

Abstract
Carnitine plays an essential role in mitochondrial fatty acid β-oxidation as a part of a cycle that transfers long-chain fatty acids across the mitochondrial membrane and involves two carnitine palmitoyltransferases (CPT1 and CPT2). Two distinct carnitine acyltransferases, carnitine octanoyltransferase (COT) and carnitine acetyltransferase (CAT), are peroxisomal enzymes, which indicates that carnitine is not only important for mitochondrial, but also for peroxisomal metabolism. It has been demonstrated that after peroxisomal metabolism, specific intermediates can be exported as acylcarnitines for subsequent and final mitochondrial metabolism. There is also evidence that peroxisomes are able to degrade fatty acids that are typically handled by mitochondria possibly after transport as acylcarnitines. Here we review the biochemistry and physiological functions of metabolite exchange between peroxisomes and mitochondria with a special focus on acylcarnitines.
Keywords: peroxisome, mitochondria, carnitine, metabolite transport, fatty acid β-oxidation
Introduction
Carnitine is a substrate for the carnitine acyltransferases, which catalyze the transesterification of an acyl-CoA to an acylcarnitine with free carnitine and free CoA as the respective co-substrate and co-product. Three types of carnitine acyltransferases are defined based on their substrate specificity; carnitine palmitoyltransferase (CPT), carnitine octanoyltransferase (COT), and carnitine acetyltransferase (CAT). Carnitine is most well-known for its essential function in mitochondrial long-chain fatty acid β-oxidation (FAO) in which it enables the translocation of fatty acid intermediates from the cytosol to the mitochondria via the carnitine cycle [1]. CPT1 and CPT2 function in the carnitine cycle. CPT1 is an integral outer mitochondrial membrane protein that converts a long-chain acyl-CoA into an acylcarnitine. The acylcarnitine is subsequently transported into the mitochondria via a specific translocase (carnitine-acylcarnitine translocase, CACT, SLC25A20). CPT2, a peripheral inner mitochondrial membrane protein, then reconverts the acylcarnitine into an acyl-CoA, which is the substrate for mitochondrial long-chain FAO (Figure 1). The acyltransferase reaction is reversible, and therefore the net flux through the enzyme will depend on the concentration of substrates and products. The role of the carnitine cycle in mitochondrial long-chain FAO has been reviewed elsewhere [2–4]. Other less well known roles of carnitine include the detoxification of a wide variety of acyl-CoA esters that may accumulate in specific inborn errors of metabolism [5–7], mediating the mobility of acyl-CoA esters between cells, tissues and body fluids [8], and mediating the mobility of acyl-CoA esters between peroxisomes and mitochondria. The latter role is the topic of this review.
Figure 1.
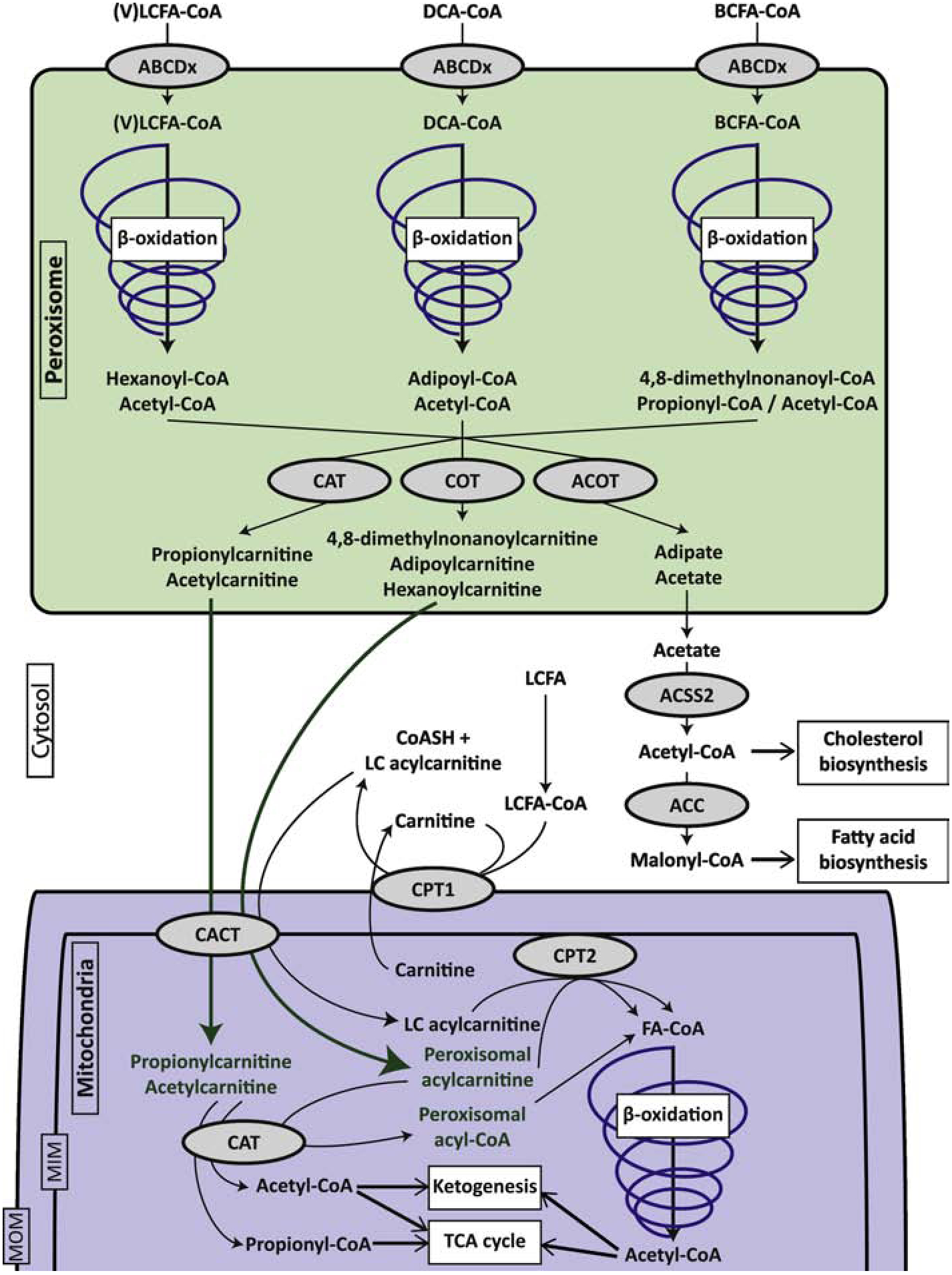
Schematic representation of acylcarnitine metabolism and transport from peroxisomes to mitochondria. The figure is focused on the substrates and products of peroxisomal fatty acid β-oxidation, but only the most abundant end-products are indicated. Details of the pathways and the individual enzyme steps are omitted. ABCD transporters are half-transporters and function as homodimers or heterodimers. For the simplicity of the figure, they have been represented as monomers. Abbreviations: ABCDx, members of the peroxisomal ABC transporter family; ACC, acetyl-CoA carboxylase; ACSS, short-chain acyl-CoA synthetase; ACOT, acyl-CoA thioesterases; BCFA, branched-chain fatty acid; CACT, carnitine acylcarnitine translocase (SLC25A20); CPT1, carnitine palmitoyltransferase 1; CPT2; carnitine palmitoyltransferase 2; DCA, dicarboxylic acid; LCFA, long-chain fatty acid; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; VLCFA, very long-chain fatty acid.
The peroxisomal carnitine acyltransferases
