

Abstract
Plasma levels of the multimeric glycoprotein von Willebrand factor (VWF) are a complex quantitative trait with a continuous distribution and wide range in the normal population (50-200%). Quantitative deficiencies of VWF (<50%) are associated with an increased risk for bleeding while high plasma levels of VWF (>150%) influence the risk for arterial and venous thromboembolism. Although environmental factors can strongly influence plasma VWF levels, it is estimated that approximately 65% of this variability is heritable. Interestingly, while variability at the VWF gene can account for ~5% of the genetic influence on plasma VWF levels, other genetic loci also strongly modify plasma VWF levels. The identification of the additional sources of VWF heritability has been the focus of recent observational trait mapping studies including genome-wide association studies (GWAS) or linkage analyses, as well as hypothesis-driven research studies. Quantitative trait loci (QTL) influencing VWF glycosylation, secretion, and clearance have been associated with plasma VWF:Ag levels in normal individuals and may contribute to quantitative VWF abnormalities in patients with a thrombotic tendency or type 1 von Willebrand disease (VWD). Identification of genetic modifiers of plasma VWF levels may allow for better molecular diagnosis of type 1 VWD and identify individuals at increased risk for thrombosis. Validation of trait-mapping studies using in vitro and in vivo methodologies has led to novel insights into the life cycle of VWF and the pathogenesis of quantitative VWF abnormalities.
Introduction
Plasma levels of the multimeric glycoprotein coagulation factor von Willebrand factor (VWF) are a continuously distributed complex quantitative trait, ranging between 50 and 200% in the normal population (Figure 1A). Low VWF or von Willebrand disease (VWD), characterized by either qualitative or quantitative VWF deficiency (VWF:Ag < 50%), is the most prevalent hereditary bleeding disorder affecting between 0.1 – 1% of individuals [1-3]. In contrast, epidemiological studies have shown elevated VWF:Ag is an independent risk factor for venous thrombosis, stroke, and ischemic heart disease [4-6]. Despite evidence that VWF plasma levels are highly heritable, variants in the VWF gene account for only a small proportion of this effect. It is increasingly recognized that the phenotypic variability in VWF plasma levels is influenced by several quantitative trait loci (QTL). Large scale studies in recent years have begun to characterize the genetic architecture that regulates VWF plasma levels. The QTL that influence VWF plasma levels in normal and pathological states and the biological mechanism(s) by which they exert their effects will be described in this review.
Figure 1. VWF:Ag is a complex quantitative trait.
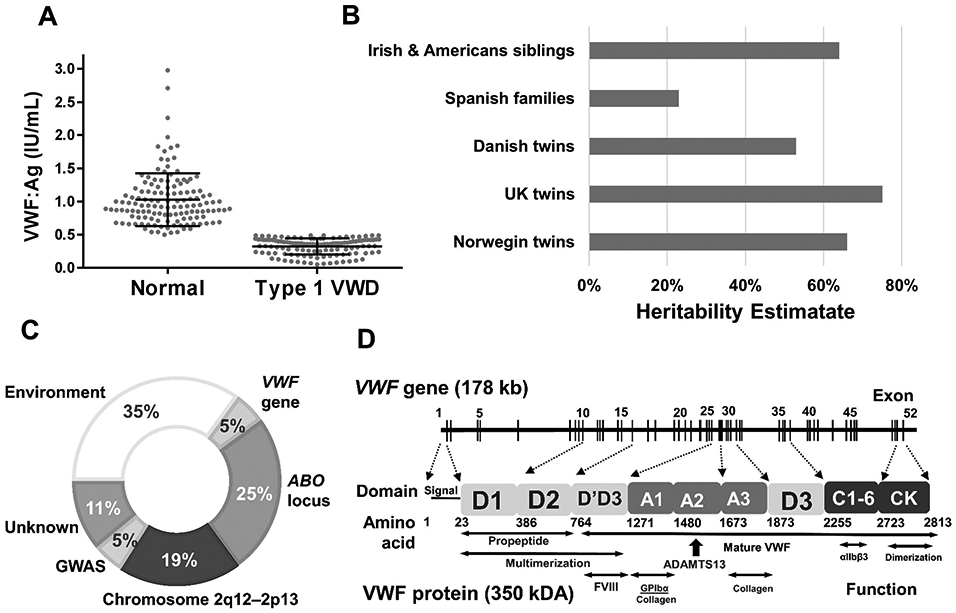
(A) Distribution of VWF:Ag levels in Canadian type 1 VWD patients and their unaffected (normal) family members [40,111]. (B) Heritability estimates of VWF:Ag [19-23]. (C) 65% of plasma VWF levels are heritable with variability at the VWF gene, ABO blood group locus, and others contributing to this phenotype [20,70]. (D) The VWF gene is comprised of 52 exons encoding a ~370 kDA mature protein that is comprised of multi-functional domains.
Genetic versus environmental influences on VWF plasma levels
Plasma VWF levels are influenced by genetic, pathological, hormonal, and environmental interactions. VWF is an acute phase protein and numerous inflammatory conditions are associated with elevated plasma levels of VWF [7-9], as pro-inflammatory mediators can stimulate VWF release from endothelial and/or platelet stores [10-12]. VWF levels may also be influenced by physiological events including the menstrual cycle, pregnancy, exercise, aging, and circadian rhythm fluctuation, or environmental exposures including cigarette smoke and air pollution [13-18].
Despite the influence of environmental factors on VWF levels, studies have demonstrated that VWF levels are also highly heritable. While a pedigree analyses has suggested that the genetic influence on VWF levels is approximately 30%, twin and sibling studies find this value is closer to 65% [19-23] (Figure 1B). The composition of the study population can influence this estimate with larger heritability found in populations with diverse genetic background and fewer environmental influences. It is estimated that variability at the VWF locus accounts for ~5% of the heritability of VWF, while the ABO blood group locus contributes approximately 25% [24]. Characterization of the sites that contribute to the unidentified heritability of VWF is thus a focus of ongoing study (Figure 1C).
Influence of variability at the VWF locus on plasma levels of VWF
The VWF gene was first cloned in the mid-1980s [25-28]; it spans 178 kb of genomic sequence and is located on the short arm of chromosome 12. The VWF coding region is comprised of 52 exons that range in size from 40 bp (exon 50) to 1.3 kb (exon 28) (Figure 1D) [29]. Analysis of the VWF gene sequence is complicated by an unprocessed pseudogene (exons 23-34) located on chromosome 22 [30]. Mechanistically, variants in the VWF gene can modify VWF plasma levels by influencing the synthesis, storage and secretion of VWF from endothelial cells or megakaryocytes/platelets, or by regulating the clearance of VWF from plasma.
Normal population:
Both the intronic regions and coding sequence of the VWF gene are known to be highly polymorphic. An analysis of the 1000 Genomes database identified a rate of ~2.5 VWF gene single nucleotide variants (SNVs) per individual, with 8.5% of individuals possessing a non-synonymous coding region variant [31]. The majority of variants (>75%) were rare or previously unreported with unknown functional consequences, although SNVs in the coding and promoter regions in normal individuals have been shown to modify VWF antigen and activity levels [32,33]. Recently, megakaryocyte VWF gene expression has been shown to be regulated by a common SNV located in a super enhancer region located ~55 kb upstream of the VWF gene [34].
Population based studies have shown that VWF levels are influenced by ethnicity, as African Americans have ~15% higher VWF levels than Caucasian populations [35]. Recent studies have highlighted the high incidence of ethnic-specific VWF sequence variations, with Africans having greater diversity in the VWF gene than non-Africans [31,36,37]. The highest level of diversity was found in the D′ and D2 domains [31], which may, in part, contribute to elevated VWF levels.
“Low VWF” or VWD populations (<0.5 IU/mL):
Type 3 VWD (VWF:Ag <0.03 U/mL), results from the inheritance of two mutant VWF alleles that cause severe deficiency of plasma VWF. Type 3 VWD can have either a recessive or co-dominant pattern of inheritance with obligate carriers displaying a type 1 VWD phenotype in 25-50% of type 3 families [38]. Type 3 VWD shows significant allelic heterogeneity, and large deletions, frameshift, missense, splice site, and nonsense mutations have been described throughout the gene. Approximately 20% of mutations are missense variants that presumably result in the expression of VWF protein whose biosynthesis and secretion is significantly disrupted. Thus, the mechanistic basis of type 3 VWD is complex, and may involve impaired synthesis, Weibel Palade body (WPB) formation, or secretion [39].
The clinical definition of partial quantitative VWF deficiency, historically termed type 1 VWD, has been variable, with a VWF:Ag cut-off level ranging between 0.03 and 0.3 – 0.5 U/mL used for studies investigating the molecular pathogenesis of this disorder. For these patients, pathogenic SNVs result in missense substitutions that occur throughout the coding region of the VWF gene, although some SNVs occur in the promoter, or at intron/exon boundaries (Figure 1D) [40-44]. While rare recessive cases of type 1 VWD have been reported, the disorder is predominantly inherited in an autosomal dominant manner [45]. Approximately 85% of type 1 VWD cases are associated with defective VWF synthesis, storage and/or secretion from the endothelium. Type 1 secretion variants tend to localize in the VWF D1, D2, and D3 domains [46], and can result in intracellular retention in the endoplasmic reticulum, irregular WPB formation or reduced secretion [47-49]. Rare cases of decreased VWF synthesis related to variants in the VWF promoter have also been described [50].
The nomenclature, type 1C VWD, while not a currently recognized ISTH classification, is often used to describe accelerated clearance variants which make up ~15% of all type 1 cases [51,52]. Studies using the VWF propeptide to antigen ratio (VWFpp/VWF:Ag) as a surrogate measure of VWF clearance have indicated that accelerated clearance occurs more frequently with more severe VWF deficient states [46]. Type 1C pathogenic variants are most frequently observed in the D3, A1, and D4 domains of VWF [46]. While little is currently known regarding the mechanistic basis by which these variants regulate VWF half-life, they may alter either the glycosylation or conformation of the VWF molecule, and likely enhance the affinity of VWF for one or more of its clearance receptors [53]. Type 2 VWD, which is predominantly characterized by platelet, collagen or FVIII-binding qualitative defects may also be complicated by impaired VWF secretion or accelerated clearance of the VWF-platelet complex [54,55]
The diagnosis of type 1 VWD may be complicated by variability in phenotypic penetrance and expressivity. For example, several studies have now documented that pathogenic variants identified in European type 1 VWD subjects can have minor allele frequencies of 10-20% in normal asymptomatic African American individuals [31,36,37]. While this phenomenon may be partially related to false attribution of variant pathogenicity, in vitro characterization demonstrates that some of these variants display minor quantitative defects. Thus, it seems likely that some mild type 1 variants co-segregate with additional ethnic-specific variants that modify their pathogenicity, or that the bleeding phenotype associated with these variants may be influenced by external environmental interactions. For example, gene-environment interactions have been shown to modify the type 1 VWD phenotype where age-related changes can normalize VWF:Ag levels in older individuals [56,57].
