

Summary
mRNA decay factors regulate mRNA turnover by recruiting non-translating mRNAs and targeting them for translational repression and mRNA degradation. How mRNA decay pathways regulate cellular function in vivo with specificity is poorly understood. Here we show that C. elegans mRNA decay factors, including the translational repressors CAR-1/LSM14 and CGH-1/DDX6, and the decapping enzymes DCAP-1/DCP1, function in neurons to differentially regulate axon development and maintenance, and axon regrowth following injury. In neuronal cell bodies, CAR-1 fully colocalizes with CGH-1, and partially colocalizes with DCAP-1, suggesting mRNA decay components form at least two types of cytoplasmic granules. Following axon injury in adult neurons, loss of CAR-1 or CGH-1 results in increased axon regrowth and growth cone formation, whereas loss of DCAP-1/2 results in reduced regrowth. To determine how CAR-1 inhibits regrowth we analyzed mRNAs bound to pan-neuronally expressed GFP::CAR-1 using a crosslinking and immunoprecipitation based approach. Among the putative mRNA targets of CAR-1, we characterized the roles of micu-1, a regulator of the mitochondrial calcium uniporter MCU-1, in axon injury. We show that loss of car-1 results increased MICU-1 protein levels, and that enhanced axon regrowth in car-1 mutants is dependent on micu-1 and mcu-1. Moreover, axon injury induces transient calcium influx into axonal mitochondria, dependent on MCU-1. In car-1 loss of function mutants and in micu-1 overexpressing animals, the axonal mitochondrial calcium influx is more sustained, which likely underlies enhanced axon regrowth. Our data uncover a novel pathway that controls axon regrowth through axonal mitochondrial calcium uptake.
Keywords: RNA binding protein, LSM14, CGH-1, DDX6, P body, MCU, MICU
Graphical Abstract
eTOC Blurb
Tang et al. dissect the roles of mRNA decay factors in C. elegans axon regeneration. Loss of function in the Lsm14 ortholog CAR-1 results in increased axon regeneration due to elevated expression of MICU-1, a mitochondrial calcium uniporter regulator that is a key target of CAR-1 repression in neurons.
Introduction
Mature mRNAs are either actively translated or exist in a translational repressed state that can be targeted for degradation by mRNA decay factors. Extensive biochemical studies show that the recognition of mRNAs by the mRNA decay factor LSM14 and DEAD-box RNA helicase DDX6 leads to translational repression, followed by irreversible removal of the 5’ cap by decapping enzymes DCP1 and DCP2, and mRNA degradation by the exonuclease XRN1 (reviewed in [1]). In multiple cell types depletion of mRNA decay factors usually leads to accumulation of stabilized mRNAs. However, increasing evidence suggests that mRNA decay factors exhibit a high degree of selectivity in mRNA stability regulation [2]. Therefore, it is crucial to identify the mRNA targets of mRNA decay factors to provide a better understanding of their cellular functions.
Neurons are polarized cells that are primed for a high degree of selective and dynamic regulation of gene expression. mRNA decay factors are widely expressed in neurons of C. elegans, Drosophila, and mammals [3–6]. For example, Drosophila Me31B/DDX6 is enriched in post-synaptic dendrites [5], and its overexpression in sensory neurons reduces higher-order dendrite arborization [3], although it remains unknown whether particular mRNA targets are involved.
C. elegans expresses many conserved mRNA decay factors, whose function have been mostly characterized in germline and early embryos [7–10]. For example, the CAR-1 protein family includes yeast Scd6, Drosophila Tral (Trailer hitch), Xenopus RAP55 and mammalian LSM14 [11]. The interaction between CAR-1/LSM14 and CGH-1/DDX6 regulates the formation of endoplasmic reticulum and anaphase spindle network; loss of function in car-1 or cgh-1 leads to sterility and embryonic lethality due to defects in germline apoptosis and embryonic cytokinesis [7, 8, 10]. Loss of function in cgh-1 has recently been shown to affect dendrite development in PVD neurons [6]. However, the neuronal mRNA targets of CAR-1 or CGH-1 have not yet been identified.
Here we show that loss of the translational repressors CAR-1 or CGH-1 results in axon breakage and branching, and increased axon regrowth after injury, whereas loss of mRNA decapping factors results in aberrant axon development and decreased axon regrowth after injury. We further identified numerous genes including the mitochondrial calcium ([Ca2+]mt) regulator micu-1 whose mRNAs are bound by CAR-1 in neurons. We present multiple lines of evidence that CAR-1 represses the expression and translation of micu-1 in neurons. Additionally, we describe a [Ca2+]mt influx induced by axon injury, and show that the dynamics of this [Ca2+]mt is regulated by CAR-1 and MICU-1 with dose-dependency. Our study reveals a novel mechanism whereby mRNA decay pathways regulate axonal mitochondrial calcium [Ca2+]mt dynamics.
Results
C. elegans mRNA decay factors are expressed in neurons and localize to distinct subcellular granules
We first asked whether key mRNA decay components were expressed in C. elegans neurons. We performed smFISH (single-molecule fluorescence in situ hybridization) for endogenous car-1 mRNAs and observed widespread expression in somatic cells including TRNs and other neurons besides the germ line (Figure 1A and Figure S1). We examined functional transgenes of full-length DCAP-1 or CGH-1 expressed under their respective promoters [12, 13] and observed expression in many neurons including touch receptor neurons (TRNs) and motor neurons, localizing to cytoplasmic puncta in neuronal cell bodies (Figure 1B and Figure S2A). To determine protein localization in single neurons, we generated transgenes expressing various mRNA decay factors using the TRN-specific mec-4 promoter. We found that in TRN cell bodies, CGH-1::GFP fully co-localized with mKate2::CAR-1 puncta (Figure 1C), whereas DCAP-1::dsRed partially co-localized with GFP::CAR-1 (Figure 1D). These results suggest mRNA decay components form at least two types of cytoplasmic granules in C. elegans neurons. The partial colocalization of CAR-1 and DCAP-1 further implies that only some CAR-1-bound mRNAs undergo mRNA decapping and decay (Figure 1E).
Figure 1. mRNA decay components show punctate localization in C. elegans neurons.
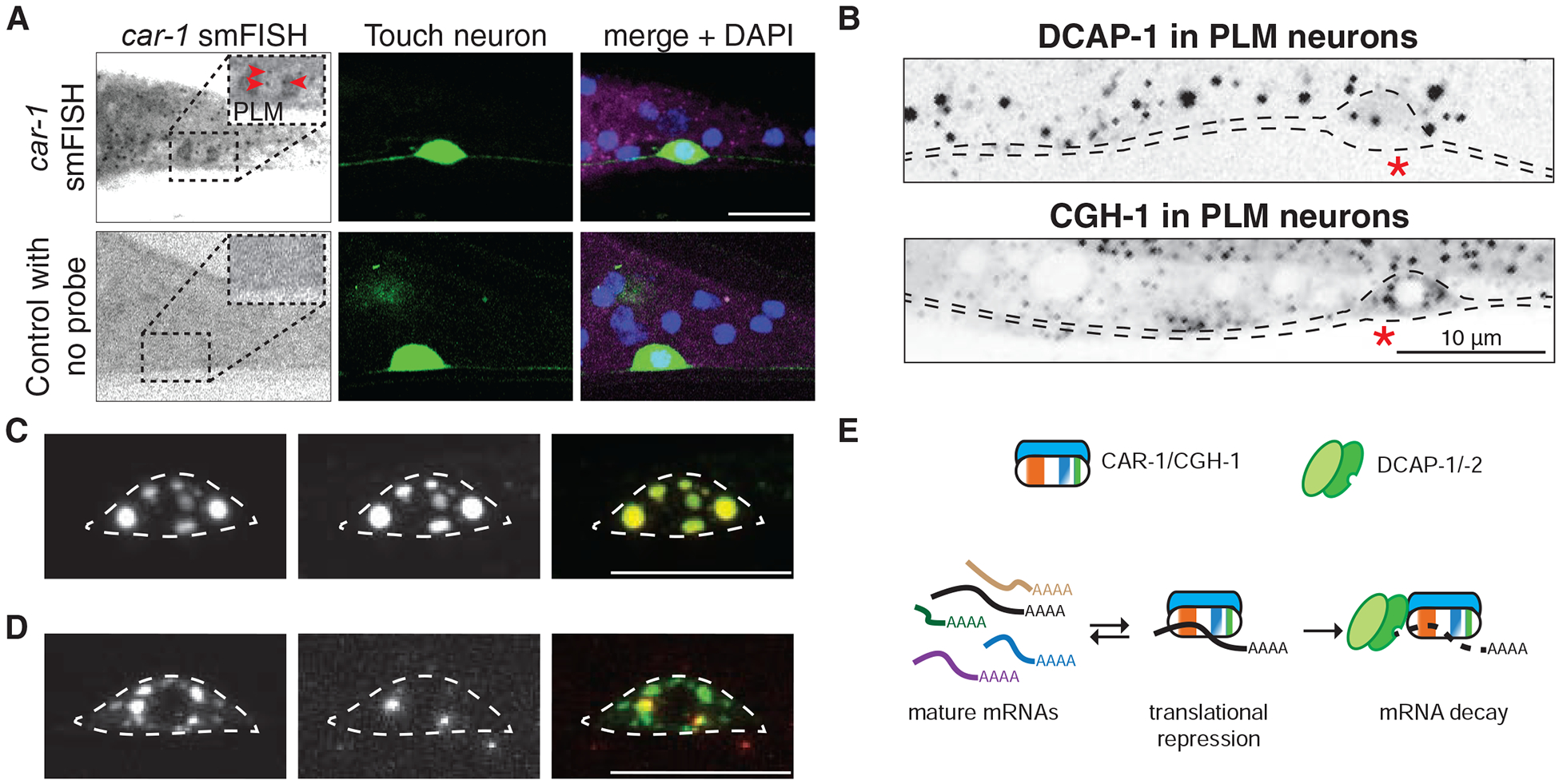
A) Single molecule fluorescence in situ hybridization (smFISH) of car-1 mRNA. Top images showing car-1 mRNAs form cytoplasmic puncta (red arrowheads) in the PLM neurons; Bottom images show control with no mRNA probe. PLM neurons are labeled with Pmec-7-GFP (muIs32). See also Figures S1 and S2.
B) C. elegans DCAP-1 [Pdcap-1-DCAP-1::dsRed(bpIs37)] or CGH-1 [Pcgh-1-CGH-1::GFP(dhIs1000)] form puncta in PLM cell bodies. The touch receptor neuron (PLM) is outlined based on TRN marker in the same image, and cell bodies marked by red *. Scale bar, 20 μm.
C) Confocal images showing co-localization of mKate2::CAR-1 [Pmec-4-mKate2::CAR-1(juEx7793)] and CGH-1::GFP (dhIs1000) in the PLM cell body (outlined).
D) Confocal images show partial co-localization of GFP::CAR-1 [Pmec-4-GFP::CAR-1(juSi338)] and DCAP-1::dsRed (bpIs37) in the PLM cell body (outlined). Scale bar, 10 μm.
E) Model of the relation between CAR-1/CGH-1 and DCAP-1/DCAP-2 functional complexes. The CAR-1/CGH-1 complex binds to mature mRNAs to repress translation. Translationally repressed mRNAs may be released to allow translation, or decapped and degraded by recruitment of the DCAP-1/DCAP-2 complex.
Assembly of mRNA decay complexes in other systems involves direct interactions such as binding of DDX6 to LSM14, and of DCP1 to the decapping activator PATL [14, 15]. To address whether punctate localization by C. elegans mRNA decay factors depended on similar interactions we examined genetic null (0) mutants. Loss of function in patr-1/PATL resulted in fewer DCAP-1 puncta in neurons, whereas loss of function in cgh-1 resulted in dimmer CAR-1 puncta (Figure S2B–S2C), indicating PATR-1 and CGH-1 promote recruitment of DCAP-1 and CAR-1, respectively. In contrast, loss of function in decapping enzymes dcap-1 or dcap-2 increased the size of CAR-1 puncta (Figure S1C). In dcap-2(0) mutants, these enlarged CAR-1 puncta showed nearly complete colocalization with DCAP-1 (Figure S1D), suggesting the lack of decapping activity may cause accumulation of granules containing CAR-1 and its bound mRNAs.
mRNA decay components have differential roles in axon morphology and regrowth after injury
To study the roles of mRNA decay components in neuronal development and maintenance, we focused on the posterior TRN, PLM. In wild type animals PLM neurons normally extend a long anterior axon and a short posterior dendrite (Figure 2A, S3A). In car-1(0) and cgh-1(0) mutant larvae PLM neurons displayed normal axon morphology (Figure S3B), but in day 1 mutant adults PLM axons displayed ectopic branching, breakage, and blebbing (Figure 2A–2B, S3A–S3B), suggesting that car-1 and cgh-1 are not essential for PLM axon development, but are necessary for maintaining their healthy morphology in adults. In contrast, dcap-1(0), dcap-2(0), or dcap-1(0) dcap-2(0) mutants at all larval and adult stages displayed low penetrance hook-shaped axon extensions, in addition to ectopic axon branching and blebbing (Figure 2A–2B, S3B), suggesting decapping enzyme activity is important for PLM development.