Both COT and CAT are peroxisomal enzymes. This finding that carnitine acyltransferases localize to the peroxisome basically established that carnitine is not only important for mitochondrial metabolism, but also for peroxisomal metabolism [9]. The CAT protein, but not COT, localizes also to the mitochondria, because the gene can encode a protein with a mitochondrial targeting sequence and/or peroxisomal targeting sequence 1. Alternative splicing in the 5’ region leads to mRNAs with different start codons, which likely affects subcellular localization [10–12]. A cytosolic localization in heart was recently suggested as well [13]. Human CRAT is ubiquitously expressed, but higher levels are observed in liver, skeletal muscle and testis [14]. Subcellular expression levels of CAT have not been systematically studied. CAT accepts various short-chain acyl-CoAs as substrate including acetyl-CoA, propionyl-CoA and butyryl-CoA [7]. CROT is also ubiquitously expressed with highest levels in thyroid [14]. COT has a peroxisomal targeting sequence 1 [15] and localizes exclusively to the peroxisome [16]. When compared to CAT, COT prefers acyl-CoAs or acylcarnitines with carbon chain lengths of 6, 8 and 10 atoms [16–18]. COT has also been demonstrated to accept 4,8-dimethylnonanoyl-CoA, a product of the peroxisomal oxidation of branched-chain fatty acids (BCFAs) [19]. More detailed studies on the substrate specificity of COT focusing on peroxisomal FAO intermediates such as dicarboxylyl-CoAs have not been reported yet.
The role of peroxisomal carnitine acyltransferases
Whereas mitochondria are thought to handle the bulk of dietary fatty acids such as palmitate and oleic acid, peroxisomes accept a more diverse spectrum of carboxylic acids including very long-chain fatty acids (VLCFAs), dicarboxylic acids (DCAs), BCFAs and bile acids (Figure 1). In order to be able to degrade these substrates, peroxisomes have two sets of FAO enzymes, a topic reviewed elsewhere [20, 21]. In contrast to mitochondrial FAO, peroxisomal FAO does not completely degrade most substrates and based on substrate specificities of isolated peroxisomes and the individual enzymes, medium-chain products with 6 carbons are thought to be the final products [22, 23]. In addition, peroxisomes do not have metabolic pathways to use acetyl-CoA, one of the major end products of FAO. Therefore, peroxisomes do not only need mechanisms to import the substrates of FAO, but also export its products [24]. Acylcarnitines theoretically could mediate both processes. However, given that CAT and COT handle short- and medium-chain acyl-CoAs, it is most likely that acylcarnitines are mainly used to export the FAO intermediates out of the peroxisome. Although there is some evidence that peroxisomes can also accept acylcarnitines as substrates for import [25], the current consensus is that peroxisomes import acyl-CoA esters. This process is mediated by peroxisomal ABC transporters (ABCD1, 2 and 3 in humans) that have intrinsic thioesterase activity leaving a free acid in the peroxisome that must be reactivated before it can undergo peroxisomal FAO [26, 27]. Other studies suggest that in human cells, VLCFA-CoA esters are transported into peroxisomes by ABCD1 independently of additional synthetase activity [28]. Specific peroxisomal transporters for acylcarnitines have not been described, and it is thought that these metabolites cross the peroxisomal membrane through unspecific channels such as PXMP2 [29].
Acylcarnitines are not the only possible products of peroxisomal FAO. Alternatively, acyl-CoA products can undergo enzymatic hydrolysis by acyl-CoA thioesterases (ACOTs) that generate a free carboxylate that can cross the peroxisomal membrane (Figure 1). It has been hypothesized that the carnitine acyltransferases and ACOTs provide complementary systems for transport of metabolites across the peroxisomal membrane [11]. This was based on the observation that in mouse tissues, the expression patterns of these genes was very different [11]. The tissue expression pattern of the human peroxisomal ACOTs (ACOT4 and ACOT8), however, is relatively ubiquitous and as such comparable to CRAT and CROT [14]. ACOT4 expression is somewhat higher in liver and kidney of both humans and mice [14, 30], but this is a commonly observed expression pattern for many peroxisomal enzymes. Expression of CAT, COT and ACOT4 is upregulated by hypolipidemic drugs such as Wy-14,643, an agonist of peroxisome proliferator-activated receptor (PPAR) α [16, 30]. Therefore, as based on tissue expression patterns, there is currently no clear evidence that the role of peroxisomal carnitine acyltransferases diverges between tissues. The availability of peroxisomal substrates, however, is expected to differ between tissues, most notably the generation of DCAs, which is restricted to liver and kidney.
