

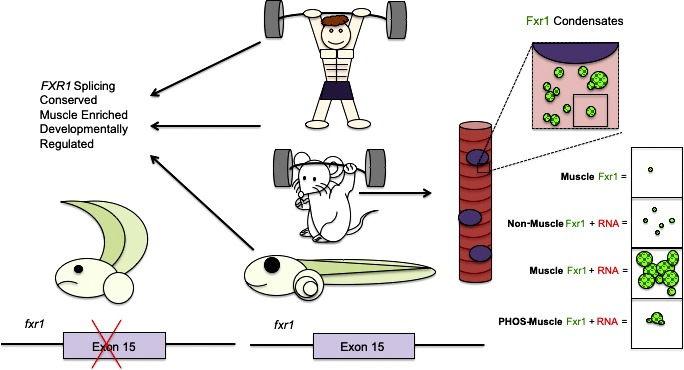
This study shows that FXR1 forms condensates in muscle cells. FXR1 alternative splicing during myogenesis alters the properties of an intrinsically disordered domain and influences the properties of condensates. This study links developmental control of RNA splicing to regulation of biomolecular condensation for muscle formation.
Abstract
Fragile-X mental retardation autosomal homologue-1 (FXR1) is a muscle-enriched RNA-binding protein. FXR1 depletion is perinatally lethal in mice, Xenopus, and zebrafish; however, the mechanisms driving these phenotypes remain unclear. The FXR1 gene undergoes alternative splicing, producing multiple protein isoforms and mis-splicing has been implicated in disease. Furthermore, mutations that cause frameshifts in muscle-specific isoforms result in congenital multi-minicore myopathy. We observed that FXR1 alternative splicing is pronounced in the serine- and arginine-rich intrinsically disordered domain; these domains are known to promote biomolecular condensation. Here, we show that tissue-specific splicing of fxr1 is required for Xenopus development and alters the disordered domain of FXR1. FXR1 isoforms vary in the formation of RNA-dependent biomolecular condensates in cells and in vitro. This work shows that regulation of tissue-specific splicing can influence FXR1 condensates in muscle development and how mis-splicing promotes disease.
Graphical Abstract
Introduction
Fragile-X mental retardation autosomal homologue-1 (FXR1) and FXR2 are vertebrate homologues of the Fragile-X mental retardation-1 (FMR1) gene. These genes encode three RNA-binding proteins (RBPs) that comprise the Fragile-X (FraX) protein family (Zarnescu and Gregorio, 2013). FraX proteins regulate mRNA transport, stability, and translation (Darnell et al., 2009). While FMR1 is associated with neuronal functions, FXR1 is highly enriched in striated muscles where it localizes to the Z-discs and costameres and is associated with muscle function in multiple organisms (Mientjes et al., 2004; Huot et al., 2005; Van’t Padje et al., 2009; Whitman et al., 2011; Zarnescu and Gregorio, 2013).
Alternative splicing results in several FXR1 isoforms with varying abundances in different cell types (Khandjian et al., 1998; Kirkpatrick et al., 1999). Isoform balance is altered in myotonic dystrophy (Orengo et al., 2008), facioscapulohumeral muscular dystrophy (Davidovic et al., 2008), and diabetes (Nutter et al., 2016), suggesting that FXR1 precursor mRNA (pre-mRNA) splicing is central to function. Supporting this, mutations causing frameshifts in muscle-specific isoforms are associated with congenital multi-minicore myopathy in humans (Estañ et al., 2019). However, it is unclear whether phenotypes arise from neomorphic frameshifts or loss of muscle-specific protein sequences (Estañ et al., 2019). So far, the phenotypes of FXR1 manipulation have been observed using methods that affect all splice isoforms. Thus, the mechanism relating FXR1 pre-mRNA splicing to its function in muscle development is not understood. Therefore, we examined the importance of muscle-specific FXR1 splicing in development.
Muscle-specific FXR1 isoforms contain a longer primary sequence than in other tissues, with a predicted 300-aa-long intrinsically disordered domain (IDD) at the C terminus. Numerous RBPs contain disordered or low-complexity sequences that are associated with biomolecular condensation or liquid–liquid phase separation (LLPS; Banani et al., 2017). Biomolecular condensation is now appreciated as a common mechanism for compartment formation in the nucleus and the cytoplasm. These assemblies are not delineated by membranes but nevertheless are discrete bodies from the surrounding cytosol or nucleoplasm (Banani et al., 2017). Depending on the composition, condensates vary in their material properties from highly dynamic liquids to more solid- or gel-like states (Berry et al., 2018). The possible functions of condensates include colocalization, regulation of biochemical reaction rates, and stress sensing (Alberti et al., 2019). Many condensates contain RNA, and in some cases, RNA is essential for the demixing process (Elbaum-Garfinkle et al., 2015; Zhang et al., 2015). We postulated that alternative splicing may regulate the IDD and thus FXR1 condensation. RNA-rich granules are prominent in large cells such as neurons where transport granules package mRNAs for local translation (Kiebler and Bassell, 2006) and in multinucleated fungi where they promote local control of the cell cycle and cell polarity (Lee et al., 2013, 2015). We hypothesized that alternative splicing events within the IDD of FXR1 regulate biomolecular condensates for patterning developing muscle.
In this study, we examined the splicing patterns of Fxr1 pre-mRNA and observed that blocking the expression of muscle-specific isoforms leads to alterations in Xenopus development in vivo and in cultured muscle cell differentiation. We further found that FXR1 forms spherical, liquid-like assemblies in both developing myotubes and cultured U2OS cells and more gel-like assemblies in vitro. Additionally, both disordered sequences and RNA binding contributed to condensate assembly and different isoforms vary in the properties of the condensates they form. In summary, this study links alternative splicing of FXR1 to LLPS in muscle development and disease.
Results
Splicing of fxr1 exon 15 impacts the development of Xenopus
Recessive mutations in muscle-specific isoforms of FXR1 are associated with congenital multi-minicore myopathy in humans (Estañ et al., 2019). These mutations result in a frameshift in FXR1 transcripts containing exon 15. It is unclear whether multi-minicore myopathy is the result of exon 15 loss, a neomorphic function conferred by the frameshift, or both. To investigate FXR1 exon 15 function in development, we removed or mutated this exon in Xenopus laevis and Xenopus tropicalis. We chose Xenopus because there are only two characterized Fxr1 splice isoforms, which differ solely by the inclusion of exon 15 (Huot et al., 2005). Furthermore, protein sequences of exon 15 from X. tropicalis and the two alloalleles of X. laevis, 5S and 5L, differ from mouse and human by only one aa (Fig. 1 A).
Figure 1.

Splicing of fxr1 exon 15 regulates Xenopus development. (A) Conservation of FXR1 exon 15-aa sequences in human (Homo sapiens), mouse (Mus musculus), X. laevis, X. tropicalis, and zebrafish (Danio rerio). Red indicates deviation relative to mouse/human sequence. Mouse exon 15 is underlined. (B) A MO targeting exon 15 (mo-e15i15) was used in X. tropicalis. Successful blocking of exon 15 inclusion was confirmed by RT-PCR. (C) Representative images of uninjected embryos or those injected with a mo-control or the mo-e15i15 (X. tropicalis) at stage 45. (D) X. tropicalis embryos at stage 45. RFP signal marks the somites of uninjected, mo-control, or mo-e15i15–treated embryos. (E) Position of mo-i14e15 and sgRNAs within the X. laevis genome. Representative images and quantification of normal or two abnormal phenotypes (1 and 2) for uninjected, mo-control, mo-i14e15, or sgRNA-treated embryos. (F and G) X. laevis embryos at stages 35–38. GFP signal marks the somites of uninjected, injected (mo-control or mo-i14e15), and Cas9 protein injected with sgRNAs targeting exon 15 or introns 14 and 15 for both 5S and 5L. White rectangles indicate the region of the somites magnified in the insets in (F). All scale bars, 1 mm. Fig. 1, E and F, is linked to Fig. S1.
To identify the role of fxr1 exon 15 inclusion in Xenopus development, delivery of a morpholino antisense oligonucleotide (MO) was chosen to block the splicing recognition of exon 15 in X. tropicalis (Fig. 1 B) as this species is diploid with a single fxr1 gene. We injected transgenic X. tropicalis embryos where the cardiac actin promoter drives RFP expression to label the developing somites (Hartley et al., 2001) at the one- or two-cell stage with a MO that binds the junction between fxr1 exon 15 and intron 15 (mo-e15i15) or control MO (mo-control). Blockage of exon 15 was confirmed by RT-PCR (Fig. 1 B). The mo-e15i15–treated embryos showed a loss of the longer exon 15–containing transcripts compared with embryos injected with the mo-control (Fig. 1 B). Compared with uninjected or mo-control–treated embryos, the mo-e15i15–injected embryos exhibited severe morphological defects including curved tails and defects in somite formation (Fig. 1, C and D), consistent with Fxr1 role in somite and muscle development in X. laevis (Huot et al., 2005).
We next sought to compare the effect of exon 15 loss to mutations that create frameshifts, which would mimic patient mutations. For this, we used two approaches: CRISPR/Cas9 editing of exon 15 and blocking the exon 15 splice site using MOs (Fig. 1 E). For these experiments, we chose X. laevis and used a second MO that targets the intron 14 and exon 15 junction (mo-i14e15) to complement the X. tropicalis results. Two single guide RNAs (sgRNAs) were designed to target the 3′ end of exon 15 for both fxr1.L and fxr1.S genes, denoted T1 and T3 (Fig. 1 E), creating a frameshift in exon 15–containing transcripts (Fig. S1). Compared with uninjected or mo-control–treated embryos, mo-i14e15– or sgRNA (T1/T3)–injected embryos differed significantly at stage 35-36 of development, displaying similar gross morphological defects (Fig. 1 E). These alterations included tail curving to varying degrees of severity, closely mimicking those observed in X. tropicalis (Fig. 1, B–D). Remarkably, the observed phenotypes are also similar to the pan-isoform knockdown of fxr1 in X. laevis (Huot et al., 2005; Gessert et al., 2010), suggesting that the majority of Fxr1 function is derived from the isoform containing exon 15.
Figure S1.

Sequences of fxr1 exon 15 from sgRNA-treated embryos. Sequences of fxr1.S and fxr1.L from five randomly selected X. laevis embryos injected with the sgRNA T1 (green) and T3 (underlined). sgRNA-injected embryos had deletions (dashed), uncallable nucleotides (N), and mismatches (red) compared with reference sequences. Deletions roughly correspond to 3–4 nt upstream of the protospacer adjacent motif (PAM) sequences (magenta). Fig. S1 is linked to Fig. 1, E and F.
We next assessed the impact of loss or frameshift of fxr1 exon 15 on somites using a transgenic line with GFP-labeled somites (Latinkić et al., 2002). We imaged GFP signal in embryos at stages 35–38 injected with sgRNAs (T1/T3) or mo-i14e15. Compared with control tadpoles, the sgRNA (T1/T3)– or mo-i14e15–injected tadpoles exhibited gross abnormalities in somite formation, including failure to segment (Fig. 1 F), similar to those observed to fxr1 pan-isoform knockdown (Huot et al., 2005) and our experiments in X. tropicalis (Fig. 1 D). We finally confirmed that deletion of exon 15 using CRISPR/Cas9 editing with four sgRNAs targeting introns 14 (T7L, T7S) and 15 (T8L, T8S) resulted in a similar phenotype (Fig. 1 G) to that obtained using mo-i14e15 (Fig. 1 F).
Together, our data show that mis-splicing of fxr1 exon 15 in Xenopus development results in somite formation defects similar to those observed when all Fxr1 isoforms were depleted in X. laevis (Huot et al., 2005). This suggests that the highly conserved protein sequence of exon 15 is essential for Xenopus development and that most of the function of Fxr1 is derived from exon 15–containing proteins.
