, resource-efficient means of optimizing biosensors and elucidating universal design rules.
Structured multivariate experimentation and statistical modeling is widely used in various engineering and process industries.15−17 This combined experimentation and modeling approach, referred to as design of experiments (DoE), is a statistical tool used to systematically explore multidimensional experimental space with the minimum number of experimental runs (Figure 1A). It allows researchers to optimize poorly understood processes and decipher nonintuitive interactions with a series of statistically designed experiments.18 DoE is commonly used to optimize environmental factors such as temperature, time, and concentration during bioprocess development. The continuous nature of these factors makes structured experimental exploration over a range of multivariate conditions facile. More recently DoE has also been used successfully in a growing number of applications in optimizing genetic factors for metabolic engineering of biosynthetic pathways;19−22 its utilization is a powerful means of dramatically improving the performance of metabolic pathways. While these studies show the utility of DoE for linear biosynthetic pathways, it has not yet been applied to more complex genetic systems consisting of multiple protein–protein and protein–DNA interactions that are more likely to display nonlinear effects. Confounding this is the challenge of converting different closely related, but discrete, genetic designs into so-called continuous factors. Without this conversion, assessment of all experimentation conditions would need to be repeated for all genetic designs, limiting the potential for a reduction in the number of required experiments. By applying a modern DoE framework23 to biosensor development we demonstrate the utility of this approach to the optimization of regulatory systems.
Figure 1.

Application of design of experiments (DoE) to modulate biosensor dose response curves. (A) An experiment can be considered as a point in multidimensional space. DoE is a statistical tool that enables the proper exploration of experimental space to understand and optimize biology. (B) Modulation of biosensor dose response curves by increasing maximum output in the ON state while minimizing output in the OFF state (vertical extension; left), increasing sensitivity (middle), and conversion of a digital response to an analogue one (horizontal extension; right).
Here we applied DoE to address the challenge of standard iterative design-build-test-learn experimental approaches for the optimization of genetic systems, which can be costly both in terms of resources and time. To assess this methodology we sought to explore and optimize the performance of various aTF-based small-molecule-responsive biosensors. Three regulatory component libraries, two promoters and one RBS, were generated and assessed for expression performance. Linear regression modeling and fractional sampling were used to explore a highly efficient, structured coarse-grained map of experimental space. This workflow was applied to optimize performance of a two-gene protocatechuic acid (PCA) responsive biosensor.24 PCA is an aromatic chemical derived from lignocellulosic biomass, and a central intermediate of lignin catabolic pathways in microorganisms, making it of biotechnological interest for the valorization of lignin into high value chemicals.25,26 An enhanced PCA biosensor was then benchmarked against the commonly used E. coli recombinant expression systems. The sensitivity of the PCA biosensor, was then increased by incorporation the pcaK transporter. The DoE concept and regulatory components were then used to engineer biosensors operating under an analogue dose response modality, by placing pcaK under the control of a PCA-responsive inverter. Finally the DoE concept and regulatory components were assessed using a more complex enzyme coupled biosensor, consisting of three functional genes that allow detection of ferulic acid,27 a major aromatic chemical building block derived from lignin.26 The results of this optimization effort suggest that this approach could also be applied to enhance the performance of other biosensors. Collectively the approach demonstrated the ability of DoE to efficiently map experimental space and develop genetic systems, with greatly enhanced output signal, basal control, dynamic range (signal-to-noise), and sensitivity.
Results and Discussion
Design of a PCA Biosensor
In previous work we constructed a two-plasmid PCA biosensor (PAB)24 composed of the PCA-responsive allosteric transcription factor (aTF), PcaV from Streptomyces coelicolor, under control of a constitutive PlacI promoter on one plasmid, and the PcaV repressible PPV promoter upstream from a reporter gene (GFP) on a second plasmid. To simplify its deployment the PAB was combined into a single plasmid (pPPV-GFP-pcaV). This single plasmid PAB displayed a good dynamic range (ON/OFF = 417; Table 1). However, only modest GFP expression was observed compared to other commonly used E. coli expression systems.28
Table 1. Definitive Screening Design of Screen Genetic Factors Constituting a PCA Biosensora.
| construct | trial | Preg | Pout | RBSout | OFF | ON | ON/OFF |
|---|---|---|---|---|---|---|---|
| pD1 | 1 | 0 | 0 | 0 | 593.9 ± 17.4 | 1035.5 ± 18.7 | 1.7 ± 0.08 |
| pD2 | 2 | 0 | 1 | 1 | 397.9 ± 3.4 | 62070.6 ± 1042.1 | 156.0 ± 1.5 |
| pD3 | 3 | –1 | –1 | –1 | 28.9 ± 0.7 | 45.7 ± 4.7 | 1.6 ± 0.16 |
| pD4 | 4 | 1 | –1 | 0 | 479.8 ± 2.0 | 860.5 ± 15.1 | 1.8 ± 0.04 |
| pD5 | 5 | –1 | 1 | 0 | 1543.3 ± 46.2 | 5546.2 ± 101.7 | 3.6 ± 0.11 |
| pD6 | 6 | 0 | –1 | –1 | 16.3 ± 4.1 | 36.0 ± 5.4 | 2.2 ± 0.68 |
| pD7 | 7 | 1 | 1 | 1 | 1282.1 ± 37.9 | 47138.5 ± 1702.8 | 36.8 ± 1.6 |
| pD8 | 8 | 1 | 0 | –1 | 41.0 ± 5.1 | 49.7 ± 2.9 | 1.2 ± 0.11 |
| pD9 | 9 | 1 | –1 | 1 | 608.8 ± 19.6 | 1032.9 ± 6.5 | 1.7 ± 0.06 |
| pD10 | 10 | –1 | 0 | 1 | 3304.9 ± 88.6 | 17212.1 ± 136.6 | 5.2 ± 0.13 |
| pD11 | 11 | 1 | 1 | –1 | 37.7 ± 4.9 | 100.0 ± 2.7 | 2.7 ± 0.29 |
| pD12 | 12 | –1 | –1 | 1 | 659.7 ± 20.6 | 1841.4 ± 113.3 | 2.8 ± 0.21 |
| pD13 | 13 | –1 | 1 | –1 | 71.9 ± 10.7 | 226.6 ± 17.7 | 3.2 ± 0.6 |
OFF and ON measurements were made in the absence or presence of 1 mM PCA, respectively. The values for OFF, ON, and OFF/ON indicate the mean of three biological replicates with ± denoting the standard deviation of those replicates. The raw data for this table can be found in Supplementary Table S1.
To explore whether biosensor performance could be improved, we sought to optimize signal output and dynamic range by refactoring the PAB and systematically varying the genetic elements making up this biosensor using DoE to guide the process (Figure 2A). By using DoE to optimize the PAB and improve the performance of various iterations, we aimed to build a statistical model describing the interaction of the genetic components, which could serve as a guide in further efforts to construct, optimize, and modulate biosensors.
Figure 2.

Configuration of PCA biosensor and construction of promoter and RBS libraries. (A) The PCA biosensor consists of the PcaV repressor, which binds to the PPV promoter controlling sfGFP expression. In the presence of PCA the system is derepressed. (B) The genetic elements regulating expression of the system components were renamed as shown and mutated with degenerate oligonucleotides to make individual libraries. pcaV was substituted with mCherry to facilitate library screening. (C–E) Transcriptional (C, D) and translational activity (E) of library variants assessed by fluorescent protein synthesis rate (upper panels). Synthesis rates were transformed into logarithmically scaled values (lower panels) and “levels” for DoE were set at −1, 0, and +1.
Refactoring of the PCA Biosensor Guided by DoE
DoE consists of distinct phases: a screening phase is carried out initially to identify those factors that are most important to the process under investigation, which is followed by an optimization phase whereby those factors are adjusted to obtain the desired optimum. These factors are set at discrete “levels” that span a defined range: the low level is coded as −1, the middle level as 0 and the high level as +1. With this in mind, to refactor the PAB and apply DoE, we first had to decide on which factors were likely to influence biosensor performance and then convert those factors into levels suitable for DoE. Three genetic regulatory components controlling the transcription and translation of the components constituting the PAB were selected and modified: (i) the constitutive proB promoter (henceforth Preg) controlling pcaV expression; (ii) the PcaV-repressible PPV promoter (henceforth Pout); and (iii) the G10 RBS (henceforth RBSout), controlling the expression of the sensor output sfGFP (Figure 2A). These three factors have all been shown to be important for the response of a biosensor1 so were selected for systematic investigation through Design of Experiments. We decided to modify these independently as RNAP binding and translation rate, set by the promoter and RBS, respectively, could have different effects on the response curve of the system.1 We kept the transcriptional terminators, gene orientation, antibiotic selection marker and plasmid copy constant throughout the initial set of experiments, although we did modify copy number in later experiments by converting a stable multicopy system to single-copy system (see below).