The metabolic fate of peroxisomal acylcarnitines and free carboxylates is likely different. Whereas further acylcarnitine metabolism is expected to be restricted to mitochondria, carboxylates, in particular acetate, can also be metabolized within the cytosol. In the cytosol, acetate can be reconverted into acetyl-CoA by acetyl-CoA synthetase (ACSS2). The expression of this enzyme is regulated by Sterol Regulatory Element-Binding Proteins (SREBPs), consistent with a role in lipid biosynthesis. Importantly, it is known that the acetyl-CoA formed by peroxisomal FAO can enter the cytosolic pool of acetyl-CoA and is used for cholesterol biosynthesis in rat hepatocytes [31] and malonyl-CoA synthesis in rat heart [32]. The tissue-specific fates of peroxisomal FAO products and the regulation of their production is clearly a topic that deserves more investigation.
Transfer of acylcarnitines from peroxisomes to mitochondria
The best studied example of metabolic crosstalk between peroxisomes and mitochondria is the oxidation of the BCFAs phytanic and pristanic acid [33–35] (Figure 1). Using fibroblasts from patients with defects in the carnitine cycle, Jakobs and Wanders first showed that propionyl-CoA, a product peroxisomal FAO of pristanic acid, is shuttled from the peroxisome to the mitochondria for complete oxidation [33]. Verhoeven et al used control and patient fibroblasts with defects in different peroxisomal proteins and enzymes or the carnitine cycle, and analyzed acylcarnitines after loading with phytanic and pristanic acid [34, 35]. They demonstrated that phytanic acid first undergoes peroxisomal α-oxidation followed by 3 cycles of peroxisomal FAO [34]. The resulting product 4,8-dimethylnonanoyl-CoA is transferred to the mitochondria as a carnitine ester (C11:0) using the carnitine shuttle and then undergoes at least one additional FAO cycle to yield 2,6-dimethylheptanoyl-CoA as one of the final mitochondrial products that is exported as a carnitine ester (C9:0) [35]. 2,6-Dimethylheptanoyl-CoA is a specific substrate for the mitochondrial long-chain acyl-CoA dehydrogenase (LCAD) [36]. The relatively poor metabolism of 2,6-dimethylheptanoyl-CoA in human fibroblasts is likely related to the poor expression of LCAD in most human cells and tissues [37].
A similar transfer of substrates must occur in the metabolism of other peroxisomal substrates. VLCFA oxidation yields multiple acetyl-CoAs as well as a medium-chain acyl-CoA, all of which require further oxidation in mitochondria or cytosolic metabolism (Figure 1). There is good evidence that peroxisomes are crucial for the metabolism of DCAs, as mitochondria are unable to handle these substrates [38–45]. The oxidation of DCAs yields acetyl-CoA units, but also a dicarboxylyl-CoA that can have different fates depending on its carbon chain length (Figure 1). Suberyl-CoA and adipoyl-CoA can be converted into suberic and adipic acid, respectively, which may be considered dead end metabolites, because they are excreted in urine. Low levels of carnitine esters of these metabolites are detected in plasma of which the physiological significance is unknown. Peroxisomal FAO of DCAs can also yield succinyl-CoA [46, 47]. Peroxisomal succinyl-CoA can be shuttled to the mitochondria as succinate or succinylcarnitine, and then serve as an anaplerotic substrate in the TCA cycle [30, 48]. Many aspects of the transfer of metabolites from peroxisomes to mitochondria have not been fully elucidated. This is likely due to the inherent difficulties of studying the origin and fate of intracellular metabolites. Studies with stable isotopes in genetically modified model organisms, as well as patient-derived cell lines, are therefore expected to yield novel insights.
Transfer of mitochondrial metabolites to peroxisomes
From a biochemical perspective, the transfer of mitochondrial acylcarnitines to peroxisomes is more difficult to explain since peroxisomes need mitochondria to adequately finish the metabolism of these molecules. Recent studies, however, have demonstrated that fatty acids and acylcarnitines that are normally destined for mitochondrial FAO can be rerouted to the peroxisome if the mitochondrial pathway is blocked [25, 49] (Figure 2). We found that fibroblasts of patients with a defect in the carnitine cycle could produce C10-carnitine, as well as shorter acylcarnitines, when loaded with lauric acid (C12), a fatty acid that normally undergoes mitochondrial FAO [49]. Subsequent knock-down of PEX13 in these cells, which essentially depletes functional peroxisomes, prevented the C10-carnitine accumulation proving that its origin was peroxisomal [49]. In a follow-up study, we performed a molecular dissection of this pathway using CRISPR-Cas9 genome editing in HEK-293 cells [25]. We found that HEK-293 cells can oxidize medium- and long-chain fatty acids in peroxisomes when the carnitine cycle is blocked. By creating double KOs with different defects in peroxisomal FAO and the carnitine cycle, we furthermore showed that ABCD3, a peroxisomal ABC transporter, and HSD17B4, a peroxisomal FAO enzyme, were essential in this process [25].
Figure 2.