Alternative splicing of Fxr1 pre-mRNA is tissue and developmental stage specific
We next confirmed the splicing patterns observed in Xenopus development are conserved in mammals. In mice and humans, the FXR1 gene contains 17 exons (Fig. 2 A) and gives rise to multiple protein isoforms (Fig. 2 B) via alternative splicing (Kirkpatrick et al., 1999). Notably, exon 16 inclusion induces a frameshift that increases protein length and alters aa composition (Fig. 2 B). Inspection of deep RNA-sequencing data from C2C12 cell differentiation (Singh et al., 2014) and mouse heart and skeletal muscle development (Giudice et al., 2014; Brinegar et al., 2017) revealed that exons 15 and 16 are regulated by alternative splicing during myogenesis and between birth and adulthood in striated muscles. Previously, alternative splicing of Fxr1 pre-mRNA in adult mouse tissues was noted, but not quantitatively analyzed (Kirkpatrick et al., 1999; Huot et al., 2005; Davidovic et al., 2008).
Figure 2.

Alternative splicing regulates FXR1 in a tissue- and developmental stage–specific manner. (A) Mouse Fxr1 gene structure. The arrow indicates transcription initiation site. Numbers indicate the exons. Alternatively spliced exons are indicated in colors. (B) Location of the alternatively spliced region within FXR1. Darker purple in isoforms A and B denotes different amino acids (AA). C, C-terminus; N, N-terminus; NES, nuclear export signal; NLS, nuclear localization signal. Blue, yellow, red, and pink colors correspond to the alternatively spliced exons shown in A. IDD is colored in purple, the RGG in green, and the KH domains in orange. (C and D) Splicing of exons 15 and 16 of Fxr1 pre-mRNA was evaluated by RT-PCR in nonstriated muscles of neonatal (P4.5) and adult (4-mo-old) mice (C). The PSI of exon 15 or exon 16 and the abundance of transcripts evaluated by densitometry (D). (E) Fxr1 pre-mRNA splicing was similarly evaluated in mouse skeletal muscles (sk muscle) and hearts at embryonic day 15.5 (E15.5), P4.5, and adulthood. (F) FXR1 pre-mRNA splicing was similarly evaluated in commercial human RNA samples. Data are means ± SEM, *P ≤ 0.05 (one-way ANOVA test with Bonferroni correction for multiple comparisons), n = 3–4 (mouse tissues), n = 2 (fetal human tissues), n = 3 (adult human tissues). Fig. 2 is linked to Fig. S2.
Here, we characterized the splicing patterns of exons 12, 13, 15, and 16 and quantitatively measured the percent spliced in (PSI) (Wang et al., 2008) of each exon at different stages of mouse development by RT-PCR. We observed that exon 15 was rarely included in kidney, spleen, pancreas, liver, lung, intestine, or brain at postnatal day 4.5 (P4.5) or adulthood (∼4 mo old; Fig. 2, C and D). By contrast, exon 15 was more included in adult hearts and skeletal muscles than in neonates or embryos (Fig. 2, D–E). While exon 16 was partially included in nonstriated muscle tissues, its inclusion increased during postnatal development only in striated muscles (Fig. 2, C–E). We further validated that splicing transitions of both exons 15 and 16 are conserved in human heart and skeletal muscle (Fig. 2 F).
The splicing events in exons 12 and 13 displayed different patterns than exons 15 and 16. All the examined tissues included the entire exon 13, while the insert in exon 12 was mostly skipped in nonmuscle tissues (Fig. S2, A and B). By contrast, in striated muscles, almost half the transcripts included the insert (Fig. S2 B). Unlike exons 15 and 16, the 87-nt insert in exon 12 was not regulated during development in striated muscles.
Figure S2.

Alternative splicing events in exons 12 and 13 of Fxr1 pre-mRNA and expression of FXR1 protein isoforms during tissue development. (A) Scheme of part of Fxr1 gene showing the alternative splicing events in exons 12 (insert of 87 nt) and exon 13 (alternative splice site leading to the inclusion or skipping of a 78-nt region). To evaluate the splicing patterns by RT-PCR, the primers designed to target the constitutive flanking exons were Fxr1-87-78-f (forward) and Fxr1-87-78-r (reverse). (B) Different tissues were collected from neonatal (P4.5) and adult (4-mo-old) mice. Alternative splicing was evaluated by RT-PCR and quantified by densitometry. n = 3–4 (P4.5 tissues) samples from two to nine pooled neonates each depending on tissue type, n = 4 (adult tissues) animals. (C) Adult mouse tissues were evaluated by Western blots using a monoclonal antibody against the N terminus of FXR1 protein that thus recognizes all of the splice variants. n = 2 animals. Data are means ± SEM, *P ≤ 0.05 (one-way ANOVA test with Bonferroni correction for multiple comparisons). sk muscle, skeletal muscle. Fig. S2 is linked to Fig. 2.
We then confirmed that the detected splice variants were translated using an antibody against the N terminus of FXR1 that recognizes all isoforms in Western blots on adult mouse tissue (Fig. S2 C). Brain and liver expressed a small protein band (∼60–65 kD) corresponding to isoforms A and B. Kidney exhibited the same band as well as an additional low molecular weight band (∼45 kD; Fig. S2 C), not corresponding to any known FXR1 isoform (Fig. 2 B). Heart and skeletal muscle expressed mostly the long FXR1 isoform of ∼80 kD (Fig. S2 C), corresponding to a combination of isoforms E and F. Overall, these data indicate that Fxr1 splicing is conserved and leads to the expression of different protein variants in specific tissues and developmental stages.
C2C12 cell differentiation recapitulates Fxr1 splicing transitions of muscle development in vivo
Given the conserved regulation of Fxr1 transcripts by splicing, we next identified a mammalian cell system, C2C12 cells, where we could examine the functional consequences of splicing. Differentiation of C2C12 myoblasts into myotubes (myogenesis) is a well-established model to study molecular aspects of skeletal muscle biology in culture (Burattini et al., 2004). A previous study implicated FXR1 in muscle cell proliferation (Davidovic et al., 2013), but its role in differentiation was not investigated. By using siRNAs, we found that FXR1-depleted cells (Fig. S3, A and B) formed fewer myotube-like cells than controls (Fig. S3 C). We further identified that this phenotype was due to an ∼50–80% decrease in cell–cell fusion (Fig. S3 D). Both siRNAs used showed this decrease, with si-Fxr1-#2 giving the strongest fusion phenotype and a reduction in the number of nuclei per field of view (Fig. S3 E). Collectively, these results demonstrate that FXR1 is required for myoblast fusion.
Figure S3.

FXR1 is required for myogenesis. (A–E) C2C12 myoblasts were transfected with two different siRNAs targeting Fxr1 (#1, #2) or a control siRNA (si-luc), and the next day differentiation was induced. Protein and RNA were collected at differentiation day 4, and FXR1 depletion was confirmed by Western blot and qPCR assays (A). Cells were evaluated by immunofluorescence to confirm knockdown. Scale bars, 100 µm (B). MYH expression was evaluated by immunofluorescence (C), and the fusion index was estimated (D). The number of nuclei per field of view was estimated from the microscopy images (E). Data are means ± SEM, *P ≤ 0.05 versus mock and si-luc (Student’s t test, two sided), n = 4–8 independent experiments. Scale bars, 200 µm. (F–I) MOs were delivered in undifferentiated myoblasts, and the next day cells were differentiated for 4 d. Scheme of the MOs used to target exons 15 (mo-1) and 16 (mo-2) where control denotes cells treated with a MO control (F). Efficiency of MO action was evaluated by RT-PCR (G) and quantified (H). Total Fxr1 mRNA levels were evaluated by qPCR (I). mo-1& 2 indicates that cells were treated with both mo-1 and mo-2 at the same time. Data are means ± SEM, *P ≤ 0.05 versus mock and control (Student’s t test, two sided), n = 3–8 independent experiments. Fig. S3 is linked to Figs. 3 and 4.
We then systematically quantified the splicing of exons 12, 13, 15, and 16 during myogenesis. Two days after differentiation induction, exons 15 and 16 were highly included (Fig. 3 A), recapitulating the findings observed in muscle development. Unlike exons 15 and 16, C2C12 cells always include exon 13 (Fig. 3 B). This is consistent with the above-mentioned RNA-sequencing dataset (Singh et al., 2014) and previous reports indicating the alternative 3′ splice site in exon 13 usage is rare (Kirkpatrick et al., 1999). The insert in exon 12 was mostly skipped in myoblasts, while it was included in ∼50% of myotube transcripts (Fig. 3 B). These findings suggest that myotubes express isoforms E and F at a 1:1 ratio. Finally, we confirmed that during cell differentiation smaller protein isoforms A (539 aa) and B (568 aa) are replaced by the largest protein isoforms E (677 aa) and F (648 aa; Fig. 3 C). Collectively, these data suggest that C2C12 cell differentiation reproduces Fxr1 splicing transitions that take place during striated muscle development in vivo, leading us to use this culture model for our molecular studies.
Figure 3.

C2C12 cell differentiation reproduces Fxr1 splicing transitions. RNA or protein was extracted at undifferentiated stage (undiff) and differentiating (D0 to D8) C2C12 cells. (A) Splicing of exons 15 and 16 was evaluated by RT-PCRs. The PSI was determined by densitometry. (B) Inclusion or skipping of the 87-nt insert in exon 12 of Fxr1 pre-mRNA. (C) Protein lysates were analyzed by Western blotting. Data are means ± SEM, *P ≤ 0.05 versus undifferentiated stage (one-way ANOVA test with Bonferroni correction), n = 3–5 experiments. Fig. 3 is linked to Fig. S3, A–E.
Fxr1 splicing produces distinct intrinsically disordered proteins that form condensates
What is the functional consequence of producing splice isoforms of FXR1 with C-terminal extensions in myotubes? To answer this question, we examined the disorder tendency of the C termini of isoforms A versus E via the intrinsically unstructured proteins predicted disorder (IUPRED) algorithm (Dosztányi et al., 2005a,b). We found that while the C termini of both isoforms have IDDs, isoform E has a predicted IDD of ∼300 aa, roughly twice as long as isoform A (∼160 aa; Fig. 4 A). Because a frameshift occurs when exon 16 is included, the different isoform IDDs also vary by aa composition; however, both are rich in serine and arginine residues. In serine/arginine-rich splicing factors, these regions are subject to posttranslational modifications (PTMs), modulating their propensity for disorder (Haynes and Iakoucheva, 2006; Xiang et al., 2013; Wang et al., 2014). Thus, both the IDD length and the potential for PTMs may regulate FXR1 activity during development.
Figure 4.

Alternative splicing results in disordered protein isoforms that are capable of phase separation. (A) Disorder predictions based on the primary aa sequence of FXR1 isoforms A and E using the IUPRED algorithm. (B) Endogenous FXR1 protein was visualized by immunofluorescence in myotubes using an antibody against the N terminus of FXR1 (recognizes all splice variants). Representative images depicting FXR1 puncta. Scale bar, 5 µm. Inset shows droplets magnified fourfold. Dapi, 6′-diamidino-2-phenylindole. (C) Time-lapse acquisition of human isoform 2 FXR1-EGFP (pAGB828). Two condensates (red and blue arrows) fuse together to form one (purple arrow). Scale bar, 1 µm. (D) C2C12 myoblasts were differentiated for 1, 2, or 3 d (D1, D2, D3). Cell lysates and pellets were analyzed by Western blot using an FXR1 antibody specific to the C terminus of isoform E. (E–G) MOs targeting exons 15 (mo-1) and/or 16 (mo-2) were delivered in myoblasts, and the next day cells were differentiated for 4 d. Cells were analyzed by Western blotting and immunofluorescence using an antibody against FXR1 N terminus (E). The CV was calculated as a measurement of FXR1 condensation. P > 0.5 mo-1 versus mo-control, P < 0.001 mo-2 and mo-1 & 2 versus mo-control (F). Representative images of MO-treated cells showing FXR1 puncta. Scale bar, 5 µm. Bottom row shows droplets magnified fivefold (G). Data are standard box and whisker plot. Fig. 4 is linked to Fig. S3, F–I.