Recent recommendations from the UK Haemophilia Centre Doctors Organization and the US National Heart, Lung, and Blood Institute (NHLBI) have further sub-classified partial VWF quantitative deficiency into type 1 VWD (0.03 – 0.3 U/mL) and a “Low VWF” phenotype (0.3 – 0.5 U/mL) [58,59]. Overall, approximately 35% patients with partial quantitative VWF deficiency do not have an identified pathogenic variant in their VWF coding region or consensus splice sites with a lower proportion of coding region variants identified in “Low VWF” individuals [40-44,60,61]. While some the pathogenic variants have been hypothesized to be found in distal regulatory regions or deep intronic sequences of the VWF gene, linkage analysis performed on two separate, cohorts demonstrated that the proportion of families that show linkage to the VWF locus is ~0.44 [62,63]; thus, non-VWF gene QTL may contribute to the “Low VWF” phenotype in the absence of a pathogenic VWF gene variant.
Thrombosis population:
Epidemiological studies have demonstrated that elevated VWF levels are a risk factor for venous and arterial thrombosis, and that patients diagnosed with type 1 VWD have a decreased incidence of thrombosis [64]. SNVs found throughout the VWF gene, including the promoter, coding regions, and introns, have been associated with either elevated VWF:Ag levels, or risk for venous thrombosis or coronary heart disease [20,65-68] (reviewed in detail by Van Schie et al.[69]). For example, the common VWF non-synonymous variant c.2365A>G (p.Thr789Ala), which is in strong linkage disequilibrium (r=99%) with the synonymous variant c.2385T>C (p.Tyr795=), associates with both increased VWF levels and risk for VTE (OR=1.2) [67,70]. These variants influence VWF synthesis/secretion in a heterologous expression system and also demonstrate evidence of increased VWF half-life [71,72].
Identification of non-VWF Loci that influence VWF levels
Non-VWF gene QTL contribute to the majority of the heritability of VWF plasma levels in normal individuals. They may also modify the quantitative VWF abnormalities that contribute to either the type 1 VWD phenotype, or to a hypercoagulable state. Identification of these additional loci has been the focus of both candidate gene-driven studies where knowledge of VWF (patho)biology is used to identify genes that modify VWF levels, and observational trait mapping studies (such as genome-wide association studies (GWAS) or linkage analyses) where variants that influence VWF levels are assessed in an unbiased manner. Trait-based association studies have thus far provided an initial insight into the architecture of the genetic regulation of VWF plasma levels, and have documented a number of QTL, estimated their effect size, and investigated their potential to mediate epistatic and pleiotropic effects on VWF levels (Table 1). They may also support or bring into question previous hypothesis driven studies.
Table 1. Non-VWF and ABO-variants associate with plasma levels of VWF in normal individuals.
MAF = minor allele frequency. N/A=not available. For some genes the most significant SNVs only were chosen.
| Gene | rfSNP ID | HGVS nomenclature | Position | MAF | Putative Mechanism |
Study type |
Refs |
|---|---|---|---|---|---|---|---|
| ACE | rs 1799752 | c.644-119_644-118insG | Intron | 2.59x10−5 | Secretion? | Gene candidate | 87 |
| AVPR2 | rs2071126 | c.35G>A p.Gly12Glu | Exon 2 | 0.0455 | Secretion | Gene candidate | 85 |
| BAI3 | rs9363864 | g.68182664G>A | 3’ UTR | Unknown | GWAS | 68 | |
| CLEC4M | rs868875 | c.631+73A>G | Intron 4 | 0.262 | Clearance | GWAS | 63 |
| FUT1 | rs104894686 | c.948C>G p.Tyr316Ter | Exon 5 | 8.24x10−6 | Glycosylation | Gene candidate | 77 |
| FUT2 | rs601338 | c.461G>A p.Trp154Ter | Exon 2 | 0.39 | Glycosylation | Gene candidate | 77 |
| LRP1 | rs34577247 | c.6238G>A p.Asp2080Asn | Exon 39 | 0.0149 | Glycosylation | Gene candidate | 101, 102 |
| LRP1 | rs1800127 | c.650C>T p.Ala217Val | Exon 6 | 0.017 | Clearance | Gene candidate | 101, 102 |
| SCARA5 | rs2726953 | c.242-21543C>T | Intron 3 | 0.309 | Clearance | GWAS | 63 |
| ST3GAL4 | rs2186717 | c.−60-14159G>A | Intron 1 | 0.488 | Glycosylation | Gene candidate and GWAS | 83 |
| STAB2 | rs4981022 | c.6987+378G>A | Intron 63 | 0.315 | Clearance | GWAS | 63 |
| STAB2 | rs141041254 | c.7129G>A p.Glu2377Lys | Exon 65 | 8.5x10−4 | Clearance | GWAS | 107 |
| STX2 | rs7978987 | c.786+1576C>T | Intron 9 | 0.346 | Secretion? | GWAS | 63 |
| STXBP5 | rs9390459 | c.2445A>G p.Leu815= | Exon 23 | 0.442 | Secretion | GWAS | 63 |
| TC2N | rs 10133762 | c.−57+9946A>C | Intron 1 | 0.444 | Unknown | GWAS | 63 |
| UFM1 | rs17057285 | g.38737821 A>C | 200 kb upstream | 0.036 | Unknown | GWAS | 67 |
To date, VWF modifying QTL have been identified in two genome-wide linkage analyses. The Genetic Analysis of Idiopathic Thrombophilia (GAIT) study [23], performed in 21 Spanish families (342 individuals) identified six QTL with LOD scores >1 that associate with plasma VWF levels, including the ABO locus at 9q34, as well as regions on chromosomes 1p36.13, 2q23.2, 5q31.1, 6p22.3, and 22q11.1. Importantly, the 2q12 locus was replicated in a separate genome wide linkage analysis of young siblings [20], and three of these regions show synteny with QTL linked to VWF levels in mice [73].
Linkage analysis data has now been complemented with a number of GWAS studies, the largest of which is the CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) consortium GWAS meta-analysis [70]. Plasma levels of VWF and/or FVIII were correlated with ~2.6 million alleles in an initial discovery population of over 23,000 normal subjects of European ancestry collected from five studies, and replication analysis was performed in 7,600 additional subjects. VWF levels associated with over 400 SNVs at eight loci with a genome-wide significance threshold of 5.0x10−8: VWF, ABO, STAB2, SCARA5, TC2N, STXBP5, STX2, and CLEC4M. Together, these loci explained 12.8% of the variation in plasma VWF levels. The majority of these variants were located within introns and may mediate their influence through a number of potential mechanisms: altering mRNA splicing patterns or efficiency, modifying levels of gene expression, or at least as likely, may be in linkage disequilibrium with coding region variants that modify protein function. Non-synonymous variants may also influence protein translation through splicing effects, codon use bias or by altering mRNA stability. To date, the association between VWF and the novel loci identified in the CHARGE study has been replicated in several additional GWAS analyses, with a number of new candidate loci identified [74-76].
An updated GWAS meta-analysis from the CHARGE consortium, recently described in abstract form, included results from a discovery cohort of >46,000 normal individuals of European, African, East Asian, and Hispanic descent [77]. Eleven additional QTLs were found to associate with plasma VWF levels: ARSA, C2CD4B, DAB2IP, FCHO2, GIMAP7, HLA-DGA1, OR13C5, PDHB, RAB5C, ST3GAL4, TAB1/SYNGR1. The putative function of the majority of these novel loci appears to involve the regulation of VWF secretion from endothelial cells and/or megakaryocytes, however these associations await further confirmation and validation.
Both observational and hypothesis-driven genetic studies have the capability of identifying common variants that have both small and large influences on VWF plasma levels, and rare variants that impart a large influence (Figure 2). Variants external to the VWF gene may modify the severity or penetrance of type 1 VWD, or conversely, independently associate with elevated VWF levels or an increased incidence of thrombosis (Table 2). Complementary experimental studies are required to identify the mechanistic basis by which the GWAS-identified variants convey their effects, to understand the biological pathway that results in altered VWF plasma levels, and to rule out the possibility of false positive results. The majority of VWF QTL fall into three broad categories: genes that modify VWF glycosylation, secretion from endothelial cells or platelets, and clearance from the plasma.
Figure 2. Rare and common genetic variants influence VWF levels.

VWF plasma levels are influenced by both rare and common variants in the VWF gene, ABO blood group locus, and additional loci. LOF = loss of function.
Table 2. Influence of non-VWF and ABO-variants on quantitative VWF pathologies.