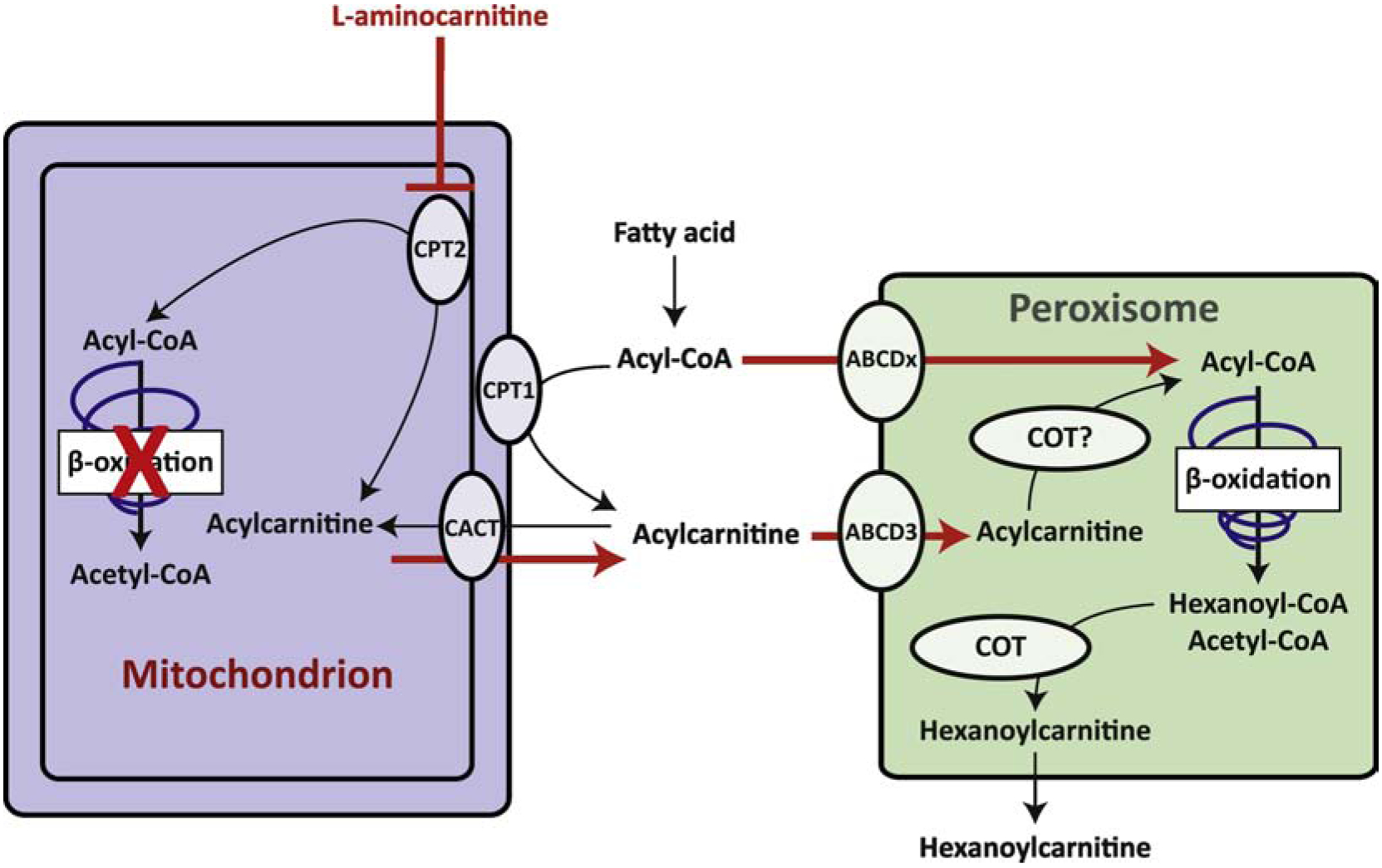
Schematic representation of the interactions between mitochondria and peroxisomes for the β-oxidation of medium- and long-chain fatty acids. The figure highlights the relevance of this interaction in the case of mitochondrial fatty acid β-oxidation dysfunction due to an enzyme deficiency or inhibition of CPT2 by L-aminocarnitine (red arrows). Details of the pathways and the individual enzymes are omitted. Abbreviations: ABCD3, ATP Binding Cassette Subfamily D Member 3. The remaining abbreviations are specified in the legend of figure 1.
Plasma acylcarnitine profiles of patients with defects in the carnitine cycle may provide evidence that a similar pathway also occurs in vivo. Patients with CPT2 and CACT deficiency are identified by the accumulation of plasma long-chain acylcarnitines (C16 and C18) in particular an elevated (C16+C18:1)/C2 ratio. Interestingly, shorter acylcarnitines such as C12-carnitine are sometimes also reported as elevated in a diagnostic plasma sample (i.e. before starting any treatment [50–58]). The defect in the carnitine cycle and the absence of C12 fatty acids from regular diets suggests that these acylcarnitines may have a peroxisomal origin. In an effort to prove this, we treated mice with L-aminocarnitine [25], a potent, versatile and specific inhibitor of CPT2 [59]. Treated animals not only showed a pronounced increase in C16 and C18 acylcarnitine species, but also several other acylcarnitines, including C10:1-, C10-, C12:1-, C12-, C14:1-, and C14-carnitine. We found that the accumulation of most of these chain-shortened species was decreased in the HSD17B4 KO mice [25]. This result should be confirmed in another model system with potentially larger effects such as the ABCD3 KO mouse. Our work has demonstrated that peroxisomal FAO can oxidize mitochondrial substrates when the mitochondrial pathway is blocked (Figure 2). A more general interpretation of these results would be that peroxisomes always oxidize a small portion of the “mitochondrial” substrates and that blocking mitochondrial FAO at the level of the carnitine cycle unveils this contribution because the subsequent and final mitochondrial metabolism of peroxisomal intermediates is blocked. This interpretation is consistent with early biochemical studies that demonstrated a contribution of peroxisomes to the oxidation of canonical mitochondrial substrates such as palmitate [23, 31, 32, 60–65].