Figure 2. mRNA decay components affects axon development, maintenance and regrowth after injury of PLM neurons.
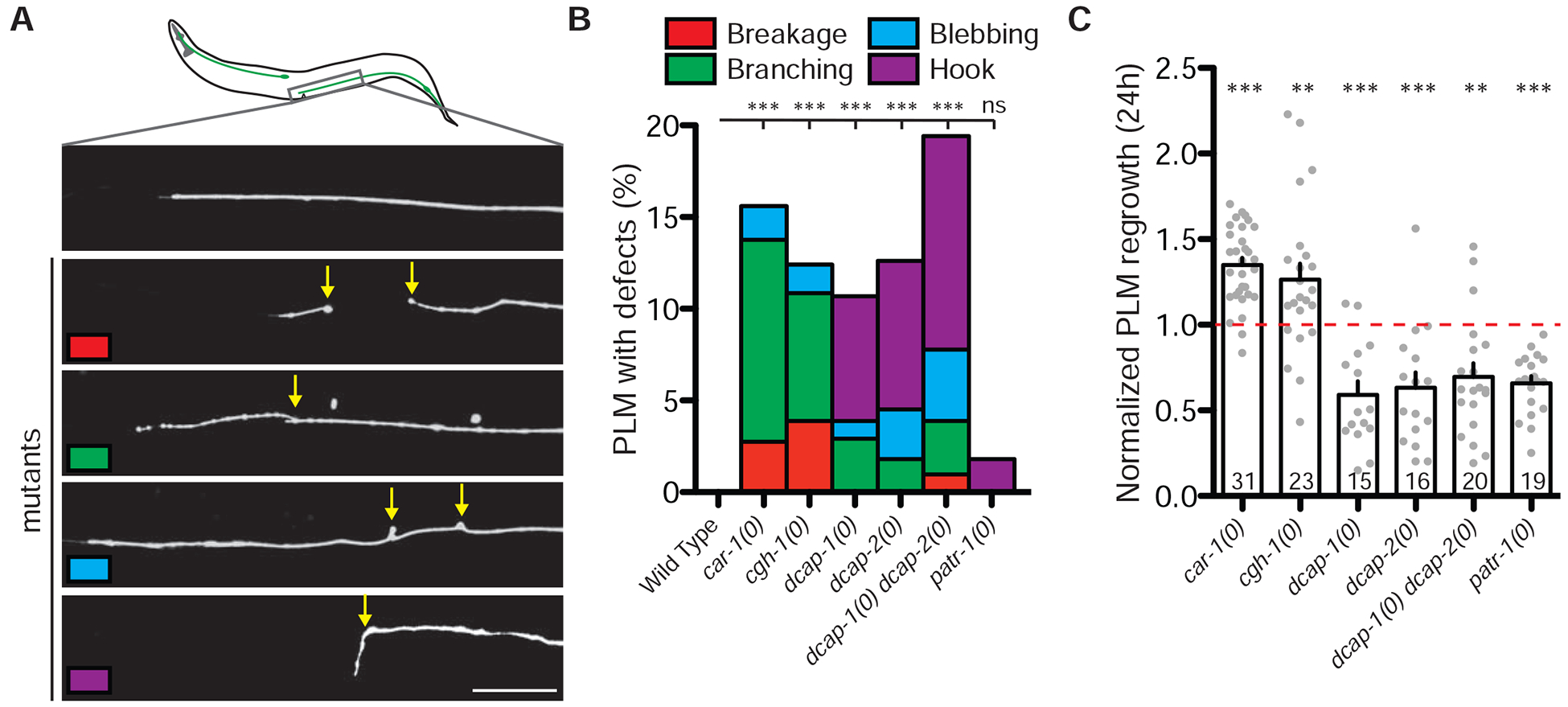
A) Top image shows the anterior end of PLM neurons in wild type animals. Bottom images show abnormal axon morphology observed in mRNA decay mutants (yellow arrows). Scale bar, 20 μm. See also Figure S3.
B) Quantification of abnormal PLM phenotypes observed in wild type and mRNA decay mutants. Day 1 adults were observed and quantified. Color correlates to images in A. N≥100. Statistics: Fisher’s exact test; ** p<0.01, *** p<0.001; ns, not significant (P>0.05).
C) Quantification of PLM regrowth length 24 h post-axotomy, normalized to the same-day control animals. Bars indicate mean ± SEM. Statistics: one-way ANOVA with Bonferroni’s post test; **p<0.01; ***p<0.001. Sample size is indicated in the bar.
Adult PLM axons display robust regrowth following femtosecond laser surgery [16]. We next examined how the mRNA decay factors affected axon regrowth. We severed morphologically normal PLM axons in larval (L4) animals lacking specific mRNA decay components and imaged axon regrowth 24 hours post-axotomy (hpa). car-1(0) and cgh-1(0) mutants showed increased regrowth after injury, whereas injured PLM axons in single null mutants for dcap-1, dcap-2, or patr-1 and in dcap-1(0) dcap-2(0) double mutants all showed decreased axon regrowth (Figure 2C). This analysis implies that although these mRNA decay factors can form protein complex and partly colocalize within the same cell, they exert differential functions in axon development, maintenance and regrowth. Below we focus on CAR-1, which is not required for PLM development but is a strong inhibitor of PLM axon regrowth.
CAR-1 is a cell-intrinsic inhibitor of axon regrowth
Three independent car-1(0) mutants all exhibited enhanced PLM axon regrowth after injury in the L4 stage, as well as adult-onset axon morphology defects (Figure 3A–3C and Figure S3C). The enhanced regrowth in car-1(0) mutants was restored to wild type levels by single-copy transgenes expressing car-1 using endogenous or TRN-specific promoters but not using a muscle-specific promoter (Figure 3B), indicating that CAR-1 regulates axon regrowth cell-autonomously. Axon regrowth involves initial growth cone formation followed by axon extension [17–19]. We observed higher rate of growth cone formation in car-1(0) mutants at multiple time points after axonal injury (Figure 3D–3E), suggesting CAR-1 inhibits growth cone formation throughout the process of regrowth. Moreover, overexpression of CAR-1 in TRNs resulted in severe axonal defects in adult animals and strongly impaired axon regrowth and growth cone formation after injury (Figure 3B–3C and Figure S3C), indicating that axon maintenance and regrowth are sensitive to CAR-1 levels.
Figure 3. CAR-1 is a cell-intrinsic inhibitor of axon regrowth.
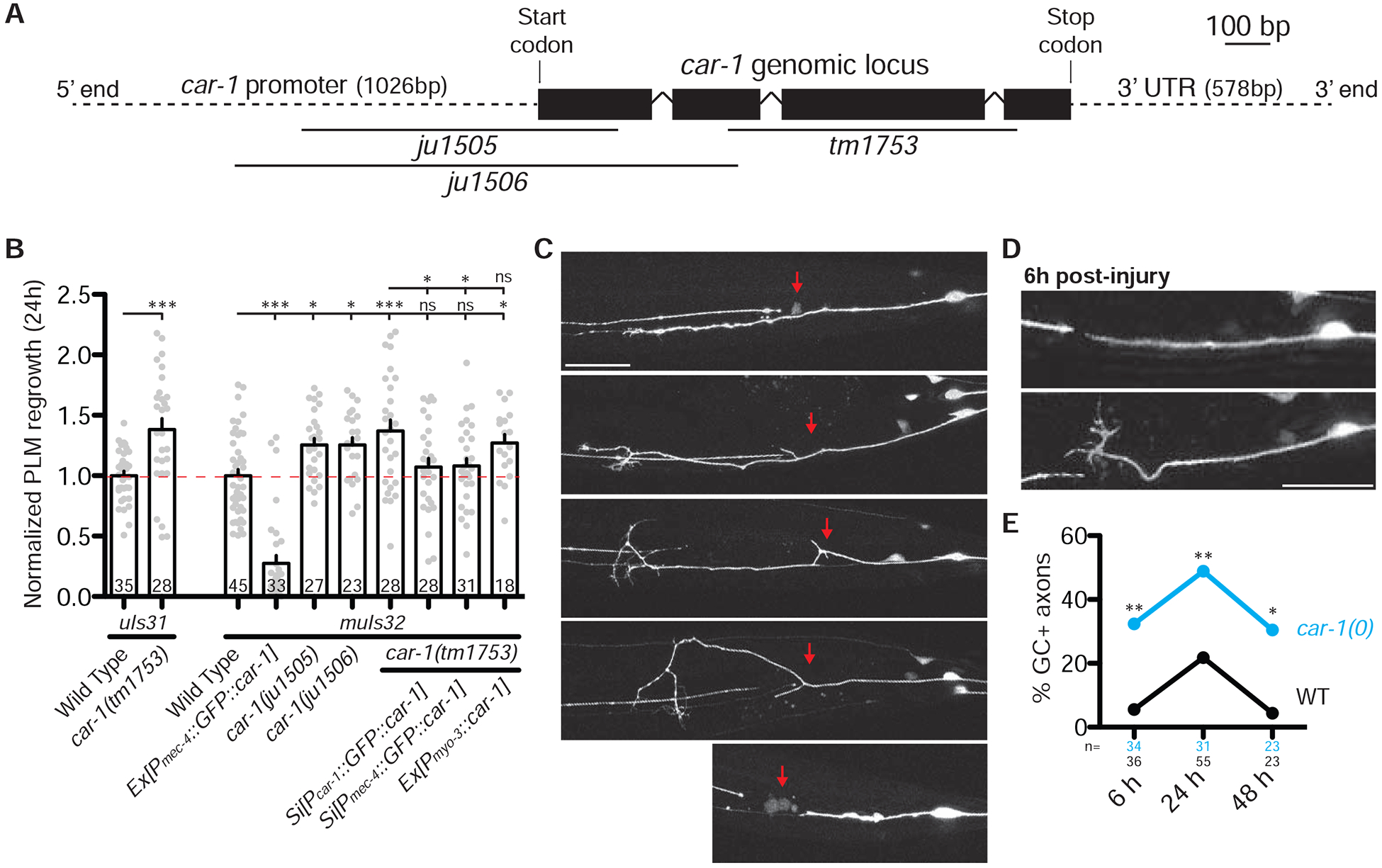
A) Map of car-1 gene showing deletion mutations. See also Table S2.
B) PLM axon regrowth length 24 h post-axotomy in genotype indicated. Bars indicate mean ± SEM. Statistics: one-way ANOVA with Bonferroni’s post test; *p<0.05; ***p<0.001, ns, not significant (P>0.05). Sample size is indicated in the bar.
C) Representative images of axon regrowth 24 h post-axotomy in wild type, car-1(0) mutants and Pmec-4-GFP::CAR-1(juEx7280). Anterior is to the left. Red arrows indicate site of axotomy.
D) Representative images of regrowing axons 6 h post-axotomy in genotype indicated. PLM in car-1(0) mutants has a more persistent regenerative growth cone. Scale bar, 20 μm.
E) Quantitation of % of growth cones at 6, 24 and 48 h post-axotomy. Statistics: Fisher’s exact test; *p<0.05; **p<0.01.
CAR-1 Sm and FDF domains are required for inhibition of axon regrowth
CAR-1 contains three conserved domains (Figure 4A): an N-terminal Sm domain, an FDF (phenylalanine-aspartate-phenylalanine) motif, and a C-terminal RGG (arginine-glycine-glycine) motif [7, 8]. The Sm domain in yeast Scd6 is essential for mRNA translational repression and stimulation of mRNA decay [20], and the FDF motif of C. elegans CAR-1 can interact with human DDX6 [14]. We next asked which domain is relevant for CAR-1 cytoplasmic puncta formation and for its function in axon regrowth.
Figure 4. The Sm domain and FDF motif are required for CAR-1 function in axon regrowth.