DVT = deep vein thrombosis. CHD = coronary heart disease.
| Study design | Population | Gene(s) investigated |
Major Finding(s) | Ref |
|---|---|---|---|---|
| Association of CHARGE SNVs with VTE | 656 women with DVT, 710 controls | CHARGE SNVs | -SNVs in STXBP5 and VWF associate with DVT | 60 |
| SNVs associated with VTE, plasma VWF levels | 1166 VTE patients, 1408 healthy subjects, 5 extended families | Linkage analysis, GWAS | -ABO locus, STAB-2, BAI3, 2p12-13 regions associate with risk for VTE -BAI3 SNV variants associate with VWF levels |
68 |
| Association between CHARGE SNVs and VWD (type 1& 2) | 364 type 1 VWD, 240 type 2 Netherland VWD patients | CHARGE SNVs | -SNVs in STXBP5 and CLEC4M associate with low VWF levels in type 1 VWD | 90 |
| Association of CLEC4M variants with type 1 VWD | 318 type 1 VWD patients and 173 unaffected family members | CLEC4M VNTR polymorphism and CHARGE SNV | -association of CLEC4M VNTR with unaffected individuals, VWF:RCo | 103 |
| Association of STXBP2 and STX2 variants with arterial thrombosis | 463 arterial thrombosis patients, 406 controls | STXBP5 and STX3 CHARGE SNVs | -SNVs in STXBP5 and STX2 associate with VWF:Ag in patients with arterial thrombosis -SNVs in STX2 associate with risk for arterial thrombosis |
92 |
| Association of STXBP2 and STX2 variants with type 1 VWD | 158 type 1 VWD patients | STXBP5 and STX2 CHARGE SNVs | -SNVs in STX2 associate with VWF:Ag in type 1 VWD -SNVs in STXBP5 associate bleeding score in females with type 1 VWD | 91 |
| Association between variants in VWF gene and CHD in younger individuals | 421 young CHD patients and 409 healthy controls | VWF gene | -SNVs in VWF are associated with elevated VWF levels and risk for cardiovascular disease | 59 |
| Association between VWF promoter SNV and CHD | 352 subjects with CHD, 736 controls | VWF promoter | -a SNVs in the VWF promoter is associated an increased risk of CHD in subjects with advanced atherosclerosis. | 58 |
| Association between CHARGE SNVs and VT | 1744 VT patients and 1389 healthy controls | CHARGE SNVs | -TC2N varient associates with increased risk for VTE | 117 |
Genes influencing VWF glycosylation
VWF is produced in endothelial cells and megakaryocytes in a complex biosynthetic process that involves C-terminal dimerization, and cleavage of an N-terminal pro-peptide, followed by N-terminal multimerization, with subsequent N- and O-linked glycosylation. VWF possesses 13 potential N-linked and 10 O-linked glycan sites that constitute approximately 20% of the molecular mass of the mature protein (Figure 3A). Glycomic analysis has revealed the majority of VWF N-linked glycans to be of complex type, with ABO(H) expressed as terminal sugars [78,79]. Mutagenesis studies have demonstrated that both N- and O-linked glycans on the VWF molecule are involved in regulating its secretion in a heterologous cell system and its clearance from the plasma [80,81].
Figure 3. VWF:Ag levels are influenced by VWF glycosylation.
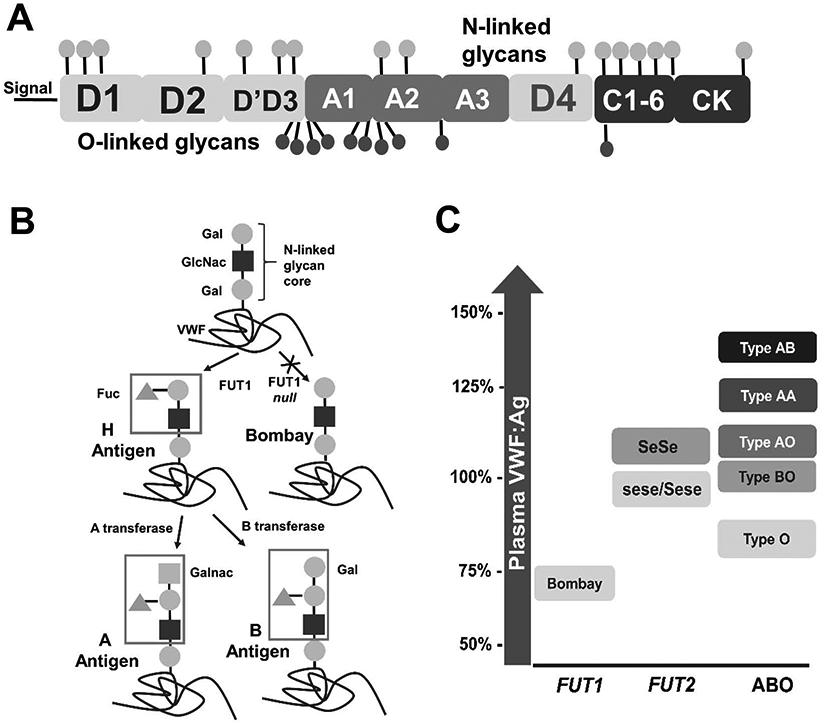
(A) Post-translational modification of VWF includes the addition of 16 putative N-linked and 10 putative O-linked glycans. (B) ABO(H) blood group antigens are added to the VWF N- and O-linked glycans. (C) Dysfunction of FUT1/2 and ABO blood group status can influence VWF plasma levels. Bombay = FUT1 and FUT1/FUT2 null, Se=FUT2, se=FUT2 null.
FUT1/2:
FUT1/2 are fucosyltransferases that produce the H antigen by transferring a fucose to the glycan core structure (Figure 3B). FUT1 encodes the H enzyme that regulates H antigen formation on VWF, while FUT2 encodes the Se enzyme, and regulates H antigen formation on red blood cells, and secretions in the mucosa and gastrointestinal tract respectively. The core H antigen structure is then either modified by additional glycosyltransferases to create complex N-linked glycans. Individuals with both Bombay phenotype (FUT1/2 deficient), and para-Bombay phenotype (FUT1 deficient) have no VWF H antigen, and VWF plasma levels of ~70% (Figure 3C), suggesting that FUT1 fucosyltransferase activity is a significant regulator of plasma VWF levels [82]. While conflicting reports exist, FUT2 variants have been shown to also associate with VWF plasma levels [19,83,84]; the mechanism by which this effect is exerted is unknown, but may involve FUT2 modification of pathways that regulate VWF secretion or clearance.
ABO:
The ABO blood group locus is located on chromosome 9 and is highly polymorphic. The locus encodes either A or B glycosyltransferase alleles, which modify the core H antigen by adding N-acetylgalactosamine or galactose respectively to the core galactose (Figure 3B). Type O individuals do not encode a functional AB glycosyltransferase and thus express only H antigen. ABO was first identified as a modifier of VWF levels in the 1980s [24], and has been repeatedly identified as the locus with the strongest influence on plasma VWF levels in trait-mapping studies [20,70,74]. Approximately 25% of the variability in plasma VWF levels can be accounted for by the ABO system, with blood type O (H-antigen) associating with ~25% lower VWF:Ag levels than non-O individuals [24]. Concordantly, individuals heterozygous for O and either A or B glycosyltransferase alleles (AO or BO) have intermediate levels of VWF, while AB and AA individuals have high levels of VWF:Ag (Figure 3C).
The ABO blood group genotype can modify partial quantitative VWF deficiency, with type O individuals more likely to be diagnosed with either type 1 VWD or “low VWF”, and more prone to severe bleeding diatheses than non-O type 1 VWD patients [24], thus contributing to the variable expressivity of this condition. Conversely, the elevated levels of VWF:Ag observed in non-O individuals in part explains the increased risk for thrombosis associated with these blood types. While type O normal individuals and patients with the type 1C VWD “Vicenza” variant have a decreased VWF half-life in response to DDAVP treatment [85,86] the clearance pathway(s) that underlies this effect is currently unknown. Recent VWF half-life studies performed in type 3 patients suggest that non-VWF ABO(H) glycans may also contribute mechanistically to the VWF clearance phenotype [87].
ST3GAL4:
ST3GAL4 encodes ST3 beta-galactoside alpha-2,3-sialyltransferase 4, which facilitates the transfer of terminal sialic acid sugars to the N- and O-linked glycans of VWF. Desialylation of VWF has been associated with increased VWF clearance by the asialoglycoprotein receptor (ASGPR) (Figure 4B) and results in the decreased VWF half-life in humans post DDAVP administration [88,89]. In mice, enzymatically desialylated VWF also has a significantly decreased half-life [89], and ST3GAL4-deficient mice have low VWF levels and a bleeding phenotype [90]. Sequencing of the ST3GAL4 gene in normal individuals has identified six clustered SNVs in the first intron that associate with plasma levels of VWF [91] suggesting that polymorphisms in the ST3GAL4 gene can regulate the degree of VWF sialylation.
Figure 4. VWF:Ag levels are influenced by processes that regulate VWF synthesis/secretion and mechanisms that regulate VWF clearance.


(A) VWF synthesis/secretion can be influenced by variants in or adjacent to the VWF gene that regulate VWF transcriptional activity, mRNA stability, codon use, protein folding, and Weibel Palade Body packaging and secretion. Variants at additional loci including SNARE proteins can influence the secretion of VWF from endothelial WPB or platelet α-granules. (B) VWF clearance can be influenced by variants in the VWF amino acid sequence and/or glycome that modify the affinity of VWF for one or more clearance receptors. Additionally, variants in the clearance receptors for VWF that alter ligand binding or expression can modify VWF plasma levels. HL = hepatic lectin, CBD = carbohydrate binding domain, EGF = endothelial growth factor, VNTR = variable number of tandem repeats, SRCR = scavenger receptor cysteine-rich.
Genes influencing VWF secretion:
Upon synthesis, mature VWF is packaged into intracellular storage vesicles termed Weibel-Palade bodies (WPB) in endothelial cells or α-granules in platelets and released constitutively or on-demand upon hemostatic challenge (Figure 4A). Platelet activation and WPB secretion can be stimulated by a variety of pro-inflammatory secretagogues including thrombin and desmopressin that signal through endothelial cell surface receptors including PAR-1 and vasopressin receptors. Secretion of VWF from platelets and endothelial cells is regulated by SNARE (soluble NSF attachment protein receptor) complexes that direct the fusion of secretory vesicles with the cell plasma membrane.
AVPR2:
Stimulation of the arginine vasopressin 2 receptor (AV2R) with arginine vasopressin induces the exocytosis of WPB, releasing VWF into the circulation in an on-demand fashion [92]. The AVPR2 ligand desmopressin (or DDAVP) is used in clinical practice to transiently increase circulating VWF in type 1 VWD. The gain-of-function p.Gly12Glu variant in AVPR2, that enhances binding of arginine vasopressin, is associated with elevated plasma VWF levels in normal individuals [93]. Thus, patients with inherited nephrogenic diabetes insipidus associated with pathogenic variants in the AVPR2 gene are unresponsive to VWF release upon DDAVP administration [94].
ACE:
Angiotensin-converting enzyme (ACE) regulates blood pressure by converting angiotensin I to angiotensin II, a potent vasoconstrictor. An insertion/deletion variant in ACE has been described to increase the risk of cardiovascular disease, potentially through endothelial dysfunction and modification of plasma levels of coagulation factors, including VWF, in elderly patients with hypertension [95]. As high blood pressure is associated with increased VWF plasma levels, it is likely that dysfunction of ACE promotes the release of VWF from the damaged endothelium, although this mechanism has yet to be confirmed.