FXR1 is an RBP (Siomi et al., 1995; Zhang et al., 1995), and numerous RBPs contain IDDs that aid in LLPS (Wang et al., 2014; Uversky et al., 2015; Pak et al., 2016; Banani et al., 2017). We therefore hypothesized that FXR1 may form condensates in the myotube cytosol, with isoform E having a highest propensity due to its extended IDD. To determine whether FXR1 undergoes LLPS, we visualized FXR1 in C2C12 cells. Immunolabeling of endogenous FXR1 revealed punctate structures throughout the myotube cytosol (Fig. 4 B). To examine the properties of these structures, Fxr1 tagged with GFP (Fxr1-GFP) was transiently transfected into C2C12 myoblasts that were then differentiated for 2 d. We observed FXR1-GFP spherical structures in cells that readily fused, suggesting that FXR1 forms liquid-like assemblies (Fig. 4 C). These droplets were too small and dynamic for partial FRAP studies to accurately assess exchange and rearrangement dynamics. Using an FXR1 antibody against the C terminus that does not detect isoform A or B, Western blots were performed on differentiating C2C12 cells (Fig. 4 D). We found that after 2 d of differentiation, a portion of FXR1 is visible in the pellet, which may correspond to assembly into insoluble higher order structures. These data indicate that, like other intrinsically disordered RBPs, FXR1 forms higher order assemblies and liquid-like droplets in cells.
We next asked whether FXR1 condensates are dependent upon alternative splicing. We used MOs to redirect endogenous splicing of exon 15 (mo-1) and/or exon 16 (mo-2) in C2C12 cells. MOs were designed to target the splice sites and block exon recognition, thus promoting exon skipping (Fig. S3 F). MOs were delivered to naive C2C12 cells that were then differentiated for 4 d. Both mo-1 and mo-2 blocked exon 15 or 16 inclusion, respectively (Fig. S3, G and H), without altering total mRNA levels (Fig. S3 I). These MOs also produced smaller protein isoforms excluding these exons (Fig. 4 E). To determine whether altering endogenous Fxr1 splicing in myotubes caused morphological differences in condensation, we analyzed cells treated with the MOs by immunofluorescence with antibodies against endogenous FXR1. We found that cells lacking exons 15 and 16 formed smaller and fewer FXR1 puncta than control cells expressing the long isoforms. We quantified this effect by measuring the variability in fluorescence intensity of endogenous FXR1 within individual cells and determining the coefficient of variation (CV) of the fluorescent signal (Fig. 4, F and G). Consistent with having fewer, smaller FXR1 structures, cells with both exons 15 and 16 targeted have the smallest CV, indicative of less heterogeneity in the localization due to more soluble, homogeneously distributed protein (Fig. 4, F and G). We conclude that alternative splicing of Fxr1 during myogenesis functions in part to produce isoforms more capable of condensation.
FXR1 assemblies are protein concentration dependent
If FXR1 is undergoing phase separation to form puncta in muscle cells, assembly should be protein concentration dependent. To titrate FXR1 expression, isoform E was placed under the control of a tetracycline (Tet)-inducible promoter in U2OS human osteosarcoma fibroblast cells that had FXR1, FXR2, and FMRP genes knocked out by CRISPR/Cas9 editing (U2OSΔFFF). It was important to use the triple knockout to ensure that endogenous FraX proteins were not acting as seeds for droplets formed with exogenous FXR1. We found that 25 ng/ml Tet-induced expression of mNeonGreen (mNG)-tagged FXR1 isoform E produces approximately endogenous levels (Fig. 5 A). We hypothesized that isoform E should exhibit increased condensation capacity compared with isoform A because of the data presented in Fig. 4. We therefore imaged live U2OSΔFFF cells expressing either mNG-tagged FXR1 isoform A or E at increasing levels of Tet induction (Fig. 5 B). Cellular condensates were observed at levels below the endogenous protein concentration for both isoforms (Fig. 5 B). In support of concentration-dependent condensation, the number of cells with droplets increased with Tet concentration (Fig. 5 C). The proportion of cells with condensates was not statistically different between isoforms A and E. However, we noticed that the condensate morphology differed between the two isoforms (Fig. 5 B). Isoform A formed large spherical droplets, while isoform E formed more irregular-shaped bodies. To evaluate droplet morphology, we assigned all the visualized cells to one of the following categories: (1) irregular or nonspherical-shaped bodies, (2) small spherical droplets, or (3) large spherical droplets (Fig. 5 D). Irregular condensates increased in both isoforms with protein concentration, with a more pronounced trend for isoform E (Fig. 5 E) than for isoform A. Unexpectedly, isoform A produced more large, spherical droplets. Consistently, both isoforms were enriched in the pellet with increasing Tet concentrations (Fig. 5, F and G). Despite having a shorter IDD, a higher proportion of isoform A was detected in the pellet at all Tet concentrations (Fig. 5, F and G), suggesting the composition rather than length of the IDD is important for the traits of the condensates. We suspect that there may be specific PTM differences between myotubes and U2OS cells that could explain the differences between isoforms A and E in these two contexts. Nevertheless, droplet formation is concentration dependent, consistent with phase-separated assembly, morphology, and droplet properties.
Figure 5.

FXR1 phase separation is concentration dependent. (A) Whole-cell extracts from WT U2OS cells or U2OSΔFFF cells transiently transfected with either an empty vector (pAGB1139) or PCMV2xTetO2-mNeonGreen-FXR1 isoform E (pAGB1162) were prepared. Extracts were analyzed by Western blot. Right: Quantification of protein levels normalized to a loading control. (B–G) U2OΔFFF cells were transiently transfected with an empty vector (pAGB1139), PCMV2xTetO2-mNeonGreen-FXR1 isoform A (pAGB1189) or isoform E (pAGB1162). (B) Representative images of cells at increasing Tet concentrations. Scale bar, 5 µm. (C) Quantification of droplet formation for individual cells. n > 720 cells from three independent experiments. (D) Zoomed-in images showing examples of irregular droplets, small spheres, and large spheres. Scale bar, 5 µm. (E) Quantification of droplet morphology. n > 185 cells from three experiments. (F–G) The cell lysate and pellet were analyzed by Western blot as described in A. The percentage of protein in the pellet was quantified as the amount in the pellet out of the amount in the whole-cell extract. Data are means ± SEM. Supe, supernatant; WCE, whole-cell extract.
We next examined the propensity of FXR1 to phase separate in vitro. To confirm RNA dependence and isoform-specific differences of FXR1 for droplet formation in vitro, we purified mouse FXR1 isoforms E and A recombinantly from BL21 cells using a 6xHis-GB1 tag (where GB1 is the B1 domain of protein G; Zhang et al., 2015). GB1 was essential to keep both isoforms A and E soluble during the purification. We observed that in the presence of luciferase RNA, both isoforms E and A had the tendency to slowly aggregate (Fig. 6 A; and Fig. S4 A) rather than form the droplets as observed in vivo (Figs. 4 and 5). Luciferase RNA was chosen as it has previously been shown to induce phase separation of FMR1, which has a similar RNA-binding domain as FXR1 (Tsang et al., 2019). There were subtle differences in the morphology of the aggregates between isoforms E and A. Isoform E formed larger aggregates at lower concentration (2 µM) than isoform A, which is consistent with isoform E containing a longer disordered sequence. Thus, RNA promotes higher order assemblies of FXR1 in vitro.
Figure 6.

RNA promotes and phosphorylation inhibits FXR1 association in vitro. (A) Representative images of FXR1 isoforms A and E aggregates (2 or 4 µM) in the presence or absence of luciferase RNA (magenta). Scale bars, 10 µm. (B) Predicted disorder (IUPRED), unspecified kinase score and CK2 kinase scores for serine are marked for each aa. (C) Representative images of isoform A and E aggregates with or without RNA and with or without phosphorylation (Phos). Scale bars, 10 µm. (D–G) Quantification of particle area (D), RNA fluorescence within particles (E), number of particles (F), and number of particles >0.3 µm2 using the images represented in C. ***P < 0.001, **P < 0.01, *P < 0.05 (Student’s t test, two sided). ns, not significant.
Figure S4.

RNA addition and protein phosphorylation influence FXR1 aggregation in vitro. (A) Luciferase RNA accelerates isoform E aggregation. Representative images for either 4 or 2 µM isoform E with or without luciferase RNA after 3, 4, 5, 6, and 24 h. Scale bars, 10 µm. (B) FMR1 Predicted disorder (IUPRED) and unspecified kinase score for serine are marked for each aa. Predicted phosphorylated serines are uniformly distributed across the protein. (C) Coomassie staining of the in vitro–purified FXR1 isoforms A and E. Phosphorylation (Phos) results in a band shift with respect to the control (Ctrl) or mock-treated samples. (D) Representative images of 2 µM FXR1 isoform E (shown in white) with or without RNA and with or without phosphorylation (Phos) at 3, 4, 5, 6, and 24 h. Scale bars, 10 µm. Fig. S4 is linked to Fig. 6.
We postulated that a PTM not present in the bacteria was responsible for inducing the droplet (rather than aggregate) behavior in mammalian cells. To this end, we characterized potential phosphorylation sites in FXR1 sequences. Strikingly, a number of serine phosphorylation sites were predicted in the IDD of isoforms A and E, with more sites in the longer IDD of isoform E (Fig. 6, B and C). Interestingly, a similar concentration of phosphorylation sites in the IDD was not observed in the FMR1 sequence (Fig. S4 B). To test the importance of FXR1 phosphorylation in vitro, we treated isoforms A and E with casein kinase 2 (CK2) either in the presence or absence of ATP and then incubated the resulting protein either in the presence or absence of luciferase RNA. We confirmed that phosphorylation induced a gel shift in FXR1 protein bands (Fig. S4 C), with a more significant shift in size for isoform E, which had more predicted sites for CK2 phosphorylation (Fig. 6 B).
Consistent with our previous results, RNA promoted aggregation in both control samples of isoform A and E (Fig. S4 D). However, phosphorylation tended to reduce (isoform E) or block entirely (isoform A) the aggregation of FXR1 (Fig. 6 D; and Fig. S4 D). Following 24 h of incubation with RNA, control-treated isoform E formed larger aggregates than isoform A (Fig. 6 E), whereas phosphorylation blocked aggregate formation for isoform A or substantially reduced aggregation for isoform E (Fig. 6 D). Phosphorylation had a minimal but significant effect in the observed RNA signal within the particles at 24 h, likely due to the reduced large particle formation (Fig. 6 G). This observation is consistent with few predicted phosphorylation sites within the RNA-binding domains of FXR1 (N terminus) shared between isoforms E and A (Fig. 6 F). Taken together, our studies revealed that at the same protein concentration in vitro, isoform E is more prone to form aggregates than isoform A, that RNA accelerates and increases the size of aggregates formed by both isoforms, and that phosphorylation reduces aggregation of both isoforms A and E. This evidence suggests that PTMs such as phosphorylation are critical to the regulation of FXR1 functions in vivo.
FXR1 assemblies are RNA-binding domain dependent
The in vitro data suggest RNA is critical for the formation of FXR1 assemblies. To determine which FXR1 domains are required for condensation, we created mutations in the K Homology (KH) domains (Fxr1KH1, Fxr1KH2, Fxr1KH1/2), the arginine-glycine-glycine containing motif (RGG) box (Fxr1ΔRGG), and both a partial truncation (Fxr11-490) and complete truncation of the IDD (Fxr11-374) of the Fxr1 coding sequence in the Tet-inducible mNG-tagged isoform E construct (Fig. 7 A). We then imaged live U2OSΔFFF cells transiently transfected with each mutant induced to endogenous levels and quantified the proportion of cells with droplets (Fig. 7, B and C). When the three RNA-binding domains were mutated (Fxr1ΔRGG KH1/2), no condensates were detected. Intermediate phenotypes were observed for the single RNA-binding mutations, implying that the RNA-binding domains are partially redundant for LLPS. All of the five RNA-binding–deficient constructs had droplet formation significantly different from WT.