Having selected three factors for study, we converted them into continuous variables by generating, screening and ranking the performance of libraries for each of these components (Preg, Pout, and RBSout). This step facilitates statistical-model based optimization by converting categorical variables, in this case a particular promoter or RBS, into continuous variables that span a wide expression range. Rather than use previously published libraries29 we decided to generate new libraries to provide greater confidence of the component performance within the genetic context of the biosensor, and to ensure that the expression level of the library was finely resolved and covered a broad range. The libraries were constructed in the pSEVA131 vector containing sfGFP as the biosensor output and mCherry substituted for pcaV to serve as a proxy for regulator expression. Following library construction and performance assessment mCherry was replaced with pcaV to reconstitute a functional biosensor (see below). The genes encoding sfGFP and mCherry were arranged in a divergent configuration to prevent transcriptional read-through and separated by a ∼150 bp spacer. Pout and RBSout were used to control expression of sfGFP and Preg and a strong RBS (gaaataaggaggtaatacaa) were used for to control expression of mCherry, yielding a construct termed p131B, which served as the starting point for library generation. To generate the individual libraries, we chose to randomize the nucleotides at the following positions: (i) for Preg, 3 Ns were introduced at the −10 hex-box to make Preg-lib (Figure 2B); (ii) for Pout, 3 Ns were introduced at both the −10 and −35 hex-boxes to make Pout-lib (Figure 2B); and (iii) for RBSout, 6 Ns were introduced at the core RBS binding region to make RBSout-lib (Figure 2B). This produced total theoretical library sizes of 64 (43), 4096 (46), and 4096 (46) for Preg-lib, Pout-lib, and RBSout-lib, respectively. The mutant libraries were screened for sfGFP fluorescence in E. coli for Pout-lib and RBSout-lib and for mCherry fluorescence for Preg-lib. Following the initial screen, 22 members from each library were selected to span a wide range of fluorescence values. Promoter and RBS activity was calculated by determining the rate of fluorescent protein (FP) production according to previously published work29,30 with the following equation:
| 1 |
The constructed libraries spanned a wide range of FP synthesis rates (Figure 2C,D,E) with Pout-lib having a 46-fold range (maximum of 17 340 and minimum 377), RBSout-lib having a 160-fold range (maximum of 15 860 and a minimum of 99), and Preg-lib having a 46-fold range (maximum of 8101.3 and minimum of 177). The expression data from libraries generated were rescaled using a linlog transformation described previously19 with the following equation:
| 2 |
P = Pmax, X = 1
P = Pmin, X = −1
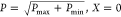
The linlog transformation was previously found to be essential for successful application of a DoE-based optimization process as logarithmic variables better reflect the cellular biophysics of transcription and translation,19 hence its implementation here. Library members were rank-ordered from −1 to +1 with the strongest member of each library recoded as +1, the weakest as −1, and the midpoint level 0 the geometric average of level +1 and −1 (Figure 2C,D,E).
Given the size of the screened libraries, a total of 10 648 (22 × 22 × 22) combinations would be needed to fully explore the gene expression space. DoE aims to reduce the number of combinations needed to properly explore an experimental space (Figure 1A) and determine the importance of different factors by using structured screening designs.18 A range of screening designs are available in a DoE methodology, here definitive screening design (DSD) was selected as it allows the identification of main (linear) factors and two-factor interactions with a relatively small number of experimental runs while avoiding confounding of pairs of second-order effects.23 DSD designs use 3 levels instead of 2 levels, thereby permitting some estimation of curvature (nonlinearity) in a factor-response relationship, which are likely to be found in biological systems. Here using DSD was employed to reduce the total experimental configurations from 10 648 to 13, a compression ratio of 819:1 (Table 1).
Statistical Modeling of PCA Biosensor Variants
The constructs were designed according to the DSD shown in Table 1 (see Supplementary Table S1 for the raw data), which was generated with statistical software (Materials and Methods). Additional runs over the minimum required (2n + 1 = total run number, n (number of factors) = 3) were included to account for the predicted high number of statistically significant factors and interactions. Following the replacement of mCherry with pcaV, all 13 constructs (Figure 3 and Table 1) were assembled correctly and transformed into E. coli. Next, we assessed the performance of each of the different permutations of the PAB by measuring end-point sfGFP fluorescence when uninduced (OFF) and induced with 1 mM PCA (ON), and calculated the biosensor dynamic range (ON/OFF). The results from these trials are shown in Figure 4A,B and Table 1 and give a broad range of values for the measured responses. The best performing candidate (pD2), displayed an excellent maximum signal (62 071 RFU/OD) and good dynamic range (156-fold) while maintaining tight basal control, whereas the poorest performer (pD8) produced negligible output signal (50 RFU/OD) and was barely responsive, with a dynamic range of 1.2-fold, highlighting the importance of a library-based optimization approach.
Figure 3.
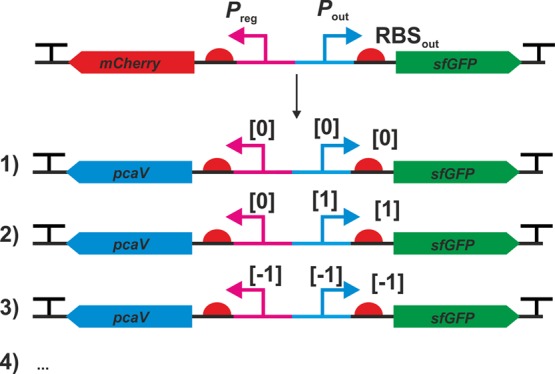
Genetic configuration of biosensor designs conforming to definitive screening design. Following library construction pcaV was reinstalled to create a functioning biosensor and regulatory elements were cloned at the appropriate levels. Three out of 13 constructs are shown, and the full table can be found in Table 1.
Figure 4.
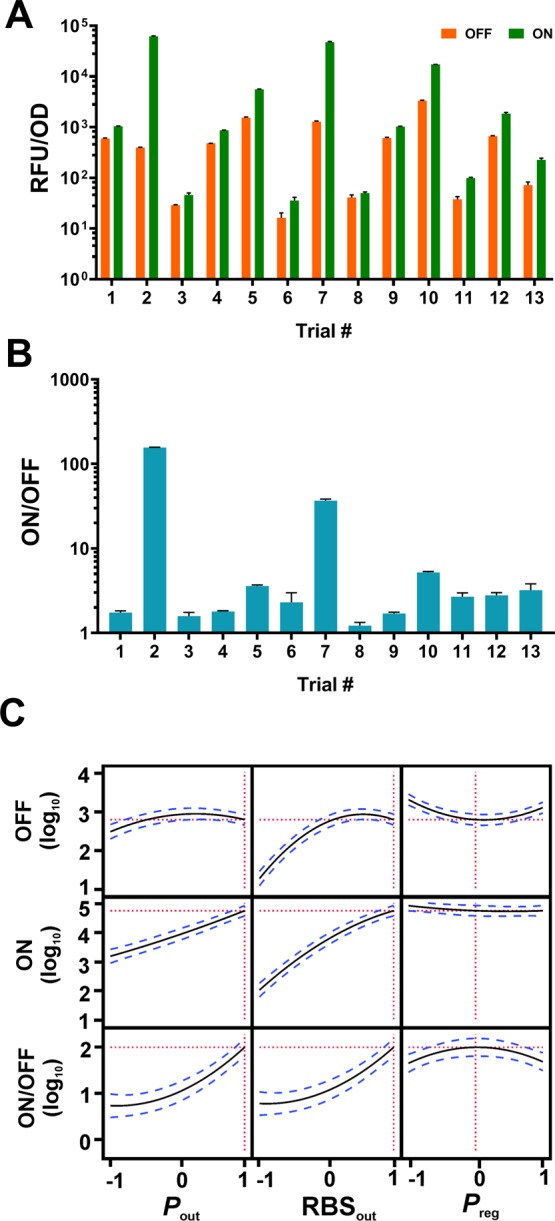
Experimental trials and statistical modeling. (A) GFP fluorescence for PCA biosensor variants in the ON state (1 mM PCA; green bars) and OFF state (no PCA; orange bars). (B) The dynamic range for PCA biosensor variants (ON/OFF). Error bars represent the standard deviation of three biological replicates. Each experiment was repeated a minimum of two times and typical results are shown. (C) Prediction profile of standard least-squares regression model based on experimental data.
Factor screening analysis was used to assess the importance of each main effect and their interactions. Significant factors were selected based on half-normal plots, which allows interpretation of factor effect on each of the three responses (OFF, ON, ON/OFF). Factor screening analysis revealed that the strongest significant effects for dynamic range (ON/OFF) were from Pout (p < 0.0001), Pout × RBSout (p < 0.0001), and Preg (p = 0.0004). For both the ON and OFF biosensor output responses, Pout, RBSout and Pout × RBSout showed the strongest significant effect (Supplementary Figure S1). Using those factors shown to be significant (p < 0.05) for biosensor performance, we carried out statistical modeling of the data using a standard least-squares regression (SLSR) model and analysis of variance (ANOVA) (Materials and Methods).