SNARE proteins:
STX2 (syntaxin 2), STXBP5 (syntaxin binding protein 5), and STXBP1 are members of the SNARE family expressed in endothelial cells and/or platelets. These proteins have all been implicated in regulating plasma VWF levels in both GWAS (STX2 and STXBP5) and hypothesis-driven studies (STXBP1) (Figure 4A). In endothelial cells, STXBP5 functions as a negative regulator of VWF exocytosis [96,97], while in platelets the same protein promotes the release of VWF from α-granules. Interestingly, STXBP5-deficient mice have elevated levels of VWF, but counterintuitively display prolonged bleeding times and impaired arterial thrombosis, that may be related to abnormal platelet granule formation and secretion [96]. Variants in STXBP5 have been associated with VWF levels and bleeding severity in type 1 VWD [98,99], as well as elevated VWF levels in patients with arterial thrombosis [100]. STXBP5 variants can also modify the risk for venous thrombosis [67], although this association may involve both regulation of VWF levels as well as additional procoagulant factors associated with WPB or α-granule secretion.
Similar to STXBP5, variants in STX2 have been shown to influence VWF levels in normal individuals [67] and arterial thrombosis patients, and independently associates with an increased risk for arterial thrombosis [70,100]. However, conflicting reports exist on the association between variants in STX2 and plasma VWF levels in type 1 VWD [98,99]. In endothelial cells, syntaxin-2 forms a complex at the cell membrane with STXBP1 and Slp4-a, a Rab27a effector that localizes to WPBs and mediates their exocytosis [101]. STX2 is also involved in vesicle exocytosis in other cell types. However, STX2-defiency does not influence VWF release from platelet α-granules [102] and the direct influence of syntaxin-2 on VWF secretion from endothelial cells has yet to be described.
While common variants in STXBP1 have not been identified in GWAS of VWF levels, rare variants in this gene may contribute to a “low VWF” phenotype. STXBP1 loss-of-function is associated with the rare, severe epileptic disorder, EIEE4 (early infantile epileptic encephalopathy-4), related to impaired neurotransmitter release [101]. Studies of endothelial progenitor cells derived from an EIEE4 patient demonstrated decreased VWF secretion (~50% of wild-type), and low VWF plasma levels, but no reported association with a bleeding phenotype.
Genes influencing VWF clearance:
Clearance of VWF and FVIII proceeds through carbohydrate, amino acid and protein conformation-recognition based pathways, making this a semi-selective process that features numerous receptor-ligand interactions (Figure 4B). A growing number of VWF clearance receptors have been identified through a range of genetic, molecular and cellular approaches including LRP1, the macrophage galactose type lectin, Siglec-5, galactin-3 and −5, CLEC4M, stabilin-2, and the asialoglycoprotein receptor [89,103-108]. In this review, only clearance receptors with genetic variation that modify VWF plasma levels will be described.
LRP1:
The macrophage-expressed LRP1 (low-density lipoprotein receptor related protein) is a member of the LDLR superfamily of receptors, and is comprised of repetitive clustered repeats (I–IV) containing ligand binding and EGF homology region sequences that bind VWF under conditions of shear [104]. In mice, macrophage LRP-1 deficiency increases plasma VWF and FVIII levels, while both the LRP-1 ligand RAP and macrophage LRP-1 deficiency increase VWF half-life [104]. Although conflicting data exists, the rare, non-synonymous SNVs in the LRP-1 gene, p.Asp2080Asn and p.Ala217Val, have been shown to be associated with plasma levels of VWF and FVIII [109,110]. Additional receptors in the LDLR superfamily may also regulate VWF clearance, however, to date, genetic variants at these loci that modify this phenotype have not been described.
CLEC4M:
The CHARGE GWAS identified two receptors, CLEC4M and stabilin-2, expressed on the sinusoidal endothelium of the liver that associate with VWF plasma levels [70]. CLEC4M (C-type lectin domain family 4 member M) was originally characterized as an adhesive receptor involved in regulating pathogen infection. CLEC4M is comprised of a mannose-binding lectin domain, and a polymorphic neck region comprised of a variable number of tandem repeats (VNTRs) that mediates homotetramerization of the molecule and is in strong linkage-disequilibrium with the CHARGE SNV. We have characterized CLEC4M as a lectin receptor that mediates the endocytosis of VWF and FVIII via interactions with N-linked glycans [111,112]. Moreover, variants in CLEC4M including the VNTR polymorphism and the CHARGE SNV associate with plasma VWF levels in type 1 VWD populations [98,111].
STAB2:
Stabilin-2 is a scavenger receptor comprised of a repeating series of EGF-like, and fasciclin-1 (FAS-1) domains with an X-link domain located proximal to its transmembrane region that is C-type lectin-like. Interestingly, while human and murine stabilin-2 have retained an approximately 80% amino acid identity, stabilin-2 deficient mice exhibit normal VWF levels and normal half-life of murine VWF [113]. However, the half-life of recombinant or plasma-derived human VWF is increased by approximately 2-fold in stabilin-2 deficient mice, suggesting that stabilin-2 regulates the clearance of human but not murine VWF.
The CHARGE STAB2 SNVs associate with VWF:Ag levels in the Canadian type 1 VWD population, and with low plasma VWF levels in a separate GWAS [74,113]. In contrast, neither STAB2 SNV associated with type 1 VWD in a Dutch cohort [98]; this observation may be linked with differences in the composition of the study populations. Pathogenic variants in the STAB2 gene have been found to associate with large elevations in plasma VWF:Ag levels (>33%) in both association and exome sequencing studies [114,115], and modify the ability of stabilin-2 expressing cells to bind and internalize VWF in vitro [113]. Pathogenic STAB2 variants have also been shown to associate with the incidence of VTE [117], however given the scavenger function of stabilin-2, altered clearance of non-VWF plasma coagulation factors or inflammatory regulators may also contribute to the pathogenic basis of this observation.
SCARA5:
SCARA5 (Scavenger Receptor Class A Member 5) is a pattern recognition receptor expressed on murine epithelial cells and interstitial fibroblasts in the spleen [118]. SCARA5 assembles at the cell surface as a homo-trimer and contains an extracellular collagenous and cysteine rich scavenger domain. SCARA5 can bind a range of ligands including ferritin, polyanions, and microbes [119,120]. Solid phase binding assays have confirmed that VWF binds to SCARA5, and HEK 293 cells transfected with the SCARA5 cDNA are capable of binding and endocytosing both VWF and FVIII in a VWF-dependent manner [121]. However, to date, there is little available knowledge regarding SCARA5 expression in humans, and the in vivo location of VWF-SCARA5 interaction is unknown.
Genes influencing VWF multimerization:
VWF circulates in the plasma as a series of multimers ranging in size between 0.5–20 mDa with larger multimers possessing increased hemostatic activity. Modification of VWF multimer size by ADAMTS13 and thrombospondin-1 has been proposed to regulate VWF clearance, but animal models have failed to demonstrate a conclusive influence of proteolysis on VWF clearance in vivo [122]. In human studies, SNVs in the ADAMTS13 gene have been associated with plasma VWF levels [20], however this signal has been attributed to the ABO locus, as both genes are located within 136 kb on chromosome 9. Interestingly, variants in both ADAMTS13 and TSP-1 genes have been associated with an increased risk for cardiovascular disease, presumably through the regulation of VWF multimeric size and hemostatic activity [123,124].
Unknown Mechanisms
For several of the novel genetic loci that are associated with plasma levels of VWF, the functional relationship between VWF and the gene product is currently unknown, highlighting the challenge of mapping intergenic variants to the correct locus and identifying of causal variants associated with a genetic signal. Associations with the chromosome region 2q12–2p13, and the TC2N gene have been replicated by independent studies, increasing the probability that these two loci represent true genotype-phenotype associations and are not artifacts. Other associations, including BAI3 and UFM1 have not been replicated to date and additional work is required to test their influence on VWF [74,75].
Chromosome 2q12–2p13:
Two linkage analysis studies have identified signals at chromosome 2q12–2p13 that associate with VWF levels and contributed to 19.2% of its variability [20,75]. This locus has not been detected in GWAS, which may be related to the presence of rare pathogenic variants in this region that strongly regulate VWF levels. The identified linkage interval on chromosome 2q12-2p13 contains over 100 genes in a region ~34 Mb in size, and VWF modifying candidates in this region include SNARE proteins and glycosyltransferases. A subsequent GWAS and linkage analysis of the VWF propeptide in the same study population revealed no association in the 2q12-2p13 region [72], suggesting that variants in this genomic region modify VWF clearance rather than synthesis/secretion.
TC2N:
The association between VWF/FVIII levels and variants in TC2N (tandem C2 domains, nuclear), were first identified in the CHARGE study and subsequently replicated in additional cohorts [70,75]. The C2 domains of TC2N feature a basic cluster that imparts nuclear localization capabilities [125], however, to date, the mechanistic basis by which TC2N modulates VWF plasma levels is unknown. Importantly, TC2N variants may associate with an increased risk for venous thrombosis [126].
Variants influencing FVIII plasma levels
VWF circulates in the plasma in a non-covalent complex with the coagulation cofactor FVIII. Binding of VWF to FVIII protects FVIII from accelerated proteolysis and clearance, and both the plasma levels of VWF, and the binding constant between VWF and FVIII strongly influence plasma FVIII levels. Therefore, QTL that modify plasma levels of VWF tend to concomitantly modify FVIII levels, and the pathophysiological influence of these QTL on bleeding or thrombotic tendencies may therefore also involve altered plasma FVIII levels. Qualitative VWF abnormalities that involve pathogenic variants in the VWF D’D3 region that impair FVIII binding result in type 2N VWD involving isolated FVIII deficiency.