Figure 7.

Phase separation is RNA binding dependent. (A) Location of mutations in the domains of isoform E. (B and C) U2OSΔFFF cells were transfected with WT PCMV2xTetO2-mNeonGreen-FXR1 isoform E (pAGB1162), plasmids with mutated KH1 (pAGB1171), KH2 (pAGB1172), KH1/2 (pAGB1173), RGG (pAGB1174), RGG and KH1/2 (pAGB1175), complete IDD (pAGB1176), or partial IDD (pAGB1177). All constructs were induced with 25 ng/ml Tet to endogenous levels. Black Xs indicate mutated regions. (B) Quantification of droplet formation. n > 350 cells from three experiments. (C) Representative images of cells expressing the different mutants. Scale bar, 5 µm. (D) Whole-cell extracts (WCE), lysate, and pellets were analyzed by Western blotting using an antibody against FXR1. Right: percent protein in the pellet quantified as in Fig. 5. (E) Quantification of droplet morphology using the same three categories that were used in Fig. 5. Data are means ± SEM.
We next investigated the role of the IDD in droplet formation. When the IDD was truncated to the RGG (Fxr11-490), we observed significantly more cells with droplets than WT, implying that the longer IDD may limit LLPS nucleation. Truncation of the entire IDD (Fxr11-374) did not abolish droplet formation, suggesting that the disordered sequence is not essential for phase separation even in cells with no WT FraX proteins (Fig. 7, B and C). These data show that the relationship between IDD length and FXR1 droplet formation is nonlinear. The whole-cell extract from transfected cells showed that none of the mutant proteins have reduced expression (Fig. 7 D). Western blots comparing the lysate to the pellet confirmed our observations of condensation from microscopy, with FXR1ΔRGG and FXR11-490 showing the most protein in the pellet and FXR1KH1/2 and FXR1ΔRGG KH1/2 showing the least. While the morphologies of FXR1KH1/2 and FXR1ΔRGG KH1/2 are different (Fig. 7 C), they show similar results in the Western blot assay (Fig. 7 D).
Droplet morphologies were also affected by some of these mutations. Because the Fxr1ΔRGG KH1/2 mutation abolished droplet formation, we did not include this mutant in these analyses. Isoform E showed mostly irregular droplets with some small spherical droplets (Fig. 7 E), suggesting that small droplets are coalescing incompletely into irregularly shaped assemblies. Mutation of the KH domains individually or together increased the percentage of cells with small spherical droplets. Without the KH domains, RNA binding is presumably mediated through the RGG box, which may associate with a different set of RNAs. These RGG-mediated associations still enable small droplets to form, but they have limited or slowed fusion. Deletion of the RGG box alone did not alter droplet formation or morphology, implying that the KH domains are the major RNA-binding domains for phase separation. Interestingly, truncation of the IDD back to the RGG box (Fxr11-490) did not significantly alter droplet morphology, while deletion of the entire IDD (Fxr11-374) caused most cells to have large spherical droplets. This is surprising given that IDDs in other proteins are crucial for this phenotype. These results raise the possibility that RNA interactions are more critical in driving the phase separation of FXR1 than the IDD and that the IDD primarily impacts the material properties of the condensates.
Discussion
FXR1 is hypothesized to regulate mRNA translation, localization, and stability (Mientjes et al., 2004; Vasudevan and Steitz, 2007; Davidovic et al., 2013; Zarnescu and Gregorio, 2013; Cook et al., 2014; Majumder et al., 2016; Patzlaff et al., 2017); however, there is little known about the mechanisms of function of FXR1 in physiological or pathological contexts. Here, we show quantitative differences in FXR1 isoforms arising from tissue-specific and developmentally regulated alternative splicing and a functional role for fxr1 splicing in myotube formation and Xenopus development. Alternative splicing of FXR1 pre-mRNA leads to the expression of distinct protein isoforms that vary substantially in the length of the C-terminal IDD. FXR1 can condense into liquid-like droplets in an RNA-dependent manner. We observed that the different splice isoforms vary in droplet morphology. These data provide a role for developmentally controlled splicing in regulating the formation of biomolecular condensates.
We hypothesized that FXR1 isoforms specifically expressed in myotubes might be important for proper function of muscle cells. Our in vivo studies in Xenopus revealed that fxr1 exon 15 mis-splicing resulted in somite formation defects, while blocking exon 15 inclusion in vitro does not seem to impact myotube formation. One possible explanation of this discrepancy is that MOs are delivered using a liposomal reagent (endoporter) in the cells that we know helps cell fusion. However, we did observe FXR1 condensation in myotubes, and this condensation was reduced when we blocked the alternative splicing required to form the extended IDD (Fig. 4). However, condensates were still observed for the shorter isoform A and for morpholino-treated cells, which shortens IDDs. This suggests that the change in protein sequence via splicing is important for regulation of condensation with RNA, not for conferring the ability to condense. We postulate that differential IDD sequences can regulate condensation through PTMs, altered RNA targets, and/or protein–protein interactions.
FXR1 requires RNA to form droplets, such that elimination of all RNA-binding domains blocks LLPS (Fig. 6). The RNA-binding sites are partially redundant. RNA drives the phase separation of other RBPs that undergo LLPS, including WHI3, LAF-1, and multiple P-granule proteins (Elbaum-Garfinkle et al., 2015; Zhang et al., 2015; Smith et al., 2016; Langdon et al., 2018). Our in vitro experiments confirm that luciferase RNA promotes FXR1 aggregation, and future experimentations are needed to examine the effect of native RNA targets of FXR1. One experimental issue in this regard is that the RNA targets of FMR1 and likely FXR1 are quite long (Greenblatt and Spradling, 2018). To our knowledge, this fact has made them very difficult to synthesize with current techniques. We speculate that longer RNAs may be required to induce droplet formation in vitro. Only isoform E binds G-quadruplex structures (Bechara et al., 2007; Lyons et al., 2017), which promote LLPS (Fay et al., 2017). FXR1 also may have nonspecific RNA interactions via the RGG domain. It will therefore be important to understand whether specific targets of FXR1 are primarily driving phase separation and how the different modes of RNA interactions (i.e., KH versus RGG) influence droplet assembly.
One of the striking findings in this study is that a longer IDD led to more abundant and functional condensates in myotubes but limits FXR1 LLPS when analyzed in U2OS cells. While in myotubes, isoform E forms more, brighter puncta, in U2OS cells we found that the short isoform A enriches in pellets at lower protein concentrations than the long isoform E and isoform A is more likely to be found in very large and highly spherical droplets. By contrast, in U2OS cells isoform E tends to be in small or irregular droplets, which we suspect are either arrested in fusion or very slow to fuse (Fig. 5). Given that serine/arginine-rich sequences can be heavily impacted by PTMs, we hypothesize that the additional sequence provides more residues to enhance modifications that disfavor phase separation in this context.
Interestingly, isoforms A and E aggregate rather than form droplets in vitro, which is quite different than results obtained for FMR1 (Tsang et al., 2019). A potential explanation for this difference is that phosphorylation sites of FMR1 are more evenly distributed across the protein rather than strongly concentrated within the IDD, as we observed in FXR1. We found that phosphorylation strongly suppressed the FXR1 aggregation phenotype observed in vitro with a stronger impact on isoform E whose IDD contains 41 serine residues, while isoform A has 26. Phosphorylation has a well-established role in influencing biomolecular condensation (Aumiller and Keating, 2016; Hofweber and Dormann, 2019; Snead and Gladfelter, 2019) and is both rapid and reversible. Phosphorylation has previously been shown to either promote dissolution of granules (Wippich et al., 2013; Rai et al., 2018) or inhibit phase separation (Rai et al., 2018), as we found for FXR1. Interestingly, phosphorylation of the IDD of FMR1, an FXR1 homologue, is required for its interaction with another RNA-binding domain, Caprin, to tune interactions with RNA. Phosphorylation of FMR1 did not block phase separation, suggesting that phosphorylation-dependent inhibition is unique to FXR1, although the authors did not test the full-length FMR1 or native FMR1 (Kim et al., 2019). Thus, going forward, it is important to consider how splicing may regulate the patterns and levels of PTMs within IDD sequences, which may change the protein’s ability to interact with other RBPs or RNAs.
More surprising was the analysis of proteins missing the C-terminal IDD completely (Fig. 7 E). A fragment containing only the N terminus and lacking any predicted IDD robustly formed large, highly spherical droplets. The N-terminal Agenet domains may mediate interactions with other phase-separating proteins to recruit the truncated FXR1. In this case, the Agenet domains would provide multivalency to contribute to the normal LLPS process. The Agenet domains of FMR family proteins bind methylated lysine residues (Adams-Cioaba et al., 2010); therefore, PTMs of binding partners could modulate FXR1 interactions throughout development. Finally, the developmental splicing program may alter which mRNAs are available for FXR1 interaction, and depending on the identity of the bound mRNAs, the IDD role may vary from what we see in U2OS cells.
In summary, we have shown a functional role for FXR1 splicing in development, to promote extension of an IDD relevant for FXR1 condensation with RNA. The unexpected negative effect of a longer IDD on the propensity to phase separate in U2OS cells raises important possible mechanisms for how IDDs may contribute to biomolecular condensates in different cell types. Future work will determine how condensates control translation of associated mRNAs and whether FXR1 splice isoforms lead to different targets during tissue identity acquisition and maintenance. In conclusion, we link a developmental splicing program to changes in condensation in FXR1, a mechanism that may be generalizable to other proteins that undergo phase separation.
Materials and methods
Materials
Chemicals and cell culture reagents were obtained from GIBCO, Life Technologies, Thermo Fisher Scientific, and Sigma. C2C12 cells and WT U2OS cells were purchased from American Type Culture Collection. Human RNA samples were purchased from the following commercial sources: (1) heart (male, 21 yr old) from Amsbio (R1234139-50, lot no. B707146), (2) heart (male, 51 yr old) from Cell Applications Inc. (1H30-50, lot no. 1639), (3) heart (male, 30–39 yr old) from TakaRa (636532, lot no. 1610293A), (4) skeletal muscle (female, 48 yr old) from Amsbio (R1234171-50, lot no. B207200), (5) skeletal muscle (male, 58 yr old) from Cell Applications Inc. (1H60-50, lot no. 1376), (6) skeletal muscle (male and female, 20–68 yr old) from TakaRa (636534, lot no. 1609179A), (7) heart (female, 22 wk of gestation) from Cell Applications Inc. (1F30-50, lot no. 2739), (8) heart (male, 20–21 wk of gestation) from Agilent (540165, lot no. 0006262824), (9) skeletal muscle (female, 22 wk of gestation) from Cell Applications Inc. (1F60-50, lot no. 2739), and (10) skeletal muscle (female, 18 wk of gestation) from Agilent (540181, lot no. 0006260887).
Mice
FVB/NJ mice were purchased from The Jackson Laboratory. Colonies were derived by mating animals. Mice were handled following the National Institutes of Health (NIH) Guidelines for Use and Care of Laboratory Animals that were approved by the Institutional Animal Care and Use Committee at The University of North Carolina at Chapel Hill.
Mouse tissue isolation
Adult (4–5 mo old) and neonatal (P4.5) mice were euthanized using isoflurane. After cervical dislocation (adult) or decapitation (P4.5), tissues were removed and frozen in liquid nitrogen. WT C57BL/6J mouse embryos at day 15.5 were donated from Dr. Stephanie Gupton’s laboratory (University of North Carolina at Chapel Hill, Chapel Hill, NC). Embryos were rinsed with PBS, the thoracic cavity was opened, and the hearts were dissected. The epidermal layer was then removed, and the fore- and hind limbs were excised after removing the paws. Embryonic tissue was blotted dry and snap frozen in liquid nitrogen.