Comparison of effect sizes in the SLSR shows the factors and interactions with the greatest impact upon the three responses (Supplementary Figure S1, Supplementary Table S2 and S3). A prediction profile of the model is shown in Figure 4C. As expected, for maximum output (ON) Pout and RBSout are predicted to have the greatest effect and should be set at +1 for maximum signal output, while for basal output (OFF) RBSout is the strongest determinant and should be reduced if the basal output signal is too high. Increasing Pout and RBSout improves ON/OFF, but in a nonlinear manner when changing the expression level from midpoint to maximal (0 vs +1). Here the model indicates an interesting trade-off shown when comparing the effect of RBSout at −1 vs 0: both OFF and ON are scaled proportionality leading to no significant change in ON/OFF. At high expression levels of RBSout (+1) the OFF level indicates a plateau, whereas the ON level increases leading to vertical extension and increased ON/OFF. Interestingly for the ON/OFF response a nonlinear effect is also observed from changing the level of Preg controlling the expression of pcaV (Figure 4C). The optimal level of PcaV to achieve highest dynamic range lies near the middle level (0) and the system operates with poorer dynamic range at high (+1) and low levels (−1) for Preg. A lower biosensor dynamic range at low levels of aTF expression is unsurprising as there is insufficient transcription factor in the system to fully interfere with RNAP-promoter complex formation.31 However, increased output signal at high levels of PcaV was unexpected and suggests that excessive PcaV interferes with stable regulator-promoter complex formation. Collectively, these findings highlight the importance of a 3-level DSD as the nonlinear effect of Preg level would have been overlooked in a two level design consisting solely of a high and low level.19,20 Through the use of the DSD we were able to confidently identify nonlinear effects within the design space and assign the nonlinear effects to the RBSout and Preg levels. This assignment of nonlinear effects is not possible with traditional DoE screening designs due to heavy aliasing between nonlinear effect terms within the designed data structure. This means that while traditional screening designs can indicate the presence of a nonlinear effect, it is not possible to assign this nonlinearity to a causative factor, without augmenting the DoE design with additional experimental data, which would require further use of time and resources. This highlights a significant advantage of the definitive screening design employed here. The reliable resolution of this nonlinear effect removes the need for further rounds of experimentation to identify the cause of the nonlinear response.
Following identification of nonlinearity within the explored expression space we sought to further resolve the curvature within the promoter activity landscape of Preg. To do so we carried out additional trials in which Pout and RBSout were set at the highest level (+1) and the level of Preg was set at 4 different levels (−0.56, −0.28, 0.36, and 0.67) to explore the landscape around the Preg midpoint (Figure 5A). The responses for these iterations of the PAB were measured and their dynamic range is displayed. The results pointed to an optimum for dynamic range between level 0 and 0.36, so a final construct, p131C–B10, was created in which Preg was set at 0.14 with Pout and RBSout kept at +1. This construct gave the best performance of all tested with a dynamic range of 276-fold (Figure 5A and Supplementary Table S14).
Figure 5.

Optimization of PCA biosensor and effect of copy number. (A) The level of aTF was tuned to determine optimal dynamic range. Shown is the dynamic range (ON/OFF) for the PCA biosensor when induced with 1 mM PCA with Preg set at different levels. The blue circles represent the initial trials, the red squares represent the first iteration with Preg set at different levels and the green triangle represents the final iteration of Preg level. (B) Prediction profile of standard least-squares regression model based on data from new trials. (C) Comparison of original PCA biosensor with the optimized version (p131C–B10) in an end point assay. Cells were induced with varying concentration of PCA for 3 h at 37 °C then measured for GFP fluorescence. (D) Performance of PCA biosensor when present as one-copy in the genome. The level of repressor was tuned to determine optimal dynamic range when present as a single copy. Shown is the dynamic range (ON/OFF) for the PCA biosensor when induced with 1 mM PCA with Preg set at different strengths. Error bars represent the standard deviation of three biological replicates. Each experiment was repeated a minimum of two times and typical results are shown.
The results of the validation trials were used to modify the model to generate a new prediction profile describing the data (Figure 5B). From this model we are able to elucidate some design rules that should be applicable to other repression based aTF biosensor systems: (i) Construct the strongest chimeric promoter-operator and RBS combination possible, then (ii) fine-tune the level of regulator with a wide range of expression levels. If a satisfactory dynamic range cannot be met after tuning the regulator, then (iii) weaken the RBS driving signal output. Importantly, we were able to map the optima and develop this statistical model in a small number of experimental runs (18 constructs). Also, as the experimental space has been efficiently mapped through this DoE approach, we can be confident that an optimal configuration of the PAB has been achieved. We carried out a titration of the best variant (p131C–B10) and the original PAB with full induction at 4 mM PCA (Figure 5C and Supplementary Table S5), which showed that we had improved the output signal over 30-fold (3121 to 97 099 RFU/OD) and the dynamic range by 25% (417- to 521-fold).
Copy Number Effects upon Biosensor Performance
Next, we investigated the effect of copy number on the performance of the PAB by transferring the plasmid-based multicopy biosensor system into single-copy system on the chromosome. Different permutations of the biosensor were cloned into a pKIKO vector and inserted into the arsB locus (Materials and Methods). As before, Pout and RBSout were set at the highest level and the level of Preg was varied. The responses of the chromosomal PABs were assessed. We found that the maximum level of output signal was reduced ∼10-fold from the plasmid-based biosensor (Supplementary Table S6), consistent with the copy number reduction from the pBBR1 origin, which is reported to have 5–10 copies per E. coli cell.32,33 As shown in Figure 5D, the level Preg needed for the optimal dynamic range was increased from 0.14 to 0.61 (Figure 5A,D) and the overall dynamic range of the system was reduced (276-fold to 42-fold; Supplementary Table S6). It is well-known that expression correlates proportionally with gene-dosage;34,35 however, this relationship is complex, nonlinear,36 and copy reduction is believed to perturb the equilibrium of aTF-based systems due to a reduction in the steady state aTF concentration.37 Mechanistic based approaches have attempted to rationalize these observations and indicate that increasing strength of the RBS or promoter controlling the aTF is required to restore steady-state levels to functionality.37 This explains the requirement for a stronger promoter for pcaV to decrease basal expression of sfGFP from the biosensor when implemented as a single-copy system. The wide range of expression space covered by the calibrated regulatory component libraries enabled us to quickly refactor the biosensor in order to retune biosensor performance of the genome-integrated PAB.
Enhancing the Sensitivity of the PCA Biosensor and Modulating the Dose–Response Curve
Having demonstrated the applicability of DoE to improve the fold change and maximum, we turned our attention to modifying the sensitivity and slope of response curve. Using the DoE approach and the associated genetic libraries none the PAB variants demonstrated a marked difference in sensitivity (EC50) or slope of the response curve (nH); therefore we first investigated whether the sensitivity of the PAB could be attenuated by altering the internal PCA concentration. E. coli cannot metabolize PCA and has no known transport mechanisms. Therefore, we inserted the high affinity PCA transporter, pcaK from Pseudomonas putida, downstream of pcaV to form a synthetic operon (Figure 6A). Expression of pcaK led to increased sensitivity of the PAB to PCA of over 1500-fold (EC50 from 557 μM to 0.335 μM; Figure 6B and Supplementary Table S7).
Figure 6.
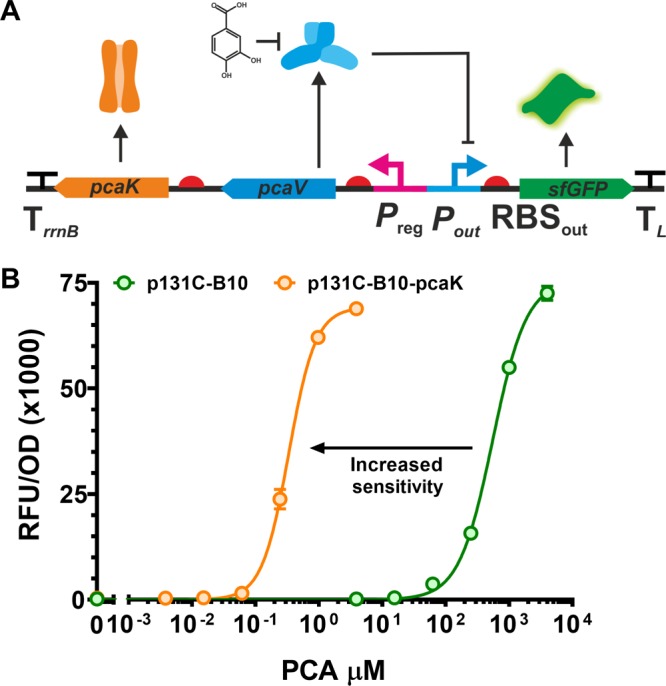
Increasing the sensitivity of the PCA biosensor. (A) The pcaK gene from Pseudomonas putida was inserted downstream of the Preg promoter and strong G10 RBS (+1) and pcaV. (B) The expression of a high-affinity, PCA permease leads to a reduction in EC50 of the PCA biosensor, as shown here. We observed a shift of the dose response curve to the left when pcaK is expressed (orange), compared to the p131C–B10 biosensor (green). Error bars represent the standard deviation of three biological replicates.