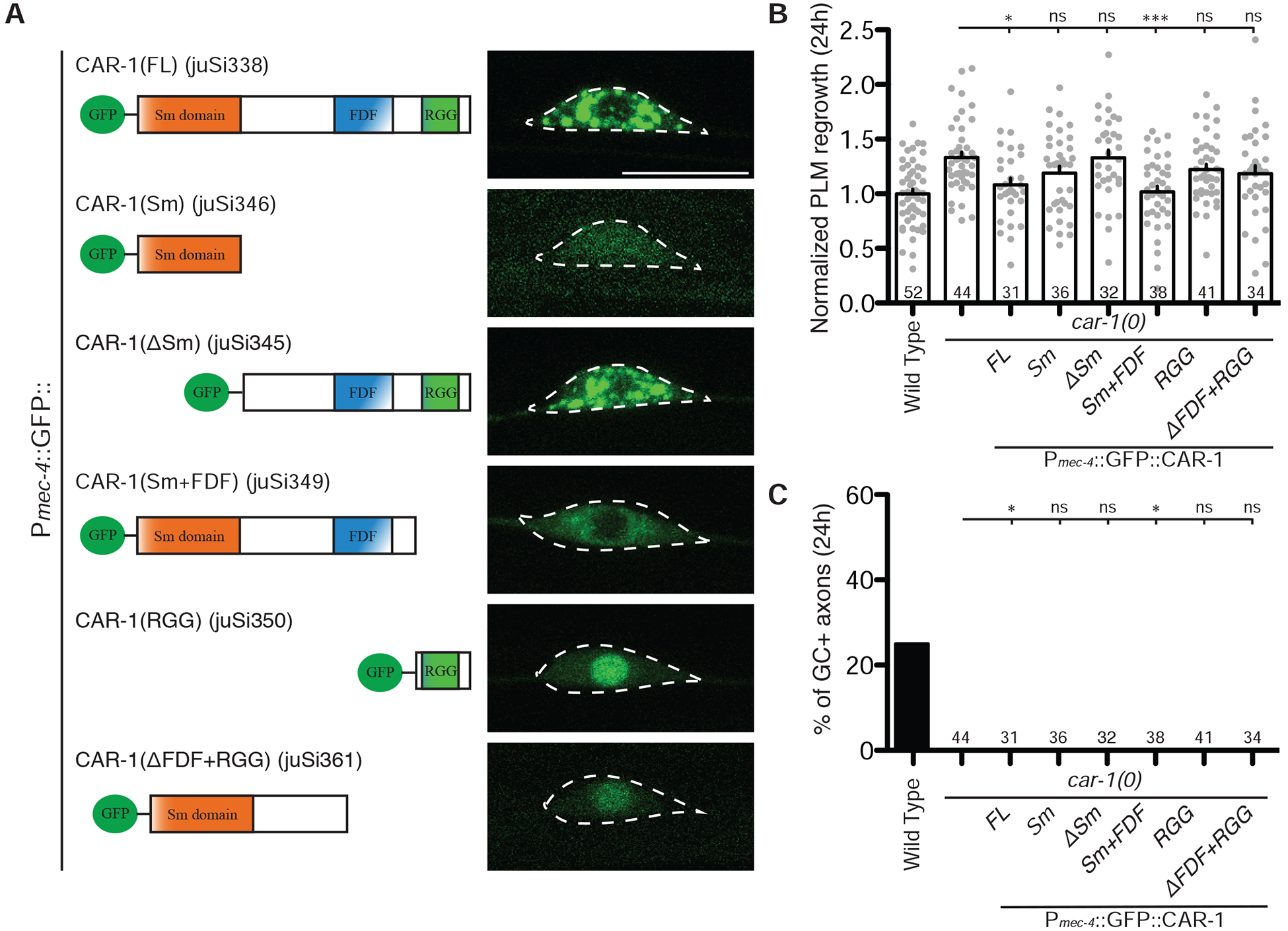
A) Puncta formation by truncated CAR-1 proteins. Left: schematic of truncated CAR-1 fragments fused to GFP in the N-terminus. Right: confocal images showing localization of GFP::CAR-1 constructs.
B) Increased PLM axon regrowth length in car-1(0) is restored to wild type levels by Pmec-4-GFP::CAR-1-FL(juSi338) and Pmec-4-GFP::CAR-1ΔRGG(juSi349). Bars indicate mean ± SEM. Statistics: one-way ANOVA with Bonferroni’s post test; *p<0.05; ***p<0.001, ns, not significant.
C) Increased growth cone formation in car-1(0) is restored to wild type level by Pmec-4-GFP::CAR-1-FL(juSi338) and Pmec-4-GFP::CAR-1ΔRGG(juSi349). Statistics: Fisher’s exact test; *p<0.05; ns, not significant. Sample size is indicated in the bar.
We expressed truncated GFP::CAR-1 in TRNs as single-copy insertion transgenes to ensure consistency in expression level (Figure 4A). We found that the FDF and RGG motifs, but not the Sm domain, contribute to the formation of CAR-1 puncta in PLM cell bodies (Figure 4A), consistent with observations in C. elegans embryos [7]. To address the function of CAR-1 mutant proteins, we performed axon injury in car-1(0) animals expressing each transgene. Expression of CAR-1(FL) or CAR-1(Sm+FDF), but not CAR-1(Sm), CAR-1(ΔSm), CAR-1(RGG), nor CAR-1(ΔFDF+RGG), in car-1(0) mutants restored axon regrowth and growth cone formation to wild type levels (Figure 4B–4C). These data indicate that the Sm domain and FDF motif of CAR-1, hence its role in RNA translational repression, are critical for CAR-1 function in neurons.
CAR-1 regulates its mRNA expression in PLM neurons
To identify neuronal targets of CAR-1 we constructed transgenic strains overexpressing GFP::CAR-1(FL) (juIs526) or GFP::CAR-1(ΔSm) (juIs549) in all neurons using the pan-neuronal rgef-1 promoter. In axon injury assay, we found that overexpression of GFP::CAR-1(FL), but not GFP::CAR-1(ΔSm), inhibited PLM axon regrowth (Figure 5A), confirming the importance of the Sm domain in CAR-1 function. We then performed single-end enhanced crosslinking and immunoprecipitation (seCLIP) to isolate mRNA targets of CAR-1 using these transgenic animals (see Methods for details) [21, 22]. We reasoned that mRNAs repressed by CAR-1 should be more stably bound to non-functional GFP::CAR-1(ΔSm) than to GFP::CAR-1(FL). We analyzed seCLIP data to identify peaks specifically enriched in GFP::CAR-1(ΔSm) samples (Methods) and selected about 30 or so potential targets (Table S1). Among them, we found that car-1 itself was specifically enriched in GFP::CAR-1(ΔSm) co-immunoprecipitates (Figure 5B), suggesting CAR-1 might repress its own expression. To test this, we performed smFISH for car-1 in animals expressing Pmec-4::GFP::CAR-1 and found that car-1 mRNA puncta partly colocalized with GFP::CAR-1 cytoplasmic puncta in wild type and dcap-2(0) animals (Figure 5C). Since cgh-1(0) mutants display dimmer GFP::CAR-1 puncta (Figure S1C), we visualized car-1 mRNAs in cgh-1(0) mutants and observed that cgh-1(0) mutants displayed more car-1 mRNAs (Figure 5D–5E), suggesting car-1 mRNA levels are normally limited by CAR-1/CGH-1 activity.
Figure 5. CAR-1 regulates its mRNA expression in PLM neurons.
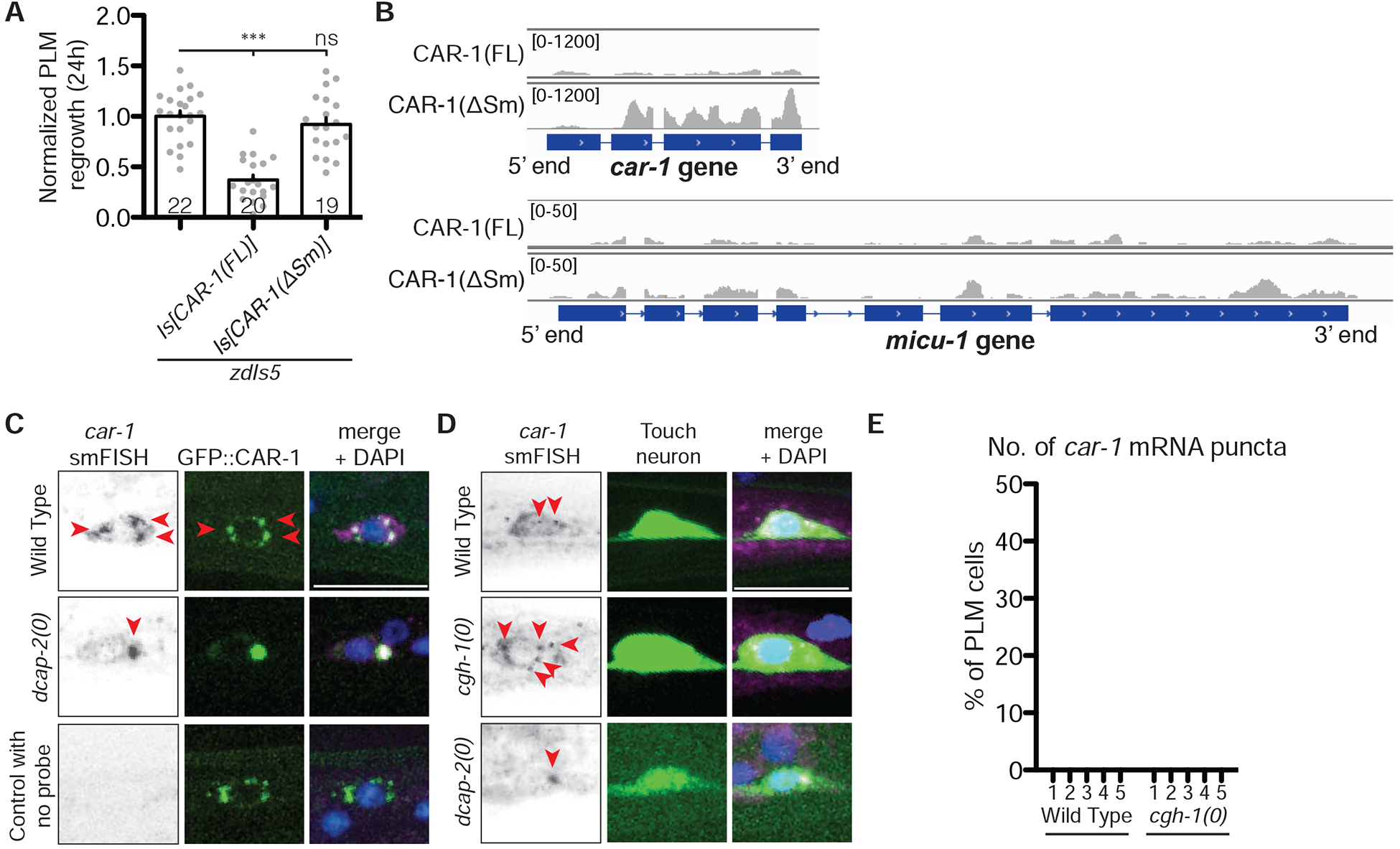
A) PLM axon regrowth length 24 h post-axotomy in transgenic animals overexpressing CAR-1(FL)(juIs526) or CAR-1(ΔSm)(juIs549) driven by the pan-neuronal promoter Prgef-1. Bars indicate mean ± SEM. Statistics: one-way ANOVA with Bonferroni’s post test; ***p<0.001, ns, not significant.
B) seCLIP read density track on car-1 and micu-1 gene for CAR-1(FL) and CAR-1(ΔSm) samples. Distinct peaks were enriched in the CAR-1(ΔSm) sample. See also Table S1.
C) smFISH shows colocalization of car-1 mRNA (red arrowheads) with GFP::CAR-1 protein in the PLM cell body. Cytoplasmic puncta of car-1 mRNAs co-localize with CAR-1 protein in wild type, and enlarged in dcap-2(0) backgrounds.
D) smFISH of car-1 mRNAs (red arrowheads). The number of car-1 mRNA puncta increases in cgh-1(0), whereas in dcap-2(0) mutants car-1 mRNAs accumulate in a large punctum.
E) Quantification of car-1 mRNA puncta in PLM cell bodies of wild type and cgh-1(0) mutant. Representative images shown in d. N≥30 for each strain.