Cell culture
C2C12 undifferentiated cells (myoblasts) were maintained at 37°C under 5% CO2 in DMEM supplemented with 10% FBS (Gemini Bio-Products), 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin under low confluence conditions (<40%). For differentiation of myoblasts into myotubes, cells were washed with PBS and then cultured in DMEM supplemented with 2% horse serum, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. U2OS cells were maintained at 37°C under 5% CO2 in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. For plasmid transfection, cells were transfected at ∼60–70% confluence with Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s protocols. In Tet-experiments, 24 h after transfection, cells were induced with Tet for 16 h and subsequently used for the assays.
Generation of U2OSΔFFF
To create the U2OSΔFFF cell line, FXR2 was first knocked out using a guide RNA with the sequence 5′-CCCCATAGGTTCGAGTCGCA-3′ in the U2OS Tet-inducible cells. Several clones were selected, and FXR2 protein expression was evaluated by immunofluorescence and Western blotting. Clone 6 was selected for subsequent experiments. This cell line was then cotransfected with Cas9 and guide RNAs targeting FXR1 (5′-TTCCTAGGAATCTCGTTGGT-3′) and FMR1 (5′-AAGAGGCGGCACATAAGGAT-3′). Clones were selected and screened in a similar manner, and finally clone 24 was selected. All loci were sequenced to confirm deletions in the DNA.
Plasmids
Restriction enzymes and competent cells were from New England Biolabs (NEB). Oligonucleotides used for plasmid generation were synthesized at Integrated DNA Technologies. PCRs were performed using iProof high-fidelity polymerase (Bio-Rad). Sequencing was performed by Genewiz. The sequences of the primers used for cloning are detailed in Table S1.
The mNG-tagged constructs were created by first making a pCMV-mNeonGreen plasmid (pAGB1103). The mNG ORF was amplified from pAGB1070 (pNCS-mNeonGreen AmpR; Allele Biotechnology) with the primers AGO2195 and AGO2196 via PCR. The resulting product was cloned into pAGB879 (PCMV-Fxr1isoE-tGFP AmpR; Origene) cut with BglII and SacI (to remove Fxr1-turboGFP from the plasmid) using the NEBuilder HiFi DNA assembly reaction (NEB). To make pAGB1111, Fxr1 isoform E was then amplified with primers AGO2202 and AGO2203 via PCR and cloned via NEBuilder HiFI DNA assembly reaction into pAGB1103 previously cut with NotI and SacII to make an N-terminally tagged construct. To create the C-terminal truncations where the IDD was either truncated back to the RGG (Fxr11-490) or completely truncated (Fxr11-374), the same approach was taken using different reverse primers to only amplify the first 1,122 bp of the ORF (pAGB1176 = Fxr11-374, reverse primer was AGO2206) or the first 1,470 bp of the ORF (pAGB1177 = Fxr11-490, reverse primer was AGO2207). All of the mutations were created in the Tet-inducible mNG-tagged isoform E construct.
To mutate the KH domains, point mutations were created in the conserved GxxG protein loop required for nucleic acid binding in either the first (Fxr1KH1), second (Fxr1KH2), or both domains (FXR1KH1/2). These mutations should abrogate nucleic acid binding to the KH domain without destabilizing protein structure (Hollingworth et al., 2012; Wong et al., 2013; Wang et al., 2015) These point mutations and deletions were created via site-directed mutagenesis using the following primers for each mutation: T236D H237D (KH1), AGO1869 and AGO1870 and K299D N300D (KH2), AGO1871 and AGO1872. To analyze the effect of loss of the RGG box, this region was completely removed (Δ464–490) to create Fxr1ΔRGG using site-directed mutagenesis with primers AGO1873 and AGO1874.
To move all mNG-tagged mutations and truncations into the Tet-inducible plasmid, pAGB1139 (PCMV2xTetO2 AmpR ZeoR) was cut with BamHI and EcoRI so that each insert could be cloned into the plasmid via NEBuilder HiFi DNA assembly reaction. The primers AGO2350 and AGO2351 were used to amplify the following mNG-tagged ORFs for assembly into pAGB1139: WT (creating pAGB1162), KH1 (creating pAGB1171), KH2 (creating pAGB1172), KH1/2 (creating pAGB1173), ΔRGG (creating pAGB1174), and ΔRGG KH1/2 (creating pAGB1175). Different reverse primers were used to amplify mNeonGreen-Fxr1isoE1-374 (AGO2374, creating pAGB1166) and mNeonGreen-Fxr1isoE1-490 (AGO2375, creating pAGB1167) for assembly into pAGB1139. To create the mNG-tagged isoform A construct, isoform A was first created in the pCMV isoform E background (creating pAGB936) using primers AGO1863 and AGO1864 to remove aa 569–677 and change aa 564–568 from DDSEK to GKRCD, and primers AGO1867 and AGO1868 to remove internal aa 308–408. To move this ORF into the Tet-inducible system with an N-terminal mNG tag, mNG was amplified from pAGB1103 with primers AGO2350 and AGO2486, and Fxr1isoA was amplified from pAGB936 with primers AGO2487 and AGO2488. Both inserts were integrated into pAGB1139 cut with BamHI and EcoRI via NEBuilder HiFi DNA assembly reaction to create pAGB1189. All constructs and mutations were verified by sequencing.
Delivery of siRNAs into C2C12 cells
C2C12 cells were seeded into six-well plates (8 × 104 cells/well) in DMEM supplemented with 10% FBS and 2 mM glutamine. The next day, transfections were performed using Lipofectamine RNAiMax transfection reagent (Invitrogen) and Stealth siRNAs (Invitrogen) following the manufacturer’s protocols. The siRNA sequences were as follows: (1) si-luc (luciferase reporter control, no. 12935146), 5′-GCACUCUGAUUGACAAAUACGAUU-3′ (sense) and 5′-AAAUCGUAUUUGUCAAUCAGAGUGC-3′ (antisense); (2) si-Fxr1-#1 (MSS204455), 5′-GCAAUCCAUACAGCUUACUUGAUAA-3′ (sense) and 5′-UUAUCAAGUAAGCUGUAUGGAUUGC-3′ (antisense); and (3) si-Fxr1-#2 (MSS204457), 5′-GAAGUUGAUGCUUAUGUCCAGAAAU-3′ (sense) and 5′-AUUUCUGGACAUAAGCAUCAACUUC-3′ (antisense). The next day, cells were washed with PBS and differentiation was induced for 4–5 d.
Delivery of MOs into C2C12 cells
C2C12 myoblasts were seeded (8 × 104 cells/well) in DMEM supplemented with 10% FBS and 2 mM glutamine into six-well plates or six-well plates with 12-mm coverslips pretreated with 40 µg/ml PureCol (Advanced BioMatrix). The following day, 10–20 µM MOs (Gene Tools, LLC) were delivered using 6–10 µM aqueous Endo-Porter (Gene Tools, LLC) following the suggested protocol from the manufacturer. The MO oligo sequences were as follows: (1) control, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; (2) Fxr1 mo-1, 5′-AACAGGCCCACTCAAGTTACCTGGC-3′; and (3) Fxr1 mo-2, 5′-GCAACTGTGACTGTTAAAGATGAGA-3′. The day after delivery, cells were washed with PBS and differentiated in DMEM supplemented with 2% horse serum and 2 mM glutamine for 3–5 d.
Injection of MOs and sgRNAs in X. tropicalis and X. laevis
Xenopus were handled following the NIH Guidelines for Use and Care of Laboratory Animals that were approved by the Institutional Animal Care and Use Committee at the Marine Biological Laboratory, Woods Hole, MA. In Fig. 1, C and D, Xtr.Tg(pax6:GFP;cryga:RFP;act1:RFP)Papal (RRID:NXR_1021) embryos were injected with 20 ng of mo-e15i15 or standard mo-control and analyzed at stage 45. In Fig. 1, E and F, Xla.Tg(actc1:GFP)Mohun (RRID:NXR_007) embryos were injected with 10 ng of standard mo-control, 10 ng of mo-i14e15 (targeting both L and S genomes) or coinjected with 1,500 pg of Cas9 protein plus 500 pg of each sgRNA (T1, T3) and analyzed at stages 35–38. In Fig. 1 G, one cell at the two-cell stage was injected with 250 pg of each sgRNA (T7L, T7S, T8L, T8S) and 1,500 pg of Cas9 protein or with 10 ng of mo-i14e15. Phenotypes were analyzed at stage 37-38. MO and sgRNA sequence were as follows: (1) X. tropicalis mo-e15i15, 5′-GTAACTAAAGTGGGTGGAGCTATTG-3′; (2) X. laevis mo-i14e15, 5′-AAACTTTGCTGTTGCAGATGATAGT-3′; (3) mo-control, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; (4) T1 sgRNA, 5′-CCCCTGAAGCGACGCCTGCGG-3′; (5) T3 sgRNA, 5′-GGCAGAAGATAGACAGCCAGG-3′; (6) T7L sgRNA, 5′-ACTGCAGGTTGCAAACATATTGG-3′; (7) T7S sgRNA, 5′-TACTATTGCTAGTCTTCAAGAGG-3′; (8) T8L sgRNA, 5′-TCAGGACAATGGTTCTTAAATGG-3′; and (9) T8S sgRNA, 5′-AAATGATAGAATGTCTAGTGTGG-3′. All sgRNAs include a protospacer adjacent motif sequence.
RNA extraction
Frozen mouse tissues were homogenized using 1.4-mm ceramic beads (Lysing Matrix D) and a Precellys-24 homogenizer (Bertin Instruments) with the following setting: 6,500 rpm for 20-s intervals until complete homogenization. RNA was then extracted using TRIzol (Invitrogen), following the manufacturer’s protocol. For cell culture samples, TRIzol reagent was added to the plates followed by RNA extraction per the manufacturer’s suggested protocol. For Xenopus experiments, RNA was extracted from whole embryos (seven embryos per sample) using TRIzol, following the manufacturer’s protocol. RNA concentration was measured with a NanoDrop Lite spectrophotometer (ND-LITE; Thermo Fisher Scientific).
cDNA synthesis
Commercial human RNA samples or RNA extracted from mouse tissues, Xenopus embryos, or cells was used to prepare cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems) or the SuperScript IV kit (Invitrogen). The program was as follows: (1) 25°C for 10 min, (2) 37°C for 120 min, (3) 85°C for 5 min, and (4) 4°C pause.
Analysis of Fxr1 alternative splicing
The synthesized cDNA was used to perform PCR assays with the mouse (0.5 µM), X. tropicalis (0.4 µM), or human primers (0.5 µM) targeting the constitutive exons flanking the alternative regions. The sequences of the primers (Sigma or IDT) were as follows: (a) mouse Fxr1-81-92-f (5′-TGCTGTTCTGATGGATGGAC-3′) and mouse Fxr1-81-92-r (5′-GAAGCGCTAGTTGGACCATT-3′) to analyze the inclusion of exons 15 (81 nt) and 16 (92 nt; expected bands 300, 219, 208, 127 bp); (b) mouse Fxr1-87-78-f (5′-AGAAAGCATTGGGAATGTGC-3′) and mouse Fxr1-87-78-r (5′-TGTCTCGCTGATGTCGAGTC-3′) to analyze the insert within exon 12 (87 nt) and the alternative splice site in exon 13 (expected bands 377, 299, 290, 212 bp); (c) human FXR1-81-92-f (5′-TGCTGTTCTGATGGATGGAA-3′) and human FXR1-81-92-r (5′-AGCACTAGTTGGGCCGTTTA-3′) to analyze the inclusion of exons 15 (81 nt) and 16 (92 nt) (expected bands 298, 217, 206, 125 bp); (d) X. tropicalis fxr1-F1 (5′-TCTCACCACAACACAAACCG-3′) and X. tropicalis fxr1-R1 (5′-GCGGAATCTACCTAAAGAACC-3′) to analyze the inclusion of exon 15 (81 nt; expected bands 266, 185 bp); and (e) X. tropicalis eef1a1-F (5′-CCCCTCTTGGTCGTTTTGCTGTCC-3′; spans junction between exons 7 and 8) and X. tropicalis eef1a1-R (5′-TTGCCTTTCTGTGCTTTCTGAGCAG-3′; end of coding sequence in exon 8) used as a loading control (expected band 135 bp). PCRs for C2C12 cells, mouse tissues, and human samples were performed using GoTaq green master mix (Promega) and the following amplification conditions: (1) 95°C for 1 min 15 s; (2) 28 cycles of 95°C for 45 s, 57°C for 45 s, 72°C for 1 min; (3) 72°C for 10 min; and (4) 4°C pause. PCR products were separated by 6% PAGE in TBE buffer (89 mM Tris, 89 mM boric acid, and 2.5 mM EDTA, pH 8.3) for 3–4 h at 165 V. After electrophoresis, the gels were stained with 0.4 µg/ml ethidium bromide in water for 10 min and visualized using the ChemiDoc XRS+ imaging system (Bio-Rad). Quantification of gels was performed by densitometry using Image Lab 6.0.1 software (Bio-Rad) for analysis. In X. tropicalis experiments, PCRs were performed using Taq Polymerase (NEB standard protocol) and the following amplification conditions: (1) 95°C for 1 min; (2) 27 cycles of 90°C for 30 s, 60°C for 30 s, 72°C for 16 s; (3) 72°C for 5 min; and (4) 4°C pause. PCR products were separated by 2% agarose gel electrophoresis for 40 min at 135 V.