Following the dramatic increase in performance and sensitivity achieved we were satisfied that we had constructed a robust and highly functional biosensor system. Given these attributes we are confident that this biosensor is adequate for high-throughput screening method as its high sensitivity, large dynamic range and high output signal make it ideally suited for detecting low concentrations of PCA. The highly digital dose response gives an impressive signal-to-noise ratio, allowing great confidence when assigning positive analyte concentration above a required threshold for applications is screening genetic libraries and others applications in environmental monitoring and medical diagnostics.2−7 While these characteristics are ideal for primary screening, the binary nature of the dose–response means that this sensor is not well suited for determining between variants or samples with similar activities (i.e., as a result of protein engineering). For these applications it would be desirable to have a system, which gives a shallow, more analogue dose response, to allow accurate distinction between different analyte concentrations.
We hypothesized that by regulating the expression of the pcaK transporter, and therefore PCA uptake in the cell, we could expand the sensing range of the biosensor and transform the digital dose–response into an analogue response. We reasoned that by enhancing PCA uptake at low concentrations and repressing uptake at high concentrations we would be able to achieve this linear response (Figure 7A). To achieve conversion of the dose response curve from a digital to a more analogue signal a genetic system consisting of two plasmids (Figure 7B) was designed to provide negative feedback control of pcaK expression. The biosensor system consists of the lacI gene downstream of Pout and pcaK downstream of PLlac-O1. Using the negative feedback control of pcaK expression we expected that at low PCA concentrations pcaK would be maximally expressed (Pout controlling lacI repressed, PLlac-O1 controlling pcaK derepressed), giving greatest PCA uptake, accumulation inside the cell and therefore biosensor sensitivity/linearity. In contrast when PCA is at a higher concentration pcaK would be minimally expressed (lacI derepressed, pcaK repressed), leading to reduced PCA uptake and therefore an extended biosensor response linearity. As designed the negative feedback control should lead to horizontal extension of the dose response curve giving a less binary, more linear/analogue response over a large concentration range.
Figure 7.

Extending biosensor linear range through transport modulation. (A) Biosensors with analogue dose–responses have application in protein engineering as they allow more accurate identification of enzyme variants with improved function. To this end we designed a regulatory network to convert the digital-like dose response to an analogue output. (B) This circuit consists of a PcaV repressed Pout::lacI, which in turn repress pcaK expression. In the presence of high concentrations of PCA LacI is produced, leading to restricted pcaK expression, reducing ligand uptake, which ultimately reduces sfGFP output. Without PCA, lacI expression is repressed, pcaK is induced, leading to increased PCA uptake and accumulation inside the cell, thus increasing derepression of sfGFP expression. (C) Six variants of the dose–response extender circuit were designed and tested. The variants have different strength RBSs upstream of the lacI (−1, 0) and pcaK (−1, 0, +1). Expression testing under different concentrations of PCA show the different dose response performance of the construct variants. Error bars represent the standard error of three biological replicates, and the area fill denotes the 95% confidence interval for the fitted curve.
Rather than designing a single extender system ad hoc and relying on iterative redesign we chose to construct nine variants of the extender system. These constructs contain the full factorial variants of three RBS sites, upstream of pcaK ((−1), (0) and (+1)) and lacI ((−1), (0) and (+1)) each selected from the RBSout library described earlier in this study. These combinatorial plasmids were constructed using isothermal assembly of ssDNA containing the respective RBS sequences, into a linear, PCR amplified backbone (pSEVA261). During assembly we were not able to assemble the 3 constructs containing the LacI (+1) RBS variant, presumably due to the potential toxic of very high lacI expression.
Following cotransformation of the six negative feedback controller plasmids (p261_PcaK_LacI) and the sensor plasmid (p131C–B10) we measured sfGFP expression following induction with varying concentrations of PCA. Testing of the six biosensors, showed successful transformation of the digital dose response of the PcaK-sensitized PCA biosensor to an analogue system that is linearly titratable with increasing PCA concentration and gives a response over ∼4 orders of magnitude (see Figure 9C). The digital vs analogue behavior can be determined by calculation of the dynamic range of ligand response (DRLR; EC90/EC10). While all of the RBS variants showed modified dose–response behavior, the biosensor with most digital behavior, PcaK(+1)_LacI(−1), was effective at sensing PCA over a small concentration range (DRLR = 11.7, nH = 1.8) (Supplementary Table S8). Whereas, the biosensor with most analogue behavior, PcaK(−1)_LacI(0), was effective at sensing PCA over a larger concentration range (DRLR = 117.8, nH = 0.9). Demonstrating that PCA responsive inversion of pcaK expression was successful in expanding the sensing range of and converting the biosensor response into a linear signal.
Figure 9.

Optimization of a FA biosensor. (A) Schematic representation of a refactored FA biosensor. The FerC aTF (orange) represses sfGFP (green) expression by regulating the PLC2 promoter. The FerA enzyme (purple) metabolizes the sensed chemical ferulic acid into the ligand effector feruloyl-CoA, which binds to FerC derepressing sfGFP. (B) Dynamic range (ON/OFF) of the 9 DoE variants of the FA biosensor, in the first iteration, set with combinations of promoter strength levels of the FerC regulator (PregC at levels −1, 0, +1) and the FerA enzyme (PenzA at levels −1, 0, +1). (C) Performance of the 3 additional DoE variants of the FAB in the second iteration. The best variant of the first iteration pFABs9, PregC/PenzA/RBSout levels +1/+1/+1 (green circles), was compared to a group of new variants that had RBSout set at decreasing levels while the level of both PregC and PenzA was fixed at +1: pFABsG21, PregC/PenzA/RBSout levels +1/+1/+0.94 (red diamonds); pFABsG19, PregC/PenzA/RBSout levels +1/+1/+0.89 (orange triangles); and pFABsG12, PregC/PenzA/RBSout levels +1/+1/+0.81 (blue hexagons). The fluorescent signal (RFU/OD) is shown for the induction with increasing concentrations of ferulic acid. (D) The dynamic range (ON/OFF) is shown for the signal ratio of the variants at ON induced state (presence of ferulic acid at 1 mM) or OFF uninduced state (absence of ferulic acid). Error bars represent the standard deviation of three biological replicates.
The diverse performances of the different extender PAB variants, and the successful conversion of a digital to analogue dose–response curves demonstrate the importance of correctly balancing the expression of components within a regulatory system. By sparsely sampling the expression landscape using the RBS library we were able to rapidly identify a construct with the desired performance. Had we simply selected a pair of RBSs, which did not give such a pronounced conversion in dose response, it would have been easy to dismiss the system design as nonfunctional. The objective approach taken here allowed proper assessment of the expression landscape and selection of a functional extender system.
Benchmarking of the Refactored PAB against Commonly Used Expression Systems
Given the high expression levels produced from the refactored PAB we were interested in benchmarking the PAB against popular inducible expression systems in. The low sensitivity PAB (i.e., lacking the PcaK transporter) was used for benchmarking as it displayed a superior dynamic range and maximum output (Supplementary Table S7). We selected three commonly used bacterial expression systems: (i) the pET vector system utilizing a chromosome-integrated copy of T7 RNA polymerase to express the target gene, both regulated by P/Olac/LacI and induced with IPTG; (ii) the arabinose-inducible plasmid-based system in which expression of the target gene is controlled by ParaBAD/AraC; and (iii) the pCK302 rhamnose-inducible plasmid-based system in which expression of the target gene is controlled by PrhaBAD/RhaS. To benchmark the PAB against these systems, sfGFP was cloned into pET44 and pBAD expression plasmids. pCK302 already contains the sfGFP reporter so was not modified.38 The pBAD-sfGFP, pCK302, and p131C–B10 vectors were transformed into E. coli BL21 and pET44 was transformed into E. coli BL21(DE3) carrying the T7-RNA polymerase. Titrations were carried out for each of the expression systems using the appropriate inducer with samples analyzed for end-point fluorescence at 3 h and 24 h after induction. The PAB afforded comparable expression levels to the pBAD and BL21(DE3) expression systems in terms of maximum protein produced per cell (RFU/OD) after induction for 3 h (Figure 8A and Supplementary Table S9) and only the T7-RNAP system gave comparable amounts of protein at 24 h postinduction (Figure 8B and Supplementary Table S9). The PAB showed a consistently high dynamic range (>200-fold) at both time-points whereas the other systems displayed unwanted (leaky) expression during the longer induction time, presumably due to endogenous effects associated with catabolite derepression.39,40 Taken together, these findings demonstrate the utility of a properly optimized expression system from a heterologous source and highlight the potential of the PAB as tool for producing recombinant protein using PCA as a cheap, nonmetabolizable and orthogonal inducer.