Interestingly, infrequent F8 gene variants have been associated with plasma FVIII:C in normal individuals [70,115], while variability in the VWF gene and ABO blood group locus account for approximately 50% of the variability in plasma FVIII:C [109]. Recent studies have suggested that for every 1% change in plasma VWF levels, FVIII activity (FVIII:C) levels will change ~0.54% [127]. Therefore, it follows that the majority of VWF QTL also associate with FVIII activity, although the magnitude of influence may be lower [70,76,128]. Importantly, both ABO blood group status and other variants that modify VWF clearance influence FVIII replacement pharmacokinetics in hemophilia A patients [129-131]
Future directions
Trait mapping studies have thus far provided an unprecedented understanding of the genetic architecture that regulates plasma VWF levels, while cell and animal-model based confirmatory studies have provided novel insights into the life cycle of VWF as well as the pathogenetic basis of both VWD and thrombosis. To date, these data indicate that with the exception of the ABO locus, common variants associated with a number of VWF-modifying genes exert a relatively small effect on plasma VWF:Ag levels, while a smaller number of rare variants can have a larger influence on plasma VWF:Ag. However, a substantial proportion of the heritability of VWF levels currently remains uncharacterized. Future studies that will assess the influence of rare variants on plasma VWF levels through whole genome/exome analysis will increase our understanding of the genetic basis of pathological quantitative abnormalities of VWF plasma levels, including expressivity, penetrance, epistasis, and gene-environment influences. This may allow for the development of algorithms that improve the molecular diagnosis of type 1 VWD, provide further insights into the pathogenic basis of the “low VWF” phenotype, or improve the identification of individuals at-risk for thrombosis. Moreover, the development and improved accessibility of bleeding assessment tools (BATs) will increase our understanding of how these variants modify the risk for bleeding in individuals with inherited coagulopathies. These studies may also facilitate the development of personalized therapies with improved coagulation factor pharmacokinetic profiles for inherited or acquired bleeding disorders. Ultimately, technical advances in high throughout sequencing and increased availability of genomic information will improve the molecular diagnostic evaluation and clinical care of patients with quantitative VWF abnormalities.
Acknowledgements
This study was supported by funds from the NIH for the Zimmerman Program (HL081588) and by a Canadian Institutes of Health Research Foundation Grant (FDN 154285). DL is the recipient of a Canada Research Chair in Molecular Hemostasis. LLS was supported by a Canadian Institutes of Health Research fellowship.
Footnotes
Disclosure of Conflict of Interest
D. Lillicrap has received research grants from Bayer, Bioverativ, CSL, and Octapharma. L.L. Swystun declares that she has nothing to disclose.
References
- 1.Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987; 69: 454–9. [PubMed] [Google Scholar]
- 2.Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993; 123: 893–8. [DOI] [PubMed] [Google Scholar]
- 3.Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010; 8: 213–6. [DOI] [PubMed] [Google Scholar]
- 4.Koster T, Blann A, Briet E, Vandenbroucke J, Rosendall F. Role of clotting factor VIII occurrence in effect of of deep-vein thrombosis Willebrand factor. Lancet 1995; 345: 152–5. [DOI] [PubMed] [Google Scholar]
- 5.Wieberdink RG, van Schie MC, Koudstaal PJ, Hofman A, Witteman JCM, de Maat MPM, Leebeek FWG, Breteler MMB. High von Willebrand factor levels increase the risk of stroke: the Rotterdam study. Stroke 2010; 41: 2151–6. [DOI] [PubMed] [Google Scholar]
- 6.Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation 2008; 117: 1449–59. [DOI] [PubMed] [Google Scholar]
- 7.Pottinger B, Read R, Paleolog E, Higgins P, Pearson J. von Willebrand factor is an acute phase reactant in man. Thromb Res 1989; 53: 387–94. [DOI] [PubMed] [Google Scholar]
- 8.Kremer Hovinga JA, Zeerleder S, Kessler P, Romani de Wit T, van Mourik JA, Hack CE, ten Cate H, Reitsma PH, Wuillemin WA, Lammle B. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost 2007; 5: 2284–90. [DOI] [PubMed] [Google Scholar]
- 9.Guagnano MT, Romano M, Falco A, Nutini M, Marinopiccoli M, Manigrasso MR, Basili S, Davi G. Leptin increase is associated with markers of the hemostatic system in obese healthy women. J Thromb Haemost 2003; 1: 2330–4. [DOI] [PubMed] [Google Scholar]
- 10.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand-factor multimers under flow. Blood 2004; 104: 100–6. [DOI] [PubMed] [Google Scholar]
- 11.Patel KN, Soubra SH, Bellera R V, Dong J-F, McMullen CA, Burns AR, Rumbaut RE. Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler Thromb Vasc Biol 2008; 28: 2225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michels A, Albánez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, James PD, Swystun LL, Lillicrap D. Histones link inflammation and thrombosis through the induction of Weibel–Palade body exocytosis. J Thromb Haemost 2016; 14: 2274–86. [DOI] [PubMed] [Google Scholar]
- 13.Castaman G Changes of von Willebrand factor during pregnancy in women with and without von willebrand disease. Mediterr J Hematol Infect Dis 2013; 5: e2013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ribeiro J, Almeida-Dias A, Ascensão A, Magalhães J, Oliveira AR, Carlson J, Mota J, Appell HJ, Duarte J. Hemostatic response to acute physical exercise in healthy adolescents. J Sci Med Sport 2007; 10: 164–9. [DOI] [PubMed] [Google Scholar]
- 15.Timm A, Fahrenkrug J, Jørgensen HL, Sennels HP, Goetze JP. Diurnal variation of von Willebrand factor in plasma: The Bispebjerg study of diurnal variations. Eur J Haematol 2014; 93: 48–53. [DOI] [PubMed] [Google Scholar]
- 16.Al-Awadhi AM, Alfadhli SM, Mustafa NY, Sharma PN. Effects of cigarette smoking on hematological parameters and von Willebrand factor functional activity levels in asymptomatic male and female Arab smokers. Med Princ Pract 2008; 17: 149–53. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Z, Chen Y, Zhang Y, Liu H, Liu Q, Zhao J, Hu M, Huang W, Wang G, Zhu T, Zhang J, Zhu P. Changes of plasma vWF level in response to the improvement of air quality: An observation of 114 healthy young adults. Ann Hematol 2013; 92: 543–8. [DOI] [PubMed] [Google Scholar]
- 18.Kouides PA. Aspects of the laboratory identification of von Willebrand disease in women. Semin Thromb Hemost 2006; 32: 480–4. [DOI] [PubMed] [Google Scholar]
- 19.Orstavik KH, Magnus P, Reisner H, Berg K, Graham JB, Nance W. Factor VIII and factor IX in a twin population. Evidence for a major effect of ABO locus on factor VIII level. Am J Hum Genet 1985; 37: 89–101. [PMC free article] [PubMed] [Google Scholar]
- 20.Desch KC, Ozel AB, Siemieniak D, Kalish Y, Shavit JA, Thornburg CD, Sharathkumar AA, McHugh CP, Laurie CC, Crenshaw A, Mirel DB, Kim Y, Cropp CD, Molloy AM, Kirke PN, Bailey-Wilson JE, Wilson AF, Mills JL, Scott JM, Brody LC, et al. Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome-wide association. Proc Natl Acad Sci 2013; 110: 588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Lange M, Snieder H, Ariëns RAS, Spector TD, Grant PJ. The genetics of haemostasis: A twin study. Lancet 2001; 357: 101–5. [DOI] [PubMed] [Google Scholar]
- 22.Bladbjerg EM, de Maat MPM, Christensen K, Bathum L, Jespersen J, Hjelmborg J. Genetic influence on thrombotic risk markers in the elderly--a Danish twin study. J Thromb Haemost 2006; 4: 599–607. [DOI] [PubMed] [Google Scholar]
- 23.Souto JC, Almasy L, Soria JM, Buil A, Stone W, Lathrop M, Blangero J, Fontcuberta J. Genome-wide linkage analysis of von Willebrand factor plasma levels: Results from the GAIT project. Thromb Haemost 2003; 89: 468–74. [PubMed] [Google Scholar]
- 24.Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987; 69: 1691–5. [PubMed] [Google Scholar]
- 25.Ginsburg D, Handin RI, Bonthron DT, Donlon TA, Bruns GAP, Latt SA, Orkin SH. Human von Willebrand Factor (vWF): Isolation of Complementary DNA (cDNA) Clones and Chromosomal Localization. Science 1985; 228: 1401–6. [DOI] [PubMed] [Google Scholar]
- 26.Lynch DC, Zimmerman TS, Collins CJ, Brown M, Morin MJ, Ling EH, Livingston DM. Molecular cloning of cDNA for human von Willebrand factor: authentication by a new method. Cell 1985; 41: 49–56. [DOI] [PubMed] [Google Scholar]
- 27.Sadler JE, Shelton-Inloes BB, Sorace JM, Harlan JM, Titani K, Davie EW. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc Natl Acad Sci 1985; 82: 6394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verweij CL, Diergaarde PJ, Hart M, Pannekoek H. Full-length von Willebrand factor (vWF) cDNA encodes a highly repetitive protein considerably larger than the mature vWF subunit. EMBO J 1986; 5: 1839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancuso DJ, Tuley EA, Westfield LA, Worrall NK, Shelton-Inloes BB, Sorace JM, Alevy YG, Sadler JE. Structure of the gene for human von Willebrand factor. J Biol Chem 1989; 264: 19514–27. [PubMed] [Google Scholar]
- 30.Mancuso DJ, Tuley EA, Westfield LA, Lester-Mancuso TL, Le Beau MM, Sorace JM, Sadler JE. Human von Willebrand factor gene and pseudogene: structural analysis and differentiation by polymerase chain reaction. Biochemistry 1991; 30: 253–69. [DOI] [PubMed] [Google Scholar]