CAR-1 binds micu-1 transcripts and represses MICU-1::GFP protein levels in PLM neurons
We identified 29 genes whose mRNAs showed enriched peaks in GFP::CAR-1(ΔSm) seCLIP analysis, including several calcium-related genes: clp-1, kcnl-1, micu-1 and unc-13 (Table S1). Several genes were previously analyzed for their roles in PLM axon regrowth [18, 23]. Given recent evidence for the function of mitochondria in axon regrowth [24–26], here we chose to focus on micu-1, which encodes a mitochondrial calcium uptake regulator of the MICU1 family (Figure S4A). Careful inspection of the seCLIP peak profiles revealed that GFP::CAR-1(ΔSm) samples were specifically enriched on micu-1 (Figure 5B). To further verify this finding, we performed smFISH of micu-1 mRNA. In wild type PLM neurons, micu-1 mRNA puncta partly colocalized with GFP::CAR-1 (Figure 6A), similar to car-1 mRNA. In car-1(0) mutants, we observed increased numbers of micu-1 mRNA puncta in PLM cell bodies (Figure 6B–6C). The increased micu-1 mRNA puncta observed in car-1(0) mutant TRNs was suppressed by expressing CAR-1(FL) or CAR-1(Sm+FDF), but not CAR-1(ΔSm) (Figure 6C), suggesting CAR-1(Sm) domain mediates translational repression. micu-1 mRNA puncta did not colocalize with GFP::DCAP-1 (Figure S5A), suggesting DCAP-1 does not regulate expression of micu-1 mRNA directly.
Figure 6. CAR-1 regulates micu-1 transcript and represses MICU-1::GFP protein levels.
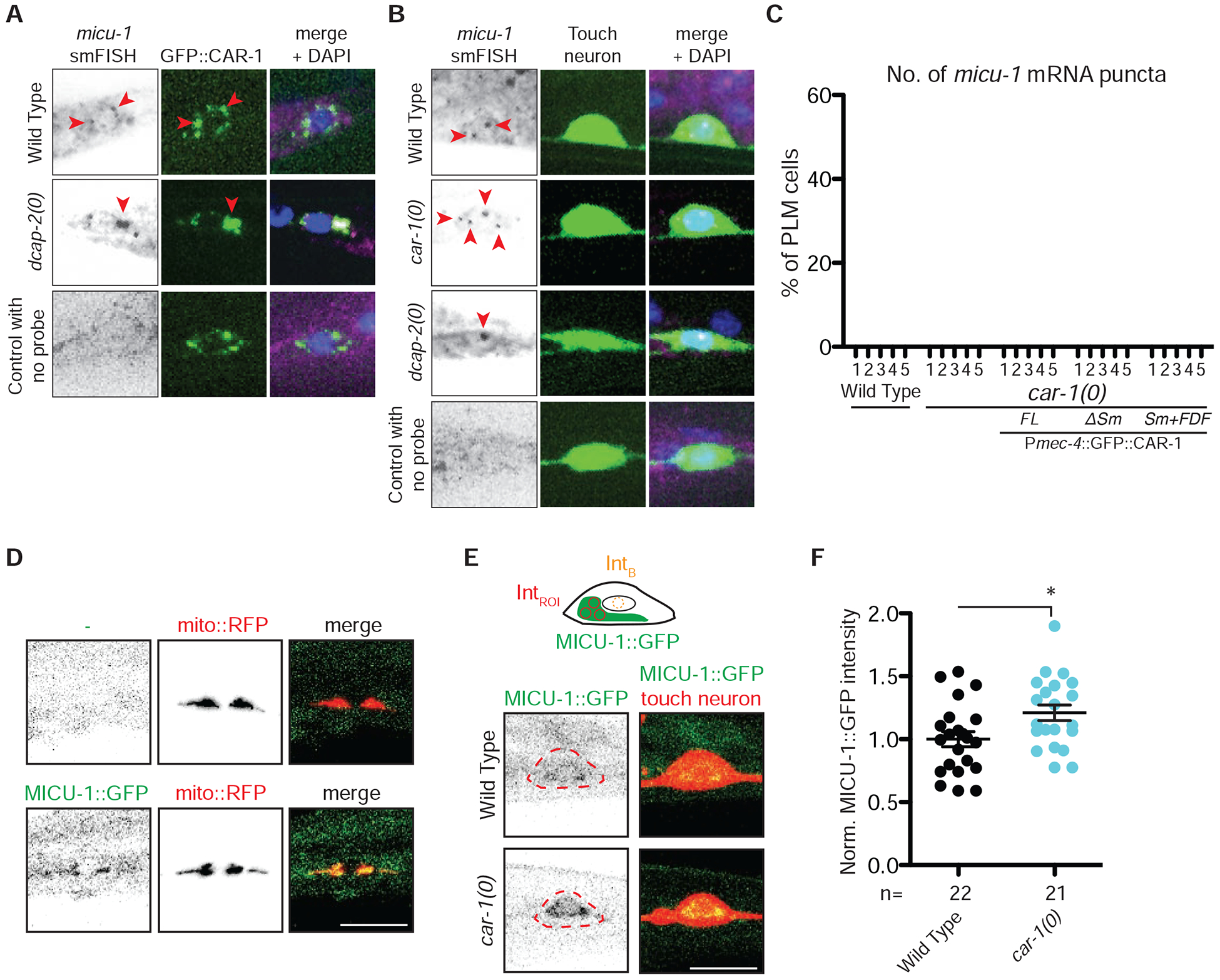
A) smFISH shows colocalization of micu-1 mRNA with GFP::CAR-1 in the PLM cell body. Cytoplasmic puncta of micu-1 mRNAs (red arrowheads) co-localize with CAR-1 protein puncta in the PLM cell body in wild type and dcap-2(0) mutant. See also Figures S4 and S5.
B) smFISH of micu-1 mRNAs (red arrowheads) shows micu-1 mRNAs form cytoplasmic puncta in PLM neurons, labeled with Pmec-7-GFP (muIs32). More micu-1 mRNA puncta in car-1(0) mutant and increased accumulation in dcap-2(0) mutant were observed.
C) Quantification of micu-1 mRNA puncta in PLM cell bodies of wild type and car-1(0) mutant. Representative images shown in g. N≥24 for each strain.
D) Top shows confocal images of mito::RFP [Pmec-7-tagRFP-mito(jsIs1073)] in the absence of MICU-1::GFP(ju1783). Bottom shows confocal images showing colocalization of MICU-1::GFP(ju1783) with mito::RFP in the PLM cell bodies.
E) Top is an illustration of the region of interest (IntROI) and background (IntB). Bottom images show MICU-1::GFP(ju1783) in PLM cell bodies (outlined), with increased expression in car-1(0). Scale bar, 10 μm.
F) MICU-1::GFP intensities in wild type and car-1(0) animals, normalized to wild type control. Statistics: unpaired Student’s t-test; *p=0.0194.
Next, we characterized endogenous MICU-1 expression by inserting GFP into the carboxy-terminus of the endogenous micu-1 gene (see Methods). MICU-1::GFP was detected at low levels in germ cells, epidermis, and some muscles (Figure S4B). MICU-1::GFP was detected at low levels in PLM neuron soma, colocalizing with a mitochondrial marker, but was below the limit of detection in axons (Figure 6D). As micu-1 transcript levels were increased in car-1(0) mutants, we examined if MICU-1::GFP signals were different in car-1(0) mutant compared to wild type. We observed a consistent 1.2-fold increase of MICU-1::GFP intensity in PLM cell bodies in car-1(0) mutant compared to wild type (Figure 6E–6F). Collectively, these observations indicate that CAR-1 binds micu-1 mRNA and represses its translation in PLM neurons.
CAR-1-mediated inhibition of axon regrowth is dependent on the mitochondrial calcium import factors MCU-1 and MICU-1
MICU1 is an EF hand protein that forms a complex with the MCU1 uniporter to regulate [Ca2+]mt uptake [27–31]. We therefore hypothesize that axonal mitochondrial calcium uptake might influence regrowth dynamics. To test this, we first asked whether micu-1, or the uniporter mcu-1, affected PLM axon regrowth. We analyzed axon regrowth in double mutants between car-1(0) and mcu-1(0) or micu-1(0). Loss of mcu-1 or micu-1 fully suppressed increased axon regrowth observed in car-1(0) mutants (Figure 7A). However, loss of mcu-1 or micu-1 did not suppress the increased growth cone formation observed in car-1(0) mutants, indicating that CAR-1 acts via other targets to regulate axon extension and growth cone formation. micu-1(0) also suppressed the adult-onset axon maintenance defects of car-1(0) mutants (Figure S5B), suggesting MICU-1 is acting downstream of CAR-1 in axon maintenance. Loss of mcu-1 or micu-1 did not rescue axon regrowth and axon morphology defects of dcap-1(0) mutants (Figure S5C–S5D), implying that micu-1 is a specific target of CAR-1 but not DCAP-1.
Figure 7. CAR-1 represses axonal mitochondrial calcium uptake after axon injury.
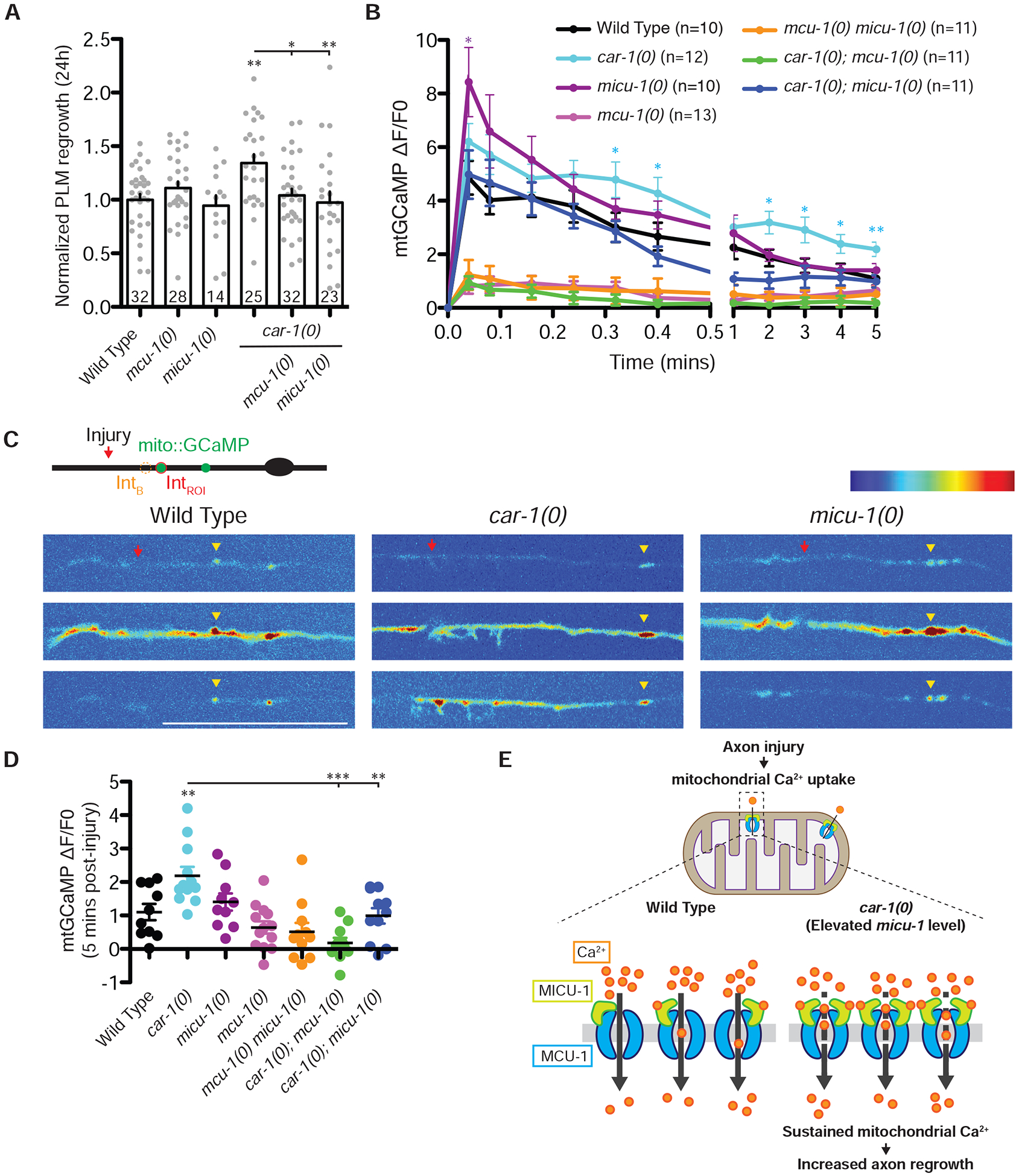
A) PLM regrowth length 24 h post-axotomy. Statistics: one-way ANOVA with Bonferroni’s posttest; *p<0.05; **p<0.01. Sample size is indicated in the bar. See also Figure S5, S6, and S7.