Figure 8.

Benchmarking of PCA biosensor with popular inducible expression systems. (A,B) The PCA biosensor (p131C–B10; green circles) was tested against three common expression systems—T7 RNAP/IPTG (pET44-sfGFP; orange squares), ParaBAD/arabinose (pBAD-sfGFP; blue triangles), and PrhaBAD/mannose (pCK302; purple diamonds)—in an end point assay. Cells were induced with varying concentrations of inducers for 3 h (A) and 24 h (B) at 37 °C then measured for GFP fluorescence. Error bars represent the standard deviation of three biological replicates. Each experiment was repeated a minimum of two times, and typical results are shown.
Optimization of a Ferulic Acid Biosensor Guided by Statistical Modeling
Given the outstanding performance achieved for the PAB (Figure 8), we applied the above design rules to improve a ferulic acid biosensor, (henceforth FAB)27 to demonstrate the wider utility of the DoE approach. The ferulic acid biosensor is a three gene system, and also differs from the PAB in that in addition to an aTF (FerC) and inducible promoter (PLC), an activating enzyme, feruloyl CoA ligase (FerA), is required to convert ferulic acid into feruloyl-CoA (FA-CoA), which is the effector able to bind to FerC leading to derepression (Figure 9A).27 To optimize this biosensor, first, the original promoter-operator PLC controlling the reporter gene was reengineered based on the strong promoter from the Anderson library (BBa_J23119)30 to generate the PLC2 aiming to improve the maximum expression level that could be achieved by the reporter gene (Supplementary Figure S3).
Following the DoE strategy used for the PAB biosensor, the original FAB design27 was refactored and combined into a single plasmid system where the expression of the reporter sfGFP, driven by the promoter-operator (PLC2) and strong RBS (RBSout + 1), was initially fixed and the expression levels of ferC and ferA were optimized using a full factorial design. The promoters controlling the production of the transcription factor FerC (PregC) and the enzyme FerA (PenzA) were set at 3 levels (−1, 0, and +1) using the promoter sequences from the Preg library, which led to 9 different designs (Figure 9A and Table 2). As described for PAB, performance of the designs was assessed by measuring end-point fluorescence when uninduced (OFF) and induced by 1 mM FA (ON), and calculating the dynamic range (ON/OFF). The expression level of PregC was the main factor determining the increase of dynamic range (ON/OFF) and expression level of PenzA had smaller influence as observed by the ON/OFF of the intermediate designs (Figure 9B and Table 2) as shown on the analysis of a full factorial model for the expression (Supplementary Figure S4). The pFABs9 construct (PregC/PenzA/RBSout levels +1/+1/+1), which had PregC and PenzA both set at the highest levels (+1) displayed the best dynamic range (52-fold), with an ON signal of 74 893 RFU/OD and OFF signal of 1433 RFU/OD (Figure 9B, Table 2; see Supplementary Table S10 for the raw data).
Table 2. Full Factorial Screening of Screen Genetic Factors Constituting a FA Biosensora.
| construct | trial | PregC | PenzA | RBSout | OFF | ON | ON/OFF |
|---|---|---|---|---|---|---|---|
| pFABs1 | 1 | –1 | –1 | 1 | 14821.8 ± 307.2 | 96497.4 ± 5257.5 | 6.5 ± 0.4 |
| pFABs2 | 2 | –1 | 0 | 1 | 7829.3 ± 497.6 | 90917.2 ± 3861.1 | 11.6 ± 0.5 |
| pFABs3 | 3 | –1 | 1 | 1 | 33501.3 ± 213.6 | 93754.5 ± 2550.6 | 2.8 ± 0.1 |
| pFABs4 | 4 | 0 | –1 | 1 | 6649.1 ± 180.7 | 88905.3 ± 1381.2 | 13.4 ± 0.5 |
| pFABs5 | 5 | 0 | 0 | 1 | 6776.3 ± 96.5 | 87954.6 ± 1154.7 | 13.0 ± 0.4 |
| pFABs6 | 6 | 0 | 1 | 1 | 6369.9 ± 286.3 | 88764.7 ± 751.5 | 13.9 ± 0.6 |
| pFABs7 | 7 | 1 | –1 | 1 | 2140.5 ± 55.7 | 82976.5 ± 2964.9 | 38.8 ± 0.5 |
| pFABs8 | 8 | 1 | 0 | 1 | 1960.9 ± 87.3 | 77072.3 ± 1609.9 | 39.3 ± 1.4 |
| pFABs9 | 9 | 1 | 1 | 1 | 1432.8 ± 99.9 | 74892.8 ± 3048.0 | 52.4 ± 3.4 |
OFF and ON measurements were made in the absence or presence of 1 mM FA, respectively. The values for OFF, ON, and OFF/ON indicate the mean of three biological replicates with ± denoting the standard deviation of those replicates. The raw data for this table can be found in Supplementary Table S10.
However, we felt that further reduction of leakiness would be important for a high performance FAB biosensor. Therefore, following the design rules elucidated for the PAB biosensor earlier we reduced the strength of RBS controlling sfGFP expression. For this second iteration the RBS upstream of the reporter was replaced with variants from the RBSout library set at +0.94, +0.89, +0.81, generating pFABsG12 (PregC/PenzA/RBSout pattern at levels +1/+1/+0.81), pFABsG19 (PregC/PenzA/RBSout pattern at levels +1/+1/+0.89), and pFABsG21 (PregC/PenzA/RBSout pattern at levels +1/+1/+0.94). As expected, when compared to the best previous FAB variant pFABs9, these FAB variants showed a reduction of the minimum and maximum signals, and the dynamic range was increased significantly due to a greater relative reduction of the OFF signal versus the ON signal (Figure 9C, and Supplementary Table S11). In summary, we improved the performance of the FAB biosensor, relative to previously published designs,27 for both max output signal by 31-fold (992 RFU/OD to 30,783 RFU/OD) and dynamic range by 5-fold (23-fold to 118-fold) in a small number of experimental runs (12 constructs and two iterations).
In summary, by applying a DoE approach to the genetic factors constituting a biosensor, we were able to increase the maximum signal output from the PCA biosensor over 30-fold (3121 to 97 099 RFU/OD) and the dynamic range by 25% (417- to 521-fold). Further, we took advantage of a high-affinity PCA permease, PcaK, to vary the slope of the dose–response curve of the PAB and construct a whole cell biosensor with a linear responsivity to analyte concentration ∼4-orders of magnitude. We achieved coverage of the experimental space with 13 constructs and were able to fully optimize the PCA biosensor with an extra five constructs thereby demonstrating the efficient use of time and cost that a DoE approach can provide. The statistical model built from the experimental data allowed us to elucidate some design rules that we applied to improve the performance of a ferulic acid-biosensor in a small number of experimental runs. The following design rules should be applicable to other repression based aTF biosensor systems: (i) Construct the strongest chimeric promoter/RBS combination possible, then (ii) fine-tune the level of regulator with a wide range of expression levels. (iii) If a satisfactory dynamic range cannot be met after (ii), weaken the RBS driving signal output. The Preg and RBSout libraries we developed for optimizing the PAB were successfully applied to improve the FAB demonstrating the reusability of these parts in a different genetic context and abrogating the need for library construction/screening each time a biosensor is optimized. If library development is required, say for the application of DoE in a different context, we would advocate the use of empirically validated promoters/RBSs or validation of promoters/RBSs designed using predictive tools,41−43 to allow for greater confidence in the resulting data and model.
The PCA and FA whole cell biosensors we developed can detect key aromatic chemicals in the lignin biomass valorization, permitting applications for the renewable production of high value chemicals, materials and fuels from biomass.25,26 These systems can be employed for the high-throughput screening and of new enzymes,27,44 dynamic-regulation of metabolic pathways for production of target chemicals,45,46 adaptive evolution of new phenotypes,47 and the integration of regulated individual components in a whole cell bioprocess context.48 Furthermore, as demonstrated for PCA, the high performance comparable to traditional inducible systems would allow broader synthetic biology applications such as regulation of complex networks and cellular computation.49,50
By applying statistical modeling we are able to optimize biosensor performance without needing to carry out modification to the repressor protein or understanding of binding affinities of the DNA and ligand binding domains, making this objective approach ideally suited to building regulatory networks from uncharacterized genetic parts. By systematically sampling the expression space of these multipart genetic systems we can use statistical modeling approaches, which do not rely on detailed mechanistic and/or kinetic knowledge, to guide rapid iteration and performance optimization. These models also allow us to identify nonlinear effects and trade-offs, which aid selection of highly functional regulatory networks and pathways, enabling robust, data-led decision making. The DSD framework developed here shows great potential for the optimization of genetic systems by tuning component expression level in a systematic and highly efficient way.