Potential physiological role for the oxidation of mitochondrial substrates in peroxisome
The physiological importance of peroxisomes is demonstrated by the severe disease presentations in disorders caused by defects in peroxisomal metabolism and biogenesis [20, 21]. The pathophysiology of these disorders, however, can be largely explained by defects in the metabolism of canonical peroxisomal metabolites such as bile acids precursors, VLCFAs and plasmalogens, and it is unclear if a defect in the peroxisomal metabolism of mitochondrial substrates plays a role as well. As highlighted before, it is known that acetyl-CoA formed by peroxisomal FAO can enter the cytosolic pool of acetyl-CoA and is used for cholesterol biosynthesis in rat hepatocytes [31]. Consistent with this role, induction of the cholesterol biosynthesis pathway and SREBP induction has also been reported in mouse models with defects in peroxisomal metabolism and biogenesis [66–69]. Stable isotope labeling studies by Brunengraber and Des Rosiers have provided additional insights in the physiological role of this process. These studies demonstrated that peroxisomes contribute to the oxidation of long- and medium-chain fatty acids in rat heart [32, 70]. And although the rate of peroxisomal acetyl-CoA production is 100 times smaller than the mitochondrial rate, it can contribute 50% of the acetyl-CoA for the synthesis of malonyl-CoA [32]. A similar phenomenon was reported for rat liver [71], although in this case it cannot be excluded that the employed medium-chain fatty acids first underwent microsomal omega-oxidation. Malonyl-CoA is an interesting metabolite with at least two functions. It is a substrate for fatty acid synthesis and an inhibitor of CPT1 and therefore a regulator of mitochondrial long-chain FAO [72–74]. Combined these studies suggest that peroxisomes may play a role in the regulation of lipid synthesis and oxidation. Further experiments using stable isotopes in model organisms with defects in peroxisomal metabolism are necessary to further expand our understanding of these unexpected physiological roles of peroxisomal FAO.
Conclusion
Initially, when peroxisomal β-oxidation was discovered, it was thought to play a role in the oxidation of long-chain fatty acids, similar to mitochondria [23, 62, 65]. With the identification of peroxisomal diseases such as Zellweger syndrome and the characterization of the specific substrates for the peroxisomal β-oxidation system, a potential general role for peroxisomes has been forgotten and remains largely unexplored. In this review, we have tried to highlight some studies that demonstrate that such a role exists and what its function may be. It is clear that we have only just started to understand the significance of the interactions between the different cell organelles, and future research on this topic using a combination of genetic and biochemical techniques is expected to yield important new insights.
Highlights.
Peroxisomes harbor two distinct carnitine acyltransferases
Carnitine plays an important role in peroxisomal metabolism
Carnitine enables metabolite exchange between peroxisomes and mitochondria
Specific peroxisomal intermediates can be exported as acylcarnitines
Acknowledgments
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK113172. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Houten SM, Violante S, Ventura FV, Wanders RJ, The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders, Annu Rev Physiol, 78 (2016) 23–44. [DOI] [PubMed] [Google Scholar]
- [2].Longo N, Primary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle, Ann Nutr Metab, 68 Suppl 3 (2016) 5–9. [DOI] [PubMed] [Google Scholar]
- [3].Longo N, Amat di San Filippo C, Pasquali M, Disorders of carnitine transport and the carnitine cycle, Am. J. Med. Genet. C. Semin. Med. Genet, 142 (2006) 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ramsay RR, Gandour RD, van der Leij FR, Molecular enzymology of carnitine transfer and transport, Biochim. Biophys. Acta, 1546 (2001) 21–43. [DOI] [PubMed] [Google Scholar]
- [5].Violante S, IJlst L, van Lenthe H, de Almeida IT, Wanders RJ, Ventura FV, Carnitine palmitoyltransferase 2: New insights on the substrate specificity and implications for acylcarnitine profiling, Biochim. Biophys. Acta, 1802 (2010) 728–732. [DOI] [PubMed] [Google Scholar]
- [6].Violante S, IJlst L, te Brinke H, Tavares de Almeida I, Wanders RJ, Ventura FV, Houten SM, Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines, FASEB J, 27 (2013) 2039–2044. [DOI] [PubMed] [Google Scholar]
- [7].Violante S, IJlst L, Ruiter J, Koster J, van Lenthe H, Duran M, de Almeida IT, Wanders RJ, Houten SM, Ventura FV, Substrate specificity of human carnitine acetyltransferase: Implications for fatty acid and branched-chain amino acid metabolism, Biochim. Biophys. Acta, 1832 (2013) 773–779. [DOI] [PubMed] [Google Scholar]
- [8].Schooneman MG, Vaz FM, Houten SM, Soeters MR, Acylcarnitines: reflecting or inflicting insulin resistance?, Diabetes, 62 (2013) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bremer J, Carnitine--metabolism and functions, Physiol Rev, 63 (1983) 1420–1480. [DOI] [PubMed] [Google Scholar]