B) mtGCaMP fluorescence intensity (ΔF/F0) over 5 minutes post-axotomy. Statistics: Unpaired t-test against wild type; *p<0.05; **p<0.01.
C) Top is an illustration of the region of interest (IntROI) and background (IntB). Bottom images show mitochondrial calcium imaging trace in wild type, car-1(0) and micu-1(0) mutant post-axotomy. Animals expressing the mtGCaMP sensor [Pmec-4-mito-GCaMP5(juIs550)] were severed using femtosecond laser and imaged immediately. Red arrows show the cut site. Yellow arrowheads mark mitochondria in PLM neurons. Some mtGCaMP signal is in the neuronal cytoplasm likely reflecting incomplete targeting of mtGCaMP due to the high copy number of juIs550. Scale bar, 20 μm.
D) mtGCaMP fluorescence intensity (ΔF/F0) at 5 minutes post-axotomy in genotypes indicated. Each dot represents a mtGCaMP in an animal. Statistics: Unpaired t-test; **p<0.01; ***p<0.001.
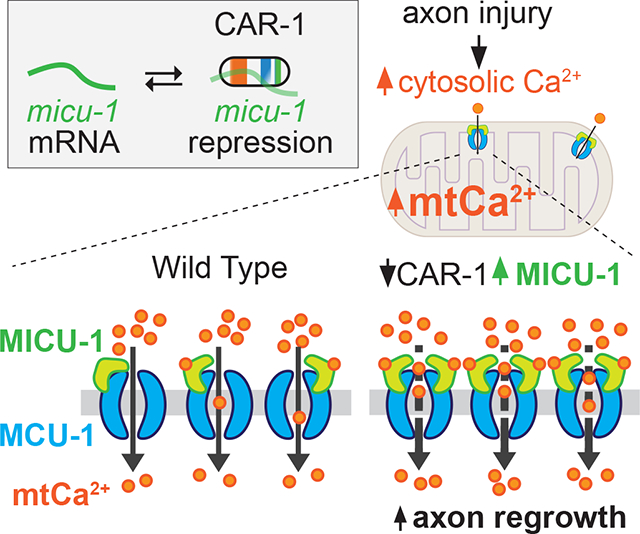
E) Model for regulation of axon regrowth by CAR-1 and [Ca2+]mt uptake. Axon injury induces transient mitochondrial Ca2+ uptake, dependent on MCU-1. Loss of CAR-1 results in elevated micu-1 mRNA expression, leading to more sustained mitochondrial Ca2+ uptake after injury, correlating with increased axon regrowth.
Axon injury triggers mitochondrial calcium uptake dependent on MCU-1 and regulated by MICU-1
We next addressed whether axon injury might affect [Ca2+]mt flux. We targeted the Ca2+ sensor GCaMP5 to the mitochondrial matrix (mtGCaMP) in TRNs. micu-1(0), mcu-1(0), and car-1(0) mutants displayed normal axonal mitochondrial distribution and baseline mtGCaMP levels (Figure S6A–S6B). We confirmed that injury causes cytosolic Ca2+ [Ca2+]c transient (Figure 7B) [32], with no significant difference between wild type, car-1(0), mcu-1(0) and micu-1(0) mutants (data not shown).
In wild type PLM axons, injury triggered a rapid increase in mtGCaMP fluorescence, consistent with axonal [Ca2+]mt uptake (Figure 7B–7C). We measured the relative change in mtGCaMP fluorescence (ΔF/F0) and found that the increased mtGCaMP fluorescence peaked within seconds of injury followed by decay to baseline levels after 5 minutes (Figure 7B–7D). Loss of function in mcu-1 abolished the axotomy-triggered mtGCaMP transient (Figure 7B). In contrast, micu-1(0) PLM axons showed a higher peak mtGCaMP fluorescence immediately after injury but decayed faster, and by 5 minutes after injury had mtGCaMP levels similar to wild type animals (Figure 7B). The increase in mtGCaMP fluorescence observed in wild type and micu-1(0) axons after injury was dependent on MCU-1, because mcu-1(0) micu-1(0) double mutants did not display any transient mtGCaMP increase following axonal injury, resembling mcu-1 single mutants (Figure 7B). Our results show that MCU-1 is essential for the transient [Ca2+]mt uptake after axon injury, whereas MICU-1 appears to inhibit initial Ca2+ overload, consistent with the gatekeeping function of mammalian MICUs [30, 33, 34].
car-1(0) mutants display more sustained axonal [Ca2+]mt uptake
We further addressed whether the axotomy-triggered [Ca2+]mt influx was regulated by CAR-1 by examining [Ca2+]mt uptake in car-1(0) animals. The mtGCaMP transients triggered immediately (2.4 s after axotomy) by injury in car-1(0) were similar to those in wild type axons (Figure 7B–7C). However, car-1(0) mutants displayed a more sustained elevation in [Ca2+]mt level compared to wild type; this elevation was completely dependent on MCU-1 (Figure 7B–7D). As expression of MICU-1 was elevated in car-1(0) mutants (Figure 6), we next tested whether loss of micu-1 was able to restore wild type [Ca2+]mt level in car-1(0) mutants. We observed wild type [Ca2+]mt level in car-1(0); micu-1(0) double mutants (Figure 7B). Further, slight elevation of micu-1 level in the TRNs resulted in increased axon regrowth and sustained axonal [Ca2+]mt uptake after injury (Figure S7A–S7C). Axon regrowth was impaired when micu-1 was overexpressed to a high level (Figure S7A), suggesting axon regrowth is sensitive to the level of MICU-1. Overall, our results suggest that mild elevation of MICU-1 expression is responsible for the more sustained [Ca2+]mt uptake and increased axon regrowth of car-1(0) mutants upon axon injury.
Discussion
In this study, we find that CAR-1 and other mRNA decay factors play multiple roles in C. elegans axon development, axon maintenance, and axon regrowth. We further dissected the mechanism by which CAR-1 inhibits axon regrowth after injury. We presented multiple lines of evidence that the mitochondrial calcium regulator MICU-1 is a major CAR-1 target in axon regrowth. We also show that the regulation of MICU-1 by CAR-1 is important for fine-tuning injury-inded [Ca2+]mt transients. Our data reveal a previously unknown molecular pathway linking mRNA decay and axonal [Ca2+]mt dynamics.
Our analyses on C. elegans mRNA decay factors are consistent with an emerging notion that these factors have distinct in vivo functions. Focusing on the PLM neurons, we show that mRNA decay factors localize to at least two main subcellular granules, as the decapping enzymes are present in a subset of granules containing translational repressors. These observations are consistent with reports that in Drosophila neurons decapping enzymes partly colocalize with the translational repressor Me31B/DDX6 [5]. Our data further show that mRNA decapping factors DCAP-1 and DCAP-2 are required for PLM developmental axon guidance and regrowth, whereas translational repressors CAR-1 and CGH-1 are not essential for PLM axon development, but inhibit PLM axon regrowth and maintain adult axon integrity. We have found that MICU-1, a member of a highly conserved family of [Ca2+]mt regulators, is a major functionally relevant target of neuronal CAR-1. micu-1 transcripts are present in CAR-1 positive, DCAP-1 negative granules, suggesting different mRNA decay factors regulate expression of distinct mRNA targets. Our observation that in the absence of DCAP-1 or DCAP-2 the mRNA targets of CAR-1 accumulate in larger CAR-1 granules suggests that increased binding of CAR-1 to its mRNA targets likely leads to translational repression, potentially accounting for the axonal developmental defects and decreased axon regrowth in decapping enzyme mutants.
We find that at least two domains of LSM14 family proteins are important for translational repression. The Sm domain is essential for binding to 4E-T, and the FDF domain binds to DDX6/CGH-1 and serves as a platform for other mRNA decay factors [14, 35]. We find that truncated CAR-1(Sm+FDF) protein can suppress the increased micu-1 transcript level of car-1(0) mutants, despite not forming distinct puncta in neurons. Our findings are consistent with prior studies showing the RGG motif is dispensable for translational repression [36, 37]. Moreover, translational repression has been observed in the absence of visible cytoplasmic puncta formation [38, 39]. Taken together, our results suggest CAR-1 may bind to and repress translation of mRNA targets even in the absence of visible puncta.
Through characterizing in vivo mRNA targets of CAR-1 in neurons, we have uncovered a previously unknown pathway involving mitochondria calcium regulation in axon injury. The functions of mitochondrial calcium uniporter MCU complex critically depend on its regulatory subunits MICU proteins and cytoplasmic calcium (reviewed in [40, 41]), and are important for human health. Mutations in human MICU genes have been linked to neurological disorders [42–44], and MCU itself is implicated in excitotoxic neuronal cell death [45]. [Ca2+]mt dynamics affects multiple aspects of cellular metabolism, including oxidative phosphorylation, Ca2+ buffering, and reactive oxygen species production, and so abnormal [Ca2+]mt dynamics could affect neuronal development and function in several ways [46]. Emerging evidence links elevated [Ca2+]mt uptake to axonal degeneration in vertebrates [47, 48] and in C. elegans [49]. Moreover increased Ca2+ levels in traumatic brain injury can be ameliorated by the inhibition of MCU1 [50], suggesting potential therapeutic benefits of mitochondrial Ca2+ regulation in brain injury. Loss of function in Drosophila MCU or MICU causes aberrant axon morphology and impairs memory formation [51]. Interestingly, expression levels of MCU and MICU are dependent on neuronal cell type and Ca2+ signaling [52]. Our results reveal that neuronal MICU-1 levels are fine-tuned by the mRNA decay machinery, adding another level of regulation to neuronal [Ca2+]mt dynamics.
Axon injury triggers a transient elevation in [Ca2+]c [17, 32, 53]. In C. elegans neurons, increased [Ca2+]c could contribute to the activation of the conserved kinase DLK-1, thereby promoting axon regrowth [54, 55]. Here, we reveal that axon injury also triggers axonal [Ca2+]mt uptake in C. elegans, reminiscent of observations in vertebrate neurons [56]. We find that this [Ca2+]mt uptake is completely dependent on MCU-1 and is regulated in complex ways by MICU-1. In cultured mammalian cells, MICU1 localizes to mitochondria [31, 57–59], and inhibits MCU function at low [Ca2+]c level (known as ‘gatekeeping’); but, at higher [Ca2+]c levels, Ca2+ binding to MICU1 to stimulate MCU1 opening [33, 34, 59]. MICU1 therefore can inhibit or promote MCU function depending on the [Ca2+]c level. Supporting this model, our data show that loss of function in micu-1 and mcu-1 have opposing effects on the amplitude of the initial [Ca2+]mt transient after axotomy. However, the enhanced axon regrowth of car-1(0) mutants is suppressed to a similar degree by loss of function in mcu-1 or micu-1. Our data suggest that the MCU-1-stimulating role of MICU-1 at lower [Ca2+]c is most relevant to the long-term outcome in axon regrowth.