Methods
Materials
Escherichia coli DH5α (NEB, #C2987U) was used for cloning, in vivo DNA assembly,51−54 plasmid propagation, promoter/RBS characterization and biosensor assays. For benchmarking studies, E. coli BL21 (NEB, #C2530H) was used with different non-T7 expression systems and E. coli BL21 (DE3) (NEB, #C2527H) with a genomic copy of T7 RNAP was used for the T7 RNAP-based expression systems. E. coli BW25113 was used as a host for the redesigned ferulic acid-responsive biosensor. E. coli strains were grown Luria–Bertani (LB) media for all experiments except for the promoter/RBS library characterization where EZ rich (EZ rich defined medium kit, Teknova, #M2105) was used. Unless noted, LB and EZ rich were supplemented with ampicillin (100 μg/mL), kanamycin (50 μg/mL for plasmid selection and 25 μg/mL for genome integration), or hygromycin (100 μg/mL). Water was from a Milli-Q filtration system (Millipore). Protocatechuic acid, isopropyl β-d-1-thiogalactopyranoside (IPTG), l-arabinose, and l-mannose stock solutions were prepared in sterile water and a ferulic acid stock solution was prepared in dimethyl sulfoxide (DMSO). Chemicals and antibiotics were purchased from Sigma, Fisher, or Formedium. DNA oligos and synthetic genes were purchased from IDT and/or GeneArt.
Molecular Cloning
Primer sequences and a list of plasmids can be found in Supplementary Table S12 and S13, respectively. Restriction enzymes were purchased from NEB and digestions were carried out according to standard protocols. Q5 polymerase (NEB, #M0491S) was used to produce DNA fragments for cloning purposes and Phire II (Thermo Fisher, #F126S) was used for genotyping of genomic insertions. Isothermal assembly55 was performed using NEBuilder (NEB, #E2621S). PCR-generated fragments were treated with DpnI (NEB). All constructs were Sanger sequenced to verify sequence identity. pSEVA131,56,57 containing the BBR1 origin and ampicillin selection marker, was used for the PCA biosensor. pSEVA 261 containing the p15A origin and kanamycin selection marker was used for the inverter system. pET28a (Novagen) containing the pBR322 origin and kanamycin selection marker was used for the ferulic acid biosensor. Full details for the molecular cloning can be found in the Supporting Information.
Promoter/RBS Library Screening
Clones from the Preg-lib, Pout-lib and RBSout-lib libraries were selected and characterized in two rounds. For the first round, 960 individual clones from each library were picked from transformation plates and arrayed into square-welled 96 deep-well plate (DWP) fitted with breathable seals, containing media (0.5 mL LB plus ampicillin) using a Hamilton Star robotic platform. The plates were grown for 16 h at 30 °C at 950 rpm, 75% humidity in a shaker-incubator (Infors HT). The next day, 2 μL of the cultures were subcultured into 198 μL of EZ rich media plus ampicillin in black, clear flat-bottomed 96-well microtiter plates (MTP; Grenier) and were incubated for 3 h at 37 °C at 1000 rpm in microtiter plate shaker (Stuart). Fluorescence and optical density (OD λ = 700 nm) were measured in a ClarioStar microplate reader (BMG) to obtain an end-point measurement. GFP fluorescence was measured at λEx/λEm = 488/520 nm and mCherry fluorescence was measured at λEx/λEm = 570/620 nm. OD700 was measured instead of OD600 to avoid bleed-through from mCherry fluorescence.58 Fluorescence was normalized to optical density and the normalized value was used to select 22 clones from each library that spanned a wide range of RFU/OD. For all RFU/OD measurements, the background signal for autofluorescence was corrected for by subtracting the RFU/OD value of the empty vector negative control.
For the second round, selected clones from each library were streaked onto ampicillin plates and grown overnight. Individual colonies were arrayed in triplicate in DWPs containing 0.5 mL LB plus ampicillin (with breathable seals) and grown for 16 h at 30 °C at 950 rpm, 75% humidity in a shaker-incubator (Infors HT). The overnight cultures were used to make “one-shot” stock solutions for cryopreservation by transferring 75ul of culture to a black microtiter plate containing 50 μL of 50% glycerol. These plates were mixed briefly in a MTP shaker (1000 rpm, 1 min) then stored at −80 °C. To determine promoter activity the cryopreserved MTPs were thawed in a MTP shaker (37 °C, 1000 rpm) for 30 min then 5 μL of each well was inoculated into a DWP containing 495 μL of LB plus ampicillin. The DWP plates were grown for 16 h at 30 °C at 950 rpm, 75% humidity in a shaker incubator. The overnight precultures were used to make the main culture by transferring 4 μL of cells into a black, clear flat-bottomed 96-well MTP containing 196 μL of EZ rich plus ampicillin and incubated in a MTP shaker at 1000 rpm at 37 °C. OD700 and FP (fluorescent protein) fluorescence was read at 2 and 3 h, which was previously determined to the period of maximum growth rate for E. coli in our experimental conditions. Promoter activity was calculated according to the literature29,30 (see eq 1). Promoter activity was transformed into a logarithmic dimensionless variable according to the literature19 (see eq 2).
Genomic Integrations
Genomic cassettes were inserted into the chromosome of E. coli DH5α using lambda red recombineering.59 Selected PAB variants were transferred to the pKIKOarsBKM integration vector,60 and then the integration cassette was amplified with primers AB 39/40 and cleaned up with a Qiagen PCR purification column. E. coli DH5α was transformed with the pSIM18 vector, grown to an OD600 of ∼0.3 and heat-shocked at 42 °C for 15 min to induce expression of the λ Red recombinase proteins. The cells were washed 5 times in ice-cold sterile water then electroporated with 300 ng of PCR product and the transformants selected on LB plates supplemented with kanamycin at 37 °C. Confirmation of cassette insertion at the correct locus was confirmed by colony PCR with primers AB 34/61. To cure the strains of pSIM18, clones were subcultured overnight on LB plus kanamycin at 42 °C and restreaked onto LB plates containing kanamycin (growth) or hygromycin (no growth) to confirm loss of pSIM18.
DoE Trials
E. coli DH5α strains bearing the plasmid or chromosome-based PAB variants were streaked on LB plus ampicillin or LB plus kanamycin plates, respectively. Individual clones from each strain were used to make “one-shot” stock solutions for cryopreservation as described above. For DoE trials the cryopreserved MTPs were thawed in a MTP shaker (37 °C, 1000 rpm) for 30 min then 10 μL of each well was inoculated into a DWP containing 190 μL of LB plus ampicillin (for plasmid-based variants) or LB only (for chromosomally integrated variants). The DWP plates were grown for 16 h at 30 °C at 950 rpm, 75% humidity in a shaker incubator to make precultures, which were subsequently used to make the main culture by transferring 5 μL of cells into a DWP containing 445 μL of LB plus ampicillin. DWPs were incubated in a MTP shaker at 1000 rpm at 37 °C for 2 h. The clones were then induced by adding 50 μL of PCA (10 mM) for a final concentration of 1 mM PCA and grown for another 3 h. The DWPs were centrifuged at 2250g for 10 min to pellet the cells and the spent medium was replaced with 500 μL PBS and mixed by pipetting. The cells were pelleted and washed again, 50 μL of cell suspension was transferred to a flat, clear-bottomed black MTP containing 150 μL of PBS, and GFP fluorescence and OD700 were measured.
Biosensor Titrations and Expression System Benchmarking
Strains containing inducible expression systems were streaked onto LB plates supplemented with ampicillin and individual clones from each strain were used to inoculate 5 mL of LB plus ampicillin in a 50 mL conical tube, which was grown for 16 h at 37 °C at 180 rpm in a shaking incubator (New Brunswick I26). The overnight cultures were diluted 1/100 into LB plus ampicillin in a DWP, incubated in a MTP shaker (Stuart) at 1000 rpm at 37 °C for 2 h then induced by adding the appropriate inducer and grown for a further 3 h. The final inducer concentrations for the titration were as follows: 4000, 1000, 250, 62.5, 15.6, 3.9 μM, and no inducer. The FA Biosensor titration was carried out in the same way, expect that concentrations were 1000, 200, 40, 8, 1.6, 0.32 μM and no inducer. The cells were pelleted, washed, and measured for OD700 and fluorescence as described above.