Real-time PCR (quantitative PCR [qPCR])
A 20-µl reaction with TaqMan Fast Advanced Master Mix (Applied Biosystems) was used to quantify 50–100 ng of cDNA with TaqMan probes for Fxr1 (Mm00484523-m1, amplicon size 73 bp; Applied Biosystems) and Hmbs (Mm01143545-m1, amplicon size 81 bp; Applied Biosystems) transcripts. An Applied Biosystems StepOnePlus Real-Time PCR System was used and the thermal cycling profile was as follows: (1) 50°C for 2 min; (2) 95°C for 20 s; and (3) 40 cycles of 95°C for 1 s, 60°C for 20 s. The cycle threshold values of quantified transcripts were normalized to that of a reference gene (Hmbs) from the same sample.
Protein extraction from mouse tissues
Frozen tissues were homogenized with 1.4-mm ceramic beads (Lysing Matrix D’; MP Biomedicals) in a Hepes/sucrose buffer (10 mM Hepes, 320 mM sucrose, and 1% SDS) supplemented with protease and phosphatase inhibitors (Sigma). The Precellys-24 homogenizer was used at 6,500 rpm for 20-s intervals until complete tissue homogenization. Samples were then sonicated in an ice water bath for 3 min (30-s bursts) at 75 V. Lysates were collected after spinning at 14,000 rpm for 10 min at 4°C, and the protein concentration was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific).
Protein extraction from cell culture
Cells were lysed in RIPA buffer (1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 150 mM NaCl, 50 mM Tris, and 5 mM EDTA, pH 8.0) supplemented with protease and phosphatase inhibitors (Roche). Cell lysates were sonicated for 3 min (30-s bursts) at 75 V. After spinning at 14,000 rpm for 10 min at 4°C, supernatants were collected and protein concentration was measured using the Pierce BCA protein assay kit. To analyze insoluble aggregates by Western blot, C2C12 or U2OS cells were lysed in NETN lysis buffer (250 mM NaCl, 5 mM EDTA, pH 8.0, 50 mM Tris-HCl, pH 8.0, and 0.5% NP-40) supplemented with protease inhibitors for 30 min on ice. Lysates were clarified by centrifugation for 10 min at 13,000 rpm at 4°C. The soluble lysate was separated from the pellet, and the pellet was resuspended in 50 µl of lysis buffer. Equivalent fractions were run on an SDS-PAGE gel for analysis via Western blotting.
Western blot assays
Protein samples (25 µg; Figs. 3 C and 4 E; and Figs. S2 C and S3 A) prepared in loading buffer (62.5 mM Tris-HCl, 10% glycerol, 2% SDS, 0.02% bromophenol blue, 143 mM β-mercaptoethanol, and 5 mM DTT, pH 6.8) were boiled for 5 min and then loaded into 10% Mini-Protean Tris-Glycine eXtended Stain-Free precast gels (Bio-Rad). Electrophoresis was performed in a buffer containing 25 mM Tris, 190 mM glycine, and 0.1% (wt/vol) SDS, pH 8.3, for 2 h at 120 V. After electrophoresis, the gels were exposed to UV light for 2.5 min to activate Tris-Glycine eXtended within the gel or counterstained with Ponceau stain solution for total protein visualization. Proteins were transferred onto 0.45-µm polyvinylidene fluoride Immobilon-FL membranes (Millipore Sigma) and visualized using the ChemiDoc XRS+ imaging system. Membranes were blocked with 5% nonfat dried milk in TBST buffer (19 mM Tris, 2.7 mM KCl, 137 mM NaCl, and 0.1% Tween 20, pH 7.4) for 1 h, washed, and incubated overnight at 4°C with primary antibodies diluted in 5% BSA in TBST. Primary antibodies were as follows: (1) rabbit monoclonal (EPR7932) anti-FXR1 (ab129089; Abcam) diluted 1:1,000; (2) mouse monoclonal anti-GAPDH (sc365062; Santa Cruz Biotechnology) diluted 1:500; (3) mouse monoclonal anti–α tubulin (sc32293; Santa Cruz Biotechnology) diluted 1:500; and (4) rabbit polyclonal anti–β tubulin (ab6046; Abcam) diluted 1:1,000. The next day, the membranes were washed three times (10 min each) with TBST and then incubated for 1–1.5 h at RT in the darkness with a goat polyclonal anti–rabbit IgG (H+L) DyLight 800 4X PEG (SA5-35571; Invitrogen) diluted (1:10,000) in 5% BSA in TBST. Membranes were washed three times (10 min each) with TBST, and fluorescent signal was detected using the Odyssey CLx Blot Imager (Li-Cor). Western blots (Figs. 4, 5, and 7) were performed as described above, with the following differences. SDS-PAGE gels were transferred to a nitrocellulose membrane and subsequently blocked for 2 d in 5% milk in 50 mM Tris, 150 mM NaCl, and 0.1% Tween 20. Membranes were washed and incubated overnight at 4°C with primary antibodies. Primary antibodies were as follows: (1) rabbit monoclonal (EPR7932) anti-FXR1 (ab129089; Abcam) diluted 1:1,000 and (2) rabbit anti-CDC2 (Santa Cruz Biotechnology) diluted 1:1,000. The membranes were then washed three times (10, 5, and 5 min) and incubated for 1 h at RT with goat anti–rabbit IgG (H+L) (Bio-Rad). Membranes were washed and signal was detected using the ChemiDoc XRS+ imaging system.
Immunofluorescence assays
Cells in plates or coverslips were washed three times with PBS and fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 20 min at RT. Cells were washed three times in PBS, incubated in blocking solution (137 mM NaCl, 2.7 mM KCl, 9.6 mM Na2HPO4, 1.5 mM KH2PO4, 1% BSA, and 0.3% Triton X-100, pH 7.4) for 1 h at RT, and then incubated with the primary antibodies diluted in blocking solution overnight at 4°C. Primary antibodies were as follows: (1) rabbit monoclonal (EPR7932) anti-FXR1 (ab129089; Abcam) diluted 1:250 and (2) mouse monoclonal (B-5) anti–myosin heavy chain (MYH; sc-376157; Santa Cruz Biotechnology) diluted 1:50. The next day, cells were washed three times (10 min each) with PBS and then incubated with the appropriate secondary antibody diluted in blocking solution for 1 h at RT in the darkness. Secondary antibodies were as follows: (1) rabbit polyclonal anti–mouse IgG (H+L) Alexa Fluor 488 (A21204; Invitrogen) diluted 1:500 and (2) goat polyclonal anti–rabbit IgG (H+L) Alexa Fluor 488 (A11008; Invitrogen) diluted 1:250. Cells were washed three times (10 min each) with PBS and then stained with 2 µM DAPI in PBS for 5 min at RT in the darkness. Cells were washed three times (5 min each) in PBS and imaged by confocal microscopy in PBS. When cells were stained with phalloidin, incubation with Alexa Fluor 647 Phalloidin (A22287, dilution 1:40; Invitrogen) was performed concurrently with the secondary antibody.
Confocal microscopy for cells
For imaging in Fig. S3, confocal microscopy was performed in the Hooker Imaging Core (University of North Carolina Chapel Hill, Chapel Hill, NC) using an LSM 880 confocal microscope (Zeiss) with a 10× Plan Apochromat objective (0.45 working distance) and Zen Black (Zeiss) software for image acquisition. Excitation parameters were as follows: argon multiline laser at 488 nm (Alexa Fluor 488; 2% power), a 405-nm diode at 30 mW (DAPI; 2% power), or a helium-neon laser at 633 nm (2% power). Emission filters were set up as follows: 490–615 nm (Alexa Fluor 488), 410–514 nm (DAPI), and 638–747 nm (Alexa Fluor 647). Cells were imaged at RT. For imaging in Fig. 4 C, live cells were imaged at RT on a Quorum WaveFX spinning disk confocal system equipped with a Plan Apochromat 60× 1.4 numerical aperture objective. Images were acquired on an electron multiplying-charged coupled device camera (C9100-13; Hamamatsu) driven by MetaMorph software version 7.8.12.0 (Molecular Devices). A 488-nm laser was used for GFP excitation using 20% laser power, 500-ms exposure, and 200 gain. Cells were imaged every 15 s through the complete depth of the cell with 0.5-µm inter-plane spacing. For Fig. 4, B and G; and Figs. 5 and 7, fixed or live cells were imaged on Yokogawa CSU-W1 spinning disk confocal system equipped with a VC Plan Apo 60× 1.49 numerical aperture oil immersion objective at RT. Fixed cells were imaged in 1× PBS, while live cells were imaged in FluoroBrite DMEM (Gibco). Images were acquired using an sCMOS 85% QE camera (Photometrics) driven by Nikon Elements Software. For Fig. 4, B and G the following parameters were used: 488-nm laser (Alexa Fluor 488) at 25% power and 200-ms exposure, 405 nm laser (Hoechst) at 15% power and 100-ms exposure, and 640-nm laser (Alexa Fluor 647 Phalloidin) at 50% power and 100-ms exposure. For Figs. 5 and 7, the following parameters were used: 488-nm laser (mNG) at 25% power and 100-ms exposure and 405-nm laser (Hoechst) at 15% power and 100-ms exposure. For Fig. 4, there is no correlation between the CV and mean fluorescence intensity of the cell. Droplets were scored as spherical or irregular based on their aspect ratio. Small clusters were composed of fewer than four droplets.
Microscopy in Xenopus experiments
Images of Xenopus tadpoles in Fig. 1 were acquired at RT in imaging media (0.1× Marc’s Modified Ringer’s Solution) on a Zeiss SteREO Discovery.V12 microscope with an Achromat S 1.25× objective and a mounted Zeiss AxioCam MRc camera controlled with the ZEN 2 lite (blue edition) software version 2.0.0.0. The filter blocks 45 Texas Red/FRFP and 57 GFP LP (long pass) were used to acquire red or green fluorescent images, respectively. Prior to imaging, the tadpoles were anesthetized until complete loss of mobility using 0.1% Tricaine-S (Western Chemical, Inc.) buffered to pH 7.4 with NaHCO3.
Protein purification
FXR1 isoforms A and E were cloned into the His-GB1 vector and Sanger sequenced to confirm insert. Plasmids were transformed into BL21 bacteria. 2xYT media containing kanamycin was inoculated with a colony of freshly transformed BL21 (DE3). Cultures were grown at 37°C overnight with shaking. The next morning, the starter culture was diluted 1:60 and was incubated at 37°C for 2 h and then at 30°C until culture reached an OD of 0.6–0.7. The culture was induced with 1 mM IPTG and then incubated at 30°C for 6 h. Cells were pelleted at 10,000 rpm for 5 min, and pellets were resuspended in 1.5 M KCl, 50 mM Hepes, pH 7.4, 20 mM imidazole, 5 mM β-mercaptoethanol, 1.5 ml of 10 mg/ml lysozyme, and protease inhibitor for 30 min at 4°C. Samples were then sonicated (10 s on, 2 min off, 10 s on) until homogenous. Samples were centrifuged at 20,000 rpm for 30 min, and the supernatant was passed through a 0.44-µm filter using a 60-ml syringe. FXR1 was purified from the supernatant using a Ni- or Co-nitrilotriacetic acid column following standard protocols for Whi3 purification (Langdon et al., 2018). Purified proteins were analyzed by SDS-PAGE and Coomassie staining.