MCU function is sensitive to the stoichiometry of MICU and other regulatory subunits. For example, liver mitochondria have a higher MICU1:MCU1 ratio than heart or muscle mitochondria and display increased cooperativity of [Ca2+]mt uptake [60]. We propose that in PLM neurons CAR-1 tightly regulates MICU-1 expression via mRNA decay machinery. car-1(0) mutants have a higher MICU-1:MCU-1 ratio, causing increased [Ca2+]mt uptake after axon injury and enhanced axon regrowth (Figure 7E). Axonal transport of mitochondria has been shown to boost regrowth in C. elegans and mammalian neurons [24–26]. Our observations suggest that more sustained [Ca2+]mt uptake also promotes axon regrowth, for example potentially through increasing ATP production required for axon repair [61]. We speculate that the mild enhancement of [Ca2+]mt uptake resulting from elevated MICU expression is insufficient to trigger deleterious consequences such as opening of the mitochondrial permeability transition pore that would lead to axonal degeneration. Precise modulation of [Ca2+]mt uptake in injured neurons may provide new therapeutic routes towards effective axon repair.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GFP Trap | ChromoTek | Cat#gtm-20 |
| Bacterial and Virus Strains | ||
| E. coli: OP50–1 | Caenorhabditis Genetics Center | RRID:WB-STRAIN:OP50–1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protein: Cas9-NLS purified protein | QB3 MacroLab, UC Berkley | N/A |
| Stellaris RNA FISH Hybridization buffer | LGC BioSearch Technologies | Cat#SMF-HB1–10 |
| Stellaris RNA FISH Wash Buffer A | LGC BioSearch Technologies | Cat#SMF-WA1–60 |
| Stellaris RNA FISH Wash Buffer B | LGC BioSearch Technologies | Cat#SMF-WB1–20 |
| ProLong Antifade Mountant with DAPI | Invitrogen | Cat#P36935 |
| TRIzol Reagent | Invitrogen | Cat#15596026 |
| DreamTaq DNA polymerases | Thermo Scientific | Cat#EP0705 |
| Phusion High-Fidelity DNA polymerases | Thermo Scientific | Cat#F530L |
| Protease Inhibitor Cocktail Tablet | Roche | Cat#05892970001 |
| Critical Commercial Assays | ||
| SuperScript III First-Strand Synthesis System | Invitrogen | Cat#18080051 |
| Gateway LR Clonase II Enzyme Mix | Thermo Scientific | Cat#11791–100 |
| Gibson Assembly Master Mix | New England BioLabs | Cat#E2611S |
| Deposited Data | ||
| Raw and Analyzed Data | This paper | GEO: GSE124714 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain wild type N2 | Caenorhabditis Genetics Center | RRID:WB-STRAIN:N2_(ancestral) |
| C. elegans: Strain Pmec-7-GFP(muIs32) II | [18] | CZ10969 |
| C. elegans: Strain Pmec-4-GFP(zdIs5) I | [18] | CZ10175 |
| C. elegans: Strain Pmec-17-GFP(uIs31) III | [62] | CZ25265 |
| C. elegans: Strain zdIs5 I; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26592 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; Pcgh-1-CGH-1::GFP(dhIs1000) | This paper | CZ26244 |
| C. elegans: Strain Pcgh-1-CGH-1::GFP(dhIs1000); Pmec-4-mKate2::CAR-1(juEx7739) | This paper | CZ25819 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(juSi338) IV; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ25293 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II | This paper | CZ11541 |
| C. elegans: Strain zdIs5 I; dcap-2(tm2470) IV | This paper | CZ25658 |
| C. elegans: Strain zdIs5 I; cgh-1(ok492) III/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III] | This paper | CZ11540 |
| C. elegans: Strain zdIs5 I; dcap-1(ok2139) IV | This paper | CZ11537 |
| C. elegans: Strain zdIs5 I; dcap-1(ok2139) dcap-2(tm2470) IV | This paper | CZ27317 |
| C. elegans: Strain zdIs5 I; patr-1(tm2402) II/mIn1 [mIs14 dpy-10(e128) II] | This paper | CZ11759 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; uIs31 III | This paper | CZ25319 |
| C. elegans: Strain muIs32 II; Pmec-4-GFP::CAR-1(juEx7280) | This paper | CZ23991 |
| C. elegans: Strain car-1(ju1505) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II | This paper | CZ25262 |
| C. elegans: Strain car-1(ju1506) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II | This paper | CZ25264 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; loxP-Pcar-1-GFP::car-1g+3’UTR-loxP (juSi343) IV | This paper | CZ25401 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ25112 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmyo-3-car-1(juEx7913) | This paper | CZ26723 |
| C. elegans: Strain Pmec-4-GFP::CAR-1 (juSi338) IV | This paper | CZ24932 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(Sm) (juSi346) IV | This paper | CZ25350 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(ΔSm) (juSi345) IV | This paper | CZ25325 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(Sm+FDF) (juSi349) IV | This paper | CZ25432 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(RGG) (juSi350) IV | This paper | CZ25433 |
| C. elegans: Strain Pmec-4-GFP::CAR-1(ΔFDF+RGG) (juSi361) IV | This paper | CZ25884 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::car-1(ΔSm) [juSi345] IV | This paper | CZ25443 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::car-1(Sm) [juSi346] IV | This paper | CZ25444 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::car-1(Sm+FDF) [juSi349] IV | This paper | CZ25566 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::car-1(RGG) [juSi350] IV | This paper | CZ25567 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-4-GFP::car-1(ΔFGF+RGG) [juSi361] IV | This paper | CZ25938 |
| C. elegans: Strain zdIs5 I; Prgef-1-GFP::car-1(juIs526) | This paper | CZ25296 |
| C. elegans: Strain zdIs5 I; Prgef-1-GFP::car-1(ΔSm) (juIs549) | This paper | CZ26634 |
| C. elegans: Strain dcap-2(tm2470) IV; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ26429 |
| C. elegans: Strain Pmec-7-tagRFP::mito(jsIs1073) | This paper | NM3573 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-7-tagRFP::mito(jsIs1073) | This paper | CZ27549 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; micu-1::GFP(ju1783) IV | This paper | CZ27519 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-7-mRFP(jsIs973) III; micu-1::GFP(ju1783) IV | This paper | CZ27538 |
| C. elegans: Strain muIs32 II; mcu-1(ju1154) IV | This paper | CZ20877 |
| C. elegans: Strain muIs32 II; micu-1(ju1155) IV/nT1 [qIs51 IV;V] | This paper | CZ26661 |
| C. elegans: Strain muIs32 II; car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; mcu-1(ju1154) IV | This paper | CZ25925 |
| C. elegans: Strain muIs32; car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; micu-1(ju1155) IV/tmC9 [In(glb-19 lgc-52 In(mec-3 unc-31)) IV] | This paper | CZ27010 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ26629 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-7-mRFP(jsIs973) III; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ26687 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; micu-1(ju1155) IV/nT1 [qIs51 IV;V]; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ26737 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; mcu-1(ju1154) IV; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ26857 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; mcu-1(ju1154) micu-1(ju1156) IV/nT1 [qIs51 IV;V]; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ26858 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-7-mRFP(jsIs973) III; ; mcu-1(ju1154) IV; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ27060 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-7-mRFP(jsIs973) III; micu-1(ju1155) IV/tmC9 [In(glb-19 lgc-52 In(mec-3 unc-31)) IV]; Pmec-4-mito::GCaMP5(juIs550) | This paper | CZ27061 |
| C. elegans: Strain Punc-17::GFP(vsIs48); [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26594 |
| C. elegans: Strain Punc-17::mCherry(nuIs321); Pcgh-1-CGH-1::GFP(dhIs1000) | This paper | CZ26802 |
| C. elegans: Strain muIs32 II; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26384 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26541 |
| C. elegans: Strain zdIs5 I; dcap-2(tm2470) IV; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26419 |
| C. elegans: Strain zdIs5 I; patr-1(tm2402) II/mIn1 [mIs14 dpy-10(e128) II]; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26425 |
| C. elegans: Strain zdIs5 I; cgh-1(ok492) III/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26511 |
| C. elegans: Strain cgh-1(ok492) III/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ25879 |
| C. elegans: Strain patr-1(tm2402) II/mIn1 [mIs14 dpy-10(e128) II]; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ26426 |
| C. elegans: Strain dcap-2(tm2470) IV; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ26429 |
| C. elegans: Strain dcap-1(ok2139) IV; Pmec-4-GFP::CAR-1(juSi338) IV | This paper | CZ26532 |
| C. elegans: Strain dcap-2(tm2470) IV; Pmec-4-GFP::CAR-1(juSi338) IV; [Pdcap-1-DCAP-1::dsRed+rol-6(su1006)](bpIs37) | This paper | CZ26632 |
| C. elegans: Strain micu-1::gfp(ju1783) IV | This paper | CZ27508 |
| C. elegans: Strain Pmec-4-GFP::DCAP-1(juEx8024) | This paper | CZ27378 |
| C. elegans: Strain zdIs5 I; mcu-1(ju1154) IV | This paper | CZ21242 |
| C. elegans: Strain zdIs5 I; micu-1(ju1155) IV/nT1 [qIs51 IV;V] | This paper | CZ27382 |
| C. elegans: Strain zdIs5 I; dcap-1(ok2139) mcu-1(ju1154) IV | This paper | CZ27368 |
| C. elegans: Strain zdIs5 I; dcap-1(ok2139)/nT1 dcap-1(ok2139) micu-1(ju1155) IV | This paper | CZ27381 |
| C. elegans: Strain muIs32 II; Pmec-7-tagRFP::mito(jsIs1073) | This paper | CZ25635 |
| C. elegans: Strain car-1(tm1753) I/hT2 [bli-4(e937) I let-?(q782) GFP(qIs48) I;III]; muIs32 II; Pmec-7-tagRFP::mito(jsIs1073) | This paper | CZ25651 |
| C. elegans: Strain muIs32 II; mcu-1(ju1154) IV; Pmec-7-tagRFP::mito(jsIs1073) | This paper | CZ26537 |
| C. elegans: Strain muIs32 II; micu-1(ju1155) IV//tmC9 [In(glb-19 lgc-52 In(mec-3 unc-31)) Pmyo-2-Venus(tmIs1221)] IV; Pmec-7-tagRFP::mito(jsIs1073) | This paper | CZ27030 |
| C. elegans: Strain zdIs5 I; Pmec-4-micu-1-gfp(juEx8048) | This paper | CZ27739 |
| C. elegans: Strain zdIs5 I; Pmec-4-micu-1-gfp(juEx8050) | This paper | CZ27756 |
| C. elegans: Strain Pmec-7-mRFP(jsIs973) III; Pmec-4-mito::GCaMP5(juIs550); Pmec-4-micu-1-gfp(juEx8048) | This paper | CZ27764 |
| C. elegans: Transgenic allele: N2 injected with 10 ng/μl of pCZGY2860 [Pmec-4-GFP::car-1(genomic)::let-858_3’UTR] and 5 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juEx7280 |
| C. elegans: Transgenic allele: N2 injected with 10 ng/μl pCZGY3372 [Pmyo-3-car-1(cDNA)::let-858_3’UTR] and 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juEx7913 |
| C. elegans: Transgenic allele: N2 injected with 10 ng/μl pCZGY3411 [Pmec-4-GFP::dcap-1(cDNA)::let-858_3’UTR] and 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juEx8024 |
| C. elegans: Transgenic allele: N2 injected with 2 ng/μl pCZGY3416 [Pmec-4-micu-1-gfp] and 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juEx8048 |
| C. elegans: Transgenic allele: N2 injected with 10 ng/μl pCZGY3416 [Pmec-4-micu-1-gfp] and 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juEx8050 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3366 [Pcar-1(1062bp upstream)-GFP::car-1(genomic+768bp 3’UTR)]. | This paper | juSi343 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3362 [Pmec-4-GFP::car-1(cDNA)::let-858_3’UTR]. | This paper | juSi338 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3367 [Pmec-4-GFP::car-1(Sm)::let-858_3’UTR]. | This paper | juSi346 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3368 [Pmec-4-GFP::car-1(ΔSm)::let-858_3’UTR]. | This paper | juSi345 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3369 [Pmec-4-GFP::car-1(Sm+FDF)::let-858_3’UTR]. | This paper | juSi349 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3370 [Pmec-4-GFP::car-1(RGG)::let-858_3’UTR]. | This paper | juSi350 |
| C. elegans: Single-copy transgenic allele: N2 injected with 50ng/μL of pCZGY3371 [Pmec-4-GFP::car-1(ΔFDF+RGG)::let-858_3’UTR]. | This paper | juSi361 |
| C. elegans: Multi-copy transgenic allele: CZ20310 [Pmex-5-his-72::miniSOG(juSi164) III unc-119(ed3) III] injected with 10 ng/μl pCZGY3363 [Prgef-1-GFP::car-1(genomic)::let-858_3’UTR] and 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juIs526 |
| C. elegans: Multi-copy transgenic allele: CZ20310 [Pmex-5-his-72::miniSOG(juSi164) III unc-119(ed3) III] injected with 10 ng/μl pCZGY3364 [Prgef-1-GFP::car-1(ΔSm)::let-858_3’UTR] 3 ng/μl pCFJ90[Pmyo-2-mCherry]. | This paper | juIs549 |
| C. elegans: Multi-copy transgenic allele: N2 injected with 40 ng/μL of Pmec-4-mito::GCaMP5 and treated with TMP/UV. | This paper | juIs550 |
| C. elegans: GFP knock-in allele: N2 injected with 50 ng/μl of pCZGY3412, 50 ng/μl of pCZGY3413, 10 ng/μl of pCZGY3414, 3 ng/μl Pmyo-2-mCherry and 3 ng/μl of Pmyo-3-mCherry. Strains were treated with heat shock as described in Dickinson et al., 2015. | This paper | ju1783 |
| Oligonucleotides | ||
| Alt-R® CRISPR-Cas9 crRNA for car-1: to make car-1(ju1505) and car-1(ju1506) deletion: /AltR1/rCrA rArGrC rUrCrG rArCrA rUrCrC rGrUrU rArCrG rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | This paper | N/A |
| Alt-R® CRISPR-Cas9 crRNA for car-1: to make car-1(ju1505) and car-1(ju1506) deletion: /AltR1/rCrU rArArU rCrUrU rGrCrU rUrCrC rGrArU rGrUrA rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | This paper | N/A |
| Alt-R® CRISPR-Cas9 crRNA for micu-1: to make micu-1(ju1155) and micu-1(ju1156) deletion: /AltR1/rGrU rGrGrG rCrGrG rUrUrU rCrUrC rGrCrG rArCrG rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | This paper | N/A |
| Alt-R® CRISPR-Cas9 crRNA for micu-1: to make micu-1(ju1155) and micu-1(ju1156) deletion: /AltR1/rArU rCrGrA rUrCrC rUrArA rCrGrA rArGrA rCrArG rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/ | This paper | N/A |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | Cat#1072532 |
| Recombinant DNA | ||
| Plasmid: Pmec-4-GFP::car-1(genomic)-let-858_3’UTR | This paper | pCZGY2860 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1(cDNA)-let-858_3’UTR for CRISPR | This paper | pCZGY3362 |
| Plasmid: Prgef-1-GFP::car-1(cDNA)-let-858_3’UTR | This paper | pCZGY3363 |
| Plasmid: Prgef-1-GFP::car-1(ΔSm)-let-858_3’UTR | This paper | pCZGY3364 |
| Plasmid: FRT-Hygro-FRT-loxP-car-1-loxP for CRISPR | This paper | pCZGY3365 |
| Plasmid: FRT-Hygro-FRT-Pcar-1(1062bp upstream)-GFP::car-1(genomic+768bp 3’UTR) for CRISPR | This paper | pCZGY3366 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1(Sm)-let-858_3’UTR for CRISPR | This paper | pCZGY3367 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1(ΔSm)-let-858_3’UTR for CRISPR | This paper | pCZGY3368 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1-(ΔRGG)-let-858_3’UTR for CRISPR | This paper | pCZGY3369 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1(RGG)-let-858_3’UTR for CRISPR | This paper | pCZGY3370 |
| Plasmid: FRT-Hygro-FRT-Pmec-4-GFP::car-1(ΔFDF+RGG)-let-858_3’UTR for CRISPR | This paper | pCZGY3371 |
| Plasmid: Pmyo-3-car-1(cDNA)-unc-54_3’UTR | This paper | pCZGY3372 |
| Plasmid: Pmec-4-GFP::dcap-1(cDNA)-let-858_3’UTR | This paper | pCZGY3411 |
| Plasmid: Peft-3::Cas9+micu-1_C1_sgRNA | This paper | pCZGY3412 |
| Plasmid: Peft-3::Cas9+micu-1_C2_sgRNA | This paper | pCZGY3413 |
| Plasmid: GFP-SEC-3xFLAG_micu-1_C1_homology_arm | This paper | pCZGY3414 |
| Plasmid: Pmec-4-micu-1-gfp | This paper | pCZGY3416 |
| Software and Algorithms | ||
| ImageJ | NIH image | RRID:SCR_003070 |
| GraphPad Prism 7 | GraphPad Software, Inc. | RRID:SCR_002798 |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/us/downloads/zen.html |
Lead Contact and Materials Availability
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Chisholm (adchisholm@ucsd.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Experimental Model and Subject Details
The nematode Caenorhabditis elegans was used as the experimental model for this study. All experiments were performed with hermaphrodite animals; males were used only for crosses. C. elegans strains were maintained at 20°C on nematode growth medium (NGM) agar plates seeded with E. coli OP50.