Dose Response Extender
E. coli DH5α bearing the p131C–B10 biosensor and p261-lacI-pcaK variants were plated onto solid LB medium containing ampicillin and kanamycin (25 μg/mL and 12.5 μg/mL, respectively) and 1 mM PCA. Single isolated colonies were inoculated into 5 mL of LB supplemented with the required antibiotics, in a 50 mL conical tube and incubated at 37 °C for 16 h shaking at 180 rpm. Cells were then diluted 100-fold in fresh LB media containing antibiotics and transferred to a 96 well DWP then incubated at 37 °C in a MTP shaker at 1000 rpm for 2 h. Following this outgrowth period the appropriate concentration of inducer was added bringing the final culture volume to 500 μL. Cells were incubated as before for a further 24 h. An extended induction time was required as the two plasmid inverter system and expression of the system components had significant deleterious effect on the growth. Final concentrations of inducer were as follows: 1000, 200, 40, 8, 1.6, 0.32, 0.064, 0.0128, and 0 μM PCA.
Data Processing and Modeling
All data processing and statistical analysis was carried out in JMP Pro 12 (SAS Institute Inc.), including design of experiments, factor screening, and standard least-squares regression. Response data were transformed to log10. The DSD data table was constructed using the DoE definitive screening function and factors were selected based on the Lenth’s t-ratio and Half-Normal plot analysis of the factor contrast and Lenth’s pseudo standard error (PSE). Factor contrasts which deviated from the half-normal distribution were deemed important for the model and so were included in SLSR fitting. Factor significance was assessed by analysis of simultaneous p-values, allowing the assessment of factor importance in the model. Effect heredity was maintained and so if a factor was not deemed significant individually but was included in a significant interaction term with another factor then both terms contained in this interaction were included in model fitting. Simultaneous p-values were generated using the PSE, which is derived from an estimation of the residual standard error using inactive terms within the model. From this PSE a 10 000 run Monte Carlo simulation was carried out to allow estimation of the p-value. Graphs were generated in PRISM 7 (GraphPad Software) and fit using a Hill fit.
Acknowledgments
A.B. was supported a by the BBSRC grant [BB/P01738X/1], R.K. was supported by the BBSRC DTP grant [BB/M011208/1]. L.F.M.M. is supported via Science without Borders/Ciências sem Fronteiras Scheme from the CNPq, Brazil (233608/2014-1) and the Presidential Doctoral Scheme at The University of Manchester. N.D. is supported by the BBSRC David Phillips Fellowship (BB/K014773/1) and BBSRC-FAPESP (FAPPA) grant (BB/L026244/1). SYNBIOCHEM, University of Manchester, is supported by the BBSRC grant “Centre for Synthetic Biology of Fine and Specialty Chemicals” [BB/M017702/1]. We thank A. Jervis and M. Dunstan (SYNBIOCHEM, University of Manchester) for help with the colony picking robot and for providing pKIKO vectors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.9b00448.
Additional Methods, Supplementary Figures S1–S4, Supplementary Tables S1–S13, Supplementary References (PDF)
Author Present Address
§ Future Biomanufacturing Research Hub, Manchester Institute of Biotechnology (MIB), The University of Manchester, Manchester M1 7DN, U.K.
Author Contributions
A.B. conceived the project. A.B., R.K., and L.F.M.M. designed and carried out all the experiments. A.B., R.K., L.F.M.M., and N.D. wrote the manuscript. All the authors read and approved the final manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Ang J.; Harris E.; Hussey B. J.; Kil R.; McMillen D. R. (2013) Tuning response curves for synthetic biology. ACS Synth. Biol. 2, 547–567. 10.1021/sb4000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe B.; Peters G.; Coussement P.; Maertens J.; De Mey M. (2017) Tailor-made transcriptional biosensors for optimizing microbial cell factories. J. Ind. Microbiol. Biotechnol. 44, 623–645. 10.1007/s10295-016-1862-3. [DOI] [PubMed] [Google Scholar]
- Lim H. G.; Jang S.; Jang S.; Seo S. W.; Jung G. Y. (2018) Design and optimization of genetically encoded biosensors for high-throughput screening of chemicals. Curr. Opin. Biotechnol. 54, 18–25. 10.1016/j.copbio.2018.01.011. [DOI] [PubMed] [Google Scholar]
- Shi S.; Ang E. L.; Zhao H. (2018) In vivo biosensors: mechanisms, development, and applications. J. Ind. Microbiol. Biotechnol. 45, 491–516. 10.1007/s10295-018-2004-x. [DOI] [PubMed] [Google Scholar]
- Liu D.; Evans T.; Zhang F. (2015) Applications and advances of metabolite biosensors for metabolic engineering. Metab. Eng. 31, 35–43. 10.1016/j.ymben.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Koch M.; Pandi A.; Borkowski O.; Cardoso Batista A.; Faulon J. L. (2019) Custom-made transcriptional biosensors for metabolic engineering. Curr. Opin. Biotechnol. 59, 78–84. 10.1016/j.copbio.2019.02.016. [DOI] [PubMed] [Google Scholar]
- Mahr R.; Frunzke J. (2016) Transcription factor-based biosensors in biotechnology: current state and future prospects. Appl. Microbiol. Biotechnol. 100, 79–90. 10.1007/s00253-015-7090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R.; Bujard H. (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210. 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti-Lafranconi P.; Dijkman W. P.; Devenish S. R.; Hollfelder F. (2013) A single mutation in the core domain of the lac repressor reduces leakiness. Microb. Cell Fact. 12, 67. 10.1186/1475-2859-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L.; Buchler N. E.; Garcia H. G.; Gerland U.; Hwa T.; Kondev J.; Phillips R. (2005) Transcriptional regulation by the numbers: models. Curr. Opin. Genet. Dev. 15, 116–124. 10.1016/j.gde.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro Y.; Kimura Y.; Furubayashi M.; Tanaka A.; Terakubo K.; Saito K.; Kawai-Noma S.; Umeno D. (2016) Directed evolution of the autoinducer selectivity of Vibrio fischeri LuxR. J. Gen. Appl. Microbiol. 62, 240–247. 10.2323/jgam.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Meyer A. J.; Segall-Shapiro T. H.; Glassey E.; Zhang J.; Voigt C. A. (2019) Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 15, 196–204. 10.1038/s41589-018-0168-3. [DOI] [PubMed] [Google Scholar]
- Ellefson J. W.; Ledbetter M. P.; Ellington A. D. (2018) Directed evolution of a synthetic phylogeny of programmable Trp repressors. Nat. Chem. Biol. 14, 361–367. 10.1038/s41589-018-0006-7. [DOI] [PubMed] [Google Scholar]
- Wang B.; Barahona M.; Buck M. (2015) Amplification of small molecule-inducible gene expression via tuning of intracellular receptor densities. Nucleic Acids Res. 43, 1955–1964. 10.1093/nar/gku1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandenius C. F.; Brundin A. (2008) Bioprocess optimization using Design-of-Experiments methodology. Biotechnol. Prog. 24, 1191–1203. 10.1002/btpr.67. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Bhalla A.; Rathore A. S. (2014) Design of Experiments applications in bioprocessing: Concepts and approach. Biotechnol. Prog. 30, 86–99. 10.1002/btpr.1821. [DOI] [PubMed] [Google Scholar]
- Franceschini G.; Macchietto S. (2008) Model-based design of experiments for parameter precision: State of the art. Chem. Eng. Sci. 63, 4846–4872. 10.1016/j.ces.2007.11.034. [DOI] [Google Scholar]
- Lendrem D. W.; Lendrem B. C.; Woods D.; Rowland-Jones R.; Burke M.; Chatfield M.; Isaacs J. D.; Owen M. R. (2015) Lost in space: design of experiments and scientific exploration in a Hogarth Universe. Drug Discovery Today 20, 1365–1371. 10.1016/j.drudis.2015.09.015. [DOI] [PubMed] [Google Scholar]
- Xu P.; Rizzoni E. A.; Sul S. Y.; Stephanopoulos G. (2017) Improving Metabolic Pathway Efficiency by Statistical Model-Based Multivariate Regulatory Metabolic Engineering. ACS Synth. Biol. 6, 148–158. 10.1021/acssynbio.6b00187. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Vonk B.; Roubos J. A.; Bovenberg R. A.; Voigt C. A. (2015) Algorithmic co-optimization of genetic constructs and growth conditions: application to 6-ACA, a potential nylon-6 precursor. Nucleic Acids Res. 43, 10560–10570. 10.1093/nar/gkv1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell P.; Jervis A. J.; Robinson C. J.; Yan C.; Dunstan M.; Swainston N.; Vinaixa M.; Hollywood K. A.; Currin A.; Rattray N. J. W.; Taylor S.; Spiess R.; Sung R.; Williams A. R.; Fellows D.; Stanford N. J.; Mulherin P.; Le Feuvre R.; Barran P.; Goodacre R.; Turner N. J.; Goble C.; Chen G. G.; Kell D. B.; Micklefield J.; Breitling R.; Takano E.; Faulon J. L.; Scrutton N. S. (2018) An automated Design-Build-Test-Learn pipeline for enhanced microbial production of fine chemicals. Commun. Biol. 1, 66. 10.1038/s42003-018-0076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. E.; Aswani A.; Han A. S.; Tomlin C. J.; Dueber J. E. (2013) Expression-level optimization of a multi-enzyme pathway in the absence of a high-throughput assay. Nucleic Acids Res. 41, 10668–10678. 10.1093/nar/gkt809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B.; Nachtsheim C. J. (2011) A Class of Three-Level Designs for Definitive Screening in the Presence of Second-Order Effects. J. Qual Technol. 43, 1–15. 10.1080/00224065.2011.11917841. [DOI] [Google Scholar]