- 31.Wang QY, Song J, Gibbs RA, Boerwinkle E, Dong JF, Yu FL. Characterizing polymorphisms and allelic diversity of von Willebrand factor gene in the 1000 Genomes. J Thromb Haemost 2013; 11: 261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campos M, Sun W, Yu F, Barbalic M, Tang W, Chambless LE, Kenneth K, Ballantyne C, Folsom AR, Boerwinkle E, Dong J, Dc W, Wu KK. Genetic determinants of plasma von Willebrand factor antigen levels : a target gene SNP and haplotype analysis of ARIC cohort. Blood 2011; 117: 5224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keightley A, Lam Y, Brady J, Cameron C, Lillicrap D. Variation at the von Willebrand factor (vWF) gene locus is associated with plasma vWF:Ag levels: identification of three novel single nucleotide polymorphisms in the vWF gene promoter. Blood 1999; 93: 4277–83. [PubMed] [Google Scholar]
- 34.Petersen R, Lambourne JJ, Javierre BM, Grassi L, Kreuzhuber R, Ruklisa D, Rosa IM, Tomé AR, Elding H, van Geffen JP, Jiang T, Farrow S, Cairns J, Al-Subaie AM, Ashford S, Attwood A, Batista J, Bouman H, Burden F, Choudry FA, et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat Commun 2017; 8: 16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller CH, Dilley A, Richardson L, Hooper WC, Evatt BL. Population differences in von Willebrand factor levels affect the diagnosis of von Willebrand disease in African-American women. Am J Hematol 2001; 67: 125–9. [DOI] [PubMed] [Google Scholar]
- 36.Bellissimo DB, Christopherson PA, Flood VH, Gill JC, Friedman KD, Haberichter SL, Shapiro AD, Abshire TC, Leissinger C, Hoots WK, Lusher JM, Ragni M V, Montgomery RR. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African-American population. Blood 2012; 119: 2135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnsen JM, Auer PL, Morrison AC, Jiao S, Wei P, Haessler J, Fox K, McGee SR, Smith JD, Carlson CS, Smith N, Boerwinkle E, Kooperberg C, Nickerson D a., Rich SS, Green D, Peters U, Cushman M, Reiner AP, NHLBI Exome Sequencing Project. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood 2013; 122: 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowman M, Tuttle A, Notley C, Brown C, Tinlin S, Deforest M, Leggo J, Blanchette VS, Lillicrap D, James P. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013; 11: 512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowman M, Casey L, Hawke L, James P. Heterogeneity Of Type 3 Von Willebrand Disease (VWD): Evidence From Patient-Derived Blood Outgrowth Endothelial Cells (BOEC). Blood (ASH Annu Meet Abstr) 2013; 122: 3517. [Google Scholar]
- 40.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, Brown C, Andrews C, Labelle A, Chirinian Y, O’Brien L, Othman M, Rivard G, Rapson D, Hough C, Lillicrap D. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood 2007; 109: 145–54. [DOI] [PubMed] [Google Scholar]
- 41.Goodeve A, Jeroen J, Castaman G, Rodeghiero F, Federici AB, Batlle JM, Mazurier C, Goudemand C, Schneppenheim J, Budde R, Ingerslev J, Vorlova D, Holmberg Z, Lethagen L, Eikenboom E. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD). Blood 2006; 109: 112–21. [DOI] [PubMed] [Google Scholar]
- 42.Yadegari H, Driesen J, Pavlova A, Biswas A, Hertfelder H-J, Oldenburg J. Mutation distribution in the von Willebrand factor gene related to the different von Willebrand disease (VWD) types in a cohort of VWD patients. Thromb Haemost 2012; 108: 662–71. [DOI] [PubMed] [Google Scholar]
- 43.Cumming A, Grundy P, Keeney S, Lester W, Enayat S, Guilliatt A, Bowen D, Pasi J, Keeling D, Hill F, Bolton-Maggs PHB, Hay C, Collins P. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemost 2006; 96: 630–41. [PubMed] [Google Scholar]
- 44.Flood VH, Christopherson PA, Gill JC, Friedman KD, Haberichter SL, Bellissimo DB, Udani RA, Dasgupta M, Hoffmann RG, Ragni M V, Shapiro AD, Lusher JM, Lentz SR, Abshire TC, Leissinger C, Hoots WK, Manco-Johnson MJ, Gruppo RA, Boggio LN, Montgomery KT, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood 2016; 127: 2481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casonato A, Cattini MG, Barbon G, Daidone V, Pontara E. Severe, recessive type 1 is a discrete form of von Willebrand disease: the lesson learned from the c.1534-3C>A von Willebrand factor mutation. Thromb Res 2015; 136: 682–6. [DOI] [PubMed] [Google Scholar]
- 46.Haberichter SL, Christopherson P, Flood V, Cox Gill J, Bellissimo DB, Friedman KD, Montgomery RR. Critical Importance Of VWF Propeptide (VWFpp) In The Diagnosis Of Type 1 Von Willebrand Disease (VWD). Blood (ASH Annu Meet Abstr) 2013; 21: 331. [Google Scholar]
- 47.Eikenboom J, Hilbert L, Ribba AS, Hommais A, Habart D, Messenger S, Al-Buhairan A, Guilliatt A, Lester W, Mazurier C, Meyer D, Fressinaud E, Budde U, Will K, Schneppenheim R, Obser T, Marggraf O, Eckert E, Castaman G, Rodeghiero F, et al. Expression of 14 von Willebrand factor mutations identified in patients with type 1 von Willebrand disease from the MCMDM-1VWD study. J Thromb Haemost 2009; 7: 1304–12. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Bouwens EAM, Pintao MC, Voorberg J, Safdar H, Valentijn KM, De Boer HC, Mertens K, Reitsma PH, Eikenboom J. Analysis of the storage and secretion of von Willebrand factor in blood outgrowth endothelial cells derived from patients with von Willebrand disease. Blood 2013; 121: 2762–72. [DOI] [PubMed] [Google Scholar]
- 49.Starke RD, Paschalaki KE, Dyer CEF, Harrison-lavoie KJ, Cutler JA, Mckinnon TAJ, Millar CM, Cutler DF, Laffan MA, Randi AM. Cellular and molecular basis of von Willebrand disease : studies on blood outgrowth endothelial cells. Blood 2015; 121: 2773–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Othman M, Chirinian Y, Brown C, Notley C, Hickson N, Hampshire D, Buckley S, Waddington S, Parker A, Baker A, James P, Lillicrap D. Functional characterization of a 13-bp deletion (c.−1522_−1510del13) in the promoter of the von Willebrand factor gene in type 1 von Willebrand disease. Blood 2010; 116: 3645–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haberichter SL, Castaman G, Budde U, Peake I, Goodeve A, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Eikenboom J, Schneppenheim R, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill FGH, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood 2008; 111: 4979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haberichter SL, Balistreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, Manco-Johnson MJ, Gill JC, Montgomery RR. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006; 108: 3344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rawley O, O’Sullivan JM, Chion A, Keyes S, Lavin M, van Rooijen N, Brophy TM, Fallon P, Preston RJS, O’Donnell JS. von Willebrand factor arginine 1205 substitution results in accelerated macrophage-dependent clearance in vivo. J Thromb Haemost 2015; 13: 821–6. [DOI] [PubMed] [Google Scholar]
- 54.Swystun LL, Georgescu I, Mewburn J, Deforest M, Nesbitt K, Hebert K, Dwyer C, Brown C, Notley C, Lillicrap D. Abnormal von Willebrand factor secretion, factor VIII stabilization and thrombus dynamics in type 2N von Willebrand disease mice. J Thromb Haemost 2017; 15: 1607–19. [DOI] [PubMed] [Google Scholar]
- 55.Casari C, Du V, Wu Y-P, Kauskot A, de Groot PG, Christophe OD, Denis C V, de Laat B, Lenting PJ. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood 2013; 122: 2893–902. [DOI] [PubMed] [Google Scholar]
- 56.Rydz N, Grabell J, Lillicrap D, James PD. Changes in von Willebrand factor level and von Willebrand activity with age in type 1 von Willebrand disease. Haemophilia 2015; 21: 636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanders Y, Giezenaar MA, Laros-van Gorkom B, Meijer K, van der Bom J, Cnossen M, Nijziel M, Ypma P, Fijnvandraat K, Eikenboom J, Mauser-Bunschoten E, Leebeek F. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost 2014; 12: 1066–75. [DOI] [PubMed] [Google Scholar]
- 58.Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, Rick ME, Sadler JE, Weinstein M, Yawn BP. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14: 171–232. [DOI] [PubMed] [Google Scholar]
- 59.Laffan MA, Lester W, Donnell JSO, Will A, Tait RC, Goodeve A, Carolyn M. The diagnosis and management of von Willebrand disease : a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Anaesth 2014; 167: 453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lavin M, Aguila S, Schneppenheim S, Dalton N, Jones KL, Sullivan JMO, Connell NMO, Ryan K, White B, Byrne M, Rafferty M, Doyle MM, Nolan M, Preston RJS, Budde U, James P, Di Paola J, O’Donnell JS. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood 2018; 130: 2344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leebeek FWG, Boender J. Low VWF : an established mild bleeding disorder? Blood 2018; 130: 2241–3. [DOI] [PubMed] [Google Scholar]
- 62.James PD, Paterson AD, Notley C, Cameron C, Hegadorn C, Tinlin S, Brown C, O’Brien L, Leggo J, Lillicrap D. Genetic linkage and association analysis in type 1 von Willebrand disease: results from the Canadian type 1 VWD study. J Thromb Haemost 2006; 4: 783–92. [DOI] [PubMed] [Google Scholar]
- 63.Eikenboom J, Van Marion V, Putter H, Goodeve A, Rodegherio F, Castaman G, Federici A, Battle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill F, et al. Linkage analysis in families diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD. J Thromb Haem 2006; 4: 774–82. [DOI] [PubMed] [Google Scholar]