CK2 in vitrophosphorylation assay
In vitro phosphorylation reactions were performed according to the manufacturer’s instructions (P6010S; NEB) using 0.9 nmol of protein with control phosphorylation performed in the absence of ATP with all other reaction components. Reactions were incubated for 50 min at 30°C. In the last 10 min of incubation, 60 µM Atto 488 dye (41051-1MG-F; Sigma) was added to the reaction. Following incubation and labeling (1 h total), samples were buffer exchanged using CENTRI SPIN-20 columns (CS-201; Princeton Separations) according to the manufacturer’s instructions to remove the buffer, excess dye, and ATP. Protein concentration was requantified using a nanodrop. Phosphorylation was confirmed by gel electrophoresis followed by Coomassie staining.
In vitro transcription
Cy5-labeled luciferase RNA was produced according to the manufacturer’s instructions (E2040S; NEB) labeled with 6% Cy5-UTP (PA53026; Millipore Sigma). Reaction was incubated at 37°C overnight and precipitated using 2.5 M LiCl. Pellets were washed three times with 70% ethanol in diethylpyrocarbonate-treated water. RNA samples were eluted in diethylpyrocarbonate-treated water and quantified using a nanodrop. In vitro–transcribed RNA (∼5 µg) was analyzed by agarose gel electrophoresis to confirm the presence of a single band with the appropriate size.
In vitro assays
Luciferase RNA (10 nM) and 2 or 4 µM FXR1 isoforms A and E (Atto 488 labeled) were mixed in droplet buffer (150 mM KCl, 20 mM Hepes, and 1 mM β-mercaptoethanol, pH 7.4) and added to 30 mg/ml fatty acid–free BSA (A8806; Sigma) in droplet buffer–blocked chamber slides (2024-03; Grace Bio-Labs) and incubated at RT for 3 h before imaging.
Image processing and quantitative analysis
Processing of microscopy images and quantitative analysis were performed using Fiji software (Schindelin et al., 2012; Schneider et al., 2012) or ImageJ software (National Institutes of Health). Images were identically contrasted with respect to black-and-white values within each figure. The fusion index was estimated as the ratio between the number of nuclei within MYH-positive cells with more than two nuclei and the total number of nuclei. To quantify the CV in Fig. 4, a region of interest was first drawn around the most focused z-stack for each cell with more than two nuclei excluding the nuclei. The mean gray value was acquired in the FXR1 channel by calculating the sum of the gray values of all the pixels in the region of interest divided by the number of pixels. This value was then background subtracted. The SD of the gray values for each cell was then quantified. The CV was then calculated by dividing the SD of a cell by its background-subtracted mean gray value. For droplet formation analysis, cells were visually scored for either having droplets or not. For morphology quantification, cells were visually scored for the three morphology categories observed. For in vitro assays, images were quantified using Fiji software as follows: Protein signal (488) was used to threshold to white level of 137 (a.u.), and the Analyze Particle plugin was used to define the area of the particles in the image. These regions of interest were then used to extract the RNA and protein fluorescent signal. Three images from three replicates were used for quantification.
Software
Protein disorder was predicted using the IUPRED algorithm (Dosztányi et al., 2005a, 2005b). The aa composition of the C-terminal IDD of FXR1 was determined using IDD Navigator with IUPRED disordered domain prediction (Patil et al., 2012) along with the ProtParam tool (https://web.expasy.org/protparam/); Wilkins et al., 1999). For phosphorylation prediction, the sequences of the His-GB1-FXR1 A/E, or FMR1 (uniprot) were analyzed using the NETphos 3.1 predictor (serine phosphorylation) and IUPRED (disorder).
Statistics
All data are presented as the mean ± SEM. The P values were estimated by a Student’s t test (two tailed) or one-way ANOVA with Bonferroni’s test for multiple comparisons. P ≤ 0.05 was considered statistically significant.
Online supplemental material
Fig. S1 shows the sequences of fxr1 exon 15 from sgRNA-treated embryos. Fig. S1 is linked to Fig. 1, E and F. Fig. S2 shows the splicing events in exons 12 and 13 of Fxr1 pre-mRNA and expression of FXR1 protein isoforms during tissue development. Fig. S2 is linked to Fig. 2. Fig. S3 shows that FXR1 is required for myogenesis. Fig. S3 is linked to Figs. 3 and 4. Fig. S4 shows that RNA addition and protein phosphorylation influence FXR1 aggregation in vitro. Fig. S4 is linked to Fig. 6. Table S1 details the primers used for cloning.
Supplementary Material
shows the sequence of the primers (IDT) used for plasmid generation. Uppercase bases denote sequences that will directly base pair with the plasmid being amplified, while lowercase bases are overhangs for NEBuilder HiFi DNA assembly reaction.
Acknowledgments
We thank the A.S. Gladfelter and J. Giudice laboratories, Nancy Kedersha, and Silvia Ramos for critical discussions; Eunice Y. Lee for technical help; Dr. Stephanie Gupton (University of North Carolina at Chapel Hill, Chapel Hill, NC) for donation of WT C57BL/6J mouse embryos; and Marcin Wlizla and National Xenopus Resource (RRID:SCR_013731) for their help in maintaining adult frogs and other important technical support.
This work has been funded by a University of North Carolina at Chapel Hill Junior Faculty Development Award (to J. Giudice); a Nutrition and Obesity Research Center, University of North Carolina at Chapel Hill, Pilot & Feasibility Research grant (P30DK056350 to J. Giudice); University of North Carolina at Chapel Hill startup funds (to J. Giudice); the March of Dimes Foundation (5-FY18-36, Basil O’Connor Starter Scholar Award to J. Giudice); and NCTraCs Pilot Grant (550KR181805) from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR002489 (to J. Giudice), National Institutes of Health National Institute of General Medical Sciences grants (R01-GM130866 to J. Giudice, R01-GM081506 to A.S. Gladfelter, R35-GM126901 to P. Anderson, K99-GM124458 to S.M. Lyons, R25-GM089569 and 2R25-GM055336-20 to E.G. Curry); Howard Hughes Medical Institute Faculty Scholars program (A.S. Gladfelter), and National Institute of Health grants R01-HD084409 and P40-OD010997 (to M.E. Horb). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
The authors declare no competing financial interests.
Author contributions: Conceptualization, S.E.R. Dundon, M.E. Horb, A.S. Gladfelter, J. Giudice; Methodology, M.E. Horb, A.S. Gladfelter, J. Giudice; Investigation, J.A. Smith, E.G. Curry, R.E. Blue, C. C. Roden, S.E.R. Dundon, A. Rodríguez-Vargas, D.C. Jordan, X. Chen, S.M. Lyons, J. Crutchley, A.S. Gladfelter, J. Giudice; Validation, J.A. Smith, E.G. Curry, R.E. Blue, C. Roden, S.E.R. Dundon, X. Chen, S.M. Lyons, J. Giudice; Formal Analysis, J.A. Smith, E.G. Curry, R.E. Blue, C. Roden, J. Giudice; Writing – Original Draft, J.A. Smith, E.G. Curry, R.E. Blue, A.S. Gladfelter, J. Giudice; Writing – Review & Editing, J.A. Smith, E.G. Curry, R.E. Blue, C. Roden, S.E.R. Dundon, A. Rodríguez-Vargas, D.C. Jordan, X. Chen, S.M. Lyons, J. Crutchley, P. Anderson, M.E. Horb, A.S. Gladfelter, J. Giudice; Visualization, J.A. Smith, R.E. Blue, J. Giudice, M.E. Horb, A.S. Gladfelter, J. Giudice.
References
- Adams-Cioaba M.A., Guo Y., Bian C., Amaya M.F., Lam R., Wasney G.A., Vedadi M., Xu C., and Min J.. 2010. Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2. PLoS One. 5:e13559 10.1371/journal.pone.0013559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S., Gladfelter A., and Mittag T.. 2019. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 176:419–434. 10.1016/j.cell.2018.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumiller W.M. Jr., and Keating C.D.. 2016. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat. Chem. 8:129–137. 10.1038/nchem.2414 [DOI] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., and Rosen M.K.. 2017. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18:285–298. 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara E., Davidovic L., Melko M., Bensaid M., Tremblay S., Grosgeorge J., Khandjian E.W., Lalli E., and Bardoni B.. 2007. Fragile X related protein 1 isoforms differentially modulate the affinity of fragile X mental retardation protein for G-quartet RNA structure. Nucleic Acids Res. 35:299–306. 10.1093/nar/gkl1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J., Brangwynne C.P., and Haataja M.. 2018. Physical principles of intracellular organization via active and passive phase transitions. Rep. Prog. Phys. 81:046601 10.1088/1361-6633/aaa61e [DOI] [PubMed] [Google Scholar]
- Brinegar A.E., Xia Z., Loehr J.A., Li W., Rodney G.G., and Cooper T.A.. 2017. Extensive alternative splicing transitions during postnatal skeletal muscle development are required for Ca 2+ handling. Elife. 6:e27192. doi: . 10.7554/eLife.27192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burattini S., Ferri P., Battistelli M., Curci R., Luchetti F., and Falcieri E.. 2004. C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur. J. Histochem. 48:223–233. 10.4081/891 [DOI] [PubMed] [Google Scholar]
- Cook D., Nuro E., Jones E.V., Altimimi H.F., Farmer W.T., Gandin V., Hanna E., Zong R., Barbon A., Nelson D.L., et al. . 2014. FXR1P limits long-term memory, long-lasting synaptic potentiation, and de novo GluA2 translation. Cell Rep. 9:1402–1416. 10.1016/j.celrep.2014.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.C., Fraser C.E., Mostovetsky O., and Darnell R.B.. 2009. Discrimination of common and unique RNA-binding activities among Fragile X mental retardation protein paralogs. Hum. Mol. Genet. 18:3164–3177. 10.1093/hmg/ddp255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovic L., Durand N., Khalfallah O., Tabet R., Barbry P., Mari B., Sacconi S., Moine H., and Bardoni B.. 2013. A novel role for the RNA-binding protein FXR1P in myoblasts cell-cycle progression by modulating p21/Cdkn1a/Cip1/Waf1 mRNA stability. PLoS Genet. 9:e1003367 10.1371/journal.pgen.1003367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovic L., Sacconi S., Bechara E.G., Delplace S., Allegra M., Desnuelle C., and Bardoni B.. 2008. Alteration of expression of muscle specific isoforms of the fragile X related protein 1 (FXR1P) in facioscapulohumeral muscular dystrophy patients. J. Med. Genet. 45:679–685. 10.1136/jmg.2008.060541 [DOI] [PubMed] [Google Scholar]
- Dosztányi Z., Csizmok V., Tompa P., and Simon I.. 2005a IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 21:3433–3434. 10.1093/bioinformatics/bti541 [DOI] [PubMed] [Google Scholar]
- Dosztányi Z., Csizmók V., Tompa P., and Simon I.. 2005b The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 347:827–839. 10.1016/j.jmb.2005.01.071 [DOI] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S., Kim Y., Szczepaniak K., Chen C.C.-H., Eckmann C.R., Myong S., and Brangwynne C.P.. 2015. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA. 112:7189–7194. 10.1073/pnas.1504822112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estañ M.C., Fernández-Núñez E., Zaki M.S., Esteban M.I., Donkervoort S., Hawkins C., Caparros-Martin J.A., Saade D., Hu Y., Bolduc V., et al. . 2019. Recessive mutations in muscle-specific isoforms of FXR1 cause congenital multi-minicore myopathy. Nat. Commun. 10:797 10.1038/s41467-019-08548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay M.M., Anderson P.J., and Ivanov P.. 2017. ALS/FTD-associated C9ORF72 repeat RNA promotes phase transitions in vitro and in cells. Cell Rep. 21:3573–3584. 10.1016/j.celrep.2017.11.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessert S., Bugner V., Tecza A., Pinker M., and Kühl M.. 2010. FMR1/FXR1 and the miRNA pathway are required for eye and neural crest development. Dev. Biol. 341:222–235. 10.1016/j.ydbio.2010.02.031 [DOI] [PubMed] [Google Scholar]