- Machado L. F. M.; Currin A.; Dixon N. (2019) Directed evolution of the PcaV allosteric transcription factor to generate a biosensor for aromatic aldehydes. J. Biol. Eng. 13, 91. 10.1186/s13036-019-0214-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Lei P.; Zhai R.; Wen Z.; Jin M. (2019) Recent advances in lignin valorization with bacterial cultures: microorganisms, metabolic pathways, and bio-products. Biotechnol. Biofuels 12, 32. 10.1186/s13068-019-1376-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham G. T.; Johnson C. W.; Karp E. M.; Salvachua D.; Vardon D. R. (2016) Opportunities and challenges in biological lignin valorization. Curr. Opin. Biotechnol. 42, 40–53. 10.1016/j.copbio.2016.02.030. [DOI] [PubMed] [Google Scholar]
- Machado L. F.; Dixon N. (2016) Development and substrate specificity screening of an in vivo biosensor for the detection of biomass derived aromatic chemical building blocks. Chem. Commun. (Cambridge, U. K.) 52, 11402–11405. 10.1039/C6CC04559F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra R.; Shankar J.; Robinson C. J.; Halliwell S.; Butler L.; Upton M.; Hay S.; Micklefield J.; Dixon N. (2016) Dual transcriptional-translational cascade permits cellular level tuneable expression control. Nucleic Acids Res. 44, e21 10.1093/nar/gkv912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. H.; Rubin A. J.; Sauer R. T. (2011) Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39, 1131–1141. 10.1093/nar/gkq810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. R.; Rubin A. J.; Davis J. H.; Ajo-Franklin C. M.; Cumbers J.; Czar M. J.; de Mora K.; Glieberman A. L.; Monie D. D.; Endy D. (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzer M.; Bujard H. (1988) Promoters largely determine the efficiency of repressor action. Proc. Natl. Acad. Sci. U. S. A. 85, 8973–8977. 10.1073/pnas.85.23.8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach M. E.; Elzer P. H.; Hill D. S.; Robertson G. T.; Farris M. A.; Roop R. M. 2nd; Peterson K. M. (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176. 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- Jahn M.; Vorpahl C.; Hubschmann T.; Harms H.; Muller S. (2016) Copy number variability of expression plasmids determined by cell sorting and droplet digital PCR. Microb. Cell Fact. 15, 211. 10.1186/s12934-016-0610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier T.; Jones K. L.; Keasling J. D. (1998) mRNA stability and plasmid copy number effects on gene expression from an inducible promoter system. Biotechnol. Bioeng. 59, 666–672. 10.1002/(SICI)1097-0290(19980920)59:6<666::AID-BIT2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Kittleson J. T.; Cheung S.; Anderson J. C. (2011) Rapid optimization of gene dosage in E. coli using DIAL strains. J. Biol. Eng. 5, 10. 10.1186/1754-1611-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileyko Y.; Joh R. I.; Weitz J. S. (2008) Small-scale copy number variation and large-scale changes in gene expression. Proc. Natl. Acad. Sci. U. S. A. 105, 16659–16664. 10.1073/pnas.0806239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W.; Gyorgy A.; Cameron D. E.; Pyenson N.; Choi K. R.; Way J. C.; Silver P. A.; Del Vecchio D.; Collins J. J. (2016) Creating single-copy genetic circuits. Mol. Cell 63, 329–336. 10.1016/j.molcel.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C. L.; Liu Z.; Yoshihara A.; Jenkinson S. F.; Wormald M. R.; Otero J.; Estevez A.; Kato A.; Marqvorsen M. H.; Fleet G. W.; Estevez R. J.; Izumori K.; Heap J. T. (2016) Synthetic chemical inducers and genetic decoupling enable orthogonal control of the rhaBAD promoter. ACS Synth. Biol. 5, 1136–1145. 10.1021/acssynbio.6b00030. [DOI] [PubMed] [Google Scholar]
- Terpe K. (2006) Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 72, 211–222. 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- Balzer S.; Kucharova V.; Megerle J.; Lale R.; Brautaset T.; Valla S. (2013) A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microb. Cell Fact. 12, 26. 10.1186/1475-2859-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H. M.; Mirsky E. A.; Voigt C. A. (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharanikumar R.; Premkumar K. A. R.; Palaniappan A. (2018) Promoter Predict: sequence-based modelling of Escherichia coli sigma(70) promoter strength yields logarithmic dependence between promoter strength and sequence. PeerJ 6, e5862 10.7717/peerj.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jervis A. J.; Carbonell P.; Taylor S.; Sung R.; Dunstan M. S.; Robinson C. J.; Breitling R.; Takano E.; Scrutton N. S. (2019) SelProm: A Queryable and Predictive Expression Vector Selection Tool for Escherichia coli. ACS Synth. Biol. 8, 1478–1483. 10.1021/acssynbio.8b00399. [DOI] [PubMed] [Google Scholar]
- Ho J. C. H.; Pawar S. V.; Hallam S. J.; Yadav V. G. (2018) An improved whole-cell biosensor for the discovery of lignin-transforming enzymes in functional metagenomic screens. ACS Synth. Biol. 7, 392–398. 10.1021/acssynbio.7b00412. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Carothers J. M.; Keasling J. D. (2012) Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 30, 354–359. 10.1038/nbt.2149. [DOI] [PubMed] [Google Scholar]
- Lo T. M.; Chng S. H.; Teo W. S.; Cho H. S.; Chang M. W. (2016) A two-layer gene circuit for decoupling cell growth from metabolite production. Cell Syst 3, 133–143. 10.1016/j.cels.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Williams T. C.; Pretorius I. S.; Paulsen I. T. (2016) Synthetic evolution of metabolic productivity using biosensors. Trends Biotechnol. 34, 371–381. 10.1016/j.tibtech.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Alvarez-Gonzalez G.; Dixon N. (2019) Genetically encoded biosensors for lignocellulose valorization. Biotechnol. Biofuels 12, 246. 10.1186/s13068-019-1585-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Dalchau N.; Wang B. (2018) Scaling up genetic circuit design for cellular computing: advances and prospects. Nat. Comput. 17, 833–853. 10.1007/s11047-018-9715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltepe B.; Kehribar E. S.; Su Yirmibesoglu S. S.; Safak Seker U. O. (2018) Cellular biosensors with engineered genetic circuits. ACS Sens 3, 13–26. 10.1021/acssensors.7b00728. [DOI] [PubMed] [Google Scholar]
- Beyer H. M.; Gonschorek P.; Samodelov S. L.; Meier M.; Weber W.; Zurbriggen M. D. (2015) AQUA cloning: A versatile and simple enzyme-free cloning approach. PLoS One 10, e0137652 10.1371/journal.pone.0137652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nafria J.; Watson J. F.; Greger I. H. (2016) IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly. Sci. Rep. 10.1038/srep27459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobus A. P.; Gross J. (2015) Optimal cloning of PCR fragments by homologous recombination in Escherichia coli. PLoS One 10, e0119221. 10.1371/journal.pone.0119221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev M.; Otwell A. E.; Richardson R. E.; Suzuki Y. (2015) Cloning should be simple: Escherichia coli DH5 alpha-mediated assembly of multiple DNA fragments with short end homologies. PLoS One 10, e0137466. 10.1371/journal.pone.0137466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia E.; Aparicio T.; Goni-Moreno A.; Fraile S.; de Lorenzo V. (2015) SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 43, D1183–1189. 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Rocha R.; Martinez-Garcia E.; Calles B.; Chavarria M.; Arce-Rodriguez A.; de Las Heras A.; Paez-Espino A. D.; Durante-Rodriguez G.; Kim J.; Nikel P. I.; Platero R.; de Lorenzo V. (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41, D666–D675. 10.1093/nar/gks1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A.; Endy D.; Salit M.; Munson M. S. (2016) When wavelengths collide: bias in cell abundance measurements due to expressed fluorescent proteins. ACS Synth. Biol. 5, 1024–1027. 10.1021/acssynbio.6b00072. [DOI] [PubMed] [Google Scholar]
- Datsenko K. A.; Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri S.; Steen J. A.; Bongers M.; Nielsen L. K.; Vickers C. E. (2013) Knock-in/Knock-out (KIKO) vectors for rapid integration of large DNA sequences, including whole metabolic pathways, onto the Escherichia coli chromosome at well-characterised loci. Microb. Cell Fact. 12, 60. 10.1186/1475-2859-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.