- 64.Sanders Y V, Eikenboom J, de Wee EM, van der Bom JG, Cnossen MH, Degenaar-Dujardin MEL, Fijnvandraat K, Kamphuisen PW, Laros-van Gorkom B a P, Meijer K, Mauser-Bunschoten EP, Leebeek FWG. Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost 2013; 11: 845–54. [DOI] [PubMed] [Google Scholar]
- 65.van der Meer IM, Brouwers G-JJ, Bulk S, Leebeek FWG, van der Kuip DAM, Hofman A, Witteman JCM, Gomez Garcia EB, Gómez García EB. Genetic variability of von Willebrand factor and risk of coronary heart disease: the Rotterdam Study. Br J Haematol 2004; 124: 343–7. [DOI] [PubMed] [Google Scholar]
- 66.van Schie MC, De Maat MPM, Isaacs A, Van Duijn CM, Deckers JW, Dippel DWJ, Leebeek FWG. Variation in the von Willebrand factor gene is associated with von Willebrand factor levels and with the risk for cardiovascular disease. Blood 2011; 117: 1393–400. [DOI] [PubMed] [Google Scholar]
- 67.Smith NL, Rice KM, Bovill EG, Cushman M, Bis JC, McKnight B, Lumley T, Glazer NL, van Hylckama Vlieg A, Tang W, Dehghan A, Strachan DP, O’Donnell CJ, Rotter JI, Heckbert SR, Psaty BM, Rosendaal FR. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood 2011; 117: 6007–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Loon JE, Kavousi M, Leebeek FWG, Felix JF, Hofman A, Witteman JCM, De Maat MPM. von Willebrand factor plasma levels, genetic variations and coronary heart disease in an older population. J Thromb Haemost 2012; 10: 1262–9. [DOI] [PubMed] [Google Scholar]
- 69.van Schie MC, van Loon JE, de Maat MPM, Leebeek FWG. Genetic determinants of von Willebrand factor levels and activity in relation to the risk of cardiovascular disease: a review. J Thromb Haemost 2011; 9: 899–908. [DOI] [PubMed] [Google Scholar]
- 70.Smith NL, Chen M-H, Dehghan A, Strachan DP, Basu S, Soranzo N, Hayward C, Rudan I, Sabater-Lleal M, Bis JC, de Maat MPM, Rumley A, Kong X, Yang Q, Williams FMK, Vitart V, Campbell H, Mälarstig A, Wiggins KL, Van Duijn CM, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation 2010; 121: 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mufti A, Ogiwara K, Swystun LL, Eikenboom JCJ, Budde U, Hopman WM, Halldén C, Goudemand J, Peake I, Goodeve AC, Lillicrap D, Hampshire DJ. The common VWF single nucleotide variants c.2365A>G and c.2385T>C modify VWF biosynthesis and clearance. Blood Adv 2018; 2: 1585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ozel AB, McGee B, Siemieniak D, Jacobi PM, Haberichter SL, Brody LC, Mills JL, Molloy AM, Ginsburg D, Li JZ, Desch KC. Genome-wide studies of von Willebrand Factor Propeptide Identify Loci Contributing to Variation in Propeptide Levels and von Willebrand Factor Clearance. J Thromb Haemost 2016; 14: 1888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hemmerhirt H, Broman K, Shavit J, Ginsburg D. Genetic regulation of plasma von Willebrand factor levels : quantitative trait loci analysis in a mouse model. J Thromb Haem 2007; 5: 329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Loon J, Dehghan A, Weihong T, Trompet S, McArdle WL, Asselbergs FFW, Chen M-H, Lopez LM, Huffman JE, Leebeek FWG, Basu S, Stott DJ, Rumley A, Gansevoort RT, Davies G, Wilson JJF, Witteman JCM, Cao X, de Craen AJM, Bakker SJL, et al. Genome-wide association studies identify genetic loci for low von Willebrand factor levels Eur J Hum Genet Nature Publishing Group; 2016; 24: 1035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Antoni G, Morange P, Luo Y, Saut N, Burgos G, Heath S, Germain M, Biron-Andreani C, Schved J, Pernod G, Galan P, Zelenika D, Alessi M, Drouet L, Visvikis-Siest S, Wells PS, Lathrop M, Emmerich J, Tregouet D, Gagnon F. A multi-stage multi-design strategy provides strong evidence that the BAI3 locus is associated with early-onset venous thromboembolism. J Thromb Haemost 2010; 8: 2671–9. [DOI] [PubMed] [Google Scholar]
- 76.Antoni G, Oudot-Mellakh T, Dimitromanolakis A, Germain M, Cohen W, Wells P, Lathrop M, Gagnon F, Morange P-E, Tregouet D-A. Combined analysis of three genome-wide association studies on vWF and FVIII plasma levels. BMC Med Genet 2011; 12: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sabater-Llea M, de Vries P, Huffman J, Martin J, Martinez A, Morange P, Kleber M, Hayward C, Guo X, de Haan H, Trompet S, Yanek L, Smith V, Ozel A, Tang W, Pankratz N, Strachan D, Lowenstein C, O’Donnell CJ, Smith N, et al. Multi-ethnic Genome-wide Association Study Identifies New Loci Regulating Factor VIII and von Willebrand Factor. Res Pract Thromb Haemost (ISTH Congr Abstr) 2017; 1: 102. [Google Scholar]
- 78.Canis K, McKinnon TAJ, Nowak A, Haslam SMM, Panico M, Morris HRR, Laffan MAA, Dell A. Mapping the N-glycome of human von Willebrand factor. Biochem J 2012; 447: 217–28. [DOI] [PubMed] [Google Scholar]
- 79.Lenting PJ, Pegon JN, Christophe OD, Denis C V. Factor VIII and von Willebrand factor--too sweet for their own good. Haemophilia 2010; 16 Suppl 5: 194–9. [DOI] [PubMed] [Google Scholar]
- 80.McKinnon TAJ, Goode EC, Birdsey GM, Nowak AA, Chan ACK, Lane DA, Laffan MA. Specific N-linked glycosylation sites modulate synthesis and secretion of von Willebrand factor. Blood 2010; 116: 640–8. [DOI] [PubMed] [Google Scholar]
- 81.Badirou I, Kurdi M, Legendre P, Rayes J, Bryckaert M, Casari C, Lenting PJ, Christophe OD, Denis C V. In vivo analysis of the role of o-glycosylations of von Willebrand factor. PLoS One 2012; 7: e37508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Donnell JS, McKinnon TAJ, Crawley JTB, Lane DA, Laffan MA. Bombay phenotype is associated with reduced plasma-VWF levels and an increased susceptibility to ADAMTS13 proteolysis. Blood 2005; 106: 1988–91. [DOI] [PubMed] [Google Scholar]
- 83.Green D, Jarrett O, Ruth K, Folsom A, Liu K. Relationship among Lewis phenotype, clotting factors, and other cardiovascular risk factors in young adults. J Lab Clin Med 1995; 125: 334–9. [PubMed] [Google Scholar]
- 84.O’Donnell J, Boulton FE, Manning RA, Laffan MA. Genotype at the Secretor blood group locus is a determinant of plasma von Willebrand factor level. Br J Haematol 2002; 116: 350–6. [DOI] [PubMed] [Google Scholar]
- 85.Gallinaro L, Cattini MG, Sztukowska M, Padrini R, Sartorello F, Pontara E, Bertomoro A, Daidone V, Pagnan A, Casonato A. A shorter von Willebrand factor survival in O blood group subjects explains how ABO determinants influence plasma von Willebrand factor. Blood 2008; 111: 3540–5. [DOI] [PubMed] [Google Scholar]
- 86.Castaman G, Tosetto A, Eikenboom JC, Rodeghiero F. Blood group significantly influences von Willebrand factor increase and half-life after desmopressin in von Willebrand disease Vicenza. J Thromb Haemost 2010; 8: 2078–80. [DOI] [PubMed] [Google Scholar]
- 87.Groeneveld DJ, van Bekkum T, Cheung KL, Dirven RJ, Castaman G, Reitsma PH, van Vlijmen B, Eikenboom J. No evidence for a direct effect of von Willebrand factor’s ABH blood group antigens on von Willebrand factor clearance. J Thromb Haemost 2015; 13: 592–600. [DOI] [PubMed] [Google Scholar]
- 88.Millar CM, Riddell AF, Brown SA, Starke R, Mackie I, Bowen DJ, Jenkins PV, Van Mourik JA. Survival of von Willebrand factor released following DDAVP in a type 1 von Willebrand disease cohort: influence of glycosylation, proteolysis and gene mutations. Thromb Haemost 2008; 99: 916–24. [DOI] [PubMed] [Google Scholar]
- 89.Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, Marth JD. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med 2008; 14: 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, Marth JD. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci 2002; 99: 10042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song J, Xue C, Preisser JS, Cramer DW, Houck KL. Association of Single Nucleotide Polymorphisms in the ST3GAL4 Gene with VWF Antigen and Factor VIII Activity. PLoS One 2016; 11: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaufmann JE, Oksche A, Wollheim CB, Günther G, Rosenthal W, Vischer UM. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J Clin Invest 2000; 106: 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nossent AY, Robben JH, Deen PMT, Vos HL, Rosendaal FR, Doggen CJM, Hansen JL, Sheikh SP, Bertina RM, Eikenboom HCJ. Functional variation in the arginine vasopressin 2 receptor as a modifier of human plasma von Willebrand factor levels. J Thromb Haemost 2010; 8: 1547–54. [DOI] [PubMed] [Google Scholar]
- 94.Bichet DG, Razi M, Lonergan M, Arthus M-F, Papukna V, Kortas C, Barjon J-N. Hemodynamic and coagulation responses to 1-desamino(8-D-Arginine) vasopressin in patients witih congenital nephrogenic diabetes insipidus. N Engl J Med 1988; 318: 881–7. [DOI] [PubMed] [Google Scholar]
- 95.Kario K, Matsuo T, Kobayashi H, Kanai N, Hoshide S, Mitsuhashi T, Ikeda U, Nishiuma S, Matsuo M, Shimada K. Endothelial cell damage and angiotensin-converting enzyme insertion/deletion genotype in elderly hypertensive patients. J Am Coll Cardiol 1998; 32: 444–50. [PubMed] [Google Scholar]
- 96.Ye S, Huang Y, Joshi S, Zhang J, Yang F, Zhang G, Smyth SS, Li Z, Takai Y, Whiteheart SW. Platelet secretion and hemostasis require syntaxin-binding protein STXBP5. J Clin Invest 2014; 124: 4517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu Q, Yamakuchi M, Ture S, Garcia-hernandez MDL, Ko KA, Modjeski KL, Lomonaco MB, Johnson AD, Donnell CJO, Takai Y, Morrell CN, Lowenstein CJ. Syntaxin-binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest 2014; 124: 4503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanders Y V, van der Bom JG, Isaacs A, Cnossen MH, de Maat MPM, Laros-van Gorkom BAP, Fijnvandraat K, Meijer K, van Duijn CM, Mauser-Bunschoten EP, Eikenboom J, Leebeek FWG. CLEC4M and STXBP5 gene variations contribute to von Willebrand factor level variation in von Willebrand disease. J Thromb Haemost 2015; 13: 956–66. [DOI] [PubMed] [Google Scholar]