- [10].Corti O, DiDonato S, Finocchiaro G, Divergent sequences in the 5’ region of cDNA suggest alternative splicing as a mechanism for the generation of carnitine acetyltransferases with different subcellular localizations, Biochem J, 303 (Pt 1) (1994) 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Westin MA, Hunt MC, Alexson SE, Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of beta-oxidation products out of peroxisomes, Cell Mol.Life Sci, 65 (2008) 982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Robic A, Faraut T, Liaubet L, Milan D, The carnitine acetyltransferase gene (CRAT): a characterization of porcine transcripts with insights into the 5’-end variants of mammalian transcripts and their possible sub-cellular localization, Cell Mol Biol Lett, 14 (2009) 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Altamimi TR, Thomas PD, Darwesh AM, Fillmore N, Mahmoud MU, Zhang L, Gupta A, Al Batran R, Seubert JM, Lopaschuk GD, Cytosolic carnitine acetyltransferase as a source of cytosolic acetyl-CoA: a possible mechanism for regulation of cardiac energy metabolism, Biochem J, 475 (2018) 959–976. [DOI] [PubMed] [Google Scholar]
- [14].GTEx Consortium, Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans, Science, 348 (2015) 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choi SJ, Oh DH, Song CS, Roy AK, Chatterjee B, Molecular cloning and sequence analysis of the rat liver carnitine octanoyltransferase cDNA, its natural gene and the gene promoter, Biochim Biophys Acta, 1264 (1995) 215–222. [DOI] [PubMed] [Google Scholar]
- [16].Farrell SO, Bieber LL, Carnitine octanoyltransferase of mouse liver peroxisomes: properties and effect of hypolipidemic drugs, Arch Biochem Biophys, 222 (1983) 123–132. [DOI] [PubMed] [Google Scholar]
- [17].Miyazawa S, Ozasa H, Osumi T, Hashimoto T, Purification and properties of carnitine octanoyltransferase and carnitine palmitoyltransferase from rat liver, J Biochem, 94 (1983) 529–542. [DOI] [PubMed] [Google Scholar]
- [18].Farrell SO, Fiol CJ, Reddy JK, Bieber LL, Properties of purified carnitine acyltransferases of mouse liver peroxisomes, J Biol Chem, 259 (1984) 13089–13095. [PubMed] [Google Scholar]
- [19].Ferdinandusse S, Mulders J, IJlst L, Denis S, Dacremont G, Waterham HR, Wanders RJ, Molecular cloning and expression of human carnitine octanoyltransferase: evidence for its role in the peroxisomal beta-oxidation of branched-chain fatty acids, Biochem. Biophys. Res. Commun, 263 (1999) 213–218. [DOI] [PubMed] [Google Scholar]
- [20].Wanders RJ, Waterham HR, Biochemistry of mammalian peroxisomes revisited, Annu. Rev. Biochem, 75 (2006) 295–332. [DOI] [PubMed] [Google Scholar]
- [21].Waterham HR, Ferdinandusse S, Wanders RJ, Human disorders of peroxisome metabolism and biogenesis, Biochim Biophys Acta, 1863 (2016) 922–933. [DOI] [PubMed] [Google Scholar]
- [22].Ferdinandusse S, Denis S, Hogenhout EM, Koster J, van Roermund CW, IJlst L, Moser AB, Wanders RJ, Waterham HR, Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency, Hum. Mutat, 28 (2007) 904–912. [DOI] [PubMed] [Google Scholar]
- [23].Lazarow PB, Rat liver peroxisomes catalyze the beta oxidation of fatty acids, J Biol Chem, 253 (1978) 1522–1528. [PubMed] [Google Scholar]
- [24].Visser WF, van Roermund CW, Ijlst L, Waterham HR, Wanders RJ, Metabolite transport across the peroxisomal membrane, Biochem J, 401 (2007) 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Violante S, Achetib N, van Roermund CWT, Hagen J, Dodatko T, Vaz FM, Waterham HR, Chen H, Baes M, Yu C, Argmann CA, Houten SM, Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4, FASEB J, 33 (2019) 4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De Marcos Lousa C, van Roermund CW, Postis VL, Dietrich D, Kerr ID, Wanders RJ, Baldwin SA, Baker A, Theodoulou FL, Intrinsic acyl-CoA thioesterase activity of a peroxisomal ATP binding cassette transporter is required for transport and metabolism of fatty acids, Proc Natl Acad Sci U S A, 110 (2013) 1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Carrier DJ, van Roermund CWT, Schaedler TA, Rong HL, L IJ, Wanders RJA, Baldwin SA, Waterham HR, Theodoulou FL, Baker A, Mutagenesis separates ATPase and thioesterase activities of the peroxisomal ABC transporter, Comatose, Sci Rep, 9 (2019) 10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, Berger J, Impaired very long-chain acyl-CoA beta-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction, J Biol Chem, 288 (2013) 19269–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Antonenkov VD, Hiltunen JK, Transfer of metabolites across the peroxisomal membrane, Biochim Biophys Acta, 1822 (2012) 1374–1386. [DOI] [PubMed] [Google Scholar]
- [30].Westin MA, Hunt MC, Alexson SE, The identification of a succinyl-CoA thioesterase suggests a novel pathway for succinate production in peroxisomes, J Biol Chem, 280 (2005) 38125–38132. [DOI] [PubMed] [Google Scholar]
- [31].Kondrup J, Lazarow PB, Flux of palmitate through the peroxisomal and mitochondrial beta-oxidation systems in isolated rat hepatocytes, Biochim Biophys Acta, 835 (1985) 147–153. [DOI] [PubMed] [Google Scholar]