Method Details
C. elegans genetics
car-1(0), cgh-1(0) and patr-1(0) mutants display maternal effect lethality, and micu-1(0) mutants display sterility. There mutants were kept as balanced strains. Homozygous car-1(0), cgh-1(0), patr-1(0) and micu-1(0) mutants produced from heterozygous mothers were analyzed. car-1(0) and micu-1(0) mutants do not display overt behavioral or developmental phenotypes. We used the following transgenes: Pmec-7-GFP(muIs32), Pmec-4-GFP(zdIs5), Pmec-17-GFP(uIs31) and Pmec-7-mRFP(jsIs973) to visualize touch receptor neurons (TRNs), Punc-17-mCherry(nuIs321) and Punc-17-GFP(vsIs48) to visualize cholinergic motor neurons, and Pmec-7-mito-tagRFP(jsIs1073) to visualize mitochondria in TRNs. Different transgenes were used when possible to confirm that phenotypes are not transgene dependent. Transgenes were introduced to various mutants by standard genetic crossing.
Molecular cloning
To generate car-1 rescue constructs, we cloned ~3kb of car-1 genomic DNA into pCR8 vector. A full-length car-1 cDNA was generated by RT-PCR from wild type N2 total RNA. Expression constructs for tagged or truncated CAR-1 were generated from cDNA by PCR and cloned into destination vectors containing Pmec-4 or Pmyo-3 promoter using the Gateway system (Life Technologies). We expressed the Ca2+ sensor GCaMP5 [63] to the mitochondrial matrix (mtGCaMP) in TRNs using the mec-4 TRN-specific promoter. We used Gibson assembly (New England Biolabs) to generate clones for single-copy transgenes.
Transgenic strain construction
For standard extrachromosomal transgenes, plasmid DNAs were used at 10 ng/μl, co-injection marker Pttx-3-RFP at 90 ng/μl or Pmyo-2-mCherry at 3–5 ng/μl. At least 2 independent transgenic lines were analyzed per construct. Single-copy transgenes were inserted into the transposon site cxTi10882 on chromosome IV using CRISPR/Cas9. Three plasmids were injected into wild type hermaphrodites: a plasmid encoding HygR+GFP-car-1-cDNA flanked by homology arms, a plasmid encoding Cas9+sgRNA, and a plasmid encoding co-injection marker. Animals were selected for genomic insertion based upon resistance to hygromycin (HygR), the absence of co-injection markers and transgene was confirmed by PCR genotyping. We used optogenetic mutagenesis to generate chromosomal integration of pan-neuronal GFP-CAR-1 constructs [Prgef-1-GFP::CAR-1(FL)(juIs527) and Prgef-1-GFP::CAR-1(ΔSm)(juIs549)] [64]. Briefly, plasmids were injected into CZ20310 [Pmex-5-HIS::miniSOG(juSi164); unc-19(ed3)] L4 hermaphrodites, and 6 hours after injection, these animals were exposed to blue light for 30 min at 4 Hz. F1 progeny with co-injection marker were singled onto seeded plates. F2 progeny from F1 plates with > 75% transmission were singled, and those showing 100% transmission of the co-injection maker in the following generation were outcrossed with wild type males to confirm chromosomal integration. We also determined by quantitative PCR the copy number of the expression construct in juIs526 and juIs549 to be 52 and 78 copies respectively. Pmec-4-mito-GCaMP5(juIs550) was integrated using UV-trimethylpsoralen and outcrossed 4 times before use.
CRISPR-mediated deletion alleles and GFP knock-in
We generated car-1 deletions using two CRISPR RNA (crRNAs), 5’-CTAATCTTGCTTCCGATGTA-3’ and 5’-CAAGCTCGACATCCGTTACG-3’ (Integrated DNA Technologies) targeting the coding sequence in the first exon of car-1. The crRNAs were injected into wild type hermaphrodites with purified Cas9 (MacroLabs, University of California, Berkeley), trans-activating crRNA (tracrRNA) and dpy-10 crRNA, as described [65]. Among 147 Dpy F1 worms, we identified two worms heterozygous for car-1 deletions ju1505 and ju1506. Like car-1(tm1753), eggs laid by homozygous car-1(ju1505) and car-1(ju1506) worms failed to hatch, indicating all three alleles are null mutants. car-1 alleles were balanced over hT2 I; III [bli-4(e937) I let-?(q782) GFP(qIs48) I;III].
We generated micu-1 deletion alleles as described [66]. We designed two subgenomic RNAs (sgRNAs): GTGGGCGGTTTCTCGCGACG and ATCGATCCTAACGAAGACAG. pU6::micu-1 sgRNAs were generated by primer annealing and injected into wild type or mcu-1(ju1154) worms as mixtures of 40 ng/μl of each pU6::micu-1 sgRNA, 100 ng/μl of Peft-3-Cas9-SV40 NLS::tbb-2_3’UTR, and 20 ng/μl of Pcol-19-GFP as coinjection marker. We obtained worms heterozygous for micu-1(ju1155) in N2 background and micu-1(ju1156) in mcu-1(ju1154) background. micu-1 alleles were balanced over nT1 IV; V [qIs51] or tmC9 [In(glb-19 lgc-52 In(mec-3 unc-31)) IV [67].
We inserted GFP at the C-terminus of micu-1 using the self-excising drug selection cassette [68]. We designed two subgenomic RNAs (sgRNAs): CTAATAAAATGGAAGAGGAC and ATGGCCACTGTAAATACTTG. We injected 50 ng/μl of each micu-1 sgRNA, 10 ng/μl of homology arm repair template, 3 ng/μl Pmyo-2-mCherry and 3 ng/μl of Pmyo-3-mCherry into wild type worms. New alleles are in Table S2.
Fluorescence microscopy and laser axotomy
Fluorescence images were collected using Zeiss LSM510, LSM710, or LSM800 confocal microscopes, unless specified. Laser axotomy was performed as described [16, 18]. Images shown were projected as z-stack. For live imaging of GCaMP fluorescence, we collected images every 240 ms using the spinning-disk confocal. Femtosecond laser power was 140 to180 mW with a shutter time of 1.2–1.5 ms. The mitochondrion closest to the axotomy site in the proximal axon was set as the region of interest (ROI). We outlined the mitochondrion and measured its integrated density using ImageJ and subtracted the integrated density from background using an identical outline within the axon to obtain net fluorescence (F). Baseline fluorescence (F0) was obtained from the ROI one frame before axotomy. The change in fluorescence ΔF was expressed as the ratio of change with respect to the baseline.
Single-end enhanced crosslinking and immunoprecipitation (seCLIP)
We performed single-end enhanced crosslinking and immunoprecipitation (seCLIP) as previously described [21] with a few modifications. Worms expressing pan-neuronal GFP::CAR-1 [Prgef-1-GFP::CAR-1(juIs527)] or pan-neuronal GFP::CAR-1(ΔSm) [Prgef-1-GFP::CAR-1(ΔSm)(juIs549)] were UV-crosslinked at 3 kJ/m2 then resuspended in lysis buffer [50 mM Tris-HCl pH7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, supplemented with one tablet of protease inhibitor cocktail tablet (Roche) in 20 ml]. Subsequent steps were performed as described [21]. Immunoprecipitation was performed using GFP-Trap (ChromoTek). Washes were performed using lysis buffer instead of high salt wash buffer. The quality of sequencing reads from fastq files was evaluated by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Bioinformatic analysis of sequencing reads was performed as previously described [22].
43,905,923 and 64,702,729 reads were obtained from GFP::CAR-1(FL) and GFP::CAR-1(ΔSm) sample respectively, of which 4,022,150 [CAR-1(FL)] and 5,470,606 [CAR-1(ΔSm)] reads uniquely mapped to C. elegans genome, after removing PCR duplicates and repeats. We identified 1190 genes with peaks > 2× enriched in CAR-1(ΔSm) over CAR-1(FL) (Table S1) using CLIPper peak calling [69]. Fold enrichment was calculated using the number of eCLIP reads overlapping CLIPper-identified peaks, after normalizing to total usable read counts in each sample. We also analyzed fastq files using HISAT2 [70], followed by transcript assembly using Cufflinks [71] and differential analysis using Cuffdiff (p-value < 0.05) [72]. 29 genes were enriched in CLIPper and in Cufflinks-Cuffdiff analyses (Table S1). We inspected all genes enriched in CAR-1(ΔSm) and focused on micu-1, which showed 2x Peak enrichment in exon 7 at ChrIV:13299691–13299814.
Single molecule fluorescence in situ hybridization (smFISH)
Forty-eight 20-nt probes for car-1 or micu-1 mRNAs were designed and purchased from LGC BioSearch Technologies and conjugated to Cal Fluor 610. We performed smFISH using mixed stage animals according to a modified Stellaris protocol [73]. We used Pmec-7-GFP(muIs32) or Pmec-4-GFP(zdIs5) animals expressing GFP in TRNs as a reference for mRNA localization in PLM neurons. For imaging we used Leica DMi8 microscope, Andor spinning disk confocal (CSU-W1) and iXon ultra 888 EMCCD camera. All images shown are z-stack projections.