- Giudice J., Xia Z., Wang E.T., Scavuzzo M.A., Ward A.J., Kalsotra A., Wang W., Wehrens X.H.T., Burge C.B., Li W., and Cooper T.A.. 2014. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 5:3603 10.1038/ncomms4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt E.J., and Spradling A.C.. 2018. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science. 361:709–712. 10.1126/science.aas9963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley K.O., Hardcastle Z., Friday R.V., Amaya E., and Papalopulu N.. 2001. Transgenic Xenopus embryos reveal that anterior neural development requires continued suppression of BMP signaling after gastrulation. Dev. Biol. 238:168–184. 10.1006/dbio.2001.0398 [DOI] [PubMed] [Google Scholar]
- Haynes C., and Iakoucheva L.M.. 2006. Serine/arginine-rich splicing factors belong to a class of intrinsically disordered proteins. Nucleic Acids Res. 34:305–312. 10.1093/nar/gkj424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M., and Dormann D.. 2019. Friend or foe-Post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem. 294:7137–7150. 10.1074/jbc.TM118.001189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth D., Candel A.M., Nicastro G., Martin S.R., Briata P., Gherzi R., and Ramos A.. 2012. KH domains with impaired nucleic acid binding as a tool for functional analysis. Nucleic Acids Res. 40(14):6873–6886. 10.1093/nar/gks368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot M.-E., Bisson N., Davidovic L., Mazroui R., Labelle Y., Moss T., and Khandjian E.W.. 2005. The RNA-binding protein fragile X-related 1 regulates somite formation in Xenopus laevis. Mol. Biol. Cell. 16:4350–4361. 10.1091/mbc.e05-04-0304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandjian E.W., Bardoni B., Corbin F., Sittler A., Giroux S., Heitz D., Tremblay S., Pinset C., Montarras D., Rousseau F., and Mandel J.. 1998. Novel isoforms of the fragile X related protein FXR1P are expressed during myogenesis. Hum. Mol. Genet. 7:2121–2128. 10.1093/hmg/7.13.2121 [DOI] [PubMed] [Google Scholar]
- Kiebler M.A., and Bassell G.J.. 2006. Neuronal RNA granules: movers and makers. Neuron. 51:685–690. 10.1016/j.neuron.2006.08.021 [DOI] [PubMed] [Google Scholar]
- Kim T.H., Tsang B., Vernon R.M., Sonenberg N., Kay L.E., and Forman-Kay J.D.. 2019. Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science. 365:825–829. 10.1126/science.aax4240 [DOI] [PubMed] [Google Scholar]
- Kirkpatrick L.L., McIlwain K.A., and Nelson D.L.. 1999. Alternative splicing in the murine and human FXR1 genes. Genomics. 59:193–202. 10.1006/geno.1999.5868 [DOI] [PubMed] [Google Scholar]
- Langdon E.M., Qiu Y., Ghanbari Niaki A., McLaughlin G.A., Weidmann C.A., Gerbich T.M., Smith J.A., Crutchley J.M., Termini C.M., Weeks K.M., et al. . 2018. mRNA structure determines specificity of a polyQ-driven phase separation. Science. 360:922–927. 10.1126/science.aar7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latinkić B.V., Cooper B., Towers N., Sparrow D., Kotecha S., and Mohun T.J.. 2002. Distinct enhancers regulate skeletal and cardiac muscle-specific expression programs of the cardiac α-actin gene in Xenopus embryos. Dev. Biol. 245:57–70. 10.1006/dbio.2002.0639 [DOI] [PubMed] [Google Scholar]
- Lee C., Zhang H., Baker A.E., Occhipinti P., Borsuk M.E., and Gladfelter A.S.. 2013. Protein aggregation behavior regulates cyclin transcript localization and cell-cycle control. Dev. Cell. 25:572–584. 10.1016/j.devcel.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C., Occhipinti P., and Gladfelter A.S.. 2015. PolyQ-dependent RNA-protein assemblies control symmetry breaking. J. Cell Biol. 208:533–544. 10.1083/jcb.201407105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons S.M., Gudanis D., Coyne S.M., Gdaniec Z., and Ivanov P.. 2017. Identification of functional tetramolecular RNA G-quadruplexes derived from transfer RNAs. Nat. Commun. 8:1127 10.1038/s41467-017-01278-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M., House R., Palanisamy N., Qie S., Day T.A., Neskey D., Diehl J.A., and Palanisamy V.. 2016. RNA-binding protein FXR1 regulates p21 and TERC RNA to bypass p53-mediated cellular senescence in OSCC. PLoS Genet. 12:e1006306 10.1371/journal.pgen.1006306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mientjes E.J., Willemsen R., Kirkpatrick L.L., Nieuwenhuizen I.M., Hoogeveen-Westerveld M., Verweij M., Reis S., Bardoni B., Hoogeveen A.T., Oostra B.A., and Nelson D.L.. 2004. Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum. Mol. Genet. 13:1291–1302. 10.1093/hmg/ddh150 [DOI] [PubMed] [Google Scholar]
- Nutter C.A., Jaworski E.A., Verma S.K., Deshmukh V., Wang Q., Botvinnik O.B., Lozano M.J., Abass I.J., Ijaz T., Brasier A.R., et al. . 2016. Dysregulation of RBFOX2 is an early event in cardiac pathogenesis of diabetes. Cell Rep. 15:2200–2213. 10.1016/j.celrep.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo J.P., Chambon P., Metzger D., Mosier D.R., Snipes G.J., and Cooper T.A.. 2008. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 105:2646–2651. 10.1073/pnas.0708519105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C.W., Kosno M., Holehouse A.S., Padrick S.B., Mittal A., Ali R., Yunus A.A., Liu D.R., Pappu R.V., and Rosen M.K.. 2016. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell. 63:72–85. 10.1016/j.molcel.2016.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A., Teraguchi S., Dinh H., Nakai K., and Standley D.M.. 2012. Functional annotation of intrinsically disordered domains by their amino acid content using IDD Navigator. Pac Symp Biocomput. 164–175. . [PubMed] [Google Scholar]
- Patzlaff N.E., Nemec K.M., Malone S.G., Li Y., and Zhao X.. 2017. Fragile X related protein 1 (FXR1P) regulates proliferation of adult neural stem cells. Hum. Mol. Genet. 26:1340–1352. 10.1093/hmg/ddx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A.K., Chen J.X., Selbach M., and Pelkmans L.. 2018. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 559:211–216. 10.1038/s41586-018-0279-8 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. . 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9(7):676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., and Eliceiri K.W.. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 9(7):671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.K., Xia Z., Bland C.S., Kalsotra A., Scavuzzo M.A., Curk T., Ule J., Li W., and Cooper T.A.. 2014. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol. Cell. 55:592–603. 10.1016/j.molcel.2014.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi M.C., Siomi H., Sauer W.H., Srinivasan S., Nussbaum R.L., and Dreyfuss G.. 1995. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J. 14:2401–2408. 10.1002/j.1460-2075.1995.tb07237.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Calidas D., Schmidt H., Lu T., Rasoloson D., and Seydoux G.. 2016. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. eLife. 5:e21337 10.7554/eLife.21337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead W.T., and Gladfelter A.S.. 2019. The control centers of biomolecular phase separation: how membrane surfaces, PTMs, and active processes regulate condensation. Mol. Cell. 76:295–305. 10.1016/j.molcel.2019.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang B., Arsenault J., Vernon R.M., Lin H., Sonenberg N., Wang L.Y., Bah A., and Forman-Kay J.D.. 2019. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. USA. 116:4218–4227. 10.1073/pnas.1814385116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V.N., Kuznetsova I.M., Turoverov K.K., and Zaslavsky B.. 2015. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 589:15–22. 10.1016/j.febslet.2014.11.028 [DOI] [PubMed] [Google Scholar]
- Van’t Padje S., Chaudhry B., Severijnen L.-A., van der Linde H.C., Mientjes E.J., Oostra B.A., and Willemsen R.. 2009. Reduction in fragile X related 1 protein causes cardiomyopathy and muscular dystrophy in zebrafish. J. Exp. Biol. 212:2564–2570. 10.1242/jeb.032532 [DOI] [PubMed] [Google Scholar]
- Vasudevan S., and Steitz J.A.. 2007. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 128:1105–1118. 10.1016/j.cell.2007.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., and Burge C.B.. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature. 456:470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.T., Smith J., Chen B.C., Schmidt H., Rasoloson D., Paix A., Lambrus B.G., Calidas D., Betzig E., and Seydoux G.. 2014. Regulation of RNA granule dynamics by phosphorylation of serine-rich, intrinsically disordered proteins in C. elegans. eLife. 3:e04591 10.7554/eLife.04591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Boevink P., McLellan H., Armstrong M., Bukharova T., Qin Z., and Birch P.R.J.. 2015. A Host KH RNA-Binding Protein Is a Susceptibility Factor Targeted by an RXLR Effector to Promote Late Blight Disease. Mol Plant. 8(9):1385–1395. 10.1016/j.molp.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman S.A., Cover C., Yu L., Nelson D.L., Zarnescu D.C., and Gregorio C.C.. 2011. Desmoplakin and talin2 are novel mRNA targets of fragile X-related protein-1 in cardiac muscle. Circ. Res. 109:262–271. 10.1161/CIRCRESAHA.111.244244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins M.R., Gasteiger E., Bairoch A., Sanchez J.C., Williams K.L., Appel R.D., and Hochstrasser D.F.. 1999. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112:531–552. 10.1385/1-59259-584-7:531 [DOI] [PubMed] [Google Scholar]
- Wippich F., Bodenmiller B., Trajkovska M.G., Wanka S., Aebersold R., and Pelkmans L.. 2013. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 152:791–805. 10.1016/j.cell.2013.01.033 [DOI] [PubMed] [Google Scholar]
- Wong A.G., McBurney K.L., Thompson K.J., Stickney L.M., and Mackie G.A.. 2013. S1 and KH domains of polynucleotide phosphorylase determine the efficiency of RNA binding and autoregulation. J. Bacteriol. 195(9):2021–2031. 10.1128/JB.00062-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S., Gapsys V., Kim H.Y., Bessonov S., Hsiao H.H., Möhlmann S., Klaukien V., Ficner R., Becker S., Urlaub H., et al. . 2013. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure. 21:2162–2174. 10.1016/j.str.2013.09.014 [DOI] [PubMed] [Google Scholar]
- Zarnescu D.C., and Gregorio C.C.. 2013. Fragile hearts: new insights into translational control in cardiac muscle. Trends Cardiovasc. Med. 23:275–281. 10.1016/j.tcm.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., O’Connor J.P., Siomi M.C.M.C., Srinivasan S., Dutra A., Nussbaum R.L., Dreyfuss G., and O’Connor J.P.. 1995. The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 14:5358–5366. 10.1002/j.1460-2075.1995.tb00220.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Elbaum-Garfinkle S., Langdon E.M., Taylor N., Occhipinti P., Bridges A.A., Brangwynne C.P., and Gladfelter A.S.. 2015. RNA controls PolyQ protein phase transitions. Mol. Cell. 60:220–230. 10.1016/j.molcel.2015.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
shows the sequence of the primers (IDT) used for plasmid generation. Uppercase bases denote sequences that will directly base pair with the plasmid being amplified, while lowercase bases are overhangs for NEBuilder HiFi DNA assembly reaction.