- [32].Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL, Brunengraber H, Des Rosiers C, Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart, J Biol Chem, 279 (2004) 19574–19579. [DOI] [PubMed] [Google Scholar]
- [33].Jakobs BS, Wanders RJ, Fatty acid beta-oxidation in peroxisomes and mitochondria: the first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria, Biochem Biophys Res Commun, 213 (1995) 1035–1041. [DOI] [PubMed] [Google Scholar]
- [34].Verhoeven NM, Jakobs C, ten Brink HJ, Wanders RJ, Roe CR, Studies on the oxidation of phytanic acid and pristanic acid in human fibroblasts by acylcarnitine analysis, J Inherit Metab Dis, 21 (1998) 753–760. [DOI] [PubMed] [Google Scholar]
- [35].Verhoeven NM, Roe DS, Kok RM, Wanders RJ, Jakobs C, Roe CR, Phytanic acid and pristanic acid are oxidized by sequential peroxisomal and mitochondrial reactions in cultured fibroblasts, J Lipid Res, 39 (1998) 66–74. [PubMed] [Google Scholar]
- [36].Wanders RJ, Denis S, Ruiter JP, IJlst L, Dacremont G, 2,6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation, Biochim. Biophys. Acta, 1393 (1998) 35–40. [DOI] [PubMed] [Google Scholar]
- [37].Chegary M, te Brinke H, Ruiter JP, Wijburg FA, Stoll MS, Minkler PE, van Weeghel M, Schulz H, Hoppel CL, Wanders RJ, Houten SM, Mitochondrial long chain fatty acid beta-oxidation in man and mouse, Biochim. Biophys. Acta, 1791 (2009) 806–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vamecq J, Draye JP, Brison J, Rat liver metabolism of dicarboxylic acids, Am. J. Physiol, 256 (1989) G680–G688. [DOI] [PubMed] [Google Scholar]
- [39].Vamecq J, Draye JP, Peroxisomal and mitochondrial beta-oxidation of monocarboxylyl-CoA, omega-hydroxymonocarboxylyl-CoA and dicarboxylyl-CoA esters in tissues from untreated and clofibrate-treated rats, J. Biochem, 106 (1989) 216–222. [DOI] [PubMed] [Google Scholar]
- [40].Suzuki H, Yamada J, Watanabe T, Suga T, Compartmentation of dicarboxylic acid beta-oxidation in rat liver: importance of peroxisomes in the metabolism of dicarboxylic acids, Biochim. Biophys. Acta, 990 (1989) 25–30. [DOI] [PubMed] [Google Scholar]
- [41].Pourfarzam M, Bartlett K, Products and intermediates of the beta-oxidation of [U-14C]hexadecanedionoyl-mono-CoA by rat liver peroxisomes and mitochondria, Biochem. J, 273(Pt 1) (1991) 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bergseth S, Poisson JP, Bremer J, Metabolism of dicarboxylic acids in rat hepatocytes, Biochim. Biophys. Acta, 1042 (1990) 182–187. [DOI] [PubMed] [Google Scholar]
- [43].Houten SM, Denis S, Argmann CA, Jia Y, Ferdinandusse S, Reddy JK, Wanders RJ, Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids, J. Lipid Res, 53 (2012) 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ferdinandusse S, Denis S, van Roermund CW, Wanders RJ, Dacremont G, Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids, J. Lipid Res, 45 (2004) 1104–1111. [DOI] [PubMed] [Google Scholar]
- [45].Dirkx R, Meyhi E, Asselberghs S, Reddy J, Baes M, Van Veldhoven PP, Beta-oxidation in hepatocyte cultures from mice with peroxisomal gene knockouts, Biochem. Biophys. Res. Commun, 357 (2007) 718–723. [DOI] [PubMed] [Google Scholar]
- [46].Tserng KY, Jin SJ, Metabolic conversion of dicarboxylic acids to succinate in rat liver homogenates. A stable isotope tracer study, J Biol Chem, 266 (1991) 2924–2929. [PubMed] [Google Scholar]
- [47].Jin SJ, Tserng KY, Biogenesis of dicarboxylic acids in rat liver homogenate studied by 13C labeling, Am J Physiol, 261 (1991) E719–724. [DOI] [PubMed] [Google Scholar]
- [48].Jin Z, Bian F, Tomcik K, Kelleher JK, Zhang GF, Brunengraber H, Compartmentation of Metabolism of the C12-, C9-, and C5-n-dicarboxylates in Rat Liver, Investigated by Mass Isotopomer Analysis: ANAPLEROSIS FROM DODECANEDIOATE, J Biol Chem, 290 (2015) 18671–18677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Violante S, IJlst L, te Brinke H, Koster J, Tavares de Almeida I, Wanders RJ, Ventura FV, Houten SM, Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient, Biochim Biophys Acta, 1831 (2013) 1467–1474. [DOI] [PubMed] [Google Scholar]
- [50].Albers S, Marsden D, Quackenbush E, Stark AR, Levy HL, Irons M, Detection of neonatal carnitine palmitoyltransferase II deficiency by expanded newborn screening with tandem mass spectrometry, Pediatrics, 107 (2001) E103. [DOI] [PubMed] [Google Scholar]
- [51].Wieser T, Carnitine Palmitoyltransferase II Deficiency, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE (Eds.) GeneReviews® [Internet], University of Washington, Seattle, Place Published, 2004. [PubMed] [Google Scholar]
- [52].Yamada K, Bo R, Kobayashi H, Hasegawa Y, Ago M, Fukuda S, Yamaguchi S, Taketani T, A newborn case with carnitine palmitoyltransferase II deficiency initially judged as unaffected by acylcarnitine analysis soon after birth, Mol Genet Metab Rep, 11 (2017) 59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Anichini A, Fanin M, Vianey-Saban C, Cassandrini D, Fiorillo C, Bruno C, Angelini C, Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency, Neurological research, 33 (2011) 24–32. [DOI] [PubMed] [Google Scholar]
- [54].Hurvitz H, Klar A, Korn-Lubetzki I, Wanders RJ, Elpeleg ON, Muscular carnitine palmitoyltransferase II deficiency in infancy, Pediatr Neurol, 22 (2000) 148–150. [DOI] [PubMed] [Google Scholar]