Quantification and Statistical analysis
Statistical tests were performed using GraphPad Prism. Two-way comparisons of categorical data were performed using Fisher’s exact test. For multiple comparisons, we used one-way ANOVA with Bonferroni’s post test. All data compared using unpaired Student’s t-test passed the D’Agostino & Pearson omnibus normality test. To compare regrowth between experiments with different control means, we normalized each experimental data point by dividing it by its control means. Sample sizes are indicated in Figures (dot plots) or in Figure legends.
Data and Code Availability
The raw and analyzed seCLIP datasets have been deposited at the Gene Expression Omnibus (accession number: GSE124714).
Supplementary Material
Table S1. Genes enriched in CAR-1(ΔSM) seCLIP samples. Related to Figure 5. Column A shows genes enriched >2× using CLIPper. Column B shows genes enroched >2× using Cufflinks; 28 genes in bold face are enrched in both sets.
Highlights.
C. elegans mRNA decay factors affect axon regeneration and maintenance
The Lsm14 ortholog CAR-1 is a repressor of axon regeneration
CAR-1 represses expression of the mitochondrial calcium regulator MICU-1
Axon injury triggers mitochondrial calcium uptake regulated by MICU-1
Acknowledgments
We thank Yao Yao and Xuefeng Meng for assistance in strain construction, Zhiping Wang for CasSCI vectors, and members of the Jin and Chisholm laboratories for discussions. Some mutants were provided by the Japan National Bioresource Project or by the Caenorhabditis Genetics Center (funded by the NIH office of research infrastructure programs P40 OD010440). K.W.K. received Hallym University research funds (HRF-201809-014). This work was supported by NIH grant R01 GM054657 to A.D.C., R01 NS093588 to A.D.C. and Y.J., and R01 HG004659 to G.W.Y.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
G.W.Y. is a co-founder and a member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locana and Eclipse BioInnovations. G.W.Y. is a visiting professor at the National University of Singapore, with the terms of this arrangement reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The other authors declare no competing financial interests.
References
- 1.Decker CJ, and Parker R (2012). P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol 4, a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grudzien-Nogalska E, and Kiledjian M (2017). New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip. Rev. RNA 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbee SA, Estes PS, Cziko AM, Hillebrand J, Luedeman RA, Coller JM, Johnson N, Howlett IC, Geng C, Ueda R, et al. (2006). Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 52, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cougot N, Bhattacharyya SN, Tapia-Arancibia L, Bordonne R, Filipowicz W, Bertrand E, and Rage F (2008). Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. J. Neurosci 28, 13793–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hillebrand J, Pan K, Kokaram A, Barbee S, Parker R, and Ramaswami M (2010). The Me31B DEAD-Box Helicase Localizes to Postsynaptic Foci and Regulates Expression of a CaMKII Reporter mRNA in Dendrites of Drosophila Olfactory Projection Neurons. Front. Neural Circuits 4, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonacci S, Forand D, Wolf M, Tyus C, Barney J, Kellogg L, Simon MA, Kerr G, Wells KL, Younes S, et al. (2015). Conserved RNA-binding proteins required for dendrite morphogenesis in Caenorhabditis elegans sensory neurons. G3 (Bethesda) 5, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Audhya A, Hyndman F, McLeod IX, Maddox AS, Yates JR 3rd, Desai A, and Oegema K (2005). A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J. Cell Biol 171, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boag PR, Nakamura A, and Blackwell TK (2005). A conserved RNA-protein complex component involved in physiological germline apoptosis regulation in C. elegans. Development 132, 4975–4986. [DOI] [PubMed] [Google Scholar]
- 9.Lall S, Piano F, and Davis RE (2005). Caenorhabditis elegans decapping proteins: localization and functional analysis of Dcp1, Dcp2, and DcpS during embryogenesis. Mol. Biol. Cell 16, 5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Squirrell JM, Eggers ZT, Luedke N, Saari B, Grimson A, Lyons GE, Anderson P, and White JG (2006). CAR-1, a protein that localizes with the mRNA decapping component DCAP-1, is required for cytokinesis and ER organization in Caenorhabditis elegans embryos. Mol. Biol. Cell 17, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marnef A, Sommerville J, and Ladomery MR (2009). RAP55: insights into an evolutionarily conserved protein family. Int. J. Biochem. Cell Biol 41, 977–981. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, Yang P, Zhang Y, Bao X, Li J, Hou W, Yao X, Han J, and Zhang H (2011). A genome-wide RNAi screen identifies genes regulating the formation of P bodies in C. elegans and their functions in NMD and RNAi. Protein Cell 2, 918–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammell CM, Lubin I, Boag PR, Blackwell TK, and Ambros V (2009). nhl-2 Modulates microRNA activity in Caenorhabditis elegans. Cell 136, 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandmann T, Fakim H, Padamsi Z, Youn JY, Gingras AC, Fabian MR, and Jinek M (2018). Molecular architecture of LSM14 interactions involved in the assembly of mRNA silencing complexes. EMBO J. 37, e97869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teixeira D, and Parker R (2007). Analysis of P-body assembly in Saccharomyces cerevisiae. Mol. Biol. Cell 18, 2274–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, and Chisholm AD (2007). Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc. Natl. Acad. Sci. USA 104, 15132–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradke F, Fawcett JW, and Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci 13, 183–193. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O’Rourke S, Bowerman B, Wu Z, Jin Y, and Chisholm AD (2011). Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron 71, 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tedeschi A, Dupraz S, Curcio M, Laskowski CJ, Schaffran B, Flynn KC, Santos TE, Stern S, Hilton BJ, Larson MJE, et al. (2019). ADF/Cofilin-Mediated Actin Turnover Promotes Axon Regeneration in the Adult CNS. Neuron 103, 1073–1085 e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeidan Q, He F, Zhang F, Zhang H, Jacobson A, and Hinnebusch AG (2018). Conserved mRNA-granule component Scd6 targets Dhh1 to repress translation initiation and activates Dcp2-mediated mRNA decay in vivo. PLoS Genet. 14, e1007806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Nostrand EL, Nguyen TB, Gelboin-Burkhart C, Wang R, Blue SM, Pratt GA, Louie AL, and Yeo GW (2017). Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). Methods Mol. Biol 1648, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim KW, Tang NH, Piggott CA, Andrusiak MG, Park S, Zhu M, Kurup N, Cherra Iii SJ, Wu Z, Chisholm AD, et al. (2018). Expanded genetic screening in C. elegans identifies new regulators and an inhibitory role for NAD(+) in axon regeneration. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, and He Z (2016). The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han SM, Baig HS, and Hammarlund M (2016). Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou B, Yu P, Lin MY, Sun T, Chen Y, and Sheng ZH (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Stefani D, Raffaello A, Teardo E, Szabo I, and Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- 30.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, and Chisholm AD (2010). Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci 30, 3175–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, et al. (2016). MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 16, 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimura T, Padamsi Z, Fakim H, Milette S, Dunham WH, Gingras AC, and Fabian MR (2015). The eIF4E-Binding Protein 4E-T Is a Component of the mRNA Decay Machinery that Bridges the 5’ and 3’ Termini of Target mRNAs. Cell Rep. 11, 1425–1436. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka KJ, Ogawa K, Takagi M, Imamoto N, Matsumoto K, and Tsujimoto M (2006). RAP55, a cytoplasmic mRNP component, represses translation in Xenopus oocytes. J. Biol. Chem 281, 40096–40106. [DOI] [PubMed] [Google Scholar]
- 37.Nissan T, Rajyaguru P, She M, Song H, and Parker R (2010). Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol. Cell 39, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon S, Zhang Y, and Matthias P (2007). The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes & Dev. 21, 3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchan JR, Muhlrad D, and Parker R (2008). P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol 183, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giorgi C, Marchi S, and Pinton P (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev. Mol. Cell Biol 19, 713–730. [DOI] [PubMed] [Google Scholar]
- 41.Kamer KJ, and Mootha VK (2015). The molecular era of the mitochondrial calcium uniporter. Nature reviews. Mol. Cell Biol 16, 545–553. [DOI] [PubMed] [Google Scholar]
- 42.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, et al. (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nature Genet. 46, 188–193. [DOI] [PubMed] [Google Scholar]
- 43.Shamseldin HE, Alasmari A, Salih MA, Samman MM, Mian SA, Alshidi T, Ibrahim N, Hashem M, Faqeih E, Al-Mohanna F, et al. (2017). A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain 140, 2806–2813. [DOI] [PubMed] [Google Scholar]
- 44.Lewis-Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, Titov D, Duff J, Pyle A, Taylor RW, Yu-Wai-Man P, et al. (2016). Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet 2, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, and Hardingham GE (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Stefani D, Rizzuto R, and Pozzan T (2016). Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem 85, 161–192. [DOI] [PubMed] [Google Scholar]
- 47.Barrientos GC, Feng W, Truong K, Matthaei KI, Yang T, Allen PD, Lopez JR, and Pessah IN (2012). Gene dose influences cellular and calcium channel dysregulation in heterozygous and homozygous T4826I-RYR1 malignant hyperthermia-susceptible muscle. J. Biol. Chem 287, 2863–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villegas R, Martinez NW, Lillo J, Pihan P, Hernandez D, Twiss JL, and Court FA (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci 34, 7179–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarasija S, Laboy JT, Ashkavand Z, Bonner J, Tang Y, and Norman KR (2018). Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Wang H, Zhou X, Mao L, Ding K, and Hu Z (2019). Role of mitochondrial calcium uniporter-mediated Ca(2+) and iron accumulation in traumatic brain injury. J. Cell. Mol. Med 23, 2995–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drago I, and Davis RL (2016). Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep. 16, 2763–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markus NM, Hasel P, Qiu J, Bell KF, Heron S, Kind PC, Dando O, Simpson TI, and Hardingham GE (2016). Expression of mRNA Encoding Mcu and Other Mitochondrial Calcium Regulatory Genes Depends on Cell Type, Neuronal Subtype, and Ca2+ Signaling. PloS One 11, e0148164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho Y, Sloutsky R, Naegle KM, and Cavalli V (2013). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155, 894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan D, Wu Z, Chisholm AD, and Jin Y (2009). The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138, 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan D, and Jin Y (2012). Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron 76, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vargas ME, Yamagishi Y, Tessier-Lavigne M, and Sagasti A (2015). Live Imaging of Calcium Dynamics during Axon Degeneration Reveals Two Functionally Distinct Phases of Calcium Influx. J. Neurosci 35, 15026–15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gottschalk B, Klec C, Leitinger G, Bernhart E, Rost R, Bischof H, Madreiter-Sokolowski CT, Radulovic S, Eroglu E, Sattler W, et al. (2019). MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat. Commun 10, 3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, et al. (2013). MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 5, 1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, and Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spat A, et al. (2017). Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca(2+) Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 18, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu D, Lee S, Luo J, Xia H, Gushchina S, Richardson PM, Yeh J, Krugel U, Franke H, Zhang Y, et al. (2018). Intraneural Injection of ATP Stimulates Regeneration of Primary Sensory Axons in the Spinal Cord. J. Neurosci 38, 1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Hagan R, Chalfie M, and Goodman MB (2005). The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat. Neurosci 8, 43–50. [DOI] [PubMed] [Google Scholar]
- 63.Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, et al. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. The J. Neurosci 32, 13819–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Noma K, and Jin Y (2015). Optogenetic mutagenesis in Caenorhabditis elegans. Nat. Commun 6, 8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paix A, Folkmann A, Rasoloson D, and Seydoux G (2015). High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein Complexes. Genetics 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, and Calarco JA (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dejima K, Hori S, Iwata S, Suehiro Y, Yoshina S, Motohashi T, and Mitani S (2018). An Aneuploidy-Free and Structurally Defined Balancer Chromosome Toolkit for Caenorhabditis elegans. Cell Rep. 22, 232–241. [DOI] [PubMed] [Google Scholar]
- 68.Dickinson DJ, Pani AM, Heppert JK, Higgins CD, and Goldstein B (2015). Streamlined Genome Engineering with a Self-Excising Drug Selection Cassette. Genetics 200, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. (2013). Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol 20, 1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotech 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim KW, Tang NH, Andrusiak MG, Wu Z, Chisholm AD, and Jin Y (2018). A Neuronal piRNA Pathway Inhibits Axon Regeneration in C. elegans. Neuron 97, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes enriched in CAR-1(ΔSM) seCLIP samples. Related to Figure 5. Column A shows genes enriched >2× using CLIPper. Column B shows genes enroched >2× using Cufflinks; 28 genes in bold face are enrched in both sets.
Data Availability Statement
The raw and analyzed seCLIP datasets have been deposited at the Gene Expression Omnibus (accession number: GSE124714).