- 99.van Loon JE, Sanders Y V., de Wee EM, Kruip MJHA, de Maat MPM, Leebeek FWG. Effect of genetic variation in STXBP5 and STX2 on von willebrand factor and bleeding phenotype in type 1 von willebrand disease patients. PLoS One 2012; 7: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Loon JE, Leebeek FWG, Deckers JW, Dippel DWJ, Poldermans D, Strachan DP, Tang W, O’Donnell CJ, Smith NL, De Maat MPM. Effect of genetic variations in syntaxin-binding protein-5 and syntaxin-2 on von willebrand factor concentration and cardiovascular risk. Circ Cardiovasc Genet 2010; 3: 507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Breevoort D, Snijders AP, Hellen N, Weckhuysen S, Van Hooren KWEM, Eikenboom J, Valentijn K, Fernandez-borja M, Ceulemans B, De Jonghe P, Voorberg J, Hannah M, Carter T, Bierings R. STXBP1 promotes Weibel-Palade body exocytosis through its interaction with the Rab27A effector Slp4-a. Blood 2016; 123: 3185–95. [DOI] [PubMed] [Google Scholar]
- 102.Ye S, Karim ZA, Hawas R Al, Pessin JE, Filipovich AH, Whiteheart SW. Syntaxin-11 , but not syntaxin-2 or syntaxin-4 , is required for platelet secretion. Blood 2012; 120: 2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pegon JN, Kurdi M, Casari C, Odouard S, Denis C V, Christophe OD, Lenting PJ. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica 2012; 97: 1855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rastegarlari G, Pegon JN, Casari C, Odouard S, Navarrete A-M, Saint-Lu N, van Vlijmen BJ, Legendre P, Christophe OD, Denis C V, Lenting PJ. Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood 2012; 119: 2126–34. [DOI] [PubMed] [Google Scholar]
- 105.Bovenschen N, Mertens K, Hu L, Havekes LM, van Vlijmen BJM. LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood 2005; 106: 906–12. [DOI] [PubMed] [Google Scholar]
- 106.Bovenschen N, Rijken DC, Havekes LM, van Vlijmen BJM, Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. J Thromb Haemost 2005; 3: 1257–65. [DOI] [PubMed] [Google Scholar]
- 107.Wohner N, Muczynski V, Mohamadi A, Legendre P, Aymé G, Christophe OD, Lenting PJ, Denis C V, Casari C. Macrophage scavenger-receptor SR-AI contributes to the clearance of von Willebrand factor. Haematologica 2018; 103: 728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ward SE, Sullivan JMO, Drakeford C, Aguila S, Jondle CN, Sharma J, Fallon PG, Brophy TM, Preston RJS, Smyth P, Sheils O, Chion A, Donnell JSO. A novel role for the macrophage galactose-type lectin receptor in mediating von Willebrand factor clearance. Blood 2018; 131: 911–7. [DOI] [PubMed] [Google Scholar]
- 109.Morange PE, Tregouet DA, Frere C, Saut N, Pellegrina L, Alessi MC, Visvikis S, Tiret L, Juhan-Vague I. Biological and genetic factors influencing plasma factor VIII levels in a healthy family population: Results from the Stanislas cohort. Br J Haematol 2005; 128: 91–9. [DOI] [PubMed] [Google Scholar]
- 110.Vormittag R, Bencur P, Ay C, Tengler T, Vukovich T, Quehenberger P, Pabinger I, Mannhalter C. Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Polymorphism 663 C> T Affects Clotting Factor VIII Activity and Increases the Risk of Venous Thromboembolism. J Thromb Haemost 2006; 108: 497–502. [DOI] [PubMed] [Google Scholar]
- 111.Rydz N, Swystun LL, Notley C, Paterson AD, Riches JJ, Sponagle K, Boonyawat B, Montgomery RR, James PD, Lillicrap D. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood 2013; 121: 5228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Swystun LL, Notley C, Sponagle K, James PD, Lillicrap D. The endothelial lectin CLEC4M is a novel clearance receptor for factor VIII. J Thromb Haemost (ISTH Congr Abstr) 2013; 11: 98. [Google Scholar]
- 113.Swystun L, Lai J, Notley C, Georgescu I, Paine A, Mewburn J, Nesbitt K, Schledzewski K, Géraud C, Kzhyshkowska J, Goerdt S, Hopman W, Montgomery R, James P, Lillicrap D. The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J Clin Invest 2018; 128: In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Desch KC, Ozel A, Halvorsen M, Michels A, Swystun L, Mokry L, Richards B, Germain M, Tregouet DA, Reitsma PH, Kearon K, Li JZ, Goldstein D., Lillicrap D, Ginsburg D. Exome Sequencing Studies Identify Mutations in STAB2 As a Genetic Risk for Venous Thromboembolic Disease. Blood (ASH Annu Meet Abstr) 2017; 126. [Google Scholar]
- 115.Huffman JE, De Vries PS, Morrison AC, Sabater-lleal M, Kacprowski T, Auer PL, Brody JA, Chasman DI, Chen M, Guo X, Lin L, Marioni RE, Martina M, Cushman M, Wiggins KL, Qi L, Sennblad B, Harris SE, Polasek O, Riess H, et al. Rare and low-frequency variants and their association with plasma levels. Blood 2015; 126: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Swystun LL, Lai J, Notley C, Georgescu I, Paine A, Mewburn J, Nesbitt K, Schledzewski K, Géraud C, Kzhyshkowska J, Goerdt S, Hopwman W, Montgomery RR, James P, Lillicrap D. The endothelial receptor stabilin-2 regulates von Willebrand factor-factor VIII complex half-life and immunogenicity. Submitt J Clin Investig 2017; . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Desch KC, Ozel AB, Halvorsen M, Jacobi PM, Germain M, Tregouet DA, Reitsma PH, Goldstein D, Ginsburg D. Exome Sequencing in Venous Thromboembolic Disease Identifies Excess Mutation Burden in PROS1, PROC, SERPINC1, STAB2. Blood (ASH Annu Meet Abstr) 2016; 128: 3794. [Google Scholar]
- 118.Ojala JRM, Pikkarainen T, Elmberger G, Tryggvason K. Progressive reactive lymphoid connective tissue disease and development of autoantibodies in scavenger receptor A5-deficient mice Am J Pathol American Society for Investigative Pathology; 2013; 182: 1681–95. [DOI] [PubMed] [Google Scholar]
- 119.Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, Drexler IR, Chen X, Sanna-Cherchi S, Mohammed F, Williams D, Lin CS, Schmidt-Ott KM, Andrews NC, Barasch J. Scara5 Is a Ferritin Receptor Mediating Non-Transferrin Iron Delivery. Dev Cell 2009; 16: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang Y, Oliver P, Davies KE, Platt N. Identification and characterization of murine SCARA5, a novel class A scavenger receptor that is expressed by populations of epithelial cells. J Biol Chem 2006; 281: 11834–45. [DOI] [PubMed] [Google Scholar]
- 121.Ogiwara K, Swystun L, Brown C, Notley C, Ojala J, Tryggvason K, Lillicrap D. Scavenger receptor class A member 5 (SCARA5) binds and internalizes VWF in vitro: a novel candidate VWF clearance receptor. J Thromb Haemost 2015; 13: 222. [Google Scholar]
- 122.Badirou I, Kurdi M, Rayes J, Legendre P, Christophe OD, Lenting PJ, Denis C V. von Willebrand factor clearance does not involve proteolysis by ADAMTS-13. J Thromb Haemost 2010; 8: 2338–40. [DOI] [PubMed] [Google Scholar]
- 123.Schettert IT, Pereira AC, Lopes NH, Hueb WA, Krieger JE. Association between ADAMTS13 polymorphisms and risk of cardiovascular events in chronic coronary disease. Thromb Res 2010; 125: 61–6. [DOI] [PubMed] [Google Scholar]
- 124.Stenina OI, Topol EJ, Plow EF. Thrombospondins, their polymorphisms, and cardiovascular disease. Arterioscler Thromb Vasc Biol 2007; 27: 1886–94. [DOI] [PubMed] [Google Scholar]
- 125.Fukuda M, Mikoshiba K. Tac2-N, an atypical C-type tandem C2 protein localized in the nucleus. FEBS Lett 2001; 503: 217–8. [DOI] [PubMed] [Google Scholar]
- 126.Morange P-E, Saut N, Antoni G, Emmerich J, Trégouët D. Impact on venous thrombosis risk of newly discovered gene variants associated with FVIII and VWF plasma levels. J Thromb Haemost 2011; 9: 229–31. [DOI] [PubMed] [Google Scholar]
- 127.Song J, Chen F, Campos M, Bolgiano D, Houck K, Chambless LE, Wu KK, Folsom AR, Couper D, Boerwinkle E, Dong JF. Quantitative influence of ABO blood groups on factor VIII and its ratio to von willebrand factor, novel observations from an ARIC study of 11,673 subjects. PLoS One 2015; 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hermanns MI, Grossmann V, Spronk HMH, Schulz A, Jünger C, Laubert-Reh D, Mazur J, Gori T, Zeller T, Pfeiffer N, Beutel M, Blankenberg S, Münzel T, Lackner KJ, Cate-Hoek AJT, Cate H Ten, Wild PS. Distribution, genetic and cardiovascular determinants of FVIII:c - Data from the population-based Gutenberg Health Study Int J Cardiol Elsevier B.V.; 2015; 187: 166–74. [DOI] [PubMed] [Google Scholar]
- 129.Vlot A, Mauser-Bunschoten E, Zarkova A, Haan E, Kruitwagen C, Sixma J, van den Berg H. The half-life of infused factor VIII is shorter in hemophiliac patients with blood group O than in those with blood group A. Thromb Haemost 2000; 83: 65–9. [PubMed] [Google Scholar]
- 130.Kepa S, Horvath B, Reitter-Pfoertner S, Schemper M, Quehenberger P, Grundbichler M, Heistinger M, Neumeister P, Mannhalter C, Pabinger I. Parameters influencing FVIII pharmacokinetics in patients with severe and moderate haemophilia A. Haemophilia 2015; 21: 343–50. [DOI] [PubMed] [Google Scholar]
- 131.Ogiwara K, Swystun L, Georgescu I, Brown C, Tuttle A, Tinlin S, Leggo J, Albanez S, Carcao M, Blanchette V, Tom C, Male K, Lillicrap D. Clearance and Genetic Variability of Von Willebrand Factor Are Major Determinants of the Pharmacokinetic Behavior of Factor VIII Concentrates in the Treatment of Pediatric Hemophilia A. Blood (ASH Annu Meet Abstr) 2014; 123. [Google Scholar]