- [55].Fontaine M, Briand G, Largilliere C, Degand P, Divry P, Vianey-Saban C, Mousson B, Vamecq J, Metabolic studies in a patient with severe carnitine palmitoyltransferase type II deficiency, Clinica chimica acta; international journal of clinical chemistry, 273 (1998) 161–170. [DOI] [PubMed] [Google Scholar]
- [56].Al-Sannaa NA, Cheriyan GM, Carnitine-acylcarnitine translocase deficiency. Clinical course of three Saudi children with a severe phenotype, Saudi Med J, 31 (2010) 931–934. [PubMed] [Google Scholar]
- [57].Vitoria I, Martin-Hernandez E, Pena-Quintana L, Bueno M, Quijada-Fraile P, Dalmau J, Molina-Marrero S, Perez B, Merinero B, Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature, JIMD Rep, 20 (2015) 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Roschinger W, Muntau AC, Duran M, Dorland L, L IJ, Wanders RJ, Roscher AA, Carnitine-acylcarnitine translocase deficiency: metabolic consequences of an impaired mitochondrial carnitine cycle, Clinica chimica acta; international journal of clinical chemistry, 298 (2000) 55–68. [DOI] [PubMed] [Google Scholar]
- [59].Chegary M, te Brinke H, Doolaard M, IJlst L, Wijburg FA, Wanders RJ, Houten SM, Characterization of L-aminocarnitine, an inhibitor of fatty acid oxidation, Mol. Genet. Metab, 93 (2008) 403–410. [DOI] [PubMed] [Google Scholar]
- [60].Shindo Y, Hashimoto T, Acyl-Coenzyme A synthetase and fatty acid oxidation in rat liver peroxisomes, J Biochem, 84 (1978) 1177–1181. [DOI] [PubMed] [Google Scholar]
- [61].Mannaerts GP, Debeer LJ, Thomas J, De Schepper PJ, Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats, J Biol Chem, 254 (1979) 4585–4595. [PubMed] [Google Scholar]
- [62].Lazarow PB, De Duve C, A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug, Proc Natl Acad Sci U S A, 73 (1976) 2043–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Veerkamp JH, van Moerkerk HT, Peroxisomal fatty acid oxidation in rat and human tissues. Effect of nutritional state, clofibrate treatment and postnatal development in the rat, Biochim Biophys Acta, 875 (1986) 301–310. [DOI] [PubMed] [Google Scholar]
- [64].Veerkamp JH, Van Moerkerk HT, Glatz JF, Van Hinsbergh VW, Incomplete palmitate oxidation in cell-free systems of rat and human muscles, Biochim Biophys Acta, 753 (1983) 399–410. [DOI] [PubMed] [Google Scholar]
- [65].Lazarow PB, Three hypolipidemic drugs increase hepatic palmitoyl-coenzyme A oxidation in the rat, Science, 197 (1977) 580–581. [DOI] [PubMed] [Google Scholar]
- [66].Martens K, Ver Loren van Themaat E, van Batenburg MF, Heinaniemi M, Huyghe S, Van Hummelen P, Carlberg C, Van Veldhoven PP, Van Kampen A, Baes M, Coordinate induction of PPAR alpha and SREBP2 in multifunctional protein 2 deficient mice, Biochim Biophys Acta, 1781 (2008) 694–702. [DOI] [PubMed] [Google Scholar]
- [67].Kovacs WJ, Tape KN, Shackelford JE, Wikander TM, Richards MJ, Fliesler SJ, Krisans SK, Faust PL, Peroxisome deficiency causes a complex phenotype because of hepatic SREBP/Insig dysregulation associated with endoplasmic reticulum stress, J Biol Chem, 284 (2009) 7232–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kovacs WJ, Shackelford JE, Tape KN, Richards MJ, Faust PL, Fliesler SJ, Krisans SK, Disturbed cholesterol homeostasis in a peroxisome-deficient PEX2 knockout mouse model, Mol Cell Biol, 24 (2004) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kovacs WJ, Charles KN, Walter KM, Shackelford JE, Wikander TM, Richards MJ, Fliesler SJ, Krisans SK, Faust PL, Peroxisome deficiency-induced ER stress and SREBP-2 pathway activation in the liver of newborn PEX2 knock-out mice, Biochim Biophys Acta, 1821 (2012) 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bian F, Kasumov T, Thomas KR, Jobbins KA, David F, Minkler PE, Hoppel CL, Brunengraber H, Peroxisomal and mitochondrial oxidation of fatty acids in the heart, assessed from the 13C labeling of malonyl-CoA and the acetyl moiety of citrate, J Biol Chem, 280 (2005) 9265–9271. [DOI] [PubMed] [Google Scholar]
- [71].Kasumov T, Adams JE, Bian F, David F, Thomas KR, Jobbins KA, Minkler PE, Hoppel CL, Brunengraber H, Probing peroxisomal beta-oxidation and the labelling of acetyl-CoA proxies with [1-(13C)]octanoate and [3-(13C)]octanoate in the perfused rat liver, Biochem J, 389 (2005) 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].van Weeghel M, Abdurrachim D, Nederlof R, Argmann CA, Houtkooper RH, Hagen J, Nabben M, Denis S, Ciapaite J, Kolwicz SC Jr., Lopaschuk GD, Auwerx J, Nicolay K, Des Rosiers C, Wanders RJ, Zuurbier CJ, Prompers JJ, Houten SM, Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B, Cardiovascular research, 114 (2018) 1324–1334. [DOI] [PubMed] [Google Scholar]
- [73].Stanley WC, Morgan EE, Huang H, McElfresh TA, Sterk JP, Okere IC, Chandler MP, Cheng J, Dyck JR, Lopaschuk GD, Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia, American journal of physiology. Heart and circulatory physiology, 289 (2005) H2304–2309. [DOI] [PubMed] [Google Scholar]
- [74].McGarry JD, Mannaerts GP, Foster DW, A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis, J. Clin. Invest, 60 (1977) 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]